

Abstract
Within-species contamination is a major issue in sequencing studies, especially for mitochondrial studies. Contamination can be detected by analyzing the nuclear genome or by inspecting polymorphic sites in the mitochondrial genome (mtDNA). Existing methods using the nuclear genome are computationally expensive, and no appropriate tool for detecting sample contamination in large-scale mtDNA data sets is available. Here we present haplocheck, a tool that requires only the mtDNA to detect contamination in both targeted mitochondrial and whole-genome sequencing studies. Our in silico simulations and amplicon mixture experiments indicate that haplocheck detects mtDNA contamination accurately and is independent of the phylogenetic distance within a sample mixture. By applying haplocheck to The 1000 Genomes Project Consortium data, we further evaluate the application of haplocheck as a fast proxy tool for nDNA-based contamination detection using the mtDNA and identify the mitochondrial copy number within a mixture as a critical component for the overall accuracy. The haplocheck tool is available both as a command-line tool and as a cloud web service producing interactive reports that facilitates the navigation through the phylogeny of contaminated samples.
The human mitochondrial DNA (mtDNA) is an extranuclear DNA molecule of ∼16.6 kb in length (Andrews et al. 1999). It is inherited exclusively through the maternal line, facilitating the reconstruction of the human maternal phylogeny and female (pre-)historical demographic patterns worldwide. The strict maternal inheritance of mtDNA results in a natural grouping of haplotypes into monophyletic clusters, referred to as haplogroups (Kivisild et al. 2006; Kloss-Brandstätter et al. 2011). Furthermore, second-generation sequencing enables the detection of heteroplasmy over the complete mitochondrial genome. Heteroplasmy is the occurrence of at least two different haplotypes of mtDNA in the investigated biological samples (e.g., cells or tissues). Depending on the sequencing coverage, heteroplasmic positions are reliably detectable down to the 1% variant level (Ye et al. 2014; Weissensteiner et al. 2016a).
It has been shown that external or cross-contamination (Yao et al. 2007; Just et al. 2014, 2015; Yin et al. 2019; Brandhagen et al. 2020), artificial recombination (Bandelt et al. 2004), or index hopping (Van Der Valk et al. 2019) can generate polymorphic sites that can be erroneously interpreted as heteroplasmic sites (He et al. 2010; Bandelt and Salas 2012; Just et al. 2014, 2015; Ye et al. 2014).
Sample contamination is still a major issue in both nuclear DNA (nDNA) and mtDNA sequencing studies that must be prevented to avoid mistakes as they occurred with Sanger sequencing studies in the past (Salas et al. 2005). Because of the accuracy and sensitivity of second-generation sequencing combined with the availability of improved computational models, within-species contamination is traceable down to the 1% level in whole-genome sequencing (WGS) studies (Jun et al. 2012).
Several approaches exist to detect contamination in mtDNA sequencing studies. We and others previously showed that a contamination approach based on the coexistence of phylogenetically incompatible mitochondrial haplotypes observable as polymorphic sites is feasible (Li et al. 2010, 2015; Avital et al. 2012; Weissensteiner et al. 2016a). The method of Dickins et al. (2014) facilitates the check for contamination by building neighbor joining trees. Mixemt (Vohr et al. 2017) incorporates the mitochondrial phylogeny and estimates the most probable haplogroup for each sequence read. The implemented algorithm reveals advantages for contamination detection by detecting several haplotypes within one sample and is independent of variant frequencies. However, it is too computationally expensive when applied to thousands of samples. For ancient DNA studies, schmutzi (Renaud et al. 2015) uses sequence deamination patterns and fragment-length distributions to estimate contamination. Additionally, specific laboratory protocols were designed for eliminating contamination, for example, double-barcode sequencing approaches (Yin et al. 2019).
For contamination detection in mitochondrial studies, mostly DNA cross-contamination is investigated (Ding et al. 2015; Wei et al. 2019; Yuan et al. 2020) by applying VerifyBamID (Jun et al. 2012; Zhang et al. 2020). Nevertheless, it becomes apparent that a tool for mitochondrial studies that rapidly and accurately detects contamination in thousands of samples is still missing. Because mtDNA is also present hundredfold to several thousandfold per cell depending on the cell type, also WGS data sets specifically targeting the autosomal genome result in a high coverage over the mitochondrial genome.
In this study, we systematically evaluate the approach of using the mtDNA phylogeny for contamination detection and present haplocheck, a tool to report contamination in mtDNA targeted sequencing and WGS studies. In general, haplocheck works by identifying polymorphic sites down to 1% within an input sample. By grouping polymorphic sites into haplotypes, haplocheck identifies contamination using the mitochondrial phylogeny and the concept of haplogroups. Overall, this work should show the merits of the mitochondrial genome as an instrument for additional quality control in sequencing studies. It additionally presents haplocheck, a fast and accurate tool that takes advantage of a solid well-known mitochondrial phylogeny for detecting contamination.
Methods
Haplocheck takes as input BAM or VCF files. For BAM files, an initial variant calling step based on a maximum likelihood (ML) function (Ye et al. 2014) is performed. Detected polymorphic variants are then reported in VCF format and split by their variant allele frequency (AF) into a major and minor haplotype profile. A haplotype profile consists of all detected homoplasmic variants and the corresponding allele of each polymorphic variant. Alleles with an AF ≥ 50% are added to the major haplotype profile; otherwise, they are added to the minor haplotype profile. A haplogroup for each haplotype is then determined using HaploGrep 2 (Weissensteiner et al. 2016b). By using the mitochondrial phylogeny, the phylogenetic distance (i.e., number of nodes between the two haplogroups) is calculated. The identification of two stable haplogroups allows haplocheck to report the contamination level for each sample.
Three different scenarios need to be considered for contamination detection based on the mitochondrial phylogeny. First, two haplotypes branch into two different nodes: a major haplotype with a mutation level x and a minor haplotype with a mutation level 1 − x (Fig. 1A), whereas here H1a1 represents the last common ancestor (LCA) for both haplotypes. Second, if polymorphic sites are only identified in the major haplotype, the minor haplotype H1a1 is defined as the LCA (Fig. 1B). Third, if polymorphic sites are only present in the minor haplotype, the major haplotype H1a1 defines the LCA (Fig. 1C).
Figure 1.

All possible contamination scenarios. Here, a contamination level of 20% is shown in all three scenarios (A–C). Shared polymorphisms of two haplotypes are included in a single branch, whereas the split into two branches displays the different lineage haplotypes. (A) Shared mutations defining H1a1 (last common ancestor [LCA]) are present at 100%, whereas 7961C is present only at 20%, defining the minor haplogroup H1a1b, whereas 4639C and 10993A are present at 80%, defining the major haplogroup H1a1a1. (B) A mixture of two haplotypes within a single lineage but of different lineage depths (minor haplotype H1a1 and major haplotype H1a1a1) is observed if no minor haplotype can be found. (C) A mixture of two haplotypes within a single lineage but of different lineage depths (minor H1a1a1 and major H1a1) is detected if the minor haplotype results in a stable haplogroup. Shared homoplasmic sites facilitate the identification of the branching pattern in all three scenarios and improve the overall haplogroup quality. The used notation for variants (e.g., 1438G) includes the mtDNA position (1438) followed by the actual base change (G).
Variant calling
The overall performance of haplocheck relies on an accurate variant calling. Previously, we developed mtDNA-Server (Weissensteiner et al. 2016a) for the detection of polymorphic sites down to 1% (Ye et al. 2014) in combination with several quality-control criteria such as (1) base quality ≥ 20, (2) >10× depth per strand, (3) 1% minor AF on each strand, and (4) a log-likelihood ratio (LLR) of ≥5. LLR represents the ratio between the estimated frequency of the major allele within the ML function of the polymorphic and the homoplasmic model.
For this work, we developed a multithreaded version of mtDNA-Server and integrated it into haplocheck (https://github.com/seppinho/mutserve). As mentioned, detected polymorphic positions are reported in VCF format as heterozygous genotypes (GT) using the AF tag for the estimated contamination level. Although the term genotype applies to autosomal diploid scenarios, we use it here to refer to mtDNA variation patterns that resemble a genotype status.
For homoplasmic positions, the final genotype GT ∈ {A,C,G,T} is detected using all input reads (reads) and calculating the genotype probability P using Bayes’ theorem P(GT|reads) = P(reads|GT) × P(GT)/P(reads). To calculate the prior probability P(GT), we used The 1000 Genomes Project Consortium Phase 3 VCF file (The 1000 Genomes Project Consortium 2015) and calculated the frequencies for all sites using VCFtools (Danecek et al. 2011). To compute P(reads|GT), we calculated the sequence error rate (ei = 10−Qi/10) for each base i of a read, where Q is the reported quality value. For each genotype GT (GT ∈ {A,C,G,T}) of a read, we determined the genotype likelihood by multiplying 1 − ei in case the base of the read ri = GT and ei/3 otherwise over all reads (Ding et al. 2015). The denominator P(reads) is the sum of all four P(reads|GT).
Contamination detection model
The contamination model within haplocheck includes steps for (1) splitting homoplasmic and polymorphic sites into two haplotype profiles, (2) haplogroup classification for each haplotype profile, and (3) filtering based on quality-control criteria. Homozygous genotypes for the alternate alleles (ALT; i.e., homoplasmic sites) are added to both haplotypes, whereas heterozygous genotypes are split using the AF tag. Because mutserve always reports the AF of the nonreference allele, the split method applies the following rule: In case a GT 0/1 (e.g., Ref: G, ALT: C) with an AF of 0.20 is included, the split method defines C as the minor allele, 0.2 as the minor level, and 0.8 as the major level. Conversely, when a GT 0/1 (e.g., Ref: G, ALT: C) with an AF of 0.80 is included, the C is defined as the major allele. If no reference allele is included (e.g., 1/2), we use the first allele as the major allele and assign the included AF to that allele.
For haplogroup classification, we use HaploGrep 2 (Weissensteiner et al. 2016b) based on Phylotree 17 (van Oven and Kayser 2009), which has been refactored as a module and integrated directly into haplocheck. As a result, HaploGrep 2 reports the haplogroup of both the major and minor haplotype. For each analyzed sample, the LCA is required to estimate the final contamination level and to calculate the distance between the two haplotypes. Therefore, we traverse Phylotree from the rCRS reference to each haplotype node. The LCA is determined by starting at the final node of haplotype 1 (h1) and by iterating back until the reference (rCRS) is reached. Then, we iterate back to rCRS for haplotype 2 (h2) until the first node included in h1 is identified. This node then defines the LCA of both haplotypes. Only polymorphic positions starting from the LCA and showing a phylogenetic weight greater than five are taken into account for the subsequent filtering step. The phylogenetic weight describes the frequency of each mutation in Phylotree and is scaled from one to 10 in a nonlinear way. Variants with a high occurrence in Phylotree are assigned a small phylogenetic weight. Furthermore, back mutations (i.e., mutation changes back to the rCRS reference within a specific haplogroup) and deletions on polymorphic sites are ignored by haplocheck.
By using all previous information, we finally estimate the contamination level for samples fulfilling the following three quality-control criteria: (1) two or more polymorphic variants starting from the LCA, (2) ≥0.5 haplogroup quality for each haplotype (calculated by HaploGrep 2 using the Kulczynski metric), and (3) phylogenetic distance of two or more. The median mutation level of all detected polymorphic sites reaching the described criteria is calculated independently for both haplotypes (h1 and h2). Haplocheck reports the median level of the minor haplotype as the final contamination level.
Report
Haplocheck produces a tab-delimited text file and an interactive HTML report. For each sample, haplocheck determines the final contamination status, the contamination level, and quality metrics such as the phylogenetic distance or the coverage. Additionally, a graphical phylogenetic tree is generated dynamically for each sample, including the path from the rCRS to the two final haplotypes. This allows the user to manually inspect edge cases, visualize the contamination graphically, and analyze the source of contamination (see Supplemental Fig. S1).
Results
Haplocheck is available as a standalone command-line tool and as a cloud web service. For both scenarios, the identical computational workflow consisting of variant calling (for BAM input only), haplogroup classification, and contamination detection is applied. The Cloudgene framework (Schönherr et al. 2012) is used to provide the workflow as a service to users, which is also used for large-scale genetic services like the Michigan Imputation Server (Das et al. 2016) and the mtDNA-Server (Weissensteiner et al. 2016a), that greatly improves user experience and productivity.
Evaluation
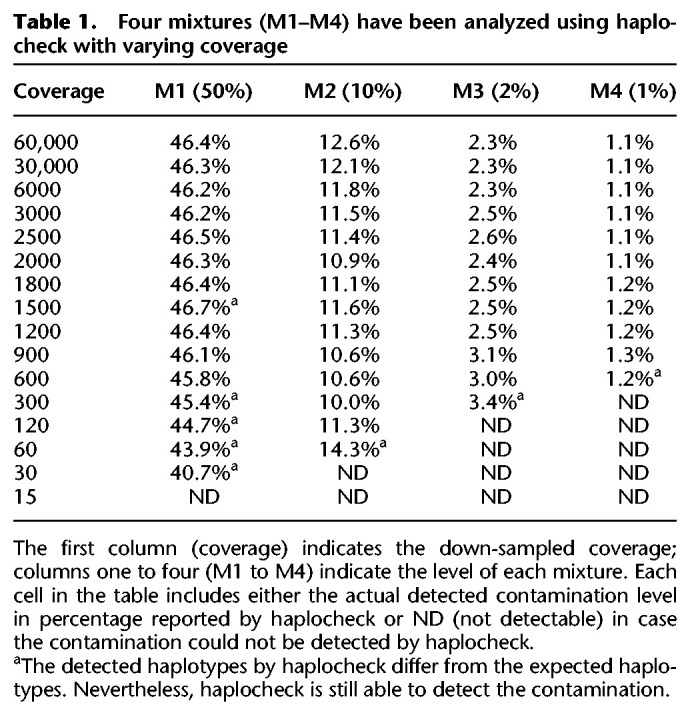
To test the performance of haplocheck within targeted mtDNA and WGS studies, we analyzed several data sets. First, we checked previously generated mtDNA mixtures of two samples including different haplotypes (Weissensteiner et al. 2016a). The mitochondrial genomes of the mixed fragments (1%–50%) were amplified by PCR and sequenced on an Illumina HiSeq system. We analyzed the original samples (coverage 60,000×) and down-sampled them accordingly. Our results show that a coverage of >100× and >600× is required to detect contamination of 10% and 1%, respectively (see Table 1). Of note, The 1000 Genomes Project Consortium low-coverage sample collection (2–4 × nDNA coverage) already includes sufficient coverage over the mitochondrial genome to detect contamination down to 1% (1800 × mtDNA coverage).
Table 1.
Four mixtures (M1–M4) have been analyzed using haplocheck with varying coverage
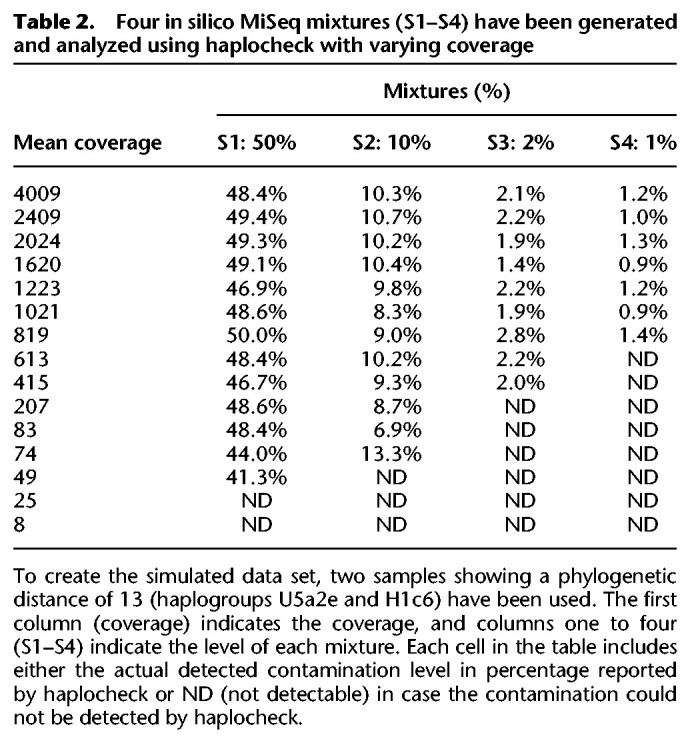
We further simulated sequencing data mixtures for different sequencing instruments by using the ART-NGS read simulator (Huang et al. 2012). The generated mixtures differ in (1) contamination level (1%–50%), (2) coverage (between 10×–5000×), and (3) phylogenetic distance between the two mixed haplotypes (three to 23 phylogenetic nodes between them). The results were highly concordant with the mixtures and show that haplocheck is able to detect contamination accurately even for samples including haplotypes with a close phylogenetic distance (see Table 2; for other phylogenetic distances, see Supplemental Table S1).
Table 2.
Four in silico MiSeq mixtures (S1–S4) have been generated and analyzed using haplocheck with varying coverage
In a second step, we created and analyzed in silico data by mixing random genotype profiles from the currently best available mtDNA phylogeny derived from Phylotree Build 17 (code on GitHub). The overall performance of haplocheck depends on a good classification of samples into haplogroups even from noisy variant calling data sets. Therefore, we initially created input profiles for each displayed haplogroup, amounting to 5426 profiles in total. Each input profile consists of a list of polymorphisms from the tree reference (rCRS) to the actual node (or haplogroup). Our test data consist of 500,000 unique mixtures of pairwise haplogroup profiles derived from the overall phylogeny comprising 5500 haplogroups (250,000 contaminated, 250,000 not-contaminated samples) and 100,000 mixtures from the haplogroup H-subtree, including 977 haplogroups. The generation of in silico data from the H-subtree allows us to test the performance of samples showing a smaller phylogenetic distance.
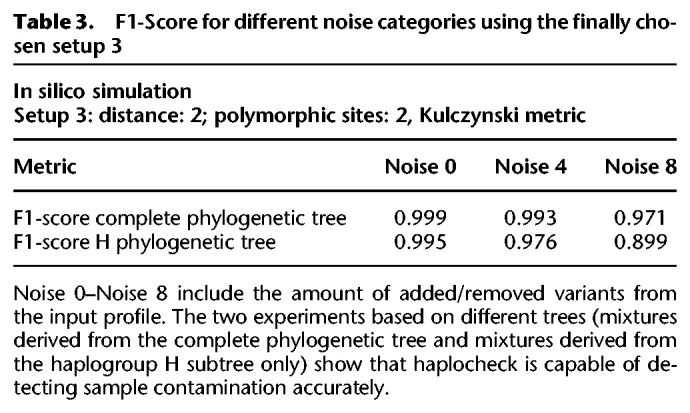
To account for noisy input data, we artificially added random variants to each input profile. This has been performed by removing expected variants from the input profile and adding random variants available within Phylotree. The amount of noise varies from zero to eight variants for each mixture. The proportion of added versus removed variants is calculated randomly. To make it further restrictive, we only added phylogenetic relevant variants from Phylotree. Variants that are not present in Phylotree (i.e., so far unknown in the phylogeny) would not affect the contamination estimation. Finally, three data sets (noise 0, 4, 8) derived from two different trees (complete tree, haplogroup H subtree) have been generated, each consisting of 500,000 and 100,000 mixtures respectively. The F1-score, defined as (2 × precision × sensitivity)/(precision + sensitivity), has been calculated for each mixture to analyze the overall accuracy of haplocheck.
To determine the best haplocheck configuration regarding accuracy, we tested different setups for all six data sets. Each setup includes a different threshold for (1) the amount of major and minor polymorphic sites, (2) the minimum allowed phylogenetic distance between two profiles, and (3) the haplogroup classification model (Kulczynski, Hamming, Jaccard). The six best setups have been tested to determine the optimal trade-off between noise, haplogroup distance, and the overall F1-score (see Supplemental Fig. S2). In our experiments, setup 3 showed the best trade-off between haplogroup distance and overall accuracy. This setup allows us to detect contamination of samples with a phylogenetic distance of at least two and has been used as the final setup for the contamination method. Table 3 summarizes the F1-score statistics for Setup 3. The result indicates that haplocheck is able to accurately detect contamination of two samples also in the case in which noise is included in the input profiles and the distance between the two haplogroups is small.
Table 3.
F1-Score for different noise categories using the finally chosen setup 3
In a last step, we also evaluated the performance of haplocheck as a tool to extrapolate the nDNA contamination level from mtDNA data. Therefore, we generated four whole-genome in silico samples from two random The 1000 Genomes Project Consortium samples showing no signs of contamination based on the VerifyBamID score (Supplemental Table S2). To analyze the impact of the mitochondrial copy number (mtCN), four samples with different amounts of mtCN were chosen from The 1000 Genomes Project Consortium sample collection. The mtCN has been inferred using the formula (mtDNA coverage)/(nDNA coverage × 2) (Ding et al. 2015). For each sample, again four different in silico whole-genome mixtures between 1% and 10% have been created and analyzed using VerifyBamID2 (for nDNA) and haplocheck (for mtDNA). Table 4 summarizes the findings, whereby each sample cell includes the average delta between the calculated and the expected value for all four different mixtures per sample. Levels obtained from VerifyBamID2 and haplocheck correlate if the copy number (CN) for each haplotype in the sample is similar (see samples 1 and 2). Values obtained from sample 3 still correlate, because the main haplotype shows a higher mtCN and is therefore less affected by the lower mtCN of haplotype 2. In a worst-case scenario (sample 4), in which the main haplotype has a lower mtCN and the minor haplotype a higher mtCN, the values obtained from haplocheck and VerifyBamID2 differ substantially.
Table 4.
Four samples including two different haplotypes, in which each haplotype shows a different amount of mtCN have been created (see mtCN ratio)

A drastic shift in the CN is atypical for a large sequencing project. In work by Zhang et al. (2017), the CN of 1500 women aged 17–85 have been analyzed and show that most samples are within a range of 100–300 (mean, 169; DNA source, whole blood). In work by Fazzini et al. (2019), the mtCN has been analyzed in a cohort of 4812 chronic kidney disease patients, also showing only moderate differences (mean, 107.2; SD, 36.4; DNA source, whole blood).
Contamination detection in The 1000 Genomes Project Consortium
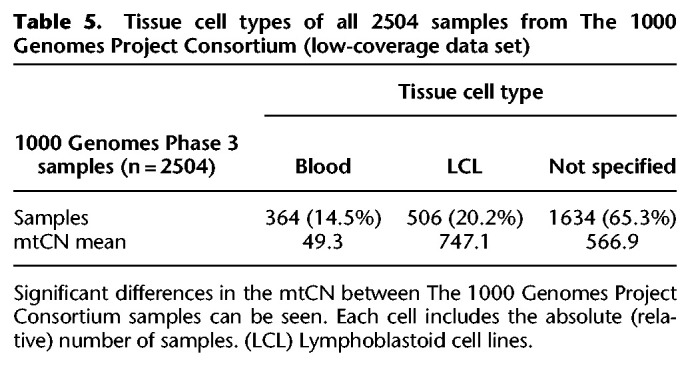
To evaluate haplocheck on a WGS study, we extracted the mtDNA genome reads (labeled as chromosome MT) from samples (Phase 3, low-coverage) from The 1000 Genomes Project Consortium (ftp://ftp.1000genomes.ebi.ac.uk/vol1/ftp/phase3/), resulting in a sample size of 2504 and a total file size of 95 GB. As an initial check, we compared variants detected by mutserve to the official The 1000 Genomes Project Consortium data release using callMom (https://github.com/juansearch/callMom) and determined the haplogroup using HaploGrep 2. Overall, 98% of the samples (n = 2504) result in an identical haplogroup (see Supplemental Fig. S3). The downloaded BAM files were then used as an input for haplocheck to test for contamination. Based on the mitochondrial genome, 5.07% (127 of 2504) of all samples show signs of contamination on mtDNA (see Supplemental Table S3). As previously shown, the performance of haplocheck as a proxy for nDNA is dependent on the mtCN. Because this is uneven for the low-coverage data from The 1000 Genomes Project Consortium, we looked at the tissue source used for DNA extraction. As depicted in Table 5 and Supplemental Figure S4, there is a significant difference in the mtCN owing to the two tissue types used within The 1000 Genomes Project Consortium (P < 2.2 × 10−16, independent t-test).
Table 5.
Tissue cell types of all 2504 samples from The 1000 Genomes Project Consortium (low-coverage data set)
Because of the different mtCN, we split The 1000 Genomes Project Consortium samples into two groups and calculated the Pearson correlation coefficient (R) separately. Group 1 (mtCN ≥ 300, n = 2004) shows a correlation of R = 0.72 between the contamination levels of VerifyBamID2 and haplocheck, with the contamination levels reported by haplocheck ranging from 0.8% to 4.8% (see Supplemental Table S4). The levels are in a very similar range as the VerifyBamID estimates for The 1000 Genomes Project Consortium, because only samples showing a VerifyBamID level of <3% are included. Group 2 (mtCN < 300, n = 500) shows a correlation of only R = 0.31, and contamination levels reported by haplocheck are between 1.8% and 25.5% (see Supplemental Table S5). Because of the higher mtCN, samples of group 1 are more stable, and contamination levels are in a similar range. Samples with a lower mtCN (group 2) differ substantially, because a contamination with a sample showing a higher amount of mtCN affects the mtDNA contamination level. Therefore, group 2 shows a much higher discrepancy in the contamination level compared with VerifyBamID2.
To verify the feasibility of haplocheck in WGS studies with only moderate differences between the samples, we downloaded the mtDNA (labeled as chromosome chrM) of the deep-sequenced The 1000 Genomes Project Consortium sample collection (30× coverage; ftp://ftp-trace.ncbi.nlm.nih.gov/1000genomes/ftp/1000G_2504_high_coverage/), amounting to 176 GB. Compared to the previously analyzed low-coverage sample collection, the mtDNA coverage is much more homogeneous for the high-coverage data (see Fig. 2). Haplocheck detected only minor mtDNA contamination in seven samples (0.9%–1.7%) (see Supplemental Table S6); all spuriously detected contamination in the low-coverage data owing to the different mtCN have vanished.
Figure 2.
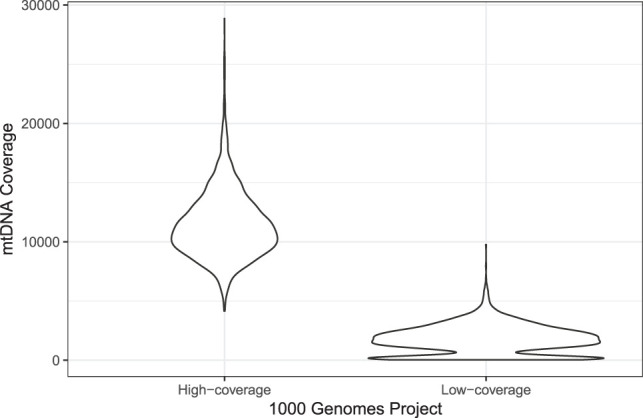
Violin plot representing the mean coverage over all 2504 samples in the two The 1000 Genomes Project Consortium data sets (high-coverage and low-Coverage). Because of different tissues in the low-coverage data, different clusters of coverage can be observed, resulting in wrong mtDNA contamination estimates for nDNA. It can be seen that the second peak within the low-coverage group vanishes for the high-coverage data, resulting in better estimates for extrapolation.
In the last step, we looked at samples that have been excluded from The 1000 Genomes Project Consortium sample collection (nDNA contamination level >3% using VerifyBamID). In total, four samples have been excluded by VerifyBamID using its sequence-only method (free-mix parameter) and seven samples using its sequence and array methods (chip-mix parameter). Haplocheck was able to identify these samples as contaminated with a correlation of 89% between the nDNA and mtDNA level (Supplemental Table S7).
nDNA of mitochondrial origin
nDNA of mitochondrial origin (NUMT) can result either in a coverage drop on mtDNA sites owing to the alignment of mitochondrial reads to NUMT or in false-positive polymorphic calls owing to the alignment of NUMT reads to the mitochondrial genome (Maude et al. 2019). Approaches exist (Goto et al. 2011; Samuels et al. 2013) that exclude reads mapping to the nDNA but overall reduce coverage and may result in false negatives (Albayrak et al. 2016). In work by Weissensteiner et al. (2016a), we annotated mitochondrial sites coming from an NUMT reference database (Li et al. 2012; Dayama et al. 2014), although limited to known NUMTs. For contamination detection with haplocheck, false-positive polymorphic sites owing to NUMTs are expected to only have a minor effect because they typically do not resemble the complete mitochondrial haplotypes. Nevertheless, sufficient coverage for the haplogroup defining variants is still required when dealing with NUMTs. In a study conducted by Maude et al. (2019), an in silico model has been set up to analyze the homology between mitochondrial variants and NUMTs. They show that 29 variants representing haplogroups A, H, L2, M, and U did not cause loss of coverage, but nevertheless, a substantial loss of coverage has been identified for specific sites (e.g., G1888A, A4769G). In a recent work, the presence of a mega-NUMT that could mimic contamination on mitochondrial haplogroup level is described (Balciuniene and Balciunas 2019). This indicates that in very rare cases, NUMTs could indeed resemble complete mitochondrial haplotypes and yield to a false-positive contamination result (Salas et al. 2020; Wei et al. 2020). Although we did not observe NUMT-related issues in the validation of The 1000 Genomes Project Consortium, we cannot entirely rule out possible NUMTs effects on contamination detection.
Runtime and performance
Haplocheck scales linearly with the data size (i.e., sequence reads). For the complete sample collection of The 1000 Genomes Project Consortium in BAM format, the contamination estimate has been calculated within 5.95 h using a single core (Intel Xeon CPU 2.30 GHz) and 1 GB RAM and 1.85 h using four cores and 4 GB RAM, respectively.
Table 6 includes the runtime for 26 samples in BAM format for VerifyBamID2 (input WGS data, varying amounts of markers and cores) and haplocheck (input mtDNA only).
Table 6.
Haplocheck v1.1.3 runtime for 26 samples of The 1000 Genomes Project Consortium low-coverage data

Contamination source
Haplocheck always reports both the major and minor haplotypes for each sample. Therefore, possible sources of contamination can be investigated. For example, sample HG00740 from The 1000 Genomes Project Consortium low-coverage data set shows a contamination level of 2.74% on nDNA (using VerifyBamID2) and 3% on mtDNA (using haplocheck). By looking at the phylogenetic tree that is created for each sample by haplocheck, the contaminating minor haplogroup B2b3a can be identified. The identical haplogroup is also assigned to sample HG01079, which has been analyzed in the same center with a similar mtCN. Such phylogenetic information provided within the interactive HTML report can help in identifying the source of contamination for all three types of contamination.
Discussion
There are many examples in the literature showing the negative impact of artifacts on mtDNA data sets in different areas of research, including medical studies, forensic genetics, and human population studies (He et al. 2010; Bandelt and Salas 2012; Just et al. 2014; Ye et al. 2014). The approach described in this paper takes advantage of the mitochondrial phylogeny and is capable of detecting sample contamination based on mitochondrial haplotype mixtures. By creating several in silico data sets and analyzing The 1000 Genomes Project Consortium samples, we show that haplocheck can be used both in studies using targeted amplification of mtDNA and in those using WGS data. We also investigated the influence of the mtCN and advise taking the mtCN into consideration when using mtDNA estimates for extrapolating nDNA levels.
Several other methods for contamination detection exist. For nDNA sequences, VerifyBamID2 (Zhang et al. 2020) offers an ancestry-agnostic DNA contamination estimation method and is widely used in WGS studies. For ancient studies, schmutzi (Renaud et al. 2015) provides a contamination estimation tool by using sequence deamination patterns; the approach presented in Fu et al. (2013) includes a likelihood-based method to estimate the frequency of present-day human mtDNA haplotypes in the contaminator population. A further approach was suggested by Dickins et al. (2014), describing a pipeline for contamination detection accessible through the Galaxy online platform (Afgan et al. 2018).
Some limitations apply to the phylogenetic-based contamination check proposed in the present investigation, previously applied in a semi-automatic manner (Li et al. 2010; Avital et al. 2012). There is currently a publication bias in favor of the European mtDNA haplogroups that provide the most phylogenetic details, whereas especially African haplogroups are underrepresented (626 African haplogroups compared to 2546 European haplogroups in Phylotree 17). Although the major changes in the phylogeny were performed during the initial growing process of the tree, the last few years showed only refinements of lineages and branches. Therefore, major changes are no longer expected in the human phylogeny, but data from upcoming sequencing studies will help to refine existing groups. Further, contamination detection based on mitochondrial genomes is not applicable in scenarios in which samples belong to the same maternal line (e.g., mother–offspring) owing to an identical haplogroup. The application of haplocheck to ancient DNA studies is limited due the required coverage for detecting polymorphic sites. Importantly, it has also been previously shown for ancient studies that the mtDNA-to-nDNA ratio influences the accuracy of extrapolating nDNA contamination levels from mtDNA estimates (Furtwängler et al. 2018).
Overall, we showed that haplogroup-based contamination detection as performed by haplocheck can be used systematically as a quality measure for mtDNA data. Such kind of analysis could become effective before data interpretation and publication of mtDNA sequencing projects.
Software availability
Haplocheck is available at GitHub (https://github.com/genepi/haplocheck) under the MIT license and requires Java 8 or higher for local execution. All generated data, scripts, and reports are available within this repository. The web service can be accessed via Mitoverse (https://mitoverse.i-med.ac.at). The complete source code from GitHub has been uploaded to the Supplemental Material as Supplemental Code.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
This research was funded by the Austrian Research Fund (FWF; W-1253 DK HOROS to F.K.). We thank all reviewers for their comments that significantly improved the manuscript. We also acknowledge the support of the IT Department from the Medical University of Innsbruck, especially Mario Bedenk, Michael Hörtnagl, Matthias Tschugg, and Dr. Christoph Wild, for providing technical support and resources for the mitoverse web-service.
Author contributions: H.W. and S.S. devised the project and implemented the software. L. Forer developed Cloudgene and was highly involved in design and implementation decisions. S.S., H.W., A.K., and L. Fendt wrote the manuscript. F.K. and A.S. supervised the project and contributed to the manuscript. All authors read and approved the final manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.256545.119.
References
- The 1000 Genomes Project Consortium 2015. A global reference for human genetic variation. Nature 526: 68–74. 10.1038/nature15393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afgan E, Baker D, Batut B, van den Beek M, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Grüning BA, et al. 2018. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res 46: W537–W544. 10.1093/nar/gky379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albayrak L, Khanipov K, Pimenova M, Golovko G, Rojas M, Pavlidis I, Chumakov S, Aguilar G, Chávez A, Widger WR, et al. 2016. The ability of human nuclear DNA to cause false positive low-abundance heteroplasmy calls varies across the mitochondrial genome. BMC Genomics 17: 1017 10.1186/s12864-016-3375-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. 1999. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet 23: 147 10.1038/13779 [DOI] [PubMed] [Google Scholar]
- Avital G, Buchshtav M, Zhidkov I, Tuval Feder J, Dadon S, Rubin E, Glass D, Spector TD, Mishmar D. 2012. Mitochondrial DNA heteroplasmy in diabetes and normal adults: role of acquired and inherited mutational patterns in twins. Hum Mol Genet 21: 4214–4224. 10.1093/hmg/dds245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balciuniene J, Balciunas D. 2019. A nuclear mtDNA concatemer (mega-NUMT) could mimic paternal inheritance of mitochondrial genome. Front Genet 10: 518 10.3389/fgene.2019.00518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt H-J, Salas A. 2012. Current next generation sequencing technology may not meet forensic standards. Forensic Sci Int Genet 6: 143–145. 10.1016/j.fsigen.2011.04.004 [DOI] [PubMed] [Google Scholar]
- Bandelt HJ, Salas A, Lutz-Bonengel S. 2004. Artificial recombination in forensic mtDNA population databases. Int J Legal Med 118: 267–273. 10.1007/s00414-004-0455-2 [DOI] [PubMed] [Google Scholar]
- Brandhagen MD, Just RS, Irwin JA. 2020. Validation of NGS for mitochondrial DNA casework at the FBI laboratory. Forensic Sci Int Genet 44: 102151 10.1016/j.fsigen.2019.102151 [DOI] [PubMed] [Google Scholar]
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, et al. 2011. The variant call format and VCFtools. Bioinformatics 27: 2156–2158. 10.1093/bioinformatics/btr330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das S, Forer L, Schönherr S, Sidore C, Locke AE, Kwong A, Vrieze SI, Chew EY, Levy S, McGue M, et al. 2016. Next-generation genotype imputation service and methods. Nat Genet 48: 1284–1287. 10.1038/ng.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayama G, Emery SB, Kidd JM, Mills RE. 2014. The genomic landscape of polymorphic human nuclear mitochondrial insertions. Nucleic Acids Res 42: 12640–12649. 10.1093/nar/gku1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickins B, Rebolledo-Jaramillo B, Su MS-W, Paul IM, Blankenberg D, Stoler N, Makova KD, Nekrutenko A. 2014. Controlling for contamination in re-sequencing studies with a reproducible web-based phylogenetic approach. BioTechniques 56: 134–141. 10.2144/000114146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding J, Sidore C, Butler TJ, Wing MK, Qian Y, Meirelles O, Busonero F, Tsoi LC, Maschio A, Angius A, et al. 2015. Assessing mitochondrial DNA variation and copy number in lymphocytes of ∼2000 Sardinians using tailored sequencing analysis tools. PLoS Genet 11: e1005306 10.1371/journal.pgen.1005306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazzini F, Lamina C, Fendt L, Schultheiss UT, Kotsis F, Hicks AA, Meiselbach H, Weissensteiner H, Forer L, Krane V, et al. 2019. Mitochondrial DNA copy number is associated with mortality and infections in a large cohort of patients with chronic kidney disease. Kidney Int 96: 480–488. 10.1016/j.kint.2019.04.021 [DOI] [PubMed] [Google Scholar]
- Fu Q, Mittnik A, Johnson PLF, Bos K, Lari M, Bollongino R, Sun C, Giemsch L, Schmitz R, Burger J, et al. 2013. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr Biol 23: 553–559. 10.1016/j.cub.2013.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furtwängler A, Reiter E, Neumann GU, Siebke I, Steuri N, Hafner A, Lösch S, Anthes N, Schuenemann VJ, Krause J. 2018. Ratio of mitochondrial to nuclear DNA affects contamination estimates in ancient DNA analysis. Sci Rep 8: 14075 10.1038/s41598-018-32083-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto H, Dickins B, Afgan E, Paul IM, Taylor J, Makova KD, Nekrutenko A. 2011. Dynamics of mitochondrial heteroplasmy in three families investigated via a repeatable re-sequencing study. Genome Biol 12: R59 10.1186/gb-2011-12-6-r59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Wu J, Dressman DC, Iacobuzio-Donahue C, Markowitz SD, Velculescu VE, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N. 2010. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature 464: 610–614. 10.1038/nature08802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Li L, Myers JR, Marth GT. 2012. ART: a next-generation sequencing read simulator. Bioinformatics 28: 593–594. 10.1093/bioinformatics/btr708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jun G, Flickinger M, Hetrick KN, Romm JM, Doheny KF, Abecasis GR, Boehnke M, Kang HM. 2012. Detecting and estimating contamination of human DNA samples in sequencing and array-based genotype data. Am J Hum Genet 91: 839–848. 10.1016/j.ajhg.2012.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just RS, Irwin JA, Parson W. 2014. Questioning the prevalence and reliability of human mitochondrial DNA heteroplasmy from massively parallel sequencing data. Proc Natl Acad Sci 111: E4546–E4547. 10.1073/pnas.1413478111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Just RS, Irwin JA, Parson W. 2015. Mitochondrial DNA heteroplasmy in the emerging field of massively parallel sequencing. Forensic Sci Int Genet 18: 131–139. 10.1016/j.fsigen.2015.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivisild T, Metspalu M, Bandelt H-J, Richards M, Villems R. 2006. The world mtDNA phylogeny. Nucleic Acids Mol. Biol. 18: 149–179. 10.1007/3-540-31789-9_7 [DOI] [Google Scholar]
- Kloss-Brandstätter A, Pacher D, Schönherr S, Weissensteiner H, Binna R, Specht G, Kronenberg F. 2011. HaploGrep: a fast and reliable algorithm for automatic classification of mitochondrial DNA haplogroups. Hum Mutat 32: 25–32. 10.1002/humu.21382 [DOI] [PubMed] [Google Scholar]
- Li M, Schönberg A, Schaefer M, Schroeder R, Nasidze I, Stoneking M. 2010. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am J Hum Genet 87: 237–249. 10.1016/j.ajhg.2010.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Schroeder R, Ko A, Stoneking M. 2012. Fidelity of capture-enrichment for mtDNA genome sequencing: influence of NUMTs. Nucleic Acids Res 40: e137 10.1093/nar/gks499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Schröder R, Ni S, Madea B, Stoneking M. 2015. Extensive tissue-related and allele-related mtDNA heteroplasmy suggests positive selection for somatic mutations. Proc Natl Acad Sci 112: 2491–2496. 10.1073/pnas.1419651112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maude H, Davidson M, Charitakis N, Diaz L, Bowers WHT, Gradovich E, Andrew T, Huntley D. 2019. NUMT confounding biases mitochondrial heteroplasmy calls in favor of the reference allele. Front Cell Dev Biol 7: 201 10.3389/fcell.2019.00201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renaud G, Slon V, Duggan AT, Kelso J. 2015. Schmutzi: estimation of contamination and endogenous mitochondrial consensus calling for ancient DNA. Genome Biol 16: 224 10.1186/s13059-015-0776-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas A, Yao Y-G, Macaulay V, Vega A, Carracedo A, Bandelt H-J. 2005. A critical reassessment of the role of mitochondria in tumorigenesis. PLoS Med 2: e296 10.1371/journal.pmed.0020296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas A, Schönherr S, Bandelt H-J, Gómez-Carballa A, Weissensteiner H. 2020. Extraordinary claims require extraordinary evidence in asserted mtDNA biparental inheritance. Forensic Sci Int Genet 47: 102274 10.1016/j.fsigen.2020.102274 [DOI] [PubMed] [Google Scholar]
- Samuels DC, Han L, Li J, Quanghu S, Clark TA, Shyr Y, Guo Y. 2013. Finding the lost treasures in exome sequencing data. Trends Genet 29: 593–599. 10.1016/j.tig.2013.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönherr S, Forer L, Weißensteiner H, Kronenberg F, Specht G, Kloss-Brandstätter A. 2012. Cloudgene: a graphical execution platform for MapReduce programs on private and public clouds. BMC Bioinformatics 13: 200 10.1186/1471-2105-13-200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Valk T, Vezzi F, Ormestad M, Dalén L, Guschanski K. 2019. Index hopping on the Illumina HiseqX platform and its consequences for ancient DNA studies. Mol Ecol Resour 20: 1171–1181. 10.1111/1755-0998.13009 [DOI] [PubMed] [Google Scholar]
- van Oven M, Kayser M. 2009. Updated comprehensive phylogenetic tree of global human mitochondrial DNA variation. Hum Mutat 30: E386–E394. 10.1002/humu.20921 [DOI] [PubMed] [Google Scholar]
- Vohr SH, Gordon R, Eizenga JM, Erlich HA, Calloway CD, Green RE. 2017. A phylogenetic approach for haplotype analysis of sequence data from complex mitochondrial mixtures. Forensic Sci Int Genet 30: 93–105. 10.1016/j.fsigen.2017.05.007 [DOI] [PubMed] [Google Scholar]
- Wei W, Tuna S, Keogh MJ, Smith KR, Aitman TJ, Beales PL, Bennett DL, Gale DP, Bitner-Glindzicz MAK, Black GC, et al. 2019. Germline selection shapes human mitochondrial DNA diversity. Science 364: eaau6520 10.1126/science.aau6520 [DOI] [PubMed] [Google Scholar]
- Wei W, Pagnamenta AT, Gleadall N, Sanchis-Juan A, Stephens J, Broxholme J, Tuna S, Odhams CA, Genomics England Research Consortium, NIHR BioResource, et al. 2020. Nuclear-mitochondrial DNA segments resemble paternally inherited mitochondrial DNA in humans. Nat Commun 11: 1740 10.1038/s41467-020-15336-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissensteiner H, Forer L, Fuchsberger C, Schöpf B, Kloss-Brandstätter A, Specht G, Kronenberg F, Schönherr S. 2016a. mtDNA-Server: next-generation sequencing data analysis of human mitochondrial DNA in the cloud. Nucleic Acids Res 44: W64–W69. 10.1093/nar/gkw247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissensteiner H, Pacher D, Kloss-Brandstätter A, Forer L, Specht G, Bandelt H-J, Kronenberg F, Salas A, Schönherr S. 2016b. HaploGrep 2: mitochondrial haplogroup classification in the era of high-throughput sequencing. Nucleic Acids Res 44: W58–W63. 10.1093/nar/gkw233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y-G, Bandelt H-J, Young NS. 2007. External contamination in single cell mtDNA analysis. PLoS One 2: e681 10.1371/journal.pone.0000681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Lu J, Ma F, Keinan A, Gu Z. 2014. Extensive pathogenicity of mitochondrial heteroplasmy in healthy human individuals. Proc Natl Acad Sci 111: 10654–10659. 10.1073/pnas.1403521111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C, Liu Y, Guo X, Li D, Fang W, Yang J, Zhou F, Niu W, Jia Y, Yang H, et al. 2019. An effective strategy to eliminate inherent cross-contamination in mtDNA next-generation sequencing of multiple samples. J Mol Diagn 21: 593–601. 10.1016/j.jmoldx.2019.02.006 [DOI] [PubMed] [Google Scholar]
- Yuan Y, Ju YS, Kim Y, Li J, Wang Y, Yoon CJ, Yang Y, Martincorena I, Creighton CJ, Weinstein JN, et al. 2020. Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat Genet 52: 342–352. 10.1038/s41588-019-0557-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang R, Wang Y, Ye K, Picard M, Gu Z. 2017. Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genomics 18: 890 10.1186/s12864-017-4287-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Flickinger M, Taliun SAG, InPSYght Psychiatric Genetics Consortium, Abecasis GR, Scott LJ, McCaroll SA, Pato CN, Boehnke M, Kang HM. 2020. Ancestry-agnostic estimation of DNA sample contamination from sequence reads. Genome Res 30: 185–194. 10.1101/gr.246934.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.