

Abstract
Transcriptional enhancers enable exquisite spatiotemporal control of gene expression in metazoans. Enrichment of monomethylation of histone H3 lysine 4 (H3K4me1) is a major chromatin signature of transcriptional enhancers. Lysine (K)-specific demethylase 1A (KDM1A, also known as LSD1), an H3K4me2/me1 demethylase, inactivates stem-cell enhancers during the differentiation of mouse embryonic stem cells (mESCs). However, its role in undifferentiated mESCs remains obscure. Here, we show that KDM1A actively maintains the optimal enhancer status in both undifferentiated and lineage-committed cells. KDM1A occupies a majority of enhancers in undifferentiated mESCs. KDM1A levels at enhancers exhibit clear positive correlations with its substrate H3K4me2, H3K27ac, and transcription at enhancers. In Kdm1a-deficient mESCs, a large fraction of these enhancers gains additional H3K4 methylation, which is accompanied by increases in H3K27 acetylation and increased expression of both enhancer RNAs (eRNAs) and target genes. In postmitotic neurons, loss of KDM1A leads to premature activation of neuronal activity-dependent enhancers and genes. Taken together, these results suggest that KDM1A is a versatile regulator of enhancers and acts as a rheostat to maintain optimal enhancer activity by counterbalancing H3K4 methylation at enhancers.
Transcriptional enhancers were discovered as potent gene regulatory elements that act independently of the distance and orientation to the target promoters (Banerji et al. 1981; Moreau et al. 1981). Cell type–specific and temporally controlled gene expression programs rely on coordinated actions of transcriptional enhancers (Bulger and Groudine 2011; Levine et al. 2014). Numerous studies have revealed mechanisms of how enhancers induce changes in gene expression (for review, see Long et al. 2016). Numerous genetic variants associated with various human traits have been observed at enhancers, implicating their importance in normal physiology and disease pathogenesis (Maurano et al. 2012; Smith and Shilatifard 2014; Vallianatos et al. 2018). Genome-wide identification of potential enhancer elements has been facilitated by profiling (1) binding of pioneer transcription factors (TFs), (2) chromatin accessibility, and (3) patterns of histone modifications (for review, see Ren and Yue 2015). Monomethylation of histone H3 lysine 4 (H3K4me1) can distinguish enhancers from promoters (Heintzman et al. 2007, 2009). In response to various environmental and developmental cues, TFs bind to specific DNA elements (Zaret and Carroll 2011; Spitz and Furlong 2012), and subsequent recruitment of methyltransferases KMT2C and KMT2D (also known as MLL3 and MLL4) primes the enhancers with H3K4me1 (Herz et al. 2012; Hu et al. 2013; Lee et al. 2013). Changes in the epigenetic landscape further determine the identity of enhancers as either active or poised through the acetylation or trimethylation of histone H3 lysine 27 (H3K27ac or H3K27me3), respectively (Creyghton et al. 2010; Rada-Iglesias et al. 2011).
Two H3K4 demethylases, KDM1A (also known as LSD1) and KDM5C (also known as SMCX), play critical roles in the regulation of enhancers (Whyte et al. 2012; Outchkourov et al. 2013; Respuela et al. 2016; Shen et al. 2016; Cao et al. 2018). While KDM5C removes H3K4me3/2, leaving H3K4me1 intact (Iwase et al. 2007; Tahiliani et al. 2007), KDM1A can demethylate only H3K4me2/1 (Shi et al. 2004). The substrate specificities imply that these two H3K4 demethylases may cooperate to generate and/or maintain the balance of the H3K4 methylation landscape at different classes of enhancers. During the differentiation of mouse embryonic stem cells (mESCs), KDM1A-mediated removal of H3K4me1/me2 decommissions stem-cell enhancers (Whyte et al. 2012; Respuela et al. 2016). The failure of this process contributes to aberrant stem-cell-like gene expression patterns, as observed in embryonal carcinoma cells (AlAbdi et al. 2020). Artificial tethering of KDM1A to active enhancers via TALEN or CRISPR-dCas9 impairs enhancer activity (Mendenhall et al. 2013; Kearns et al. 2015). In the present study, we investigated the roles of KDM1A in undifferentiated mESCs and postmitotic neurons to gain deeper insights into the dynamics of H3K4 methylation underlying the regulation of enhancers.
Results
KDM1A levels positively correlate with H3K4 methylation at mESC enhancers
We started by examining the genome-wide distribution of KDM1A at various regulatory elements in mESCs. We analyzed the previously published ChIP-seq data sets of KDM1A (Whyte et al. 2012), EP300 (Shen et al. 2012; Buecker et al. 2014), CTCF (Handoko et al. 2011; Shen et al. 2012), DNase I hypersensitivity (DHS) (Neph et al. 2012), and other histone modifications (Supplemental Table S1). EP300, a histone acetyltransferase, occupies both promoters and enhancers (Wang et al. 2005; Heintzman et al. 2007), whereas CTCF anchors chromatin loops (Ghirlando and Felsenfeld 2016) and insulating domains (Kim et al. 2007; Dixon et al. 2012; Hnisz et al. 2016). By examining the overlaps of KDM1A, EP300, CTCF peaks, and DHS sites, we found that most of the KDM1A peaks overlapped DHS sites (86.3%) and were present at a majority of EP300 binding sites (70.5%) (Fig. 1A; Supplemental Fig. S1). In contrast, only 7.9% of KDM1A peaks were found at non-EP300 CTCF-binding sites (Fig. 1A). A higher degree of overlap of KDM1A with EP300 compared with CTCF-only sites was observed at the promoter, genic, and intergenic regions (Supplemental Fig. S1), indicating that KDM1A occupies a large fraction of promoters and enhancers in mESCs.
Figure 1.

KDM1A occupies a large fraction of enhancers in mESCs. (A) Overlap of binding sites of EP300, CTCF, and KDM1A in mESCs. (B) Intergenic enhancers were divided into quartiles (Q1–Q4) based on the enrichment of H3K4me2 relative to H3K4me1 (left panel). Box plots show the enrichment of the indicated histone modifications, KDM1A, and EP300, as measured using ChIP-seq and eRNA levels (GRO-seq, nuclear RNA-seq, and RNA-seq) at each quartile of the intergenic enhancers. Levels of KDM1A show positive correlations with increases in H3K4me2 and eRNA expression from Q1 to Q4. In all figures, the bottom and top boxes signify the second and third quartiles, respectively, and the middle band represents the median of the population. Whiskers represent 1.5 times the interquartile range (IQR), and the notch represents the 95% confidence interval of the median. (C) The percentage of intergenic enhancers with KDM1A peaks. (D,E) Active, poised, and intermediate enhancers were classified based on the enrichment of either trimethylation or acetylation of H3K27 (D) or H3K9 (E). KDM1A occupancy at enhancers increases with higher enhancer activity.
Next, we identified putative enhancers in mESCs. We followed studies that had utilized a high H3K4me1:me3 ratio (Heintzman et al. 2007; Creyghton et al. 2010), DHS, and binding by CBP/EP300 (Visel et al. 2009; Zentner et al. 2011) to distinguish enhancers from promoters. As H3K4me2 decorates both promoters and enhancers (He et al. 2010) and exhibits sharper peaks than H3K4me1, we included our H3K4me2 data to increase the sensitivity and precision of enhancer mapping. Whereas promoters are associated with stable transcripts (Core et al. 2014), active enhancers are associated with the expression of enhancer RNA transcripts (eRNAs) (De Santa et al. 2010; Kim et al. 2010; Core et al. 2014), which are short-lived due to exosome-mediated degradation (Wyers et al. 2005; Preker et al. 2008; De Santa et al. 2010). Global Run-On (GRO)-seq (Core et al. 2008) facilitates a sensitive and quantitative evaluation of transcriptionally engaged RNA polymerase molecules and thus serves as an indirect measure of nascent transcription at promoters and enhancers, irrespective of the subsequent stability of the transcripts (Core et al. 2014). Therefore, we employed a high ratio of GRO-seq:RNA-seq signals to refine the prediction of enhancers in mESCs. We focused on only intergenic enhancers as we found it difficult to differentiate eRNAs from gene-coding and promoter-upstream (Preker et al. 2008) transcripts. In summary, intergenic enhancers were defined as ±500-base regions around EP300/DHS summits with (1) H3K4me1 enrichment (RPKM ≥ 1 and ChIP:Input > 1.5), (2) H3K4me3 lower than either H3K4me1 or H3K4me2, (3) a low rate of transcription (RNA-seq FPKM < 0.5), (4) a GRO-seq:RNA-seq ratio > 5, and (5) a high average mappability to exclude repetitive regions. This pipeline predicted 22,047 intergenic enhancers in mESCs (Supplemental Table S2).
To examine the relationship between KDM1A binding and chromatin states at enhancers, we subdivided the 22,047 predicted intergenic enhancers into quartiles (Q1–Q4) based on the enrichment of H3K4me2 relative to H3K4me1 (Fig. 1B). Similar to the previous observation in K562 cells (Core et al. 2014), H3K4me2 levels correlated positively with eRNA levels, as measured by GRO-seq, nuclear RNA-seq, and RNA-seq (Fig. 1B). Acetylation of H3K9 and H3K27 and production of eRNA are signatures of active enhancers (Creyghton et al. 2010; Rada-Iglesias et al. 2011; Ren and Yue 2015). Consistently, enhancers with higher H3K4me2/me1 ratios displayed higher acetylation levels of H3K9 and H3K27 relative to trimethylation (Supplemental Fig. S2).
We then determined the extent of KDM1A binding across various enhancer classes and found that KDM1A binds to a large fraction (63.2%) of the predicted 22,047 enhancers (Fig. 1C). As an H3K4me1/me2 demethylase, KDM1A can be expected to occupy enhancers with low H3K4me1/me2 and low eRNA expression. However, KDM1A levels correlated positively with both H3K4me2 and eRNA levels (Fig. 1B; Supplemental Figs. S3, S4A). KDM1A levels showed the highest correlations with its primary substrate, H3K4me2 (r = 0.627) (Supplemental Fig. S4B) and with H3K27ac (r = 0.604), followed by H3K4me1 (r = 0.5745) and H3K4me3 (r = 0.486). In contrast, H3K27me3 and H3K9me3 levels were inversely correlated with KDM1A levels (Supplemental Fig. S4B). We also classified the intergenic enhancers into poised, active, and intermediate enhancers based on their H3K27me3 and H3K27ac levels (Rada-Iglesias et al. 2011) or H3K9me3 and H3K9ac levels (Zentner et al. 2011). We found that KDM1A occupies substantial fractions of each class with increased occupancy of active enhancers compared with other classes (Fig. 1D,E). Similarly, we observed positive correlations between the levels of KDM1A, EP300, and permissive histone modifications at promoters and transcriptional levels of the genes (Supplemental Fig. S5).
KDM1A rarely showed binding to lineage-specific enhancers that were not bound by TFs in mESCs, indicating that KDM1A is predominantly recruited to TF-bound enhancers and may not be involved in active silencing of lineage-specific enhancers in mESCs (Supplemental Figs. S6, S7; Supplemental Note 1). KDM1A interacts with POU5F1 (Pardo et al. 2010; van den Berg et al. 2010; Ding et al. 2012; AlAbdi et al. 2020). Consistently, we found that a majority (68.6%) of the KDM1A-bound mESC enhancers contained recognition sequences for the three pluripotency TFs: POU5F1, SOX2, and NANOG (Supplemental Table S3).
These analyses indicate that KDM1A targets TF-bound enhancers with positive correlations to H3K4me2, H3K27/H3K9 acetylation, and eRNA levels.
Loss of KDM1A results in genome-wide increase of enhancer H3K4 methylation and H3K27 acetylation
The unexpected positive correlation between KDM1A binding and H3K4me2 levels at enhancers implies that KDM1A may not be demethylating H3K4me2 at enhancers. KDM1A has been implicated in the demethylation of H3K9, when it binds to androgen receptors (Metzger et al. 2005; Wissmann et al. 2007), although a later study reported otherwise (Cai et al. 2014). H3T6 phosphorylation also interferes with KDM1A-mediated H3K4 demethylation (Metzger et al. 2010). Alternatively, the positive correlation may reflect a negative feedback mechanism, in which KDM1A or associated proteins search for and bind to genomic regions with high H3K4me2 and then KDM1A reverses this modification to maintain optimal enhancer activity.
To test whether KDM1A is involved in maintaining optimal levels of H3K4 methylation at enhancers, we investigated the previously generated Kdm1a gene-trap mESCs (Kdm1a-GT) that lack KDM1A (Macfarlan et al. 2011). Western blot analysis of WT- or Kdm1a-GT mESCs did not reveal any detectable differences in total H3K4me1, H3K4me2, H3K4me3, or H3K27ac levels (Supplemental Fig. S8). We then performed ChIP-seq to measure H3K4 methylation levels across the genomes of these two mESCs lines. Because KDM1A associates with multiple HDAC-containing corepressor complexes, including CoREST (Hakimi et al. 2002; Shi et al. 2005) and NuRD (Wang et al. 2009), we included H3K27ac and HDAC1 in the ChIP-seq analysis. The spatial distributions of these histone modifications and HDAC1 appeared highly similar between the two genotypes at most of the loci. Upon the loss of KDM1A, however, H3K4 methylations increased significantly at active, poised, and intermediate enhancers, accompanied by conspicuous increases in H3K27ac (Fig. 2A; Supplemental Fig. S9). The increases in H3K4 methylation and H3K27ac at many of the poised and intermediate enhancers suggest a shift in their identity toward active enhancers. HDAC1 levels did not change significantly at poised enhancers, whereas active and intermediate enhancers showed a small but significant increase in HDAC1 binding (Fig. 2A; Supplemental Fig. S9B), likely due to either experimental variations or an unknown compensatory mechanism for KDM1A loss. The inability of HDAC1 to remove H3K27ac in Kdm1a-GT mESCs is consistent with previous observations that H3K4me interferes with HDAC activity (Lee et al. 2006; Yin et al. 2014). Representative genes Pou5f1 (Fig. 2B) and Cbln4 (Fig. 2C), which are active and poised in undifferentiated mESCs, respectively, showed higher H3K4 methylation and H3K27ac, as well as higher GRO-seq signals at both promoters and enhancers in Kdm1a-GT mESCs. These results advocate that KDM1A functions primarily as an H3K4 demethylase at enhancers and prevents runaway increases in H3K4me and H3K27ac.
Figure 2.

Loss of KDM1A results in increases in H3K4 methylation and H3K27 acetylation at enhancers. (A) H3K4me1, H3K4me2, H3K4me3, H3K27ac, and HDAC1 levels on KDM1A-bound enhancers in WT (gray boxes) and Kdm1a-GT mESCs (red boxes). Enhancers were classified into poised (P), intermediate (I), and active (A) enhancers based on the enrichment of either H3K27ac or H3K27me3. Geometric mean of ChIP:Input ratios from the two independent ChIP-seq replicates are shown. P-values (p) from Wilcoxon signed-rank tests on log2(Kdm1a-GT/WT) are denoted in blue beneath each panel. n indicates the number of enhancers in each category. (B) Dysregulation of active enhancers at the Pou5f1 locus. Several enhancers are co-occupied by EP300 and KDM1A in WT mESCs, some of which show increased H3K4me2, H3K27ac, and GRO-seq signals in Kdm1a-GT mESCs (red vs. gray). (C) Misregulation of a poised enhancer at the Cbln4 locus. This locus is decorated with a broad H3K27me3 domain and shows elevations in H3K4me1, H3K4me2, and GRO-seq signals upon the loss of KDM1A. Gray bar: Predicted enhancer, blue bar: significantly up-regulated enhancer in Kdm1a-GT mESCs compared with WT mESCs based on changes in the GRO-seq signal (see Fig. 3).
Loss of KDM1A but not KDM5C results in aberrant activation of transcriptional enhancers in mESCs
KDM5C appears to reside in the same complex as KDM1A and suppresses the overactivation of active enhancers in breast cancer cells (Shen et al. 2016). To elucidate the functional roles of KDM1A and KDM5C in mESCs, we first performed KDM5C ChIP-seq in mESCs and identified 113,166 KDM5C-binding sites (MACS2, q < 0.05). Most of the 22,047 predicted intergenic enhancers (78.3%) were bound by either KDM1A or KDM5C, and 11,483 (52.1%) were bound by both (Fig. 3A), indicating a broad cooperation between the two H3K4 demethylases. Next, we generated Kdm5c-knockout (KO) mESCs (Supplemental Fig. S10) and performed GRO-seq on the Kdm1a-GT and Kdm5c-KO mESCs. To identify misregulated enhancers, we calculated the number of GRO-seq reads mapping within ±500 bases of the center of the predicted enhancers and normalized them against the genome-wide 199,209 EP300/DHS sites using DESeq (Anders and Huber 2010). Upon the loss of KDM1A, a large fraction of intergenic enhancers (5471 of 22,047, 24.8%) or enhancers co-occupied by both KDM1A and KDM5C (3011 of 11,483, 26.2%) showed a significant elevation in associated GRO-seq transcripts (q < 0.05) (Fig. 3B), whereas only 674 (3.06%) or 543 (4.7%), respectively, displayed a reduced activity (Fig. 3B). All three enhancer classes displayed a significant increase in associated nascent transcripts (P < 2.2−16, Wilcoxon signed-rank test) (Fig. 3D). Using the same significance cutoff, we were unable to identify any misregulated enhancers in Kdm5c-KO mESCs (Fig. 3C; Supplemental Fig. S11; Supplemental Note 2). These data demonstrate that, in contrast to KDM1A's critical requirement, KDM5C is largely dispensable for suppression of aberrant activation of enhancers in mESCs.
Figure 3.

Loss of KDM1A but not KDM5C results in aberrant activation of enhancers. (A) Fractions of intergenic enhancers bound by KDM1A and/or KDM5C in mESCs. (B,C) Volcano plots of GRO-seq signals at enhancers bound by both KDM1A and KDM5C from DESeq analyses. Whereas the loss of KDM1A resulted in a large-scale increase in GRO-seq signals at enhancers, the deletion of KDM5C had a minimal impact. The x-axis and y-axis indicate the log2 fold-change and significance, respectively, of differential expression in WT and mutant mESC lines. (D) Scatterplots of GRO-seq levels at KDM1A-bound poised, intermediate, and active enhancers. Significantly up-regulated and down-regulated enhancers (q < 0.05, DESeq) are shown in blue and orange, respectively. The LOWESS curve for each class of enhancers is shown in red. The total number (n) of all, significantly up-regulated, and significantly down-regulated enhancers in each group are indicated in black, blue, and orange, respectively. Each class of enhancers shows a significant up-regulation (P < 2.2−16, Wilcoxon signed-rank test) in Kdm1a-GT mESCs compared with WT mESCs. (E) Western blot analysis to validate the re-expression of KDM1A-WT or the hypomorphic K661A mutant after inducible Kdm1a-KO in mESCs. (F) eRNA levels measured using RT-qPCR. Mean ± SEM (n = 4 biological replicates). (*) P < 0.05; Student's t-test, (n.s.) not significant. See Supplemental Table S4 for details of the enhancers.
Because GRO-seq is an in vitro transcription assay, we validated the up-regulation of enhancers in Kdm1a-GT mESCs under physiological conditions by sequencing the total cellular RNAs (RNA-seq) and nuclear RNAs (nuclear RNA-seq), but neither provided sufficiently high eRNA signals to call differentially expressed enhancers, likely due to the aforementioned exosome-mediated degradation of eRNAs. However, when we evaluated global changes in eRNA levels, RNA-seq and nuclear RNA-seq corroborated our GRO-seq results (Supplemental Fig. S11).
To elucidate the contributions of KDM1A and its catalytic activity to suppression of enhancers, we performed a cDNA complementation study using Kdm1a-KO mESCs (Kdm1aflox/flox:Cre-ERT) (Supplemental Methods; Wang et al. 2007; Macfarlan et al. 2011). Human KDM1A-WT, or its catalytically hypomorphic mutant, KDM1A-K661A (Lee et al. 2005; Kim et al. 2020), was expressed at comparable levels in the Kdm1a-KO mESCs (Fig. 3E). We selected 10 KDM1A-target enhancers that were highly up-regulated in Kdm1a-GT mESCs and measured their eRNAs levels (eRNA#1 to #10) (Supplemental Table S4) by quantitative reverse transcription PCR (RT-qPCR). Consistent with the GRO-seq results in Kdm1a-GT mESCs, each tested eRNA exhibited significant up-regulation in Kdm1a-KO mESCs (Fig. 3F), and KDM1A-WT expression led to its down-regulation. In comparison, KDM1A-K661A was less efficient in lowering the levels of six eRNAs (eRNA#1 to #6, P < 0.05, Student's t-test). The four remaining eRNAs (#7 to #10) were restored to their normal levels through the expression of KDM1A-WT or its hypomorphic mutant. These observations highlight the context-dependent requirements of KDM1A's demethylase activity at enhancers (Supplemental Note 3).
Aberrant changes in enhancer activity are associated with misregulation of physically interacting genes
To identify genes that physically interact with our set of predicted enhancers, we utilized the published HiCap data set, a high-resolution map of promoter-anchored chromatin interactions in mESCs (Sahlén et al. 2015). For instance, our enhancer prediction identified an enhancer cluster downstream from Dusp5, and the analysis of the HiCap data revealed that one of the three individual enhancers within the cluster appears to interact with the Dusp5 promoter (Fig. 4A). This enhancer cluster was significantly up-regulated in Kdm1a-GT mESCs, with pronounced increases in H3K4me2 and H3K27ac levels and increased Dusp5 transcription (Fig. 4A). A concomitant misregulation of enhancers and the anchored gene was also observed at the aforementioned mir290 cluster (Supplemental Fig. S12A).
Figure 4.

Aberrant changes in enhancer activity are associated with misregulation of physically interacting genes. (A) An example of long-range promoter-enhancer interactions (top track) obtained from the mESCs HiCap data set (Sahlén et al. 2015) at the Dusp5 locus. One of the three significantly up-regulated enhancers (blue bars) interacts with the Dusp5 promoter. Upon the loss of KDM1A, the gene and enhancers show up-regulation of H3K4me2, H3K27ac, and GRO-seq signals in Kdm1a-GT mESCs (red) compared with WT mESCs (gray). (B) Volcano plots of changes in mRNA levels (RNA-seq) of genes that physically interact with misregulated enhancers. On the basis of changes in enhancer-associated GRO-seq signals upon the loss of KDM1A, enhancers were subdivided as significantly up (q < 0.05, DESeq), significantly down, moderately up (0.05 ≤ q < 0.25), moderately down, unchanged (q ≥ 0.5 and fold-change ≤ 25%), and the rest. When multiple enhancers showed interactions with a single promoter, the assignment of the gene to an enhancer subgroup was prioritized in the aforementioned order. The total number of associated genes (n) and P-values (p) from Wilcoxon signed-rank tests on differences between mRNA levels in Kdm1a-GT and WT mESCs are shown beneath each panel. (C) χ2 test of the association of misregulated enhancers (GRO-seq) with the number of misregulated genes (RNA-seq). Significantly up- or down-regulated enhancers were more likely to be anchored to promoters of the genes that showed analogous up- or down-regulation in Kdm1a-GT mESCs. (*) P < 0.0001.
We then categorized the enhancers based on the statistical significance of their differential expression in the GRO-seq analysis of WT and Kdm1a-GT mESCs: significantly misregulated (q < 0.05); moderately misregulated (0.05 ≤ q < 0.25); unchanged (q ≥ 0.5 and fold-change ≤ 25%), and the rest. We retrieved their target promoters from the HiCap data set and plotted the changes in mRNA levels (RNA-seq) (Fig. 4B,C) or rates of nascent transcription (GRO-seq) (Supplemental Fig. S12B). Generally, the genes associated with up-regulated enhancers displayed elevated expression levels in Kdm1a-GT mESCs and vice versa (Fig. 4B; Supplemental Fig. S12B,C), and the genes anchored to the unchanged enhancers did not exhibit significant changes in expression (Fig. 5B,C). Notably, the magnitude and statistical significance of the gene expression changes correlated positively with those of enhancer activity alterations (Supplemental Fig. S12C). Alternatively, when interacting genes were called based on genomic proximity to the enhancers, we observed a similar trend (Supplemental Fig. S13). These results indicate that KDM1A at enhancers plays a vital role in precise transcription of their cognate genes.
Figure 5.

Both mESC-specific and differentiation genes are up-regulated after KDM1A loss in undifferentiated mESCs. (A) Schematic showing the number of significantly induced and repressed genes after differentiation of mESCs to epiblast stem cells with Activin A and FGF2 (Acampora et al. 2016). (B,C) Scatterplots of mRNA levels (B) and levels of nascent transcription (C), as measured using RNA-seq and GRO-seq, respectively, in WT and Kdm1a-GT mESCs. Number (n) of significantly up-regulated (q < 0.05) and down-regulated genes in each category are shown in blue and orange, respectively. Upon the loss of KDM1A in mESCs, both groups of induced and repressed genes show a significant increase (P < 2.2−16, Wilcoxon signed-rank test) in mRNA levels and nascent transcription. (D) Elevated transcription of Hmga2 and its nearby enhancers in Kdm1a-GT mESCs. Gray bar: Predicted enhancer, blue bar: significantly up-regulated enhancer in Kdm1a-GT mESCs.
KDM1A negatively regulates both pluripotency and differentiation genes in mESCs
KDM1A promotes the differentiation of mESCs by decommissioning enhancers of pluripotency genes (Whyte et al. 2012). However, its role in undifferentiated mESCs remains elusive. To understand the biological implications of the loss of KDM1A in mESCs, we identified genes that are induced or repressed during the differentiation of mESCs to epiblast stem cells, respectively. DESeq analysis of the published data set (Acampora et al. 2016) yielded 710 induced and 745 repressed genes (q < 0.01, |fold change| > 5.0) (Fig. 5A). Compared with WT mESCs, similar numbers of genes were significantly up-regulated (1493, q < 0.05) and down-regulated (1203) in Kdm1a-GT mESCs. However, both groups of the aforementioned induced and repressed genes were significantly up-regulated in Kdm1a-GT mESCs (P < 2.2−16, Wilcoxon signed-rank test) (Fig. 5B,C). For example, Hmga2 is induced during the differentiation of mESCs and promotes the exit from naive pluripotency (Navarra et al. 2016). KDM1A loss led to a marked increase in Hmga2 expression, concomitant with increased H3K4 methylation, H3K27ac, and GRO-seq signal at nearby enhancers (Fig. 5D). Loss of KDM1A resulted in similar changes at some pluripotency genes, including Pou5f1 (Fig. 2B) and mir290 (Supplemental Fig. S12A). The up-regulation of both pluripotency and differentiation genes upon the loss of KDM1A suggests that KDM1A does not instruct the fate of mESCs in a particular direction.
KDM1A suppresses inducible enhancers in neurons
To elucidate whether KDM1A-mediated regulation of enhancers affects gene expression programs of terminally differentiated cells, we knocked down (KD) Kdm1a in primary cultures of mouse cortical neurons (CNs) using lentiviral delivery of a control or two independent short hairpin RNAs (shRNAs) (Supplemental Fig. S14A). We employed BrU-seq, a nascent-RNA sequencing technique (Paulsen et al. 2013), which allows an accurate evaluation of nascent transcription of both mRNAs and eRNAs. DESeq analysis of the BrU-seq libraries indicated a significant misregulation of 1500 genes (q < 0.05, 778 down-regulated and 722 up-regulated). We noted that many well-characterized activity-regulated genes (ARGs) (Kim et al. 2010), including Arc, Fos, Fosb, Npas4, Egr1-4, and Nr4a1-3, were among the most significantly up-regulated genes upon Kdm1a-KD in unstimulated neurons (Fig. 6A). ARGs are expressed at low levels in resting neurons and are rapidly induced by depolarization of neurons via sensory inputs. Derepression of this stimulus-responsive expression program upon Kdm1a-KD inspired us to narrow our focus on these inducible genes. Using our previously published RNA-seq data set (Iwase et al. 2016), we first identified 140 ARGs induced by KCl-mediated depolarization of cortical neurons. Both Kdm1a shRNAs led to a spurious induction of many ARGs in resting neurons (Fig. 6A; Supplemental Fig. S14B,C), indicating that KDM1A suppresses the premature induction of ARGs in cortical neurons. At the Npas4 locus, we identified three putative enhancers upstream of the promoter based on DHS and H3K4me1 enrichment (Fig. 6B). The enhancers appear to be responsive to membrane depolarization, as they show activity-dependent increases in NPAS4 binding (Kim et al. 2010) and H3K27ac levels (Malik et al. 2014). Using the published KDM1A ChIP-seq data from resting and activated CNs (Wang et al. 2016), we observed that upon neuronal activation, KDM1A occupancy increases at the three Npas4 enhancers (Fig. 6B) as well as enhancers of several other ARGs, including Btg2, Fos, Fosb, and Nr4a1 (Supplemental Fig. S15). Increased nascent transcription across these enhancers in Kdm1a-KD neurons indicates that these enhancers are deregulated in the absence of KDM1A (Fig. 6B; Supplemental Fig. S15).
Figure 6.
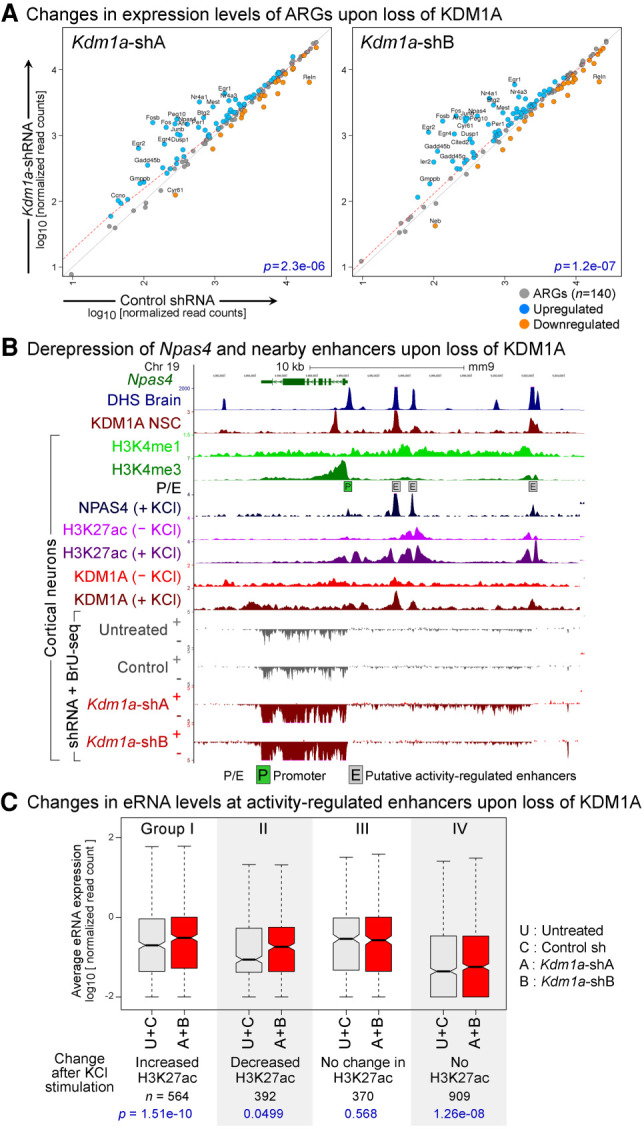
KDM1A represses inducible genes and enhancers in terminally differentiated neurons. (A) Up-regulation of activity-regulated genes (ARGs) in Kdm1a-KD cortical neurons. Scatterplots of transcription levels of ARGs (n = 140) from BrU-seq analysis in CN treated with either Kdm1a shRNAs (y-axis) or control shRNA (x-axis). Significantly up-regulated (q < 0.05, DESeq) and down-regulated ARGs are shown in blue and orange, respectively, and ARGs with greater than a twofold difference with KDM1A loss are labeled with gene symbols. P-values (p) from Wilcoxon signed-rank tests are denoted in blue. (B) Aberrant induction of Npas4, an ARG, upon Kdm1a-KD in resting CN. Boxed P: Npas4 promoter, boxed E: putative activity-regulated enhancers as evident from the presence of DHS (Neph et al. 2012), high H3K4me1, low H3K4me3 (Iwase et al. 2016), activity-dependent binding of NPAS4, and an increase in H3K27ac after KCl treatment (Malik et al. 2014). Npas4 mRNA and eRNA are up-regulated in the Kdm1a-KD neurons (red). KDM1A ChIP-seq data were obtained from previous studies using neuronal stem cells (NSC) (Wang et al. 2016) and CN (Wang et al. 2015). (C) Increased eRNA levels at activity-regulated enhancers in Kdm1a-KD CN. These enhancers were divided into four groups based on the activity-induced changes in H3K27ac (Malik et al. 2014). Three groups of enhancers show a significant increase in eRNA levels upon Kdm1a-KD (red boxes) compared with control conditions (untreated CN or control shRNA-treated CN, gray boxes). (A + B) Geometric mean of eRNA levels in CN treated with Kdm1a shRNAs A or B, (U + C) geometric mean of eRNA levels in control neurons. P-values (p) from Wilcoxon signed-rank tests are denoted in blue.
A study reported more than 10,000 putative activity-regulated enhancers based on increased CBP binding in response to membrane depolarization (Kim et al. 2010). Subsequent work categorized these candidate enhancers into four groups based on activity-dependent changes in H3K27ac (Fig. 6C, Groups I–IV) and revealed that only the enhancers that displayed activity-dependent increases in H3K27ac (Group I) promote ARG transcription (Malik et al. 2014). We found that KDM1A occupies 46.8%, 38.26%, 29.72%, and 17.6% of enhancers from Groups I–IV, respectively. Our BrU-seq analyses indicate that Kdm1a-KD significantly up-regulated eRNAs at enhancers from Groups I, II, and IV but not Group III (Fig. 6C; Supplemental Fig. S16A,B). These data indicate that KDM1A suppresses premature activation of enhancers in resting neurons.
Activation of ARGs upon Kdm1a-KD may also result from extraneous activation of signaling pathways upstream of ARG induction. Membrane depolarization induces rapid phosphorylation of extracellular signal-regulated kinases 1 and 2 (ERK1/2), which play critical roles in transcriptional responses (Greer and Greenberg 2008; Wiegert and Bading 2011). A lack of noticeable changes in the phosphorylation levels of ERK1/2 after Kdm1a-KD further supports a direct role of KDM1A in the suppression of activity-regulated enhancers and genes (Supplemental Fig. S16C).
Discussion
The early embryonic lethality of homozygous Kdm1a-KO mice indicates an essential role for KDM1A in development (Wang et al. 2007). However, the roles of KDM1A in early embryogenesis remain incompletely understood. KDM1A decommissions pluripotency enhancers, aiding the silencing of pluripotency genes during the differentiation of mESCs (Whyte et al. 2012). The study employed tranylcypromine (TCP), a pharmacological agent, to block KDM1A enzymatic activity. However, TCP also inhibits the H3K4 demethylase activity of KDM1B (Karytinos et al. 2009), the paralog of KDM1A involved in transcriptional elongation (Fang et al. 2010). Thus, it remains unclear whether the observed effects were a result of TCP-mediated inhibition of KDM1A, KDM1B, or both. In contrast, we utilized genetic ablation of KDM1A.
Contradictory observations have been made regarding the role of KDM1A in pluripotency. Loss of KDM1A in early embryos results in the dedifferentiation of the pluripotent mESCs toward the totipotent two-cell state (Macfarlan et al. 2012). In contrast, KDM1A deficiency also causes premature differentiation of human ESCs to endodermal and mesodermal lineages (Adamo et al. 2011) or interferes with mESC self-renewal (Yin et al. 2014). These conflicting observations could be reconciled by our findings that KDM1A is required for the suppression of both pluripotency and differentiation genes in mESCs (Fig. 5). Pathway analysis of our RNA-seq data detected an up-regulation of genes involved in cellular responses to a broad range of extracellular stimuli, including hormones, growth factors, and radiation (Supplemental Table S5). Additionally, KDM1A-deficient postmitotic neurons exhibited premature activation of ARGs (Fig. 6). Thus, KDM1A is critical in suppressing spurious transcriptional responses in the absence of developmental or environmental cues.
We provide several lines of evidence that KDM1A plays an essential role in genome-wide homeostasis of transcriptional enhancers. KDM1A recruitment was positively correlated with the levels of H3K4me2, H3K27ac, and eRNA transcription at enhancers (Fig. 1) and occurred mostly at TF-bound enhancers (Supplemental Figs. S6, S7). Loss of KDM1A up-regulated numerous enhancers, as demonstrated by increased H3K4 methylation, H3K27ac (Fig. 2), and eRNA transcription (Fig. 3), concomitant with an up-regulation of the target genes (Fig. 4). These results suggest the following model for the dynamics and KDM1A-mediated homeostasis of the epigenetic landscape during the life cycle of an enhancer (Fig. 7). TF binding and subsequent recruitment of KMT2C/D (Herz et al. 2012; Hu et al. 2013; Lee et al. 2013) prime the enhancers with H3K4me1/me2, which in turn attract KDM1A, irrespective of whether the enhancers are destined to be “active” or “poised.” KDM1A then counteracts KMT2-mediated methylation to maintain optimal H3K4 methylation. Enhancers with relatively low H3K4me2 may represent the early stages of priming and may recruit little KDM1A, which is not detectable with ChIP-seq (Q1 in Fig. 1B). When gene expression needs to be increased, recruitment of additional TFs and chromatin modifiers may further activate the enhancer with higher H3K4 methylation and H3K27ac, in turn requiring higher levels of KDM1A. Our model of KDM1A functioning as a rheostat for the suppression of enhancers in undifferentiated mESCs through active H3K4 demethylation and H3K27 deacetylation is not mutually exclusive to KDM1A-mediated decommissioning of enhancers. During ES cell differentiation, pluripotency enhancers may lose ES-specific TFs and KMT2-mediated H3K4 methylation. KDM1A and associated HDACs then remove remnant H3K4me2/me1 and H3K27ac, respectively, to completely disengage the enhancer from active regulation. In the absence of H3K4me, KDM1A is then evicted from such decommissioned enhancers. As differentiation requires or dictates the activation of a new set of genes, the processes of priming and activation are repeated at a different set of enhancers, resulting in the recruitment of KDM1A at these new genomic loci. To test this part of our model, we differentiated mESCs for 24 h and performed ChIP-seq of H3K4me2, KDM1A, and KMT2D in undifferentiated and differentiated mESCs. Consistent with our model, we found new H3K4me2 peaks in proximity to many genes that are up-regulated during differentiation (Supplemental Fig. S17), including both well-known differentiation genes and “induced” genes, as identified earlier. To ascertain the validity of our model of recruitment or eviction of KDM1A in response to changes in H3K4me2 levels, we compared the changes in levels of H3K4me2 with changes in either KDM1A or KMT2D. Indeed, changes in H3K4me2 levels at promoter-distal sites correlated positively with changes in KDM1A levels (0.783, P < 2.2−16) and changes in KMT2D (0.620, P < 2.2−16) (Supplemental Fig. 18A). At these putative enhancers, changes in KDM1A and KMT2D also correlated positively but showed a weaker correlation (0.397, P < 2.2−16), which could be either a result of differences in ChIP efficiency or the fact that some enhancers could be regulated by other KMT2s instead of KMT2D.
Figure 7.

A model of KDM1A-mediated homeostasis of enhancers during their life cycle. TF binding and subsequent recruitment of methyltransferases, KMT2C/D, prime an enhancer with H3K4me1 and/or H3K4me2. KDM1A is then recruited to this enhancer by yet unknown H3K4me-sensing mechanisms and cooperates with histone deacetylases (HDACs) to suppress its aberrant activation. A primed enhancer, depending on further regulatory signals, can become either “active” or “poised.” The presence of KDM1A/HDACs is required to antagonize the activities of the methyltransferases and acetyltransferases (HATs) and maintain an optimal histone modification landscape. The equilibrium of these counteractions likely defines the activity of an enhancer. When an enhancer is decommissioned upon the loss of TF binding, KDM1A may remove the remnant H3K4me1/2 before dissociating and rendering it latent.
Many nuclear receptors, including the glucocorticoid receptor, preferentially inhibit the H3K4me1-demethylase activity of KDM1A in A549, a lung adenocarcinoma cell line (Clark et al. 2019). Similar inhibition was observed for KDM1A's interaction with POU5F1 at pluripotency enhancers in embryonal carcinoma cells (AlAbdi et al. 2020). Both studies demonstrated that selective demethylation of H3K4me2 by KDM1A while leaving H3K4me1 intact can potentiate some enhancers, indicating that loss of KDM1A at some enhancers may impair their activity. These findings may help explain our earlier observation that some enhancers were significantly down-regulated in Kdm1a-GT mESCs (Fig. 3).
Our BrU-seq analyses in postmitotic neurons revealed that KDM1A suppresses premature activation of neuronal ARGs and enhancers (Fig. 6). Increased KDM1A levels at the activity-regulated enhancers after neuronal activation and up-regulation of these enhancers upon Kdm1a-KD further supports the role of KDM1A as an enhancer rheostat in differentiated cells (Fig. 6B; Supplemental Figs. S15, S16A; see Supplemental Note 4 for the role of the neuronal isoform of KDM1A in the regulation of ARGs). Several neurodevelopmental conditions involve loss-of-function KDM1A mutations (Tunovic et al. 2014; Chong et al. 2015; Pilotto et al. 2016). KDM1A-mediated homeostasis of transcriptional enhancers, therefore, underlies diverse physiological processes, including embryonic development and human cognitive function.
Methods
Cell culture, Western blot, and RT-qPCR
Kdm1a-WT, Kdm1a-GT, Kdm1aflox/flox:Cre-ERT mESCs have been described previously (Wang et al. 2007; Macfarlan et al. 2011). Kdm5c-KO mESCs were derived from the previously described mESCs that carry the floxed Kdm5c allele (Iwase et al. 2016) by Cre-mediated deletion of the JmjC domain coding exons 11 and 12. mESCs were grown on gelatin-coated plates. See Supplemental Methods for further details on culture conditions and standard molecular biology experiments.
Kdm1a knockdown in mouse cortical neurons
Lentiviral delivery of control (SHC202, Sigma-Aldrich) or Kdm1a shRNAs (A: TRCN0000071375 and B: TRCN0000071376, Sigma-Aldrich) (Moffat et al. 2006) was conducted on 7-d in vitro (DIV) cortical neurons, and 5-bromouridine incorporation was performed on DIV 11.
ChIP-seq
Antibodies used for chromatin immunoprecipitation (ChIP) were anti-H3K4me1 (Abcam ab8895 and EMD Millipore 07-436), anti-H3K4me2 (EMD Millipore 05-790), anti-H3K4me3 (EMD Millipore 04-745), anti-H3K27ac (Active Motif 39135), anti-HDAC1 (Bethyl Laboratories A300-713A and Santa Cruz Biotechnology sc-6298), anti-KDM1A (Abcam ab17721), anti-KDM5C (Iwase et al. 2016), and anti-KMT2D (Invitrogen 701869). KDM5C ChIP-seq experiments were performed as described previously (Iwase et al. 2016). Other ChIP experiments were performed as described previously (Li et al. 2003) with minor modifications, as elaborated in Supplemental Methods.
RNA-seq, nuclear RNA-seq, GRO-seq, and BrU-seq
RNA-seq libraries have been described in detail previously (Agarwal et al. 2015). For the sequencing of nuclear RNA, nuclei were isolated as described previously (Wang et al. 2011) with minor modifications. Libraries from rRNA-depleted RNA were prepared using Direct Ligation of Adapters to First-strand cDNA (DLAF) (Agarwal et al. 2015). GRO-seq was modified from the methods described previously (Core et al. 2008; Wang et al. 2011). In addition to the presence of 0.2% IGEPAL CA-630, GRO on Kdm5c mESCs, Kdm1a mESCs, and cortical neurons were performed in the presence of 0.5%, 0.25%, and 0.2% of N-lauroylsarcosine, respectively, for 8 min at 30°C. For BrU-seq, the cortical neurons were incubated with 2 mM 5-bromouridine (Sigma-Aldrich 850187) for 32 min at 37°C. To reduce the number of steps for library preparation, we developed Direct Ligation of Adaptor to the 3′ end of RNA (DLAR), a method suitable for the preparation of BrU-seq libraries. Experimental details are described in Supplemental Methods.
All sequencing experiments, except for the differentiation experiment, were conducted in biological duplicates concurrently with different genotypes to minimize technical variations.
Sequencing data analysis
Multiplexed libraries were subjected to single-end sequencing on Illumina HiSeq 2000/2500 instruments, and the reads were mapped to the mm9 genome (see Supplemental Methods for the details). We chose the mm9 assembly over the mm10 because at the time of our initial analyses, many processed data sets, including DHS peaks or HOTSPOTs, genome mappability, ENCODE blacklisted regions, and the HiCap data, were available for mm9 only. The blacklisted and repetitive regions were excluded from our analyses and only uniquely mapping reads were analyzed further. Therefore, we expect that our choice of the reference genome assembly should not significantly affect our conclusions.
For the selection of candidate EP300/DHS sites for enhancer prediction, we first scanned the genome for the strongest (with the highest MACS2 signal) EP300 or DHS site in a 1.25-kb sliding window. When both EP300 and DHS sites were present in the same window, the EP300 binding site was given higher precedence over any DHS site in its proximity. Intergenic EP300/DHS sites were defined as sites that were outside of 1.25 kb upstream of to 3 kb downstream from the genes. Enhancer predictions and other analyses are further detailed in Supplemental Methods. The Perl scripts used for the analyses are available as a Supplemental Code.
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE93952.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank Dr. Fulai Jin and Dr. Gary Hon at Ludwig Institute for Cancer Research (LICR), San Diego, for helpful discussions and guidance in bioinformatics. We also thank Dr. Nima Mosammaparast at Washington University School of Medicine in St. Louis for the gift of WT and catalytically inactive KDM1A expression plasmids. The work was supported by grants from the LICR (to B.R.), the California Institute for Regenerative Medicine (RN2-00905, to B.R.), the University of Michigan Medical School (to S.I.), Cooley's Anemia Foundation Fellowship (to S.I.), the March of Dimes Foundation (to S.I.), the National Institutes of Health (NIH) (National Institute of Neurological Disorders and Stroke: NS089896, 5R21NS104774, and NS116008 to S.I.), National Science Foundation Graduate Research Fellowship Program (#1256260, to P.M.G.), University of Michigan Career Training in Reproductive Biology (NIH T32HD079342, to K.M.B. and R.S.P.), and the Eunice Kennedy Shriver National Institute of Child Health and Human Development (HD008933, to T.S.M.).
Author contributions: S.A., B.R., and S.I. conceived the project and designed the experiments. S.A. established the protocols and performed the computational analysis. S.I. and S.A. performed the RNA-seq, nuclear RNA-seq, and GRO-seq experiments. S.A., K.M.B., E.B., and T.S.M. performed the ChIP-seq experiments. S.I., P.M.G., and S.A. performed the BrU-seq experiment. S.I. and Y.M.-N. performed the cDNA rescue experiments. K.M.B. designed and performed the differentiation experiment. Y.M.-N., P.M.G., S.A., and R.S.P. performed rest of the experiments. S.A. and S.I. designed the analysis and wrote the manuscript. All authors have read and edited the manuscript and approve its final version.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.234559.118.
References
- Acampora D, Omodei D, Petrosino G, Garofalo A, Savarese M, Nigro V, Di Giovannantonio LG, Mercadante V, Simeone A. 2016. Loss of the Otx2-binding site in the Nanog promoter affects the integrity of embryonic stem cell subtypes and specification of inner cell mass-derived epiblast. Cell Rep 15: 2651–2664. 10.1016/j.celrep.2016.05.041 [DOI] [PubMed] [Google Scholar]
- Adamo A, Sesé B, Boue S, Castaño J, Paramonov I, Barrero MJ, Izpisua Belmonte JC. 2011. LSD1 regulates the balance between self-renewal and differentiation in human embryonic stem cells. Nat Cell Biol 13: 652–659. 10.1038/ncb2246 [DOI] [PubMed] [Google Scholar]
- Agarwal S, Macfarlan TS, Sartor MA, Iwase S. 2015. Sequencing of first-strand cDNA library reveals full-length transcriptomes. Nat Commun 6: 6002 10.1038/ncomms7002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- AlAbdi L, Saha D, He M, Dar MS, Utturkar SM, Sudyanti PA, McCune S, Spears BH, Breedlove JA, Lanman NA, et al. 2020. Oct4-mediated inhibition of Lsd1 activity promotes the active and primed state of pluripotency enhancers. Cell Rep 30: 1478–1490.e6. 10.1016/j.celrep.2019.11.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji J, Rusconi S, Schaffner W. 1981. Expression of a β-globin gene is enhanced by remote SV40 DNA sequences. Cell 27: 299–308. 10.1016/0092-8674(81)90413-X [DOI] [PubMed] [Google Scholar]
- Buecker C, Srinivasan R, Wu Z, Calo E, Acampora D, Faial T, Simeone A, Tan M, Swigut T, Wysocka J. 2014. Reorganization of enhancer patterns in transition from naive to primed pluripotency. Cell Stem Cell 14: 838–853. 10.1016/j.stem.2014.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulger M, Groudine M. 2011. Functional and mechanistic diversity of distal transcription enhancers. Cell 144: 327–339. 10.1016/j.cell.2011.01.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai C, He HH, Gao S, Chen S, Yu Z, Gao Y, Chen S, Chen MW, Zhang J, Ahmed M, et al. 2014. Lysine-specific demethylase 1 has dual functions as a major regulator of androgen receptor transcriptional activity. Cell Rep 9: 1618–1627. 10.1016/j.celrep.2014.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao K, Collings CK, Morgan MA, Marshall SA, Rendleman EJ, Ozark PA, Smith ER, Shilatifard A. 2018. An Mll4/COMPASS-Lsd1 epigenetic axis governs enhancer function and pluripotency transition in embryonic stem cells. Sci Adv 4: eaap8747 10.1126/sciadv.aap8747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JX, Yu JH, Lorentzen P, Park KM, Jamal SM, Tabor HK, Rauch A, Saenz MS, Boltshauser E, Patterson KE, et al. 2015. Gene discovery for Mendelian conditions via social networking: de novo variants in KDM1A cause developmental delay and distinctive facial features. Genet Med 18: 788–795. 10.1038/gim.2015.161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EA, Wu F, Chen Y, Kang P, Kaiser UB, Fang R, Shi YG. 2019. GR and LSD1/KDM1A-targeted gene activation requires selective H3K4me2 demethylation at enhancers. Cell Rep 27: 3522–3532.e3. 10.1016/j.celrep.2019.05.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Lis JT. 2008. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science 322: 1845–1848. 10.1126/science.1162228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Martins AL, Danko CG, Waters CT, Siepel A, Lis JT. 2014. Analysis of nascent RNA identifies a unified architecture of initiation regions at mammalian promoters and enhancers. Nat Genet 46: 1311–1320. 10.1038/ng.3142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci 107: 21931–21936. 10.1073/pnas.1016071107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G. 2010. A large fraction of extragenic RNA Pol II transcription sites overlap enhancers. PLoS Biol 8: e1000384 10.1371/journal.pbio.1000384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Xu H, Faiola F, Ma'ayan A, Wang J. 2012. Oct4 links multiple epigenetic pathways to the pluripotency network. Cell Res 22: 155–167. 10.1038/cr.2011.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485: 376–380. 10.1038/nature11082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R, Barbera AJ, Xu Y, Rutenberg M, Leonor T, Bi Q, Lan F, Mei P, Yuan GC, Lian C, et al. 2010. Human LSD2/KDM1b/AOF1 regulates gene transcription by modulating intragenic H3K4me2 methylation. Mol Cell 39: 222–233. 10.1016/j.molcel.2010.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghirlando R, Felsenfeld G. 2016. CTCF: making the right connections. Genes Dev 30: 881–891. 10.1101/gad.277863.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME. 2008. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron 59: 846–860. 10.1016/j.neuron.2008.09.002 [DOI] [PubMed] [Google Scholar]
- Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R. 2002. A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes. Proc Natl Acad Sci 99: 7420–7425. 10.1073/pnas.112008599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CW, Ye C, Ping JL, Mulawadi F, et al. 2011. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat Genet 43: 630–638. 10.1038/ng.857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, et al. 2010. Nucleosome dynamics define transcriptional enhancers. Nat Genet 42: 343–347. 10.1038/ng.545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, et al. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat Genet 39: 311–318. 10.1038/ng1966 [DOI] [PubMed] [Google Scholar]
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, et al. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459: 108–112. 10.1038/nature07829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz HM, Mohan M, Garruss AS, Liang K, Takahashi YH, Mickey K, Voets O, Verrijzer CP, Shilatifard A. 2012. Enhancer-associated H3K4 monomethylation by Trithorax-related, the Drosophila homolog of mammalian Mll3/Mll4. Genes Dev 26: 2604–2620. 10.1101/gad.201327.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Day DS, Young RA. 2016. Insulated neighborhoods: structural and functional units of mammalian gene control. Cell 167: 1188–1200. 10.1016/j.cell.2016.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu DQ, Gao X, Morgan MA, Herz HM, Smith ER, Shilatifard A. 2013. The MLL3/MLL4 branches of the COMPASS family function as major histone H3K4 monomethylases at enhancers. Mol Cell Biol 33: 4745–4754. 10.1128/MCB.01181-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwase S, Lan F, Bayliss P, de la Torre-Ubieta L, Huarte M, Qi HH, Whetstine JR, Bonni A, Roberts TM, Shi Y. 2007. The X-linked mental retardation gene SMCX/JARID1C defines a family of histone H3 lysine 4 demethylases. Cell 128: 1077–1088. 10.1016/j.cell.2007.02.017 [DOI] [PubMed] [Google Scholar]
- Iwase S, Brookes E, Agarwal S, Badeaux AI, Ito H, Vallianatos CN, Tomassy GS, Kasza T, Lin G, Thompson A, et al. 2016. A mouse model of X-linked intellectual disability associated with impaired removal of histone methylation. Cell Rep 14: 1000–1009. 10.1016/j.celrep.2015.12.091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karytinos A, Forneris F, Profumo A, Ciossani G, Battaglioli E, Binda C, Mattevi A. 2009. A novel mammalian flavin-dependent histone demethylase. J Biol Chem 284: 17775–17782. 10.1074/jbc.M109.003087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns NA, Pham H, Tabak B, Genga RM, Silverstein NJ, Garber M, Maehr R. 2015. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 12: 401–403. 10.1038/nmeth.3325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TH, Abdullaev ZK, Smith AD, Ching KA, Loukinov DI, Green RD, Zhang MQ, Lobanenkov VV, Ren B. 2007. Analysis of the vertebrate insulator protein CTCF-binding sites in the human genome. Cell 128: 1231–1245. 10.1016/j.cell.2006.12.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, et al. 2010. Widespread transcription at neuronal activity-regulated enhancers. Nature 465: 182–187. 10.1038/nature09033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S-A, Zhu J, Yennawar N, Eek P, Tan S. 2020. Crystal structure of the LSD1/CoREST histone demethylase bound to its nucleosome substrate. Mol Cell 78: 903–914.e4. 10.1016/j.molcel.2020.04.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, Shiekhattar R. 2005. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 437: 432–435. 10.1038/nature04021 [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Bochar DA, Hakimi MA, Cooch N, Shiekhattar R. 2006. Functional interplay between histone demethylase and deacetylase enzymes. Mol Cell Biol 26: 6395–6402. 10.1128/MCB.00723-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Wang C, Xu S, Cho YW, Wang L, Feng X, Baldridge A, Sartorelli V, Zhuang L, Peng W, et al. 2013. H3K4 mono- and di-methyltransferase MLL4 is required for enhancer activation during cell differentiation. eLife 2: e01503 10.7554/eLife.01503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine M, Cattoglio C, Tjian R. 2014. Looping back to leap forward: transcription enters a new era. Cell 157: 13–25. 10.1016/j.cell.2014.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. 2003. A global transcriptional regulatory role for c-Myc in Burkitt's lymphoma cells. Proc Natl Acad Sci 100: 8164–8169. 10.1073/pnas.1332764100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long HK, Prescott SL, Wysocka J. 2016. Ever-changing landscapes: transcriptional enhancers in development and evolution. Cell 167: 1170–1187. 10.1016/j.cell.2016.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan TS, Gifford WD, Agarwal S, Driscoll S, Lettieri K, Wang J, Andrews SE, Franco L, Rosenfeld MG, Ren B, et al. 2011. Endogenous retroviruses and neighboring genes are coordinately repressed by LSD1/KDM1A. Genes Dev 25: 594–607. 10.1101/gad.2008511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, Firth A, Singer O, Trono D, Pfaff SL. 2012. Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487: 57–63. 10.1038/nature11244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik AN, Vierbuchen T, Hemberg M, Rubin AA, Ling E, Couch CH, Stroud H, Spiegel I, Farh KK, Harmin DA, et al. 2014. Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat Neurosci 17: 1330–1339. 10.1038/nn.3808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, et al. 2012. Systematic localization of common disease-associated variation in regulatory DNA. Science 337: 1190–1195. 10.1126/science.1222794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall EM, Williamson KE, Reyon D, Zou JY, Ram O, Joung JK, Bernstein BE. 2013. Locus-specific editing of histone modifications at endogenous enhancers. Nat Biotechnol 31: 1133–1136. 10.1038/nbt.2701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger E, Wissmann M, Yin N, Müller JM, Schneider R, Peters AH, Günther T, Buettner R, Schüle R. 2005. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 437: 436–439. 10.1038/nature04020 [DOI] [PubMed] [Google Scholar]
- Metzger E, Imhof A, Patel D, Kahl P, Hoffmeyer K, Friedrichs N, Müller JM, Greschik H, Kirfel J, Ji S, et al. 2010. Phosphorylation of histone H3T6 by PKCβI controls demethylation at histone H3K4. Nature 464: 792–796. 10.1038/nature08839 [DOI] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. 2006. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124: 1283–1298. 10.1016/j.cell.2006.01.040 [DOI] [PubMed] [Google Scholar]
- Moreau P, Hen R, Wasylyk B, Everett R, Gaub MP, Chambon P. 1981. The SV40 72 base repair repeat has a striking effect on gene expression both in SV40 and other chimeric recombinants. Nucleic Acids Res 9: 6047–6068. 10.1093/nar/9.22.6047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarra A, Musto A, Gargiulo A, Petrosino G, Pierantoni GM, Fusco A, Russo T, Parisi S. 2016. Hmga2 is necessary for Otx2-dependent exit of embryonic stem cells from the pluripotent ground state. BMC Biol 14: 24 10.1186/s12915-016-0246-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S, Vierstra J, Stergachis AB, Reynolds AP, Haugen E, Vernot B, Thurman RE, John S, Sandstrom R, Johnson AK, et al. 2012. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489: 83–90. 10.1038/nature11212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Outchkourov NS, Muiño JM, Kaufmann K, van Ijcken WF, Groot Koerkamp MJ, van Leenen D, de Graaf P, Holstege FC, Grosveld FG, Timmers HT. 2013. Balancing of histone H3K4 methylation states by the Kdm5c/SMCX histone demethylase modulates promoter and enhancer function. Cell Rep 3: 1071–1079. 10.1016/j.celrep.2013.02.030 [DOI] [PubMed] [Google Scholar]
- Pardo M, Lang B, Yu L, Prosser H, Bradley A, Babu MM, Choudhary J. 2010. An expanded Oct4 interaction network: implications for stem cell biology, development, and disease. Cell Stem Cell 6: 382–395. 10.1016/j.stem.2010.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen MT, Veloso A, Prasad J, Bedi K, Ljungman EA, Tsan YC, Chang CW, Tarrier B, Washburn JG, Lyons R, et al. 2013. Coordinated regulation of synthesis and stability of RNA during the acute TNF-induced proinflammatory response. Proc Natl Acad Sci 110: 2240–2245. 10.1073/pnas.1219192110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilotto S, Speranzini V, Marabelli C, Rusconi F, Toffolo E, Grillo B, Battaglioli E, Mattevi A. 2016. LSD1/KDM1A mutations associated to a newly described form of intellectual disability impair demethylase activity and binding to transcription factors. Hum Mol Genet 25: 2578–2587. 10.1093/hmg/ddw120 [DOI] [PubMed] [Google Scholar]
- Preker P, Nielsen J, Kammler S, Lykke-Andersen S, Christensen MS, Mapendano CK, Schierup MH, Jensen TH. 2008. RNA exosome depletion reveals transcription upstream of active human promoters. Science 322: 1851–1854. 10.1126/science.1164096 [DOI] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470: 279–283. 10.1038/nature09692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren B, Yue F. 2015. Transcriptional enhancers: bridging the genome and phenome. Cold Spring Harb Symp Quant Biol 80: 17–26. 10.1101/sqb.2015.80.027219 [DOI] [PubMed] [Google Scholar]
- Respuela P, Nikolić M, Tan M, Frommolt P, Zhao Y, Wysocka J, Rada-Iglesias A. 2016. Foxd3 promotes exit from naive pluripotency through enhancer decommissioning and inhibits germline specification. Cell Stem Cell 18: 118–133. 10.1016/j.stem.2015.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahlén P, Abdullayev I, Ramsköld D, Matskova L, Rilakovic N, Lötstedt B, Albert TJ, Lundeberg J, Sandberg R. 2015. Genome-wide mapping of promoter-anchored interactions with close to single-enhancer resolution. Genome Biol 16: 156 10.1186/s13059-015-0727-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. 2012. A map of the cis-regulatory sequences in the mouse genome. Nature 488: 116–120. 10.1038/nature11243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H, Xu W, Guo R, Rong B, Gu L, Wang Z, He C, Zheng L, Hu X, Hu Z, et al. 2016. Suppression of enhancer overactivation by a RACK7-histone demethylase complex. Cell 165: 331–342. 10.1016/j.cell.2016.02.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA. 2004. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953. 10.1016/j.cell.2004.12.012 [DOI] [PubMed] [Google Scholar]
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. 2005. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell 19: 857–864. 10.1016/j.molcel.2005.08.027 [DOI] [PubMed] [Google Scholar]
- Smith E, Shilatifard A. 2014. Enhancer biology and enhanceropathies. Nat Struct Mol Biol 21: 210–219. 10.1038/nsmb.2784 [DOI] [PubMed] [Google Scholar]
- Spitz F, Furlong EE. 2012. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 13: 613–626. 10.1038/nrg3207 [DOI] [PubMed] [Google Scholar]
- Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y. 2007. The histone H3K4 demethylase SsMCX links REST target genes to X-linked mental retardation. Nature 447: 601–605. 10.1038/nature05823 [DOI] [PubMed] [Google Scholar]
- Tunovic S, Barkovich J, Sherr EH, Slavotinek AM. 2014. De novo ANKRD11 and KDM1A gene mutations in a male with features of KBG syndrome and Kabuki syndrome. Am J Med Genet A 164: 1744–1749. 10.1002/ajmg.a.36450 [DOI] [PubMed] [Google Scholar]
- Vallianatos CN, Farrehi C, Friez MJ, Burmeister M, Keegan CE, Iwase S. 2018. Altered gene-regulatory function of KDM5C by a novel mutation associated with autism and intellectual disability. Front Mol Neurosci 11: 104 10.3389/fnmol.2018.00104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg DL, Snoek T, Mullin NP, Yates A, Bezstarosti K, Demmers J, Chambers I, Poot RA. 2010. An Oct4-centered protein interaction network in embryonic stem cells. Cell Stem Cell 6: 369–381. 10.1016/j.stem.2010.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. 2009. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457: 854–858. 10.1038/nature07730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Carroll JS, Brown M. 2005. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell 19: 631–642. 10.1016/j.molcel.2005.07.018 [DOI] [PubMed] [Google Scholar]
- Wang J, Scully K, Zhu X, Cai L, Zhang J, Prefontaine GG, Krones A, Ohgi KA, Zhu P, Garcia-Bassets I, et al. 2007. Opposing LSD1 complexes function in developmental gene activation and repression programmes. Nature 446: 882–887. 10.1038/nature05671 [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhang H, Chen Y, Sun Y, Yang F, Yu W, Liang J, Sun L, Yang X, Shi L, et al. 2009. LSD1 is a subunit of the NuRD complex and targets the metastasis programs in breast cancer. Cell 138: 660–672. 10.1016/j.cell.2009.05.050 [DOI] [PubMed] [Google Scholar]
- Wang D, Garcia-Bassets I, Benner C, Li W, Su X, Zhou Y, Qiu J, Liu W, Kaikkonen MU, Ohgi KA, et al. 2011. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature 474: 390–394. 10.1038/nature10006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Telese F, Tan Y, Li W, Jin C, He X, Basnet H, Ma Q, Merkurjev D, Zhu X, et al. 2015. LSD1n is an H4K20 demethylase regulating memory formation via transcriptional elongation control. Nat Neurosci 18: 1256–1264. 10.1038/nn.4069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Wu Q, Yang P, Wang C, Liu J, Ding W, Liu W, Bai Y, Yang Y, Wang H, et al. 2016. LSD1 co-repressor Rcor2 orchestrates neurogenesis in the developing mouse brain. Nat Commun 7: 10481 10.1038/ncomms10481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Bilodeau S, Orlando DA, Hoke HA, Frampton GM, Foster CT, Cowley SM, Young RA. 2012. Enhancer decommissioning by LSD1 during embryonic stem cell differentiation. Nature 482: 221–225. 10.1038/nature10805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiegert JS, Bading H. 2011. Activity-dependent calcium signaling and ERK-MAP kinases in neurons: a link to structural plasticity of the nucleus and gene transcription regulation. Cell Calcium 49: 296–305. 10.1016/j.ceca.2010.11.009 [DOI] [PubMed] [Google Scholar]
- Wissmann M, Yin N, Müller JM, Greschik H, Fodor BD, Jenuwein T, Vogler C, Schneider R, Günther T, Buettner R, et al. 2007. Cooperative demethylation by JMJD2C and LSD1 promotes androgen receptor-dependent gene expression. Nat Cell Biol 9: 347–353. 10.1038/ncb1546 [DOI] [PubMed] [Google Scholar]
- Wyers F, Rougemaille M, Badis G, Rousselle J-C, Dufour M-E, Boulay J, Régnault B, Devaux F, Namane A, Séraphin B, et al. 2005. Cryptic Pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121: 725–737. 10.1016/j.cell.2005.04.030 [DOI] [PubMed] [Google Scholar]
- Yin F, Lan R, Zhang X, Zhu L, Chen F, Xu Z, Liu Y, Ye T, Sun H, Lu F, et al. 2014. LSD1 regulates pluripotency of embryonic stem/carcinoma cells through histone deacetylase 1-mediated deacetylation of histone H4 at lysine 16. Mol Cell Biol 34: 158–179. 10.1128/MCB.00631-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Carroll JS. 2011. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25: 2227–2241. 10.1101/gad.176826.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zentner GE, Tesar PJ, Scacheri PC. 2011. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Res 21: 1273–1283. 10.1101/gr.122382.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.