

Abstract
Different ancestral autosomes independently evolved into sex chromosomes in snakes, birds, and mammals. In snakes and birds, females are ZW and males are ZZ; in mammals, females are XX and males are XY. Although X and Z Chromosomes retain nearly all ancestral genes, sex-specific W and Y Chromosomes suffered extensive genetic decay. In both birds and mammals, the genes that survived on sex-specific chromosomes are enriched for broadly expressed, dosage-sensitive regulators of gene expression, subject to strong purifying selection. To gain deeper insight into the processes that govern survival on sex-specific chromosomes, we carried out a meta-analysis of survival across 41 species—three snakes, 24 birds, and 14 mammals—doubling the number of ancestral genes under investigation and increasing our power to detect enrichments among survivors relative to nonsurvivors. Of 2564 ancestral genes, representing an eighth of the ancestral amniote genome, only 324 survive on present-day sex-specific chromosomes. Survivors are enriched for dosage-sensitive developmental processes, particularly development of neural crest–derived structures, such as the face. However, there was no enrichment for expression in sex-specific tissues, involvement in sex determination or gonadogenesis pathways, or conserved sex-biased expression. Broad expression and dosage sensitivity contributed independently to gene survival, suggesting that pleiotropy imposes additional constraints on the evolution of dosage compensation. We propose that maintaining the viability of the heterogametic sex drove gene survival on amniote sex-specific chromosomes, and that subtle modulation of the expression of survivor genes and their autosomal orthologs has disproportionately large effects on development and disease.
Among amniotes, snakes, birds, and mammals are all conspicuous for carrying highly differentiated sex chromosomes. Caenophidian snakes account for ∼80% of all snake species (http://www.reptile-database.org), and unlike boas or pythons, carry cytologically distinguishable sex chromosomes. In caenophidian snakes and birds, females are ZW and males are ZZ; in therian mammals, females are XX and males are XY. None of these three sets of sex chromosomes are orthologous to either of the others (Fridolfsson et al. 1998; Nanda et al. 1999; Ross et al. 2005; Matsubara et al. 2006). Sex-linked genes in one lineage are found on autosomes in the other lineages. For example, genes that are sex-linked in caenophidian snakes are autosomal in chicken (where they reside on Chromosomes 2 and 27) and humans (where they are found on Chromosomes 3, 7, 10, and 17) (Matsubara et al. 2006). Furthermore, orthologs of sex-linked genes from caenophidian snakes, birds, and mammals are found on separate autosomes in vertebrate outgroup species, like fish (Nakatani et al. 2007; Bellott et al. 2010), indicating that each set of sex chromosomes evolved independently from what were once ordinary autosomes in the common ancestor of amniotes.
The ancestral autosomes in each of these lineages followed parallel trajectories as they evolved into heteromorphic sex chromosomes. A series of events, most likely inversions on the sex-specific (W or Y) chromosome, gradually suppressed crossing over between the sex chromosomes, expanding the sex-specific region at the expense of the pseudoautosomal region, and resulting in the formation of evolutionary strata (Lahn and Page 1999; Handley et al. 2004; Schield et al. 2019). In the absence of crossing over, the sex-specific regions of each new stratum on the W and Y Chromosomes began to diverge from their counterparts on the Z and X Chromosomes and became subject to genetic decay (Charlesworth and Charlesworth 2000). In birds and mammals, Z and X Chromosomes retain >98% of the genes that were present on the ancestral autosomes (Bellott et al. 2010). In contrast, the chicken W Chromosome retains only 4% of ancestral genes (Bellott et al. 2017), and the Y Chromosomes of therian mammals retain as many as 5% of ancestral genes on the opossum Y Chromosome to as few as 1% on the mouse Y Chromosome (Bellott et al. 2014).
Previous analyses of therian Y and avian W Chromosomes revealed that the survival of ancestral genes was nonrandom. On the Y Chromosomes of therian mammals, surviving ancestral genes fall into three functional categories. First, SRY, which is responsible for testis determination (Gubbay et al. 1990; Sinclair et al. 1990). Second, testis-expressed genes and gene families retained and amplified to preserve or enhance male fertility (Lahn and Page 1997; Skaletsky et al. 2003; Hughes et al. 2010, 2012; Bellott et al. 2014). Third, broadly expressed, dosage-sensitive regulators that maintain male viability (Lahn and Page 1997; Bellott et al. 2014). Analysis of W-linked genes across chickens and 13 additional avian species also showed that survivors were enriched for broadly expressed, dosage-sensitive regulators. In the chicken, all surviving genes are broadly expressed, suggesting a focus on preserving female viability (Bellott et al. 2017).
Caenophidian snakes share a common ZW system that evolved after their divergence from boas and pythons (Gamble et al. 2017), but before the divergence of wart and file snakes from other caenophidians (Rovatsos et al. 2015; Matsubara et al. 2016), between 91 and 76 million years (Myr) ago (Kumar et al. 2017). Comparisons of male and female whole-genome shotgun sequence data from five-pacer viper (Deinagkistrodon acutus) (Yin et al. 2016), pygmy rattlesnake (Sistrurus miliarius), and mountain garter snake (Thamnophis elegans) (Vicoso et al. 2013a) yielded dozens of W-linked gene predictions from each species. Phylogenetic analyses of Z–W gene pairs indicated that any strata formed before these three lineages diverged from each other (Vicoso et al. 2013a; Yin et al. 2016) 62 Myr ago (Kumar et al. 2017). Although this is much younger than the W Chromosomes of birds (>111 Myr), or the Y Chromosomes of therian mammals (>159 Myr), genetic decay was similarly extensive; only 3% of ancestral genes were retained on the W Chromosome of the five-pacer viper (Yin et al. 2016). The recently published chromosome-level genome assembly of a male prairie rattlesnake (Crotalus viridis) includes a Z Chromosome that contains around 1500 genes—30% more than previous reconstructions (Schield et al. 2019) and more genes than were present on the ancestral autosomes that became the sex chromosomes of birds and therian mammals, combined. Thus, the sex chromosomes of caenophidian snakes provide a third and much more extensive replicate in a natural experiment in which otherwise ordinary autosomes are exposed to genetic decay and the evolutionary pressures that accompany sex chromosome evolution.
We analyzed surviving W-linked genes from three caenophidian species—five-pacer viper, pygmy rattlesnake, and mountain garter snake—to examine whether evolutionary pressures analogous to those we characterized on the male-specific Y Chromosomes of therian mammals and the female-specific W Chromosomes of birds also shaped the gene content of the caenophidian W Chromosome. Further, we wondered whether we might detect more subtle influences on the survival of ancestral genes by combining data from these three lineages. Additionally, we looked for evidence that genes on the ancestral autosomes with functions related to sex and reproduction were more likely to survive on sex-specific chromosomes.
Results
Reconstructing the ancestral autosomes
Prior to analyzing the survival of genes on the W Chromosomes of five-pacer viper, pygmy rattlesnake, and mountain garter snake, we took advantage of newer, highly contiguous snake genome assemblies to generate a more comprehensive reconstruction of the ancestral autosomes from which caenophidian sex chromosomes evolved (Table 1; Supplemental Figs. S1–S4; Supplemental Tables S1–S6; Supplemental Methods). Previous reconstructions of caenophidian sex chromosome evolution used lizard Chromosome 6 as a proxy for the ancestral autosomes and identified 1135 ancestral Z genes (Vicoso et al. 2013a; Yin et al. 2016). We began our search for ancestral Z-linked genes by compiling a list of published sex-linked genes in snakes (Supplemental Table S2; Supplemental Methods). We identified their orthologs in the human, chicken, and lizard genomes and looked for syntenic orthologs in three highly contiguous caenophidian snake assemblies—prairie rattlesnake (Schield et al. 2019), mainland tiger snake (Notechis scutatus) (https://f1000research.com/posters/7-753) (R Edwards, T Amos, J Tang, et al., pers. comm.), and five-pacer viper (Yin et al. 2016)—using orthologous autosomal scaffolds from the assembly of the Boa constrictor (Bradnam et al. 2013) as an out-group to resolve lineage-specific gains and losses within snakes (Fig. 1A; Table 1; Supplemental Table S3; Supplemental Methods). We identified 45% more genes than previous reconstructions: a total of 1648 genes in 1300 protein-coding gene families on the ancestral autosomes that became the caenophidian Z and W sex chromosomes (Fig. 1B; Supplemental Table S3). This set of ancestral genes is comprehensive, extending across the Z Chromosome to include 62 single-copy genes in the pseudoautosomal region (Fig. 1B; Supplemental Figs. S3, S4; Supplemental Table S3; Supplemental Methods). We restricted our subsequent analyses of the decay and survival of ancestral genes to the 1238 protein-coding gene families outside the pseudoautosomal region.
Table 1.
Key genomic resources used in reconstructing caenophidian ancestral autosomes

Figure 1.
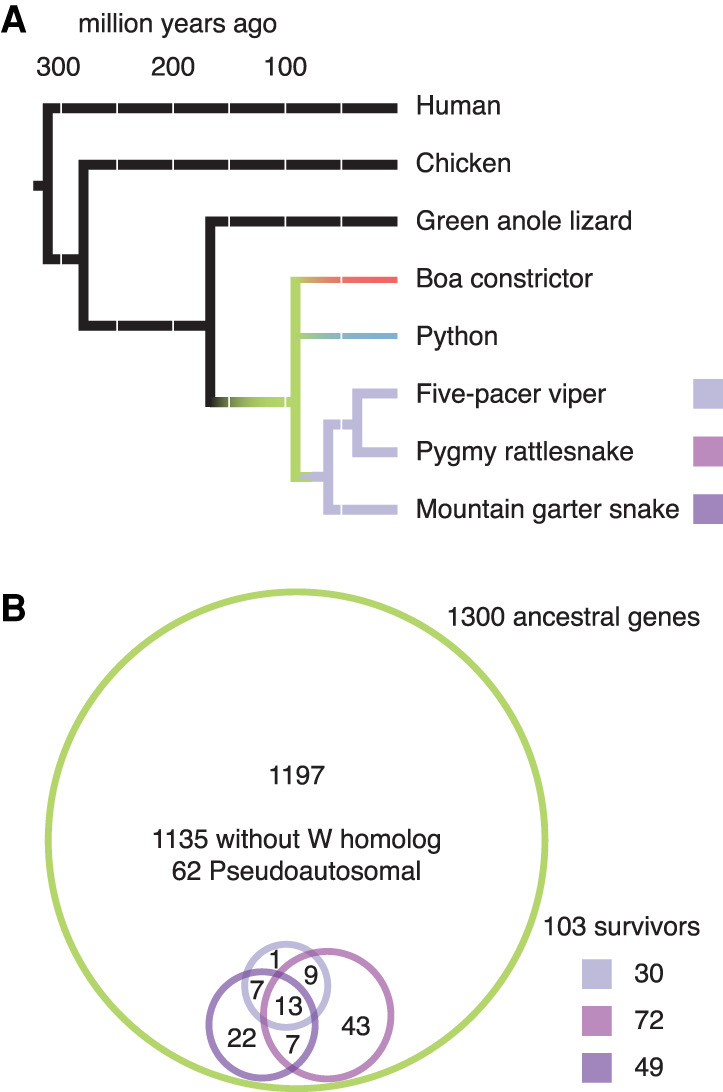
Ancestral Z–W gene pairs from three caenophidian species. (A) Phylogenetic tree of selected snake species included in this study, with branches colored to highlight relationships among species. Humans diverged from snakes 312 Myr ago. Chicken and green anole lizard diverged from snakes 280 and 167 Myr ago, respectively, and were used to resolve gene gains and losses between snakes and mammals. Snakes diverged from each other starting about 90.8 Myr ago (green). Boas (red) and pythons (blue) have independently evolved homomorphic XY sex chromosomes. Caenophidian snakes (purple) share a common ZW sex chromosome system, orthologous to the python XY. (B) Euler diagram showing overlapping sets of ancestral Z–W gene pairs identified in five-pacer viper (light purple), pygmy rattlesnake (medium purple), and mountain garter snake (dark purple) as subsets of all 1300 ancestral Z genes (green). See also Supplemental Table S3.
To analyze gene survival on caenophidian W Chromosomes, we compared our list of ancestral genes to W-linked genes from the three species with published systematic analyses of W-linked gene content: five-pacer viper (Yin et al. 2016), pygmy rattlesnake, and mountain garter snake (Supplemental Tables S1, S4; Supplemental Methods; Vicoso et al. 2013a). We included candidate W-linked genes that these studies had excluded on the basis of their chromosomal assignment in green anole lizard (Supplemental Table S5). We identified 103 ancestral genes with surviving W homologs in one of these three species (Fig. 1B; Supplemental Table S3). We compared these 103 surviving ancestral Z–W gene pairs to the other 1135 ancestral genes without surviving W homologs (Fig. 1B). Using published data sets, we searched for evidence that Z–W pair genes systematically differ from ancestral genes on the Z Chromosome that lack W homologs with regard to dosage sensitivity (Huang et al. 2010; Agarwal et al. 2015; Ruderfer et al. 2016), breadth of expression (Merkin et al. 2012; Ayers et al. 2013; Petropoulos et al. 2016; Perry et al. 2018), and intensity of purifying selection (Fig. 2; Supplemental Fig. S5; Supplemental Table S7; Yates et al. 2020).
Figure 2.

Factors in the survival of caenophidian Z–W gene pairs. Violin plots, with the median (white circle) and interquartile range (white bar) indicated, compare annotations of ancestral Z–W gene pairs identified in three species (purple) to annotations for the remainder of ancestral Z genes (green). (*) P < 0.05; (**) P < 0.01; (***) P < 0.001. P-values were obtained using one-tailed Wilcoxon rank-sum tests (Methods; Supplemental Table S24). Human orthologs of ancestral caenophidian Z–W gene pairs have greater probability of haploinsufficiency (A), deletion intolerance scores (B), duplication intolerance scores (C), and mean probabilities of conserved targeting (PCT) (D) than other ancestral Z genes. Orthologs of ancestral Z–W gene pairs are more broadly expressed than orthologs of other ancestral Z genes in a panel of seven adult eastern garter snake tissues (E). Orthologs of ancestral Z–W gene pairs are more highly expressed than orthologs of other ancestral Z genes in chicken blastocysts (F) and in human preimplantaion embryos (G). Orthologs of ancestral Z–W gene pairs have reduced dN/dS ratios compared to orthologs of other ancestral Z genes in alignments between tiger snake and green anole lizard orthologs (H).
Caenophidian Z–W gene pairs are dosage sensitive
We looked for signs of dosage sensitivity among surviving caenophidian Z–W gene pairs. We mapped various published gene-by-gene measures of dosage sensitivity from the human genome onto their snake orthologs (Supplemental Table S7). Using gene-by-gene estimates of the probability of haploinsufficiency in humans (Huang et al. 2010), we found that human orthologs of surviving Z–W gene pairs were more likely to be haploinsufficient than the human orthologs of ancestral Z-Chromosome genes that lack W-Chromosome homologs (Fig. 2A). Likewise, using deletion intolerance scores calculated from a catalog of genic copy number variation among 59,898 control human exomes (Exome Aggregation Consortium [ExAC]) (Ruderfer et al. 2016), we found that human orthologs of surviving Z–W gene pairs were less likely to tolerate deletions than the orthologs of ancestral Z-Chromosome genes that lack W-Chromosome homologs (Fig. 2B).
Of the 103 caenophidian Z–W gene pairs, at least 11 have human orthologs implicated in congenital disorders caused by heterozygous loss-of-function mutations (Table 2). We assessed the likelihood of finding 11 haploinsufficient human orthologs among these 103 Z–W pairs by examining all 299 human orthologs of ancestral caenophidian Z-linked genes linked to a phenotype with a known molecular basis in Online Mendelian Inheritance in Man (OMIM) (McKusick-Nathans Institute of Genetic Medicine 2019; Table 3). We found that Z–W pair genes were enriched fourfold for phenotypes caused by haploinsufficiency relative to Z-linked genes without surviving W homologs. Taken together, the elevated haploinsufficiency probabilities, deletion intolerance scores, and significant enrichment for human disorders caused by haploinsufficiency indicate that caenophidian Z–W pairs are enriched for haploinsufficient genes.
Table 2.
Human orthologs of caenophidian Z–W gene pairs linked to human disorders attributed to haploinsufficiency

Table 3.
Human disorders in OMIM attributed to haploinsufficiency

Using the EXAC duplication intolerance scores (Ruderfer et al. 2016), we find that, relative to other ancestral Z genes, the human orthologs of caenophidian Z–W gene pairs are more intolerant of duplication (Fig. 2C). MicroRNAs (miRNAs) are short, noncoding RNAs that fine-tune gene dosage by pairing to the 3′ untranslated region and lowering target mRNA levels (Bartel 2009). Conserved targeting by miRNAs is a feature of genes sensitive to changes in gene dosage, particularly to overexpression (Naqvi et al. 2018). We used published estimates of the probabilities of conserved targeting (PCT scores) (Friedman et al. 2009) for each gene–miRNA interaction in the human genome. We found that the human orthologs of surviving Z–W gene pairs had higher gene-average PCT scores than other ancestral Z genes (Fig. 2D). Also, across all gene–miRNA pairs, PCT scores are higher for Z–W gene pairs than for other ancestral Z genes (Supplemental Fig. S5A). Moreover, relative to other ancestral genes, the orthologs of Z–W gene pairs are targeted by more miRNA sites conserved between human and chicken (Supplemental Fig. S5B) and between human and green anole lizard (Supplemental Fig. S5C). We conclude that caenophidian Z–W pairs are similar to both therian X–Y and avian Z–W gene pairs in being more sensitive to increases and decreases in gene expression than other ancestral genes.
Caenophidian Z–W gene pairs are broadly expressed
We assessed whether surviving caenophidian Z–W pairs are more broadly expressed than other ancestral genes in snakes and whether this broad expression was ancestral. We observed that the Z homologs of Z–W gene pairs are more broadly expressed than ancestral genes that lack W homologs across a panel of adult eastern garter snake (Thamnophis sirtalis) female tissues (Fig. 2E; Perry et al. 2018). This increased breadth of expression extends to chicken (Supplemental Fig. S5D) and human (Supplemental Fig. S5E) orthologs of Z–W gene pairs. We conclude that caenophidian Z–W gene pairs maintain broad expression across adult tissues from their autosomal precursors in the amniote ancestor.
We further suggest that this breadth of expression extended to the earliest stages of embryonic development. Although gene expression data from snake embryos is not available, we found that the orthologs of ancestral caenophidian Z–W gene pairs are more highly expressed than those of ancestral Z genes that lack W homologs in both chicken blastocysts (Ayers et al. 2013; Fig. 2F) and human morulae (Fig. 2G; Petropoulos et al. 2016). We conclude that the autosomal precursors of caenophidian Z–W pairs were more broadly expressed across development as well as across adult tissues.
Caenophidian Z–W gene pairs are subject to strong purifying selection
If surviving X–Y and Z–W gene pairs are extremely dosage sensitive, alleles that alter the ancestral function of the homologs on the X or the Z Chromosomes should be detrimental to both males and females. We looked for evidence that the Z-linked homologs of caenophidian Z–W gene pairs are subject to stronger purifying selection than other ancestral Z-linked genes. To parallel our earlier analyses of Ensembl ortholog alignments in mammals and birds, we used alignments of mainland tiger snake genes, the sole snake in Ensembl version 96 (Yates et al. 2020). Compared to ancestral genes on the caenophidian Z Chromosome that lack W homologs, the Z homologs of Z–W gene pairs have a reduced ratio of nonsynonymous to synonymous substitution (dN/dS) rates when mainland tiger snake genes are compared to their orthologs in green anole lizard (Fig. 2H), chicken (Supplemental Fig. S5F), and human (Supplemental Fig. S5G). We conclude that, on caenophidian sex chromosomes, strong purifying selection preserved a subset of ancestral genes with preexisting broad expression and dosage sensitivity dating back to the common ancestor of amniotes.
Functional coherence of caenophidian Z–W gene pairs
We asked whether the set of ancestral genes that survived on caenophidian W Chromosomes carried out regulatory functions that parallel those of mammals and birds. In comparison to other ancestral genes on the caenophidian Z Chromosome, Z–W pair genes are enriched for Gene Ontology (GO) annotations such as nucleic acid binding, nucleus, and transcription (Supplemental Table S8) that suggest regulatory functions. We looked in more detail at the molecular functions of the 103 caenophidian Z–W pairs (Fig. 3; McKusick-Nathans Institute of Genetic Medicine 2019; The UniProt Consortium 2019). We observe that caenophidian Z–W pairs share functions in regulating transcription and protein stability with surviving gene pairs in human and chicken (Fig. 3). Four survivors are components of the proteasome (PSMA2, PSME3, PSMD3, and PSMD11) (Fig. 3), consistent with the idea that members of large protein complexes tend to be dosage sensitive (Papp et al. 2003; Pessia et al. 2012).
Figure 3.
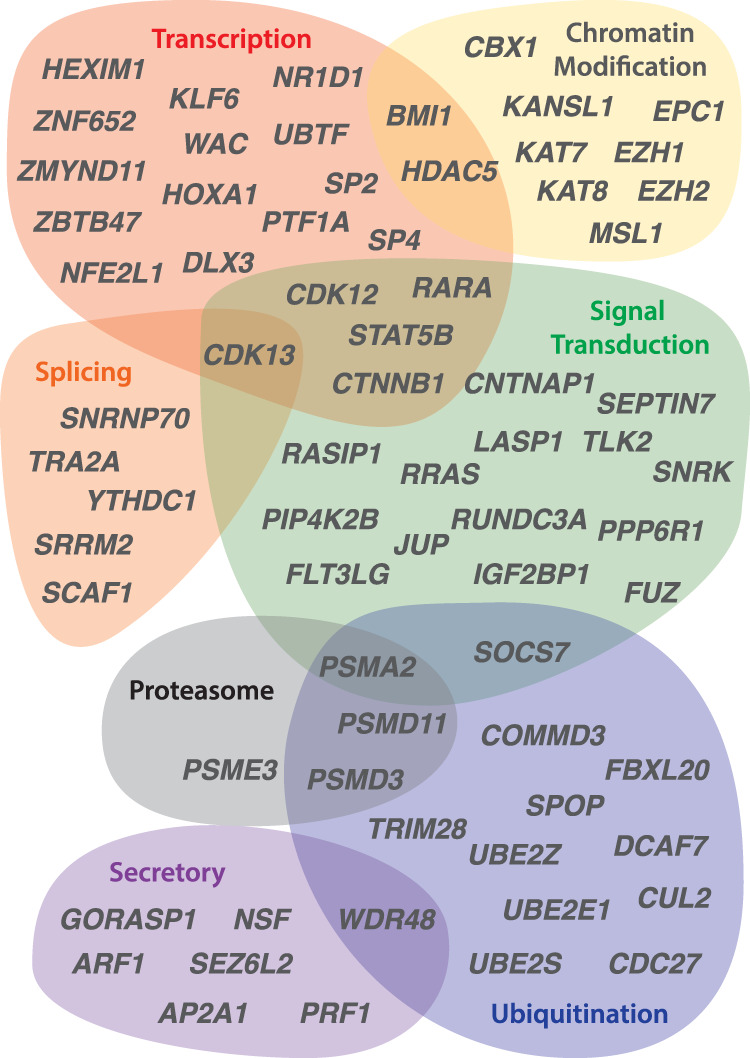
Regulatory annotations of ancestral caenophidian Z–W gene pairs. The Euler diagram depicts regulatory functions predicted for genes from selected Z–W gene pairs on the basis of UniProt annotations of human orthologs. See also Supplemental Table S8.
The annotations of caenophidian Z–W pairs also indicate overlap with functions we previously observed only in mammals or chicken; as in human, they play roles in regulating mRNA splicing, and, similar to chicken, they participate in secretory and signal-transduction pathways. We previously observed that chicken Z–W pairs included several components of the Wnt signaling pathway; we note that both junction plackoglobin (encoded by JUP) and catenin beta 1 (encoded by CTNNB1) are Z–W pairs in caenophidian snakes. This underscores the importance of the correct gene dose for proper signaling in pathways regulating growth and development.
Pressure to preserve dosage-sensitive functions in early embryonic development drove gene survival
We asked whether combining snakes with birds and mammals into a combined analysis might give us the power to detect more specific functional enrichments than we recovered from individual lineages. We therefore pooled our data on the survival of ancestral genes across all three lineages and incorporated published survivors from additional birds (Xu et al. 2019a) and mammals (Li et al. 2013; Skinner et al. 2016; Janečka et al. 2018). From 41 species—three snakes, 24 birds, and 14 mammals—we compiled a list of 324 survivors of genetic decay from a total of 2564 ancestral genes (Supplemental Fig. S1; Supplemental Tables S7, S9), representing nearly an eighth of the ancestral amniote genome. We verified that combining our data did not eliminate or reverse the enrichments we previously observed; the combined set of surviving ancestral genes is enriched for the same qualities we observed separately in each lineage (Supplemental Fig. S6).
This broad sampling of survivors allowed us to detect more specific enrichments, providing insight into the developmental processes evolving under strong purifying selection across amniotes (Supplemental Tables S7, S10–S12). Relative to nonsurvivors, survivors were enriched for GO terms that emphasize their role in regulating development: “multicellular organism growth,” “in utero embryonic development,” “regulation of anatomical structure morphogenesis,” “regulation of epithelial to mesenchymal transition,” “kidney epithelium development,” and “face development” (Supplemental Table S12).
Shortly after gastrulation, neural crest cells undertake an epithelial to mesenchymal transition, migrate through the embryo, and differentiate into a diverse array of tissues, including the bones and cartilage of the face. Neural crest cells respond to a multitude of signal-transduction pathways, and they are sensitized to TP53-mediated apoptosis (Calo et al. 2018). As a result, mutations in hundreds of genes can cause defects in the specification, migration, differentiation, and death of neural crest cells, resulting in a wide range of pathologies, including craniofacial anomalies (Trainor 2010; Vega-Lopez et al. 2018). We conclude that the enrichment for “face development” reflects the extreme sensitivity of the neural crest to changes in the dose of broadly expressed survivor genes, rather than an enrichment for face-specific expression.
The face represents a highly sensitive assay for disruptions to early human development throughout the body. The enrichment for minor physical anomalies, particularly of the head and face, in disorders of the central nervous system, like autism, intellectual disability, schizophrenia, and bipolar disorder, has been taken as evidence that these disorders are developmental in origin (Smith and Bostian 1964; Weinberg et al. 2007; Aldridge et al. 2011; Berecz et al. 2017). The sets of genes implicated in neurodevelopmental disorders overlap with each other and share characteristics with genes that survive on sex-specific chromosomes: an enrichment for chromatin modifiers and embryonic expression, as well as signs of haploinsufficiency (Iossifov et al. 2014). We therefore looked for evidence that the human orthologs of survivor genes were enriched among a set of 1919 genes identified as carrying de novo mutations in whole exome sequencing studies across two or more neurodevelopmental disorders, including autism spectrum disorders, bipolar disorder, intellectual disability, or schizophrenia (Table 4; Supplemental Table S7; Wang et al. 2019). We found that orthologs of survivors were enriched 1.7-fold for genes mutated in neurodevelopmental disorders compared with nonsurvivors. The genes implicated in neurodevelopmental disorders overlap with those implicated in congenital anomalies of other organ systems, for example, those of the heart or the kidney and urinary tract (Homsy et al. 2015; Sanna-Cherchi et al. 2018). Because survivor genes are broadly expressed, we asked whether they were also enriched among genes implicated in these disorders by using a set of 1169 genes associated with congenital heart defects (Table 5; Supplemental Table S7; Homsy et al. 2015) and 50 genes associated with congenital anomalies of the kidney and urinary tract (Table 6; Supplemental Table S7; Sanna-Cherchi et al. 2018). We found that human orthologs of survivors were enriched 1.6-fold for genes mutated in congenital heart defects compared to nonsurvivors (Table 5). Likewise, human orthologs of survivors were enriched fourfold for genes associated with congenital anomalies of the kidney and urinary tract compared to nonsurvivors (Table 6). We conclude that the early developmental role of survivors may have far-reaching consequences for fitness through their effects not only on the face, but also the brain, heart, and kidney. Close scrutiny of the human orthologs of survivors in snakes and birds will likely reveal new candidates for human disorders and insights into the developmental pathways most sensitive to changes in gene dose.
Table 4.
Survivors enriched in neurodevelopmental disorders

Table 5.
Survivors enriched in congenital heart disease

Table 6.
Survivors enriched in congenital anomalies of the kidney and urinary tract
Combining the data from all three lineages also revealed a ninefold enrichment for components of the ficolin-1-rich granule lumen (Supplemental Table S12), a cellular compartment that may play an under-appreciated role in regulating neural crest development. There were 12 survivors out of 21 ancestral genes in this GO category, including survivors from all three lineages (Supplemental Table S13). Relative to the rest of the genome, genes annotated with this GO category are more likely to be haploinsufficient (Supplemental Fig. S7). Ficolin 1 is a recognition molecule in the lectin complement pathway of the innate immune system, localized in secretory granules in the cytoplasm of neutrophils and monocytes (Liu et al. 2005). Apart from their role in innate immunity, components of the complement cascade are involved in pruning inappropriate synapses during development of the central nervous system (Stevens et al. 2007), and mutations in components upstream of the complement cascade are responsible for 3MC syndrome (also known as Craniofacial-Ulnar-Renal syndrome or Carnevale, Mingarelli, Malpuech, and Michels syndromes) in humans as a result of their role in guiding neural crest cell migration (Rooryck et al. 2011; Munye et al. 2017). Several survivors annotated as ficolin-1-rich granule components—CDK13, DDX3X, HUWE1, and JUP—are implicated in human disorders with dysmorphic facial features, cleft lip and palate, poor growth, intellectual disability, hearing loss, and cardiac anomalies that suggest a role in regulating neural crest development. We speculate that regulation of face and renal development by the complement pathway through ficolin-1-rich granules is a dosage-sensitive process, like the TGFB and Wnt pathways that are also enriched among surviving Z–W pairs in birds and snakes.
Sex determination, sex-specific expression, and gene survival on sex chromosomes
Across all three lineages, ancestral X–Y and Z–W gene pairs were preserved to maintain comparable expression between males and females, but other evolutionary strategies also contribute to gene survival on the male-specific Y Chromosome of mammals (Bellott et al. 2014). Sex-determining genes (those involved in the sex determination cascade and gonadogenesis) and genes with sex-biased functions (those with expression limited to sex-specific tissues, or with sex-biased gene expression in somatic tissues) play a large role in theoretical models of sex chromosome evolution. Linkage between sex-determining genes and genes with sex-biased functions is predicted to drive the suppression of recombination on sex chromosomes (Nei 1969; Bull 1983; Rice 1987), resulting in the formation of evolutionary strata and heteromorphic sex chromosomes. This has led to speculation that certain ancestral amniote chromosomes may have been predisposed to take on roles as sex chromosomes owing to an enrichment for genes in the sex determination pathway linked to other genes with sex-biased functions (O'Meally et al. 2012).
We looked for evidence that ancestral sex- and reproduction-related functions promoted the survival of Y- and W-linked genes across amniotes. We reanalyzed published data sets for evidence that survivors systematically differ from other ancestral genes with regard to the prevalence of genes involved in sex determination and gonadogenesis (Ayers et al. 2013), gene expression limited to sex-specific tissues (Yin et al. 2016; GTEx Consortium 2017; Perry et al. 2018; McCarthy et al. 2019), and conserved sex-biased expression (Fig. 4; Tables 7, 8; Supplemental Table S7; Naqvi et al. 2019). We were unable to detect strong associations between any of these categories and gene survival on sex-specific chromosomes.
Figure 4.

Survivors are not ancestrally specialized for expression in reproductive tract tissues. Coefficients for testis- and ovary-specific expression (circles) and 95% confidence intervals (bars) from logistic regression models of gene survival on sex-specific chromosomes in mammals (blue), birds (pink), and snakes (purple), conditional on breadth of expression. (ns) P > 0.05. After controlling for expression breadth, the regression coefficient is not significantly different from 0 for any tissue from any species.
Table 7.
Survival of sex- and reproduction-related genes

Table 8.
Survival of genes with conserved female bias in mammals

We asked whether ancestral genes with a surviving Y- or W-linked homolog were more likely than other ancestral genes to be present in a list of 117 genes implicated in sexual reproduction or gonadogenesis in vertebrate model organisms (Ayers et al. 2013). We did not detect a significant enrichment for genes involved in sexual reproduction or gonadogenesis among survivors; although seven of 324 survivors were on the list, so were 28 of 2240 nonsurvivors (Table 7).
We reasoned that genes with ancestrally sex-biased functions might be expressed more highly in sex-specific tissues, including those of the reproductive tract; therefore, we looked for evidence that the survivors of genetic decay are enriched for tissue specific expression in adult gonads in human, chicken, and snakes (Yin et al. 2016; GTEx Consortium 2017; Perry et al. 2018; McCarthy et al. 2019). For each gene-tissue pair, we calculated a tissue specificity index, dividing the expression in a tissue by the summed gene expression across all tissues (Supplemental Table S7). Most survivors showed only weak specificity in sex-specific tissues, consistent with their being expressed at nearly equal levels across adult tissues (Supplemental Table S7). Therefore, we used logistic regression to control for the effects of expression breadth while measuring the effect of tissue specific expression on survival. We analyzed each lineage separately, using the expression patterns of the autosomal orthologs in the other two lineages as a proxy for the ancestral state.
We first looked for evidence that orthologs of X–Y gene pairs from therian mammals are enriched for expression in testis. We calculated testis specificity in chicken using a panel of 15 adult male tissues (McCarthy et al. 2019) and in five-pacer viper using a panel of four adult male tissues (Yin et al. 2016). In each case, after controlling for the effects of broad expression, expression in testis had no significant effect on the odds of ancestral X genes retaining a surviving Y homolog (Fig. 4).
Likewise we searched for evidence that orthologs of Z–W gene pairs from birds were ancestrally enriched for expression in ovary. We calculated ovary specificity in human using a panel of 39 adult female tissues (GTEx Consortium 2017) and in eastern garter snake using a panel of eight adult female tissues (Perry et al. 2018). We did not detect a significant effect of ovary-specific expression in either lineage on the survival of W-linked genes in birds (Fig. 4).
Last, using the human (GTEx Consortium 2017) and chicken (McCarthy et al. 2019) ovary specificities, we found that ovary-specific expression in either lineage had no effect on the survival of W-linked genes in snakes (Fig. 4). We conclude that ancestral bias for expression in sex-specific tissues played no significant role in survival of X–Y and Z–W pairs.
We also asked whether survivors were enriched for genes that show a conserved sex bias in mammals (Naqvi et al. 2019). Because surviving X–Y gene pairs preserve ancestral gene expression levels in both sexes, the X homologs of X–Y gene pairs are not subject to X inactivation and are expressed more highly from two copies in females than they are from a single copy in males. As a result, the X homologs of X–Y gene pairs were called as having a conserved female bias in this data set; therefore, we excluded therian sex-linked genes from this analysis. Because both birds and snakes have female-specific W Chromosomes, we looked for an enrichment of genes with a conserved female bias in one or more tissues in the mammalian autosomal orthologs of surviving Z–W gene pairs, relative to other ancestral genes (Table 8). We did not observe an enrichment for conserved female-biased genes: although 54 of 275 survivors were female biased, so were 298 of 1654 nonsurvivors (Table 8).
Survival of genes that were adjacent on the ancestral autosomes
Of 324 surviving ancestral genes on present-day W and Y Chromosomes, 118 survive alongside one or more neighbors from the ancestral autosome (Supplemental Table S7). One or more biological processes could cause the frequent survival of clusters of genes from the ancestral autosomes. For example, broadly expressed genes might be adjacent to each other in the same chromatin domain. Likewise, marginal survivor genes might be less likely to be deleted from the sex-specific chromosome when they are adjacent to another dosage-sensitive survivor. Alternatively, this clustering could arise by chance, given the number of surviving genes within each evolutionary stratum in each species.
We looked for evidence of evolutionary strata in caenophidian snakes and tabulated published information about evolutionary strata in birds and mammals (Supplemental Figs. S1–S4; Supplemental Tables S6, S9, S14, S15; Supplemental Data S1, S2; Supplemental Methods). For each stratum in each species, we tallied the number of ancestral genes and the number of survivors, then calculated the expected number of adjacent survivors, assuming each gene survives independently (Supplemental Tables S16, S17; Supplemental Methods). For all 107 species-stratum pairs with more than two survivors, we tested whether we observed significantly more adjacent survivors than expected by chance (Supplemental Table S17). None were significant after correction for multiple testing (Supplemental Table S17). We conclude that the number of adjacent survivors that we observe is consistent with a simple model in which each gene survives independently from its neighbors.
Dosage sensitivity and expression breadth made independent contributions to survival
We sought to explore the interactions between dosage sensitivity, breadth of expression, and strength of purifying selection in the survival of genes on sex-specific chromosomes in our combined data set. Therefore, we set out to quantify the survival of each ancestral gene. Within a species, the sex-specific homologs of ancestral gene pairs were exposed to the forces of genetic decay for different periods of time, depending on the age of their evolutionary strata. We calculated a gene-wise “survival fraction” to control for the different numbers of sampled species and evolutionary strata of different ages when comparing across X–Y and Z–W systems. For each gene within a lineage, we calculated its total branch length across the tree of species, then divided this by the maximum branch length possible for a gene of its evolutionary stratum (Supplemental Tables S18–S23; Supplemental Methods). These values range from 0 (lost in all lineages) to 1 (survival in every possible lineage) (Supplemental Table S7).
We used principal components analysis to explore the influence of factors related to dosage sensitivity, breadth of expression, and strength of purifying selection on the survival of ancestral gene pairs across all three lineages (Fig. 5; Supplemental Figs. S8–S10). The first principal component separated genes according to their survival fraction, allowing us to visualize the contribution of each factor to survival. Consistent with the hypothesis that intense purifying selection maintains surviving gene pairs, the dN/dS ratios in mammals, birds, and reptiles were most closely aligned with the survival fraction (although they point in the opposite direction, as survivors have a reduced dN/dS ratio). Human haploinsufficiency probabilities are closely aligned with survival; these probabilities reflect the output of a model trained on known human haplosufficient genes that takes multiple factors as input, including embryonic expression levels and strength of purifying selection (Huang et al. 2010). Measures of dosage sensitivity and breadth of expression make distinct and approximately orthogonal contributions to the survival of genes on sex-specific chromosomes. This suggests that some genes may survive primarily because of dosage sensitivity, whereas others may be preserved mostly because they are required in many tissues and cell types. For example, TCF4 is a transcription factor with high scores in measures of dosage sensitivity but low scores in measures of expression breadth (Fig. 5; Supplemental Table S7). In contrast, RPL37, a component of the 60S subunit of the ribosome, is broadly expressed but has low scores in measures of dosage sensitivity (Fig. 5; Supplemental Table S7).
Figure 5.
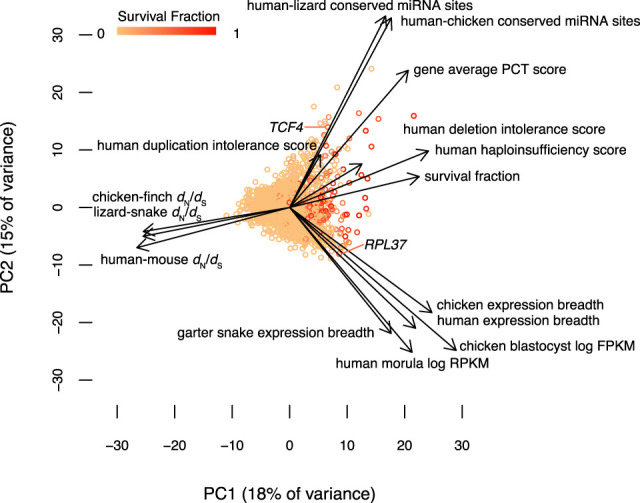
Dosage sensitivity and broad expression make independent contributions to survival. A statistical summary of survival factors from 2564 genes based on principal component axis one (PC1) and axis two (PC2). Points represent individual genes, colored by their survival fraction from orange (no survival) to red (survival in all possible lineages) (Methods; Supplemental Table S7). Arrows show the contribution of each factor to the variation in survival of ancestral genes on sex-specific chromosomes. Dosage sensitivity and breadth of expression make roughly orthogonal contributions, whereas strength of purifying selection is closely aligned with survival.
Discussion
We were unable to detect a strong influence of sex- and reproduction-related functions in the survival of ancestral genes across sex-specific chromosomes in amniotes. If genes in these functional categories are not highly dosage sensitive, broadly expressed, and subject to intense purifying selection, then this may hinder their survival on sex-specific chromosomes. Genes involved in sex determination and gonadogenesis may be poorly represented among survivors because they are less dosage sensitive. In vertebrates, development of the bipotential gonads is canalized down two pathways to generate either ovaries or testes; the regulatory cascades that govern this decision are a series of bistable switches, robust to perturbation (Munger and Capel 2012). As a consequence, the genes and networks regulating gonadal sex determination may be less affected by subtle changes in gene dose than those of developmental regulators that produce a graded response with changes in gene dose, like morphogens. Likewise, genes with expression limited to sex-specific tissues and genes with sex-biased expression are both under reduced selective constraint, which could reduce their chances of resisting genetic decay. In contrast, genes expressed across many tissues are subject to complex selection pressures that result in more intense purifying selection (Duret and Mouchiroud 2000) and greater conservation of patterns of gene expression (Yang et al. 2005). Sex-biased genes are more narrowly expressed than genes with no bias in Drosophila (Meisel 2011), birds (Mank et al. 2008), and mammals (Mank et al. 2008; Meisel 2011; Naqvi et al. 2019), and this narrow expression is accompanied by reduced selective constraint (Meisel 2011; Naqvi et al. 2019). Taken together, this suggests that the selective pressures that enriched surviving Z–W and X–Y pairs for broadly expressed, dosage-sensitive regulators under strong purifying selection may present obstacles to the survival of sex- and reproduction-related genes.
Selection for sex- and reproduction-related functions may be more influential at specific stages of sex chromosome evolution, making them more difficult to detect in our analyses of amniote sex chromosomes. The sex chromosomes of caenophidian snakes, birds, and therian mammals are all evolutionarily old and highly differentiated. As a result, our analyses will not capture the influence of factors related to sex and reproduction on gene survival during the early stages of sex chromosome evolution. In Drosophila miranda, surviving ancestral genes on Y Chromosomes are enriched for genes with ancestral male-specific expression, but this effect is strongest among the 5% of ancestral genes (109/2250 ancestral genes) that have been retained on the Y Chromosome but lost from the X Chromosome (Kaiser and Bachtrog 2014). There is no parallel to this high rate of gene loss on amniote Z and X Chromosomes, so these observations may reflect selective pressures unique to the Drosophila lineage rather than a common process in the early stages of sex chromosome evolution.
Selection for sex- and reproduction-related functions could also lead to changes in the function or expression patterns of ancestral genes later in sex chromosome evolution. We previously reported that ancestral Y-linked genes that were amplified into multicopy gene families in at least one species survived through more of therian Y Chromosome evolution than those that remained single copy, and we noted that these multicopy families were expressed exclusively or predominantly in the testis (Bellott et al. 2014). Although the evolution of testis-specific expression likely preceded gene amplification on the Y Chromosome, most of these genes were ancestrally broadly expressed. Because our analyses here focus on the ancestral expression patterns of survivors and nonsurvivors, they cannot capture changes in expression pattern that occur during the evolution of sex-specific chromosomes, and as a result they may underestimate the influence of selection for sex- and reproduction-related functions. However, specialization for reproductive function does not inevitably occur on sex-specific chromosomes, even for multicopy gene families. All of the genes on the female-specific W Chromosome of chicken, including the multicopy HINTW, retain their ancestral broad expression (Bellott et al. 2017). Studies of additional avian and caenophidian W Chromosomes will reveal how frequently ancestral genes evolve expression restricted to female reproductive tissues.
Like their counterparts on mammalian Y Chromosomes (Bellott et al. 2014) and avian W Chromosomes (Bellott et al. 2017), the surviving ancestral genes on caenophidian W Chromosomes are enriched for broadly expressed, dosage-sensitive regulators under strong purifying selection. Our findings across these three highly differentiated sets of amniote sex chromosomes have parallels in the nonrandom survival of ancestral genes on the evolutionarily young Y Chromosomes of Drosophila miranda (about 1.5 Myr) (Kaiser et al. 2011) and threespine stickleback (less than 16 Myr) (White et al. 2015). Surviving gene pairs on the Neo-X and Neo-Y Chromosomes of Drosophila miranda are expressed at higher levels and across more tissues than genes lost to decay (Kaiser et al. 2011). An abundance of protein–protein interactions is correlated with dosage sensitivity (Papp et al. 2003; Huang et al. 2010); in both threespine stickleback and Drosophila miranda, surviving X–Y gene pairs were enriched for genes encoding proteins with many partners in protein–protein interaction networks (Kaiser et al. 2011; White et al. 2015). The repeated finding that sex-specific chromosomes preferentially retain broadly expressed, dosage-sensitive genes, regardless of whether the chromosomes are female-specific or male-specific, or in the early or late stages of differentiation, shows that purifying selection is effective at preserving the correct dosage of critical ancestral genes in the absence of crossing over. We propose that the survival of ancestral genes on sex-specific W and Y Chromosomes is driven by pressure to maintain the viability of the heterogametic sex.
One corollary of this proposal is that there will be severe consequences for monosomy of sex chromosomes that carry an abundance of dosage-sensitive Z–W or X–Y gene pairs. Surviving gene pairs are enriched for roles in the epithelial to mesenchymal transition, which is critical to gastrulation. Haploinsufficiency for this process could explain why most 45,X human conceptuses spontaneously abort in the first trimester with an absent or disorganized embryo, and those that survive are often mosaic for all or part of a second sex chromosome (Hook and Warburton 1983; Hassold et al. 1988; Cockwell et al. 1991). Similarly, it would explain why chicken embryos carrying a single Z Chromosome are observed at the blastocyst stage (Fechheimer 1981), but not at 4–5 d of development (Bloom 1972). Despite nearly six decades of cytogenetic observations of the caenophidian sex chromosomes (Becak et al. 1962; Kobel 1962), including an unusual 3A:ZZW intersex twin-spotted ratsnake (Elaphe bimaculata) (Rovatsos et al. 2018), we are unaware of any reports of sex chromosome monosomy in a caenophidian snake. Given the number of dosage-sensitive genes among the surviving ancestral genes on caenophidian W Chromosomes, as well as their ancestrally broad expression, extending to early embryogenesis, we predict that sex chromosome monosomy is also embryonic lethal in caenophidian snakes.
Dosage compensation on Z and X Chromosomes also functions to maintain the viability of the heterogametic sex and is so deeply entwined with the survival of ancestral genes on W and Y Chromosomes that they cannot be studied independently. Surviving Z–W and X–Y gene pairs maintain expression in the heterogametic sex at levels similar to those of pseudoautosomal or autosomal orthologs for the same sex in close outgroups, but not necessarily equal expression between the homogametic and heterogametic sex. Depending on its intrinsic sensitivity to changes in expression level, each gene may or may not evolve dosage compensation to restore this ancestral expression level after the decay of a sex-specific W or Y-linked homolog (Naqvi et al. 2018). In genes that are more sensitive to underexpression than overexpression, autosomal paralogs derived from ancestral sex-linked genes can rescue loss of sex-specific homologs (Hughes et al. 2015). To fully account for these factors, a comprehensive characterization of the nature and extent of dosage compensation in caenophidian snakes will require the development of comparable gene expression data from a female caenophidian snake and henophidian or lizard outgroup species, paired with highly contiguous, reference-grade sequences of both Z and W sex chromosomes as well as the autosomes.
Our analyses revealed that dosage sensitivity and breadth of expression contribute independently to survival of genes on sex-specific chromosomes. This suggests that genes expressed across a broad array of tissues face complex selective pressures that make an important contribution to survival, distinct from increasing sensitivity to changes in gene dose. One possible explanation is that pleiotropic effects may impose constraints on the evolution of dosage compensation for broadly expressed genes. Models of the evolution of sex-biased gene expression predict that genes preferentially expressed in the heterogametic sex are less likely to evolve on the Z and X Chromosomes than on autosomes when they are subject to pleiotropic constraints (Connallon and Clark 2010). Ancestral genes expressed across a broad array of tissues are therefore less likely to evolve increased expression from their Z or X homologs than those expressed more narrowly. As a result, mutations that decrease expression from the sex-specific homolog will more frequently arise opposite a Z-linked or X-linked homolog that cannot compensate for their reduced dosage, increasing the stringency of selection against their loss. Under this model, even with equivalent levels of dosage sensitivity, more broadly expressed genes should prove more resistant to genetic decay than those that are more narrowly expressed.
Summed across all three lineages, nearly an eighth of the ancestral amniote genome was subjected to 2.5 billion years of sex chromosome evolution, winnowing 2564 ancestral genes to just 324 survivors on sex-specific chromosomes. These survivors are enriched for global regulators of gene activity, with broad expression across tissues and developmental time. Homologs of survivors play key roles in human biology. Previously we reported that human orthologs of survivors in birds are enriched for genes implicated in human disorders attributed to haploinsufficiency (Bellott et al. 2017), and that several X-linked intellectual disability syndromes have been mapped to the X-linked homologs of mammalian X–Y pairs (Bellott et al. 2014). Here, we extend those findings to show that the human orthologs of survivors regulate a broad range of processes in early embryonic development across multiple organ systems. Because the human homologs of survivors are depleted for variation in copy number and coding sequence, we anticipate that even subtle modulation of the transcription, splicing, translation, and stability of survivors will have disproportionately large effects on development and disease.
Methods
Functional annotation
We mapped published functional annotations onto our set of ancestral genes and their human orthologs. For expression breadth, we normalized the expression of each gene to the highest expression in any tissue and took the average expression across all tissues. We used UniProt annotations to identify caenophidian Z–W pair genes involved in regulatory processes.
OMIM
We downloaded the full text of OMIM (McKusick-Nathans Institute of Genetic Medicine 2019) and searched entries of the human orthologs of ancestral caenophidian Z-linked genes for “haploinsufficient” or “haploinsufficiency,” limiting our search to phenotypes with a known molecular basis. We examined each of the resulting entries to verify that there was evidence that the phenotype was caused by haploinsufficiency.
Gene Ontology
We used the PANTHER statistical overrepresentation test (Mi et al. 2019a,b) using Fisher's exact test with false discovery rate correction to identify functional annotations that were enriched among the human orthologs of surviving Z–W and X–Y pair genes, relative to the reference list of orthologs of other ancestral Z and X genes. We selected the orthologs of ancestral Z and X genes as a reference list, instead of all human genes, to control for any functional coherence among ancestral genes that predated the start of genetic decay on W or Y Chromosomes.
Logistic regression
Logistic regression models of survival were constructed using the function “glm” in the stats package of R 3.6.1 (R Core Team 2020), with the option family = binomial. For each species, we used kallisto and sleuth to estimate TPM values across male or female tissues. We calculated the tissue specificity index as the fraction of TPMs originating from testis or ovary across a panel of male or female tissues, respectively. We calculated expression breadth as previously described (Bellott et al. 2014).
Reconstructing evolutionary strata
We traced the age and extent of evolutionary strata by testing which phylogenetic trees were most consistent with multiple sequence alignments of orthologous coding sequences from informative surviving ancestral genes on both the W (or Y) and the Z (or X) Chromosome (Supplemental Figs. S2–S4; Supplemental Tables S6, S9, S14, S15; Supplemental Data S1, S2). For additional details, see Supplemental Methods.
Survival fraction
To calculate longevity, we summed all branch lengths in the most parsimonious tree from each of the species where a gene is present to the last common ancestor before stratum formation. We divided the longevity for each gene by the maximum possible longevity for each stratum to generate the survival fraction (Supplemental Tables S16–S21). Survival fractions range from 0 (lost in all lineages) to 1 (retained in each lineage). In cases in which a gene was transposed from the X Chromosome to the Y Chromosome after the ancestral Y gene was lost, we treated the transposition as an additional stratum. For additional details and example calculations, see Supplemental Methods.
Principal components analysis
We modeled the survival fraction as a linear combination of the functional annotations for each gene using the prcomp function in the stats package of R 3.6.1 (R Core Team 2020), with options scale = TRUE, center = TRUE, na.action = na.aggregate to normalize the annotation data and replace missing values with the mean value for each annotation. We used the 14 quantitative annotation categories we identified as enriched among survivors on sex-specific chromosomes across amniotes: human haploinsufficiency score, human deletion intolerance score, human duplication intolerance score, gene-average PCT score, the number of human–chicken conserved miRNA sites, the number of human–lizard conserved miRNA sites, human expression breadth, chicken expression breadth, garter snake expression breadth, human morula mRNA expression levels, chicken blastocyst mRNA expression levels, human-mouse dN/dS, chicken-finch dN/dS, and lizard-snake dN/dS.
Statistics
Details of all statistical tests (type of test, test statistic, and P-value) used in this article are provided in Supplemental Table S24.
Competing interest statement
The authors declare no competing interests.
Supplementary Material
Acknowledgments
We thank R. Edwards, T. Amos, J. Tang, B. Cawood, S. Rispin, D.E. Tuipulotu, and P. Waters for permission to analyze data from their unpublished assembly of the mainland tiger snake genome (accession number GCA_900518725.1); W.S. Sanders, M.A. Arick Jr., A. Thrash, D. Rhoads, J. Pummill, S. Beaupre, B. Nanduri, A. Perkins, D.G. Peterson, and R.D. Steele for permission to analyze data from their unpublished assembly of the timber rattlesnake genome (accession number GCA_001625485.1); Y. Liu, D. Hughes, H. Dinh, S. Dugan, S. Jhangiani, S. Lee, G. Okwuonu, J. Santibanez, D. Bandaranaike, H. Chao, M. Dahdouli, R. Gnanaolivu, S. Gross, W. Hale, M. Javaid, J. Jones, S. Matakis, M. Munidasa, A. Narasimhan, D. Ngo, L. Nguyen, E. Skinner, E. Ongeri, N. Osuji, R. Puazo, S. Qi, C. Qu, J. Quiroz, R. Raj, K. Rajbhandari, X. Yun, Y. Han, C. Kovar, J. Reid, H. Doddapaneni, T. Madsen, B. Ujvari, D. Muzny, K. Worley, and R. Gibbs for permission to analyze data from their unpublished assembly of the European adder genome (accession number GCA_000800605.1); A. Godfrey and S. Naqvi for assistance with the analysis of GTEx data and helpful discussions; and A. Godfrey, E. Jackson, J. Hughes, and H. Skaletsky for critical reading of the manuscript. This work was supported by the Whitehead Institute, the Howard Hughes Medical Institute, and generous gifts from Brit and Alexander d'Arbeloff and Arthur W. and Carol Tobin Brill.
Author contributions: D.W.B. and D.C.P. designed the study and wrote the paper. D.W.B. performed the analyses.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.268516.120.
References
- Agarwal V, Bell GW, Nam JW, Bartel DP. 2015. Predicting effective microRNA target sites in mammalian mRNAs. eLife 4: e05005 10.7554/eLife.05005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge K, George ID, Cole KK, Austin JR, Takahashi TN, Duan Y, Miles JH. 2011. Facial phenotypes in subgroups of prepubertal boys with autism spectrum disorders are correlated with clinical phenotypes. Mol Autism 2: 15 10.1186/2040-2392-2-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers KL, Davidson NM, Demiyah D, Roeszler KN, Grützner F, Sinclair AH, Oshlack A, Smith CA. 2013. RNA sequencing reveals sexually dimorphic gene expression before gonadal differentiation in chicken and allows comprehensive annotation of the W-chromosome. Genome Biol 14: R26 10.1186/gb-2013-14-3-r26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. 2009. MicroRNAs: target recognition and regulatory functions. Cell 136: 215–233. 10.1016/j.cell.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becak W, Becak ML, Nazareth HR. 1962. Karyotypic studies of two species of South American snakes (Boa constrictor amarali and Bothrops jararaca). Cytogenetics 1: 305–313. 10.1159/000129740 [DOI] [PubMed] [Google Scholar]
- Bellott DW, Skaletsky H, Pyntikova T, Mardis ER, Graves T, Kremitzki C, Brown LG, Rozen S, Warren WC, Wilson RK, et al. 2010. Convergent evolution of chicken Z and human X chromosomes by expansion and gene acquisition. Nature 466: 612–616. 10.1038/nature09172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellott DW, Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Cho TJ, Koutseva N, Zaghlul S, Graves T, Rock S, et al. 2014. Mammalian Y chromosomes retain widely expressed dosage-sensitive regulators. Nature 508: 494–499. 10.1038/nature13206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellott DW, Skaletsky H, Cho TJ, Brown L, Locke D, Chen N, Galkina S, Pyntikova T, Koutseva N, Graves T, et al. 2017. Avian W and mammalian Y chromosomes convergently retained dosage-sensitive regulators. Nat Genet 49: 387–394. 10.1038/ng.3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berecz H, Csábi G, Jeges S, Herold R, Simon M, Halmai T, Trixler D, Hajnal A, Tóth ÁL, Tényi T. 2017. Minor physical anomalies in bipolar I and bipolar II disorders—results with the Méhes Scale. Psychiatry Res 249: 120–124. 10.1016/j.psychres.2017.01.014 [DOI] [PubMed] [Google Scholar]
- Bloom SE. 1972. Chromosome abnormalities in chicken (Gallus domesticus) embryos: types, frequencies and phenotypic effects. Chromosoma 37: 309–326. 10.1007/BF00319873 [DOI] [PubMed] [Google Scholar]
- Bradnam KR, Fass JN, Alexandrov A, Baranay P, Bechner M, Birol I, Boisvert S, Chapman JA, Chapuis G, Chikhi R, et al. 2013. Assemblathon 2: evaluating de novo methods of genome assembly in three vertebrate species. Gigascience 2: 10 10.1186/2047-217X-2-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bull JJ. 1983. Evolution of sex determining mechanisms. Benjamin/Cummings, Advanced Book Program, Menlo Park, CA. [Google Scholar]
- Calo E, Gu B, Bowen ME, Aryan F, Zalc A, Liang J, Flynn RA, Swigut T, Chang HY, Attardi LD, et al. 2018. Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature 554: 112–117. 10.1038/nature25449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D. 2000. The degeneration of Y chromosomes. Philos Trans R Soc Lond B Biol Sci 355: 1563–1572. 10.1098/rstb.2000.0717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cockwell A, MacKenzie M, Youings S, Jacobs P. 1991. A cytogenetic and molecular study of a series of 45,X fetuses and their parents. J Med Genet 28: 151–155. 10.1136/jmg.28.3.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coe BP, Witherspoon K, Rosenfeld JA, van Bon BWM, Vulto-van Silfhout AT, Bosco P, Friend KL, Baker C, Buono S, Vissers LELM, et al. 2014. Refining analyses of copy number variation identifies specific genes associated with developmental delay. Nat Genet 46: 1063–1071. 10.1038/ng.3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connallon T, Clark AG. 2010. Sex linkage, sex-specific selection, and the role of recombination in the evolution of sexually dimorphic gene expression. Evolution 64: 3417–3442. 10.1111/j.1558-5646.2010.01136.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousineau I, Belmaaza A. 2007. BRCA1 haploinsufficiency, but not heterozygosity for a BRCA1-truncating mutation, deregulates homologous recombination. Cell Cycle 6: 962–971. 10.4161/cc.6.8.4105 [DOI] [PubMed] [Google Scholar]
- DeSanto C, D'Aco K, Araujo GC, Shannon N, Study D, Vernon H, Rahrig A, Monaghan KG, Niu Z, Vitazka P, et al. 2015. WAC loss-of-function mutations cause a recognisable syndrome characterised by dysmorphic features, developmental delay and hypotonia and recapitulate 10p11.23 microdeletion syndrome. J Med Genet 52: 754–761. 10.1136/jmedgenet-2015-103069 [DOI] [PubMed] [Google Scholar]
- Dubruc E, Putoux A, Labalme A, Rougeot C, Sanlaville D, Edery P. 2014. A new intellectual disability syndrome caused by CTNNB1 haploinsufficiency. Am J Med Genet A 164: 1571–1575. 10.1002/ajmg.a.36484 [DOI] [PubMed] [Google Scholar]
- Duret L, Mouchiroud D. 2000. Determinants of substitution rates in mammalian genes: expression pattern affects selection intensity but not mutation rate. Mol Biol Evol 17: 68–070. 10.1093/oxfordjournals.molbev.a026239 [DOI] [PubMed] [Google Scholar]
- Fechheimer NS. 1981. Origins of heteroploidy in chicken embryos. Poult Sci 60: 1365–1371. 10.3382/ps.0601365 [DOI] [PubMed] [Google Scholar]
- Fridolfsson AK, Cheng H, Copeland NG, Jenkins NA, Liu HC, Raudsepp T, Woodage T, Chowdhary B, Halverson J, Ellegren H. 1998. Evolution of the avian sex chromosomes from an ancestral pair of autosomes. Pro Natl Acad Sci 95: 8147–8152. 10.1073/pnas.95.14.8147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KKH, Burge CB, Bartel DP. 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105. 10.1101/gr.082701.108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble T, Castoe TA, Nielsen SV, Banks JL, Card DC, Schield DR, Schuett GW, Booth W. 2017. The discovery of XY sex chromosomes in a boa and python. Curr Biol 27: 2148–2153.e4. 10.1016/j.cub.2017.06.010 [DOI] [PubMed] [Google Scholar]
- Ge X, Gong H, Dumas K, Litwin J, Phillips JJ, Waisfisz Q, Weiss MM, Hendriks Y, Stuurman KE, Nelson SF, et al. 2016. Missense-depleted regions in population exomes implicate ras superfamily nucleotide-binding protein alteration in patients with brain malformation. NPJ Genom Med 1: 16036 10.1038/npjgenmed.2016.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson WT, Hood RL, Zhan SH, Bulman DE, Fejes AP, Moore R, Mungall AJ, Eydoux P, Babul-Hirji R, An J, et al. 2012. Mutations in EZH2 cause Weaver syndrome. Am J Hum Genet 90: 110–118. 10.1016/j.ajhg.2011.11.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium. 2017. Genetic effects on gene expression across human tissues. Nature 550: 204–213. 10.1038/nature24277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gubbay J, Collignon J, Koopman P, Capel B, Economou A, Münsterberg A, Vivian N, Goodfellow P, Lovell-Badge R. 1990. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature 346: 245–250. 10.1038/346245a0 [DOI] [PubMed] [Google Scholar]
- Handley LJL, Ceplitis H, Ellegren H. 2004. Evolutionary strata on the chicken Z chromosome: implications for sex chromosome evolution. Genetics 167: 367–376. 10.1534/genetics.167.1.367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassold T, Benham F, Leppert M. 1988. Cytogenetic and molecular analysis of sex-chromosome monosomy. Am J Hum Genet 42: 534–541. [PMC free article] [PubMed] [Google Scholar]
- Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et al. 2015. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350: 1262–1266. 10.1126/science.aac9396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook EB, Warburton D. 1983. The distribution of chromosomal genotypes associated with Turner's syndrome: livebirth prevalence rates and evidence for diminished fetal mortality and severity in genotypes associated with structural X abnormalities or mosaicism. Hum Genet 64: 24–27. 10.1007/BF00289473 [DOI] [PubMed] [Google Scholar]
- Huang N, Lee I, Marcotte EM, Hurles ME. 2010. Characterising and predicting haploinsufficiency in the human genome. PLoS Genet 6: e1001154 10.1371/journal.pgen.1001154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Pyntikova T, Graves TA, van Daalen SKM, Minx PJ, Fulton RS, McGrath SD, Locke DP, Friedman C, et al. 2010. Chimpanzee and human Y chromosomes are remarkably divergent in structure and gene content. Nature 463: 536–539. 10.1038/nature08700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Brown LG, Pyntikova T, Graves T, Fulton RS, Dugan S, Ding Y, Buhay CJ, Kremitzki C, et al. 2012. Strict evolutionary conservation followed rapid gene loss on human and rhesus Y chromosomes. Nature 483: 82–86. 10.1038/nature10843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JF, Skaletsky H, Koutseva N, Pyntikova T, Page DC. 2015. Sex chromosome-to-autosome transposition events counter Y-chromosome gene loss in mammals. Genome Biol 16: 104 10.1186/s13059-015-0667-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ, Ronemus M, Krumm N, Levy D, Stessman HA, Witherspoon KT, Vives L, Patterson KE, et al. 2014. The contribution of de novo coding mutations to autism spectrum disorder. Nature 515: 216–221. 10.1038/nature13908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janečka JE, Davis BW, Ghosh S, Paria N, Das PJ, Orlando L, Schubert M, Nielsen MK, Stout TAE, Brashear W, et al. 2018. Horse Y chromosome assembly displays unique evolutionary features and putative stallion fertility genes. Nat Commun 9: 2945 10.1038/s41467-018-05290-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser VB, Bachtrog D. 2014. De novo transcriptome assembly reveals sex-specific selection acting on evolving neo-sex chromosomes in Drosophila miranda. BMC Genomics 15: 241 10.1186/1471-2164-15-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser VB, Zhou Q, Bachtrog D. 2011. Nonrandom gene loss from the Drosophila miranda neo-Y chromosome. Genome Biol Evol 3: 1329–1337. 10.1093/gbe/evr103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobel HR. 1962. Heterochromosomen bei Vipera berus L. (Viperidae, Serpentes). Experientia 18: 173–174. 10.1007/BF02151711 [DOI] [PubMed] [Google Scholar]
- Koolen DA, Pfundt R, Linda K, Beunders G, Veenstra-Knol HE, Conta JH, Fortuna AM, Gillessen-Kaesbach G, Dugan S, Halbach S, et al. 2016. The Koolen-de Vries syndrome: a phenotypic comparison of patients with a 17q21.31 microdeletion versus a KANSL1 sequence variant. Eur J Hum Genet 24: 652–659. 10.1038/ejhg.2015.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Suleski M, Hedges SB. 2017. Timetree: a resource for timelines, timetrees, and divergence times. Mol Biol Evol 34: 1812–1819. 10.1093/molbev/msx116 [DOI] [PubMed] [Google Scholar]
- Lahn BT, Page DC. 1997. Functional coherence of the human Y chromosome. Science 278: 675–680. 10.1126/science.278.5338.675 [DOI] [PubMed] [Google Scholar]
- Lahn BT, Page DC. 1999. Four evolutionary strata on the human X chromosome. Science 286: 964–967. 10.1126/science.286.5441.964 [DOI] [PubMed] [Google Scholar]
- Li G, Davis BW, Raudsepp T, Pearks Wilkerson AJ, Mason VC, Ferguson-Smith M, O'Brien PC, Waters PD, Murphy WJ. 2013. Comparative analysis of mammalian Y chromosomes illuminates ancestral structure and lineage-specific evolution. Genome Res 23: 1486–1495. 10.1101/gr.154286.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Endo Y, Iwaki D, Nakata M, Matsushita M, Wada I, Inoue K, Munakata M, Fujita T. 2005. Human M-ficolin is a secretory protein that activates the lectin complement pathway. J Immunol 175: 3150–3156. 10.4049/jimmunol.175.5.3150 [DOI] [PubMed] [Google Scholar]
- Mank JE, Hultin-Rosenberg L, Zwahlen M, Ellegren H. 2008. Pleiotropic constraint hampers the resolution of sexual antagonism in vertebrate gene expression. Am Nat 171: 35–43. 10.1086/523954 [DOI] [PubMed] [Google Scholar]
- Matsubara K, Tarui H, Toriba M, Yamada K, Nishida-Umehara C, Agata K, Matsuda Y. 2006. Evidence for different origin of sex chromosomes in snakes, birds, and mammals and step-wise differentiation of snake sex chromosomes. Proc Natl Acad Sci 103: 18190–18195. 10.1073/pnas.0605274103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara K, Nishida C, Matsuda Y, Kumazawa Y. 2016. Sex chromosome evolution in snakes inferred from divergence patterns of two gametologous genes and chromosome distribution of sex chromosome-linked repetitive sequences. Zoological Lett 2: 19 10.1186/s40851-016-0056-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy FM, Pendarvis K, Cooksey AM, Gresham CR, Bomhoff M, Davey S, Lyons E, Sonstegard TS, Bridges SM, Burgess SC. 2019. Chickspress: a resource for chicken gene expression. Database 2019: baz058 10.1093/database/baz058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKusick-Nathans Institute of Genetic Medicine. 2019. Online mendelian inheritance in Man, OMIM. Johns Hopkins University, Baltimore. [Google Scholar]
- Meisel RP. 2011. Towards a more nuanced understanding of the relationship between sex-biased gene expression and rates of protein-coding sequence evolution. Mol Biol Evol 28: 1893–1900. 10.1093/molbev/msr010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB. 2012. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338: 1593–1599. 10.1126/science.1228186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, Thomas PD. 2019a. PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47: D419–D426. 10.1093/nar/gky1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Huang X, Ebert D, Mills C, Guo X, Thomas PD. 2019b. Protocol update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat Protoc 14: 703–721. 10.1038/s41596-019-0128-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirabello L, Khincha PP, Ellis SR, Giri N, Brodie S, Chandrasekharappa SC, Donovan FX, Zhou W, Hicks BD, Boland JF, et al. 2017. Novel and known ribosomal causes of Diamond-Blackfan anaemia identified through comprehensive genomic characterisation. J Med Genet 54: 417–425. 10.1136/jmedgenet-2016-104346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munger SC, Capel B. 2012. Sex and the circuitry: progress toward a systems-level understanding of vertebrate sex determination. Wiley Interdiscip Rev Syst Biol Med 4: 401–412. 10.1002/wsbm.1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munye MM, Diaz-Font A, Ocaka L, Henriksen ML, Lees M, Brady A, Jenkins D, Morton J, Hansen SW, Bacchelli C, et al. 2017. COLEC10 is mutated in 3MC patients and regulates early craniofacial development. PLoS Genet 13: e1006679 10.1371/journal.pgen.1006679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani Y, Takeda H, Kohara Y, Morishita S. 2007. Reconstruction of the vertebrate ancestral genome reveals dynamic genome reorganization in early vertebrates. Genome Res 17: 1254–1265. 10.1101/gr.6316407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanda I, Shan Z, Schartl M, Burt DW, Koehler M, Nothwang H, Grützner F, Paton IR, Windsor D, Dunn I, et al. 1999. 300 million years of conserved synteny between chicken Z and human chromosome 9. Nat Genet 21: 258–259. 10.1038/6769 [DOI] [PubMed] [Google Scholar]
- Naqvi S, Bellott DW, Lin KS, Page DC. 2018. Conserved microRNA targeting reveals preexisting gene dosage sensitivities that shaped amniote sex chromosome evolution. Genome Res 28: 474–483. 10.1101/gr.230433.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi S, Godfrey AK, Hughes JF, Goodheart ML, Mitchell RN, Page DC. 2019. Conservation, acquisition, and functional impact of sex-biased gene expression in mammals. Science 365: eaaw7317 10.1126/science.aaw7317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M. 1969. Linkage modifications and sex difference in recombination. Genetics 63: 681–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Meally D, Ezaz T, Georges A, Sarre SD, Graves JAM. 2012. Are some chromosomes particularly good at sex? Insights from amniotes. Chromosome Res 20: 7–19. 10.1007/s10577-011-9266-8 [DOI] [PubMed] [Google Scholar]
- Papp B, Pál C, Hurst LD. 2003. Dosage sensitivity and the evolution of gene families in yeast. Nature 424: 194–197. 10.1038/nature01771 [DOI] [PubMed] [Google Scholar]
- Perry BW, Card DC, McGlothlin JW, Pasquesi GIM, Adams RH, Schield DR, Hales NR, Corbin AB, Demuth JP, Hoffmann FG, et al. 2018. Molecular adaptations for sensing and securing prey and insight into amniote genome diversity from the garter snake genome. Genome Biol Evol 10: 2110–2129. 10.1093/gbe/evy157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pessia E, Makino T, Bailly-Bechet M, McLysaght A, Marais GAB. 2012. Mammalian X chromosome inactivation evolved as a dosage-compensation mechanism for dosage-sensitive genes on the X chromosome. Proc Natl Acad Sci 109: 5346–5351. 10.1073/pnas.1116763109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petropoulos S, Edsgärd D, Reinius B, Deng Q, Panula SP, Codeluppi S, Plaza Reyes A, Linnarsson S, Sandberg R, Lanner F. 2016. Single-cell RNA-seq reveals lineage and X chromosome dynamics in human preimplantation embryos. Cell 165: 1012–1026. 10.1016/j.cell.2016.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2020. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Redford-Badwal DA, Stover ML, Valli M, McKinstry MB, Rowe DW. 1996. Nuclear retention of COL1A1 messenger RNA identifies null alleles causing mild osteogenesis imperfecta. J Clin Invest 97: 1035–1040. 10.1172/JCI118495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reijnders MRF, Miller KA, Alvi M, Goos JAC, Lees MM, de Burca A, Henderson A, Kraus A, Mikat B, de Vries BBA, et al. 2018. De novo and inherited loss-of-function variants in TLK2: clinical and genotype-phenotype evaluation of a distinct neurodevelopmental disorder. Am J Hum Genet 102: 1195–1203. 10.1016/j.ajhg.2018.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice WR. 1987. The accumulation of sexually antagonistic genes as a selective agent promoting the evolution of reduced recombination between primitive sex chromosomes. Evolution 41: 911–914. 10.1111/j.1558-5646.1987.tb05864.x [DOI] [PubMed] [Google Scholar]
- Rooryck C, Diaz-Font A, Osborn DPS, Chabchoub E, Hernandez-Hernandez V, Shamseldin H, Kenny J, Waters A, Jenkins D, Kaissi AA, et al. 2011. Mutations in lectin complement pathway genes COLEC11 and MASP1 cause 3MC syndrome. Nat Genet 43: 197–203. 10.1038/ng.757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross MT, Grafham DV, Coffey AJ, Scherer S, McLay K, Muzny D, Platzer M, Howell GR, Burrows C, Bird CP, et al. 2005. The DNA sequence of the human X chromosome. Nature 434: 325–337. 10.1038/nature03440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovatsos M, Vukić J, Lymberakis P, Kratochvíl L. 2015. Evolutionary stability of sex chromosomes in snakes. Proc R Soc B 282: 20151992 10.1098/rspb.2015.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rovatsos M, Augstenová B, Altmanová M, Sloboda M, Kodym P, Kratochvíl L. 2018. Triploid colubrid snake provides insight into the mechanism of sex determination in advanced snakes. Sex Dev 12: 251–255. 10.1159/000490124 [DOI] [PubMed] [Google Scholar]
- Ruderfer DM, Hamamsy T, Lek M, Karczewski KJ, Kavanagh D, Samocha KE, Exome Aggregation Consortium, Daly MJ, MacArthur DG, Fromer M, et al. 2016. Patterns of genic intolerance of rare copy number variation in 59,898 human exomes. Nat Genet 48: 1107–1111. 10.1038/ng.3638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanna-Cherchi S, Westland R, Ghiggeri GM, Gharavi AG. 2018. Genetic basis of human congenital anomalies of the kidney and urinary tract. J Clin Invest 128: 4–15. 10.1172/JCI95300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schield DR, Card DC, Hales NR, Perry BW, Pasquesi GM, Blackmon H, Adams RH, Corbin AB, Smith CF, Ramesh B, et al. 2019. The origins and evolution of chromosomes, dosage compensation, and mechanisms underlying venom regulation in snakes. Genome Res 29: 590–601. 10.1101/gr.240952.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair AH, Berta P, Palmer MS, Hawkins JR, Griffiths BL, Smith MJ, Foster JW, Frischauf AM, Lovell-Badge R, Goodfellow PN. 1990. A gene from the human sex-determining region encodes a protein with homology to a conserved DNA-binding motif. Nature 346: 240–244. 10.1038/346240a0 [DOI] [PubMed] [Google Scholar]
- Skaletsky H, Kuroda-Kawaguchi T, Minx PJ, Cordum HS, Hillier L, Brown LG, Repping S, Pyntikova T, Ali J, Bieri T, et al. 2003. The male-specific region of the human Y chromosome is a mosaic of discrete sequence classes. Nature 423: 825–837. 10.1038/nature01722 [DOI] [PubMed] [Google Scholar]
- Skinner BM, Sargent CA, Churcher C, Hunt T, Herrero J, Loveland JE, Dunn M, Louzada S, Fu B, Chow W, et al. 2016. The pig X and Y chromosomes: structure, sequence, and evolution. Genome Res 26: 130–139. 10.1101/gr.188839.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DW, Bostian KE. 1964. Congenital anomalies associated with idiopathic mental retardation. J Pediatr 65: 189–196. 10.1016/S0022-3476(64)80519-9 [DOI] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. 2007. The classical complement cascade mediates CNS synapse elimination. Cell 131: 1164–1178. 10.1016/j.cell.2007.10.036 [DOI] [PubMed] [Google Scholar]
- Trainor PA. 2010. Craniofacial birth defects: the role of neural crest cells in the etiology and pathogenesis of Treacher Collins syndrome and the potential for prevention. Am J Med Genet A 152A: 2984–2994. 10.1002/ajmg.a.33454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The UniProt Consortium. 2019. Uniprot: a worldwide hub of protein knowledge. Nucleic Acids Res 47: D506–D515. 10.1093/nar/gky1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Lopez GA, Cerrizuela S, Tribulo C, Aybar MJ. 2018. Neurocristopathies: new insights 150 years after the neural crest discovery. Dev Biol 444: S110–S143. 10.1016/j.ydbio.2018.05.013 [DOI] [PubMed] [Google Scholar]
- Vicoso B, Emerson JJ, Zektser Y, Mahajan S, Bachtrog D. 2013a. Comparative sex chromosome genomics in snakes: differentiation, evolutionary strata, and lack of global dosage compensation. PLoS Biol 11: e1001643 10.1371/journal.pbio.1001643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Corominas R, Lin GN. 2019. De novo mutations from whole exome sequencing in neurodevelopmental and psychiatric disorders: from discovery to application. Front Genet 10: 258 10.3389/fgene.2019.00258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg SM, Jenkins EA, Marazita ML, Maher BS. 2007. Minor physical anomalies in schizophrenia: a meta-analysis. Schizophr Res 89: 72–85. 10.1016/j.schres.2006.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MA, Kitano J, Peichel CL. 2015. Purifying selection maintains dosage-sensitive genes during degeneration of the threespine stickleback Y chromosome. Mol Biol Evol 32: 1981–1995. 10.1093/molbev/msv078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willing MC, Deschenes SP, Scott DA, Byers PH, Slayton RL, Pitts SH, Arikat H, Roberts EJ. 1994. Osteogenesis imperfecta type I: molecular heterogeneity for COL1A1 null alleles of type I collagen. Am J Hum Genet 55: 638–647. [PMC free article] [PubMed] [Google Scholar]
- Wu D, Mandal S, Choi A, Anderson A, Prochazkova M, Perry H, Gil-Da-Silva-Lopes VL, Lao R, Wan E, Tang PLF, et al. 2015. DLX4 is associated with orofacial clefting and abnormal jaw development. Hum Mol Genet 24: 4340–4352. 10.1093/hmg/ddv167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Auer G, Peona V, Suh A, Deng Y, Feng S, Zhang G, Blom MPK, Christidis L, Prost S, et al. 2019a. Dynamic evolutionary history and gene content of sex chromosomes across diverse songbirds. Nat Ecol Evol 3: 834–844. 10.1038/s41559-019-0850-1 [DOI] [PubMed] [Google Scholar]
- Yang J, Su AI, Li WH. 2005. Gene expression evolves faster in narrowly than in broadly expressed mammalian genes. Mol Biol Evol 22: 2113–2118. 10.1093/molbev/msi206 [DOI] [PubMed] [Google Scholar]
- Yates AD, Achuthan P, Akanni W, Allen J, Allen J, Alvarez-Jarreta J, Amode MR, Armean IM, Azov AG, Bennett R, et al. 2020. Ensembl 2020. Nucleic Acids Res 48: D682–D688 10.1093/nar/gkz966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W, Wang ZJ, Li QY, Lian JM, Zhou Y, Lu BZ, Jin LJ, Qiu PX, Zhang P, Zhu WB, et al. 2016. Evolutionary trajectories of snake genes and genomes revealed by comparative analyses of five-pacer viper. Nat Commun 7: 13107 10.1038/ncomms13107 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.