

Abstract
The predominant methodology for DNA methylation analysis relies on the chemical deamination by sodium bisulfite of unmodified cytosine to uracil to permit the differential readout of methylated cytosines. Bisulfite treatment damages the DNA, leading to fragmentation and loss of long-range methylation information. To overcome this limitation of bisulfite-treated DNA, we applied a new enzymatic deamination approach, termed enzymatic methyl-seq (EM-seq), to long-range sequencing technologies. Our methodology, named long-read enzymatic modification sequencing (LR-EM-seq), preserves the integrity of DNA, allowing long-range methylation profiling of 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC) over multikilobase length of genomic DNA. When applied to known differentially methylated regions (DMRs), LR-EM-seq achieves phasing of >5 kb, resulting in broader and better defined DMRs compared with that previously reported. This result showed the importance of phasing methylation for biologically relevant questions and the applicability of LR-EM-seq for long-range epigenetic analysis at single-molecule and single-nucleotide resolution.
Long-read technologies have been a breakthrough in high-throughput sequencing for their abilities to phase and resolve variations and repeats over large segments of the human genome (Jain et al. 2018; Pollard et al. 2018). Phasing of methylation at single-molecule resolution represents a significant advance in addressing the mechanisms and relevance of epigenetic modifications, particularly in repeats, imprinted genes, and distant regulatory regions.
Recently, a few studies have successfully identified cytosine methylation in CpG context with increased accuracy using the ability of the Nanopore sequencer to directly “read” the modification (Flusberg et al. 2010; Rand et al. 2017; Simpson et al. 2017). By using this method, methylation can be examined over large fragments of genomic DNA. Nonetheless, because the methylation status is not preserved during amplification, only native nonamplified DNA can be used. Although enrichment strategies using Cas9 have been applied (Gilpatrick et al. 2020) for targeting specific regions in the genome (Giesselmann et al. 2019; Hafford-Tear et al. 2019), the required starting material is very high and the enrichment is relatively low.
Although a number of methodologies have been developed to study cytosine modification (Kurdyukov and Bullock 2016; Liu et al. 2019), bisulfite sequencing is still the predominant method used for methylome analysis. Bisulfite sequencing is based on the differential reactivity of cytosine (C) and 5-methylcytosine (5mC) with sodium bisulfite. Unmodified cytosines are deaminated to uracils (U's) and will be read as thymine (T) during sequencing, whereas 5mC is unchanged and will be read as “C” (Frommer et al. 1992). Nonetheless, all bisulfite-based methods introduce DNA strand breaks and result in highly fragmented DNA. This random fragmentation of the deaminated DNA remains the major roadblock to studying epigenetic modifications over large genomic regions using bisulfite sequencing. Indeed, the largest amplicons obtained and sequenced from bisulfite-deaminated DNA does not exceed 1500 bp in length (Yang et al. 2015).
Recognizing the substantial limitation of bisulfite sequencing in preserving DNA integrity, two bisulfite-free, enzyme-based methods have been recently developed. The first method, TAPS-seq, uses TET1 dioxygenase to oxidize both 5mC and 5hmC to 5-carboxylcytosine (5caC), and pyridine borane reduces 5caC to dihydrouracil (DHU), which is read as thymine (Liu et al. 2019). The modification of this method, lrTAPS-seq, was adapted for long-read sequencing (Liu et al. 2020) and achieved targeted base-resolution sequencing of several-kilobase templated DNA (for comparison between bisulfite-free methylation detection methods, see Supplemental Table S1). Recently, a second method used APOBEC3A cytidine deaminase to achieve base-resolution sequencing of 5-hydroxymethylcytosine (5hmC) while avoiding most of the DNA damage (Schutsky et al. 2018). APOBEC3A is a member of the activation induced cytidine deaminase/apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (AID/APOBEC) family of deaminases and has been shown to be critical to immunoglobulin diversification and antiretroviral defense (Salter et al. 2016). APOBEC3A preferentially deaminates cytosine and 5mC, resulting in the formation of uracil and thymine, respectively. Because unmodified cytosine and 5mC are both substrates for APOBEC3A (Schutsky et al. 2018), the identification of 5mC using APOBEC3A alone is currently not possible.
A commercial technology for the determination of 5mC called EM-seq has recently become available (Methods). This technology relies on the enzymatic treatment of DNA and thus eliminates the need for bisulfite conversion entirely. In this work, we show that such enzymatic treatment preserves the integrity of the DNA with no detectable evidence of fragmentation or damage. We therefore adapted EM-seq to long-read sequencing of amplicon using both Pacific Biosciences (PacBio) and Nanopore sequencing technologies and extended the technology to both 5mC and 5hmC detection. The resulting method, termed herein long-read-EM-seq (LR-EM-seq), use the selective enzymatic protection of 5mC and/or 5hmC before enzymatic deamination by APOBEC3A and large-fragment sequencing sample preparation to accurately profile both 5mC and 5hmC at base resolution. The preservation of DNA integrity allows the locus-specific amplification of several kilobases of genomic DNA and the long-range phasing at molecular resolution of 5mC and 5hmC. Applied to known differentially methylated regions (DMRs) in the mouse genome, LR-EM-seq accurately identifies 5mC and 5hmC in >5-kb-long amplicons, allowing the assignment of cytosine modifications to specific alleles. Compared with the existing bisulfite-free methods discussed above, LR-EM-seq provides direct and accurate measurements for both 5mC and 5hmC in an easy to use protocol.
Results
Accurate identification of 5mC and 5hmC modification
To enzymatically discriminate epigenetically important cytosine modifications, we use the overall strategy depicted in Figure 1A. The basic principle of the method consists of selectively modifying cytosine modifications in order to protect them from deamination by APOBEC3A. To protect 5hmC from deamination, 5hmC is glucosylated with DNA beta-glucosyltransferase (BGT) before deamination.
Figure 1.

5mC and 5hmC detection by enzymatic deamination method. (A) Principle of the methodology: genomic DNA can either be treated with TET2 and BGT (left) to protect both 5mC and 5hmC or with BGT alone (right) to protect 5hmC. Subsequent deamination by APOBEC3A followed by PCR amplification allows the distinction between the unprotected substrate (read as T) from the protected cytosine derivatives (read as C). The TET2 and BGT treatment results in the distinction of 5mC and 5hmC from C, whereas BGT treatment results in the distinction of 5hmC from C and 5mC. (B) Deaminated cytosines from unmethylated lambda genome display no observable sequence preference by APOBEC(5mC) deamination method. (C) False-positive methylation calling rate (nonconversion error rate) of each cytosine dinucleotides sequence context (CpA [CA], CpC [CC], CpG [CG], and CpT [CT]) estimated from the unmethylated lambda genome for the enzymatic deamination method (APOBEC(5mC), two WGBS performed in this study, i.e., BS kit 1 and BS kit 2, and six published WGBS experiments sampled from the ENCODE Project) (Supplemental Table S3). (D) Deamination of 5mC in the fully methylated XP12 genome results in no observable sequence preference by APOBEC(5hmC) enzymatic deamination method. (E) Distribution patterns of 5mCpG (blue) and 5hmCpG (red: 50 ng library; pink: 1 ng library) at various protein/DNA interaction sites. The absolute (smooth lines) and normalized (dotted lines) 5hmC and 5mC levels in the CpG context are depicted around TET1, RNA polymerase II, and CTCF binding sites, as well as at active transcription chromatin mark (H3K4me3), repressive chromatin mark (H3K27me3), active enhancer mark (H3K27ac), and general enhancer (H3K4me1 in the absence of H3K4me3) regions. Unbound sites that are randomly sampled from the reference genome server as a control. (F) Pearson's correlation between 5hmC measured using sequencing of enzymatically deaminated DNA (x-axis) versus LC-MS (y-axis) for various genomic DNA. There are two technical replicates of the APOBEC(5hmC) sequencing method for each sample. 5hmC levels are presented as 1000 percentage, and both axes use the log scale.
To discriminate 5mC from C, the 5mC needs to be protected from deamination before APOBEC3A treatment. 5mC is converted to either glucosylated hydroxymethylcytosine (5gmC) or 5caC using a combination of 5mC dioxygenase TET2 and BGT (EM-seq) (Ito et al. 2011). All these oxidative products have been shown to be protected from deamination by APOBEC3A, including 5hmC after glucosylation by BGT (EM-seq) (Schutsky et al. 2018).
We validated the APOBEC(5mC) strategy illustrated in Figure 1A using mouse embryonic stem (mES) cell (E14) genomic DNA spiked with unmethylated lambda. We also performed whole-genome bisulfite sequencing (WGBS) on the same starting material using two widely used bisulfite conversion kits, BS kit 1 and BS kit 2, (Methods) for comparison. Two technical replicates were performed for all the three protocols (APOBEC(5mC), BS kit 1, and BS kit 2) to assess reproducibility of the methods. The conversion rates and methylation results were highly consistent between all the technical replicates (Supplemental Fig. S1A,B).
Analysis of unmethylated lambda control showed a near complete C-to-U conversion rate of 99.8% with no apparent sequence preference of the enzymatic deamination from both replicates (Fig. 1B,C; Supplemental Fig. S1A). The conversion rates of bisulfite-treated lambda DNA range from 98.2%–99.6% depending on the BS kit used, which is in line with the usual conversion rates reported in the literature for WGBS (Supplemental Table S2). We also observed a higher level of residual unconverted cytosines in CpA context in the WGBS libraries made from both bisulfite conversion kits (Fig. 1C), and this CpA bias is also observed in the published WGBS data sets (Fig. 1C; Supplemental Table S3). This bias leads to sixfold to 23-fold more false-positive methylated CpA from unmethylated lambda in WGBS compared with enzymatic conversion even after binomial correction (Supplemental Fig. S1C).
Identification of CpG methylation in the mouse E14 DNA reveals consistent results between enzymatic deamination and WGBS (Supplemental Text S1). In brief, CpG methylation calls between enzymatic conversion and the two WGBS are in ∼96% agreement (Supplemental Fig. S1D). Furthermore, CpG methylation levels revealed using the enzymatic conversion method are well correlated with repressive and active chromatin markers in the mouse stem cells, showing expected depletion in the active transcription regions (H3K4me3 and H3K27ac) and promoters (RNA polymerase II binding sites) (Fig. 1E; Supplemental Fig. S1F). Genome-wide investigation of the read distribution in the mouse genome reveals that the enzymatic deamination method produces more even sequencing coverage than the bisulfite conversion–based sequencing methods (Supplemental Text S1; Supplemental Fig. S2).
To show that this strategy also results in an accurate identification of 5hmC, we prepared enzymatic 5hmC libraries using 50 ng of mouse E14 genomic DNA spiked with unmethylated lambda, cytosine methylated XP12, and hydroxymethylated T4gt phage genomic DNAs. Lambda and XP12 control DNAs were used to measure the deamination rates of APOBEC3A on C and 5mC, respectively, and T4gt DNA was used to monitor 5hmC protection by BGT. By using these controls, we calculated the nonconversion rates to be 0.2% for unmodified cytosines and 2.5% for 5mC (Supplemental Table S4). The converted methylated cytosines in XP12 showed no sequence preference (Fig. 1D), showing the lack of context bias by APOBEC3A. These nonconversion rates are in line with those reported for TAB-seq and ACE-seq (Supplemental Table S4; Yu et al. 2012; Schutsky et al. 2018). We observed a 98.3% protection rate of 5hmC by BGT, which is higher than the previously published TAB-seq (75%–92%) (Supplemental Table S4; Yu et al. 2012). Thus, our method is expected to have fewer false-negative hydroxymethylation calls compared with the widely used TAB-seq method and is in line with the performance of ACE-seq (Schutsky et al. 2018). We also made enzymatic 5hmC libraries from 1 ng genomic DNA of mES cells and show similar hydroxymethylation results as the 50-ng libraries (Supplemental Table S6). For both the 1-ng and 50-ng libraries, the identification of 5hmC relative to available mES ChIP-seq data sets reveals, as expected, deposition of 5hmC at TET1 binding sites and other epigenomic features, such as enhancers, active histone mark H3K27ac, and CTCF binding sites (Fig. 1E; Supplemental Text S2).
To measure the range of sensitivity, we made 5hmC libraries of enzymatically treated DNA derived from five mouse cell types/tissues that have been reported to have a wide range of global 5hmC levels (Globisch et al. 2010). We also included DNMT triple-knockout (TKO) J1 embryonic stem (ES) cells as a negative control (Supplemental Table S5). The average CpG hydroxymethylation level measured after sequencing correlated well with liquid chromatography with tandem mass spectrometry (LC-MS/MS) data (r = 0.99) (Fig. 1F). The correlation was linear across a wide range of global 5hmC levels, showing that the sensitivity is accurate across levels of 5hmC typically found in mammals (Globisch et al. 2010). TKO control showed 0.1%∼0.2% of false-positive 5hmC calls, indicating an exceptional low nonconversion error rate.
Conducting both enzymatic 5mC and 5hmC sequencing in parallel enabled for the first time the simultaneous investigation of 5mC and 5hmC using the same baseline enzymatic reaction (Fig. 1E; Supplemental Text S2; Supplemental Fig. S1F).
Enzymatic deamination preserves DNA integrity
Having shown the specificity and sensitivity of the enzymatic method toward the identification of 5mC and 5hmC, we investigated the ability of the method in preserving the integrity of the DNA, particularly for long genomic DNA fragments.
To directly assess the DNA integrity after bisulfite and enzymatic treatments, we separately addressed the loss of amplifiable material for small and large target sizes. For small target sizes (<1 kb), we used real-time PCR-based assay to quantify the amount of intact material. At all measured target sizes, bisulfite-treated DNA samples resulted in a lower amplification level compared with enzymatically deaminated DNA (Fig. 2A). At the largest measured target size of 809 bp, the relative amount of damage-free amplifiable template in the bisulfite-treated samples was 94% reduced compared with enzymatic deamination (Fig. 2A), which is consistent with previously reported degradation rates of bisulfite method (Grunau et al. 2001).
Figure 2.

Enzymatic deamination preserves the integrity of the DNA. (A) qPCR results show the quantities of undamaged amplifiable DNA templates of different sizes after the enzymatic deamination (green) and bisulfite treatments (orange and blue). All quantifications are normalized to the values obtained for the enzymatic deamination experiments. (B) Agilent 2100 Bioanalyzer trace on RNA 6000 pico chip comparing equal amounts of mouse E14 genomic DNA sheared to an average of 15 kb and treated with sodium bisulfite (green), APOBEC(5hmC) (red), or APOBEC(5mC) (blue) over the control ssDNA (cyan). Bisulfite treatment fragmented the DNA to an average of 800 bp, whereas enzymatically treated DNA shows no notable size differences compared with control DNA. (C) Agarose gel images of end-point PCR of six amplicons ranging from 388–4229 bp illustrating upper amplicon size limit for sodium-bisulfite-, APOBEC(5mC)-, or APOBEC(5hmC)-treated E14 genomic DNA. (D) The 731-bp amplicons from the E14 genomic DNA shown in C were cloned and sequenced, and the methylation status was determined by bisulfite treatment (left), the enzymatic deamination method for 5mC (center), and the enzymatic deamination method for 5hmC (right) (Supplemental Data S1). Open and closed circles indicate unmethylated and methylated CpG sites, respectively.
To assess the loss of material for large DNA fragments, E14 genomic DNA was fragmented to an average size of 15 kb, and fragment size distribution was profiled before and after enzymatic or bisulfite treatment. The average fragment size after bisulfite treatment dropped substantially from 15 kb to only 0.8 kb (Fig. 2B). Sharply contrasting to bisulfite conversion, enzymatic treatment of the same starting amount of DNA conserved the original 15-kb average size profile observed in the control DNA (Fig. 2B). This result shows that the enzymatic deamination method does not introduce strand breaks even in the case of large DNA fragments.
To assess the ability to amplify the DNA material described above, we designed six pairs of primers with a range of predicted amplicon sizes ranging from 388–4226 bp. In line with the DNA integrity assessment data, amplification products from bisulfite-treated DNA were only detected up to 731 bp. In contrast, all amplicon sizes were amplifiable after enzymatic deamination for both 5mC and 5hmC detection (Fig. 2C). Sanger sequencing of the 731-bp amplicons showed a nearly identical methylation profile for both enzymatic and chemical deamination methods (Fig. 2D), confirming that the enzymatic deamination method can provide the same accuracy of methylation detection as bisulfite treatment without damaging the DNA.
Lastly, the enzymatic method does not introduce additional PCR bias (Supplemental Text S3; Supplemental Fig. S3; Supplemental Methods). In comparison, bisulfite-treated DNA clearly shows overestimation of methylation after library amplification (Supplemental Fig. S3).
5mC and 5hmC phasing using long-read sequencing
Preserving the integrity of genomic DNA after enzymatic deamination offers the unique opportunity to study long-range epigenetic marks at single-base and single-molecule resolution beyond the reported 1.5-kb region achieved using single-molecule real-time (SMRT)–BS (Yang et al. 2015). As a proof of principle, we applied LR-EM-seq to a 5378-bp region of the mouse genome using DNA derived from ES cells. Two control DNA consisting of CpG methylated pUC19 and of unmethylated lambda DNA were added to the mouse genomic DNA before any enzymatic reactions. For 5hmC detection, an additional control consisting of T4gt genomic DNA was included to the spike-ins in order to monitor the 5hmC protection rate. Following enzymatic treatment, a 5378-bp mouse amplicon, a 3233-bp lambda amplicon, a 1774-bp CpG methylated pUC19 amplicon, and a 5349-bp T4gt amplicon (for 5hmC detection) were obtained and sequenced using all three major sequencing platforms: Oxford Nanopore, PacBio, and Illumina. In the case of Illumina sequencing, the amplicons were subsequently fragmented to a mean of 500 bp for compatibility with short-read sequencing.
Using Illumina data for 5mC detection, lambda amplicon shows nonconversion error rates of 0.1%, whereas the CpG methylated pUC19 amplicon shows 97.4% 5mC protection rate by the TET2/BGT enzymes. For 5hmC detection, the nonconversion error rate of cytosine is 0.1%, the nonconversion error rate of 5mC is 0.6%, and the protection rate of 5hmC measured using the T4tg amplicon is 99.4% (Table 1). These values are consistent with our WGBS data obtained with short fragments. In these experiments, the enzymatic treatment and amplification were performed on an unfragmented genomic template, showing that the enzymatic deamination method is applicable for the long DNA fragments as effectively as for the short DNA material of the WGBS applications.
Table 1.
Percentage of 5mC or 5hmC in CpG and CpH contexts (with H = A or T or C) in amplicons derived from lambda (unmethylated cytosines), CpG methylated pUC19 (CpG methylation), T4gt (hydroxymethylated cytosines), and mouse genomic DNA measured using three sequencing platforms (Illumina, PacBio, and Oxford Nanopore)
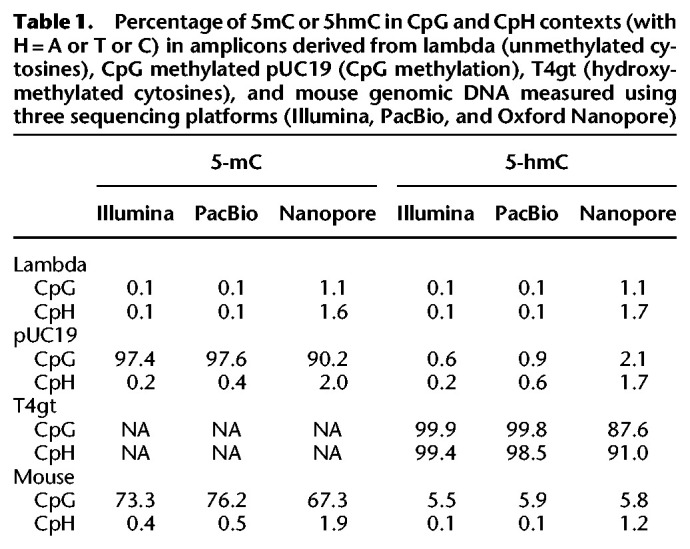
PacBio sequencing gave very similar estimates to the Illumina results. Nanopore sequencing generated slightly higher incorrect methylation calls on unmethylated cytosines in both the lambda and the CpG methylated pUC19 (CpH context). Nanopore sequencing also generated slightly lower correct 5mC calls in the CpG methylated pUC19 control (APOBEC(5mC), CpG context) and lower correct 5hmC calls in the T4gt control (APOBEC(5hmC), all context). The intrinsic higher error rate of Nanopore sequencing (Table 1), resulting in a higher base call errors at both cytosines and thymines, is presumably the explanation for these observations. At single-base resolution, both the methylation and hydroxymethylation levels at CpG sites are highly correlated across sequencing platforms, and the overall modification profiles are in good agreement across platforms (Fig. 3A,B). We also compared 5hmC results with publicly available data sets derived from Pvu-Seal-seq (Sun et al. 2015) and TAB-seq (Yu et al. 2012) from the same cell line and found consistent results with our data (Supplemental Fig. S4). These results suggest that the LR-EM-seq method is compatible with all the major sequencing platforms and produces accurate identification of both 5mC and 5hmC. Most significantly, at single-molecule resolution LR-EM-seq coupled with long-read sequencing technologies (PacBio and Nanopore) can provide complete 5mC and 5hmC information of entire molecules (Fig. 3C) and thus make it possible to study the relationships between distant cytosine sites on the same molecule as well as between individual molecules.
Figure 3.

5mC and 5hmC phasing using long-read sequencing. (A) Scatter plots and Pearson's correlations of calculated methylation (top) and hydroxymethylation (bottom) levels of all CpG sites within the 5378-bp region from the mouse E14 genome between the three sequencing platforms: PacBio, Nanopore, and Illumina. (B) Dot plots showing methylation (left) and hydroxymethylation (right) levels of individual CpG sites within the 5378-bp region calculated by the LR-EM-seq method using three major sequencing platforms: Illumina (red), Nanopore (green), and PacBio (blue). The fitted lines are drawn using the LOESS method. (C) Single-base single-molecule cytosine modification maps of the 5378-bp region generated by the LR-EM-seq method coupled with PacBio SMRT sequencing (top) and Nanopore sequencing (bottom). Methylated (left) and hydroxymethylated CpG sites are depicted by red dots, and unmodified CpG sites are depicted by beige dots.
Next, we applied LR-EM-seq using SMRT sequencing to a 4614-bp region (Chr 7: 135,829,567–135,834,180; mm9) containing a known 367-bp DMR upstream of a previously described imprinted gene, Inpp5f_v2, in the mouse brain (Choi et al. 2005). Based on the methylation call in CpC and CpT context, the overall conversion rate of the APOBEC3A-treated DNA was 99.8% (Supplemental Table S7), which is consistent with the performance of EM-seq and corresponds to about a 10-fold lower nonconversion rate compared with the previously published SMRT-BS sequencing (97.3%) (Yang et al. 2015). The methylation profiles showed a clear segregated pattern at the known DMR, confirming the differential methylation of this region (Fig. 4A). Phasing the entire 4614-bp region allowed a precise delimitation of the boundary of the DMR at molecule resolution. As a result, we report a more than twofold increase in the size of the reported DMR region from 367 bp to 1 kb (Fig. 4A,B). Moreover, when correlating long-range methylation patterns, we found two subdomains flanking both sides of the newly identified DMR (Fig. 4B). This suggests the occurrence of differentially methylated domains, whose methylation patterns do not completely follow the core DMR but are correlated with it. Whether such domains are derived from the core DMR under relaxed pressure, serve as a buffer between DMRs and non-DMRs, or indicate independent trans-acting transcription factor binding sites awaits further investigation. Another interesting observation is that the CpA methylation is missing from the DMR but displayed an oscillating pattern outside the DMR region (Supplemental Fig. S5A,B). It may suggest a role of high-level chromatin structure, for example, nucleosome positioning, in the deposition of DNA modification and gene regulation near the DMR.
Figure 4.

Phasing of 5mC and 5hmC by LR-EM-seq. (A) Single-base, single-molecule CpG methylation (middle) and hydroxymethylation (bottom) profile of a 4.6-kb region of the imprinted Inpp5f_v2 gene locus (top) in the mouse brain. Red dots represent modified sites, and beige dots represent unmodified sites. This region overlaps with the promoter for the Inpp5f_v2 gene and contains a previously reported DMR (orange box). The shaded area in the dot plots corresponds to the known DMR. (B) Correlation matrix of CpG modification state: (top) 5mC; (bottom) 5hmC. Each location on the matrix represents the correlation of any two CpG sites across the amplicon and the correlation strength is depicted by color: red indicates correlation=1; blue, correlation=−1; white, no correlation. The known DMR is indicated by a black outline.
We also successfully phased 5hmC in the same region. However, we did not observe any significant segregation pattern of hydroxymethylation (Fig. 4A,B). At the population level, the average CpG hydroxymethylation abundance significantly decreased at the DMR and generally followed the trend of 5mC across the entire region (Supplemental Fig. S5C), implying that in this region, the hydroxymethylation level may be largely determined by the substrate availability.
We used LR-EM-seq to validate previously reported allele-specific DMRs in the mouse genome of two inbred mouse strains: 129X1/SvJ (129) and Cast/EiJ (Cast) (Xie et al. 2012). We repeated the analysis performed on BALB/c strain for the Inpp5f_v2 locus and investigated three new regions (H13, Gnas [also known as Gnas1], and Peg12). For both the 129 and Cast strains, we observed similar segregations of two distinct populations of molecules according to their methylation status, that is, hypermethylated versus hypomethylated (Supplemental Fig. S5D). This result confirms the existence of DMRs for all four loci in both 129 and Cast strains. Moreover, all the DMRs are hundreds of base pairs to several kilobases larger than the reported ones, with long-read sequencing providing precise boundaries (Supplemental Fig. S5D).
To show allele-specific methylation, we used LR-EM-seq and simultaneously phased heterozygous SNPs with DNA methylation in two DMRs near the imprinted Inpp5f and Gnas genes. To acquire a large enough number of heterozygous SNPs for the identification of alleles over kilobase regions, we performed crosses between two inbred mouse strains 129X1/SvJ (129) and Cast/EiJ (Cast) as previously described (Xie et al. 2012). Consistent with the DMR results of the two inbred strains 129 and Cast, we observed larger DMRs than previously reported (Xie et al. 2012) in both Inpp5f and Gnas regions in the hybrid strain. Because we used the same strain and same organ (frontal cortex) as the reference paper, the most plausible explanation for the DMR size differences is the limitation of the original detection technique, which uses Illumina short-read sequencing.
Reliable heterozygous SNPs in both amplicons were identified with the methylation pattern segregating perfectly with the heterozygous SNP (Fig. 5A,B). In the case of the Inpp5f locus, we identified a heterozygous SNP that is ∼2 kb upstream of the DMR (Fig. 5A), showing our method's capability of phasing DMR with distal SNPs, which is especially critical for study regions with rare SNPs. And in those cases, it is necessary to have long-read sequencing to properly identify allele specific methylation.
Figure 5.

Phasing of 5mC with heterozygous variants using LR-EM-seq. (A) Phasing of 5mC with SNP of a 3.1-kb region in the imprinted Inpp5f_v2 gene promoter of the mouse cortex brain from a F1 offspring of a cross between two inbred mouse strains (129X1/SvJ male and Cast/EiJ female). Methylation state of individual CpG sites at the single-molecule level is denoted by either a beige dot (unmodified) or a red dot (methylated). The heterozygous SNP near the 5′ end of the region was either highlighted in red for paternal allele (A) or blue for maternal allele (G). The orange boxes denote previously identified DMRs. Our result not only confirmed the existence of the imprinted DMR but also revealed much extended boundaries of the imprinted DMR. (B) Phasing of 5mC and SNP in the imprinted promoter of the Gnas gene in the mouse cortex from a cross between the inbred mouse strains 129X1/SvJ (male) and Cast/EiJ (female). Methylation state of individual CpG sites at single-molecule level is denoted by either a beige dot (unmodified) or a red dot (methylated). The heterozygous SNP was highlighted in red for paternal allele (A) and blue for maternal allele (G). The orange box denotes a previously identified DMR. Our result confirmed the existence of the imprinted DMR and further extended this DMR in both directions particularly into the CpG island.
The ability to obtain large amplicons greatly expands the genomic ranges that are amenable to phasing of sequence variation with epigenetic information, thus making LR-EM-seq a convenient and promising technology for the identification of allele-specific methylation.
Discussion
In this study, we provided compelling evidence for the benefits of enzymatic deamination to identify both 5mC and 5hmC using long-read sequencing technology. Importantly, the converted genomic DNA can be amplified, and the information regarding the methylation status is preserved, allowing for locus-specific interrogation of methylation on low amounts of starting material. Although not shown here, LR-EM-seq can also be adapted to whole-genome sequencing to find de novo DMRs. In this case, genomic DNA will need to be fragmented to an average of 5-kb fragments before adaptor ligation. EM-seq treatment can be performed as described in this study, and amplified DNA could be used to prepare PacBio or Nanopore libraries.
Adapting EM-seq to long-read sequencing workflow surpasses bisulfite deamination, primarily in producing deaminated DNA without detectable damage. These advances eliminate the foremost roadblocks encountered using bisulfite sequencing for decades. As we have shown in this study, longer DNA material enabled by LR-EM-seq enables the study of the combinatorial effect of methylation over large regions at single-molecule resolution. We also showed the importance and advantage of phasing methylation on long DNA fragments on the study of imprinted genes by identifying much larger DMR regions than previously observed. These previous studies using short-read sequencing have relied on statistical methods to acquire methylation haplotype information and consequently are prone to inaccurate calls. By phasing variation with methylation on a single long-read, LR-EM-seq is more accurate and expands the fraction of the genome that can be epigenetically haplotyped.
Phasing methylation has numbers of additional applications, particularly in cancer detection, where the combinatorial methylation status of several CpG at single-molecule resolution is expected to be a much more powerful determinant of tumorigenicity compared with an average methylation level. Combined with other genomic information, phasing methylation relative to variants or epigenetic markers offers an exciting prospective empowered by LR-EM-seq.
Lastly, the ability to amplify longer amplicon provides greater flexibility in primer design, especially in encompassing or avoiding repeats or challenging to amplified regions. These results in larger covered genomic regions with less amplicons.
In summary, we described a new technology called LR-EM-seq for amplicon long-read sequencing of both 5mC and 5hmC. Our result showed the applicability of LR-EM-seq for long-range epigenetic analysis at single-molecule and single-nucleotide resolution, further expanding the range of biologically relevant questions that can be addressed.
Methods
E14 ES cell culture
ES cells were cultured as previously described (Kinney et al. 2011). Briefly, cells were grown in GMEM media (Thermo Fisher Scientific) containing 10% FBS (Gemcell), 1% nonessential amino acids (NEAA, HyClone), 1% sodium pyruvate (Thermo Fisher Scientific), 50 μM beta-mercaptoethanol (Sigma-Aldrich), and 1 × LIF (MilliporeSigma). To maintain the undifferentiated state, ES cells were grown on 0.1% gelatin-coated culture dishes (Stem Cell Technologies).
Genomic DNAs
Mouse genomic DNA from brain, spleen, heart, and liver tissues were obtained from BioChain; mouse NIH 3T3 and human Jurkat DNA were from NEB. E14 genomic DNA was extracted with a DNeasy blood and tissue kit (Qiagen). Genomic DNA from DNMT TKO J1 ES cells were obtained from Dr. Yi Zhang.
Control DNAs
Fully CpG methylated pUC19 DNA was acquired by incubating for 2 h at 37°C 3 μg of dam-dcm- plasmid DNA in a 50-μL reaction containing 20 U of M.SssI methylase (NEB), 1× NEBuffer 2, and freshly prepared 160 μM S-adenosylmethionine, followed by heat inactivation of the enzyme for 20 min at 65°C and SPRI beads purification. LC-MS/MS analysis was used to verify completeness of methylation status. T4gt (amC87(42-), amE51(56-), NB5060(ΔrllB- denB- ac), unf 39(alc)), and XP12 phage genomic DNAs were extracted as described previously (Sambrook 1989). 5mC free lambda genomic DNA was purchased from Promega.
5mC and 5hmC phasing of a 5.4-kb mouse genomic region using LR-EM-seq
Enzymatic deamination for 5mC detection
For 5mC detection, 200 ng of mouse E14 genomic DNA was mixed with 10 ng unmethylated lambda DNA, 10 ng of XP12 phage DNA, and 1 ng of CpG methylated pUC19 DNA and then incubated with 16 μg of TET2 enzyme (NEB EM-seq component E7130A) for 30 min at 37°C in 50 μL 1× reaction buffer (EM-seq TET2 Reaction Buffer [reconstituted], E7128A, and E7131A diluted) followed by a 30-min incubation with 20 U of T4 BGT (NEB EM-seq component E7129A) in the same buffer at 37°C. Oxidized genomic DNA was incubated additional 30 min with 0.8 U of Proteinase K (NEB) at 37°C and subsequently purified with a Genomic DNA Clean & Concentrator kit (Zymo Research). Purified DNA was then denatured at 90°C in presence of 29% of formamide for 10 min and deaminated with 100 U of APOBEC3A (NEB EM-seq components E7133AA and E7134AA) in 100 μL reaction volume for 3 h. Three microliters of deaminated genomic DNA and control DNAs was used without further purification for PCR amplifications with Phusion U hot start DNA Polymerase (Thermo Fisher Scientific) using primer pairs listed in Supplemental Table S9. For some amplicons (Supplemental Table S9, eight to 11 primer pairs), we used the enzymatic EM-seq conversion module (NEB E7125) for 5mC detection.
Enzymatic deamination for 5hmC detection
For 5hmC detection, 200 ng of mouse E14 genomic DNA was mixed with 10 ng unmethylated lambda DNA, 10 ng of T4gt phage DNA, and 1 ng of CpG methylated pUC19 DNA and then incubated with 20 U of BGT (NEB) in 1× NEBuffer 2 for 2 h at 37°C. Glucosylated genomic DNA was incubated additional 30 min with 0.8 U of Proteinase K (NEB) at 37°C and purified with a Genomic DNA Clean & Concentrator kit (Zymo Research). Purified DNA was then denatured at 90°C in presence of 29% of formamide for 10 min and deaminated with 100 U of APOBEC3A (EM-seq components E7133AA and E7134AA) in 100 μL reaction volume for 16 h. Three microliters of deaminated genomic DNA and control DNAs were used without further purification for PCR amplifications with Phusion U hot start DNA Polymerase (Thermo Fisher Scientific) using primer pairs listed in Supplemental Table S9.
High-throughput sequencing of the enzymatically deaminated amplicons
5mC and 5hmC amplicons were pooled with the control amplicons, respectively, and were sequenced using the Illumina, Nanopore, and PacBio platforms (Supplemental Methods). In brief, for Illumina sequencing, 50 ng of each amplicon pool was fragmented to an average size of 500 bp using the Covaris S2 instrument in 50 μL of 0.1× TE buffer. Sonicated DNA was used to construct libraries with a NEBNext ultra DNA library prep kit (NEB) and sequenced on an Illumina MiSeq instrument. For Nanopore sequencing, the 1D Native barcoding genomic DNA kit (Oxford Nanopore Technologies EXP-NBD103 and SQK-LSK108 kits) was used for library preparation. Five hundred nanograms of each 5mC and 5hmC amplicon pool was barcoded and sequenced on the same flow cell (FLO-MIN106 Rev D) for a total of 11 h on a MinION (Oxford Nanopore Technologies). Raw fast5 data were generated using MinKNOW version 18.12 and base-called using Guppy base caller version 2.1.3. For PacBio sequencing, 400 ng of the 5mC and 5hmC amplicon pools was respectively prepared and sequenced on a PacBio Sequel platform following the manufacturer's protocols (Pacific Biosciences Sequencing primer v3 and Sequel binding Kit 2.1, Sequel sequencing Kit 2.1). One SMRT cell (SMRT Cell 1M v2) was used for each library with a 600-min movie. Circular consensus sequences (CCSs) were generated using the SMRT Link (version 6.0.0.47841).
5mC and 5hmC phasing of mouse DMRs using LR-EM-seq
Mice
Three different mice strains are used for this project: (1) Cast/EiJ (Cast), (2) 129 × 1/SvJ (129), and (3) the F1 offspring of Cast (female) X129 (male). The crosses of Cast and 129 mice were performed by the Jackson Laboratory. The frontal cortex samples of the male mice F1 offspring and a male mouse of each parental strain were collected at 8–10 wk at the Jackson Laboratory and were shipped to the investigator at NEB on dry ice (compliant with the provisions of the Public Health Service Policy on Humane Care and Use of Laboratory Animals).
Genomic DNA extraction and purification
The genomic DNA was extracted from ∼10-mg frozen brain cortex samples using the NEB Monarch genomic DNA purification kit (NEB). Four microliters of RNase A (100 mg/mL) has been added to the tissue lysate (and incubated 5 min at room temperature) in both protocols to prevent the inhibition of APOBEC3A by RNA during the deamination process. The extracted genomic DNA was purified again using AMPure XP beads.
Preparation of LR-EM-seq long amplicons
Twenty nanograms of purified genomic DNA was glucosylated by incubating with 20 U of BGT (NEB) for 2 h at 37°C (for 5hmC detection). Glucosylated genomic DNA was incubated an additional 30 min with 0.8 U of Proteinase K at 37°C and subsequently purified with Genomic DNA Clean & Concentrator kit (Zymo Research, USA Research). For 5mC detection, mouse brain genomic DNA (200 ng) was oxidized by incubating with 16 µg of TET2 (EM-seq TET2 reaction buffer [reconstituted], E7128A, E7131A diluted, and E7130A) for 30 min at 37°C followed 30-min incubation with BGT (NEB EM-seq component E7129A) in the same buffer at 37°C. Oxidized brain genomic DNA was incubated an additional 30 min with 0.8 U of Proteinase K at 37°C and subsequently purified with Genomic DNA Clean & Concentrator kit (Zymo Research, USA Research). Purified DNA was denatured at 80°C in presence of 66% of formamide and was deaminated with 0.3 µg of APOBEC3A in 100 mL reaction volume (EM-seq components E7133AA and E7134AA) for 16 h for 5hmC detection and 3 h for 5mC detection. We then purified DNA with Genomic DNA Clean & Concentrator kit (Zymo Research, USA Research). Targeted DMRs were amplified from each of the purified deaminated DNA using custom designed primers (Supplemental Table S9).
SMRT sequencing
The purified long amplicons were prepared for PacBio SMRT sequencing (Pacific Biosciences) following the “amplicon template preparation and sequencing” protocol. One library was prepared for each region and for each modification type and was loaded onto the SMRT cell using the MagBead method. The LR-EM-seq libraries were sequenced on a PacBio RSII machine with 5.5-h movie. Consensus sequences of individual sequenced molecules (read of insert) were generated by the “RS_ReadsOfInsert” protocol using the SMRT portal (version 2.3.0.140893). Reads that were shorter than the expected amplicon size were removed from downstream analysis. We then corrected the quality scores of the SMRT consensus sequences using the BBmap tools (Bushnell B; sourceforge.net/projects/bbmap/) and then conducted phasing analysis (see below “5mC and 5hmC phasing analysis”).
Bioinformatics analysis
Data processing and 5mC, 5hmC calling of Illumina libraries
Raw reads were first trimmed by the Trim Galore software (https://github.com/FelixKrueger/TrimGalore) to remove adapter sequences and low-quality bases from the 3′ end. Unpaired reads owing to adapter/quality trimming were also removed during this process. The trimmed read sequences were C-to-T converted and were then mapped to a composite reference sequence including the mouse genome (mm9) and the complete sequences of lambda, pUC19, phage XP12, and T4 controls using the Bismark program (Krueger and Andrews 2011) with the default Bowtie 2 setting (Langmead and Salzberg 2012). We used the GRCm37 (mm9) assembly to map all sequencing reads from mouse origin in this study because many annotations were not available or were incomplete for the GRCm38 (mm10) build when we started our study. GRCm38 is not known to have a significant difference from mm9 in base composition nor in cytosine content. In addition, because our study compares results between different methods or compares to the previous work, which also used mm9 as reference sequence, mapping the reads to GRCm38 (mm10) should not significantly affect our conclusions.
The aligned reads were then subjected to three postprocessing QC steps: First, alignment pairs that shared the same alignment start positions (5′ ends) were regarded as PCR duplicates and were discarded. This deduplication step was skipped for loci-specific amplicon libraries. Second, the first 5 bp at the 5′ end of R2 reads were removed to reduce end-repair errors, and third, reads that contained excessive cytosines in non-CpG context (e.g., more than five for 5mC libraries and more than three for 5hmC libraries) were removed to reduce nonconversion errors. CpH-based filtering for bisulfite experiments of mammalian samples have been used to reduce nonconversion errors (Lister et al. 2009). This filtering method is not applicable to organisms that have appreciable levels of non-CpG methylation (e.g., most plants). The remaining good quality alignments were then used for cytosine methylation and hydroxymethylation calling by a Bismark methylation extractor.
For additional 5mC and 5hmC analysis of the Illumina libraries, see the Supplemental Methods.
5mC and 5hmC phasing analysis
We used Bismark (Krueger and Andrews 2011) to map full-length reads from PacBio SMRT sequencing and Oxford Nanopore sequencing to the mouse reference genome (mm9) with the following parameters: ‐‐bowtie2 -N1 -L15 ‐‐score_min L,0,-0.6. The modification states of individual CpG sites were called by the bismark_methylation_extrator program. We then extracted the context-specific methylation information of individual molecules and plotted in R (R Core Team 2017). SNPs were called from the same PacBio sequencing reads using the SAMtools package (Li et al. 2009). We used the SNPs that are consistent with the previously reported heterozygous SNPs (Xie et al. 2012) to distinguish paternal and maternal copies in the F1 sample for phasing analysis. The conversion rates were calculated using all the cytosines in CpC and CpT context by following formula: converted C(C/T)/total C(C/T). We exclude CpA from the calculation of nonconversion error rate because it was previously reported that brain DNA has a high level of CpA modification (Xie et al. 2012; Lister et al. 2013; Guo et al. 2014).
Data access
All raw and processed sequencing data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE141908.
Competing interest statement
Reagents described in this paper are products of New England Biolabs, Inc. The authors are employed by New England Biolabs, Inc., a commercial supplier of molecular biology reagents.
Supplementary Material
Acknowledgments
We thank Yi Zhang and Hao Wu for DNMT TKO J1 ES cell genomic DNA, Peter Weigele for providing XP12 phage DNA, and Laurie Mazzola, Joanna Bybee, and Danielle Rivizzigno for sequencing. This study is supported by New England Biolabs, Inc.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.265306.120.
Freely available online through the Genome Research Open Access option.
References
- Choi JD, Underkoffler LA, Wood AJ, Collins JN, Williams PT, Golden JA, Schuster EF, Loomes KM, Oakey RJ. 2005. A novel variant of Inpp5f is imprinted in brain, and its expression is correlated with differential methylation of an internal CpG island. Mol Cell Biol 25: 5514–5522. 10.1128/MCB.25.13.5514-5522.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7: 461–465. 10.1038/nmeth.1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. 1992. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci 89: 1827–1831. 10.1073/pnas.89.5.1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesselmann P, Brändl B, Raimondeau E, Bowen R, Rohrandt C, Tandon R, Kretzmer H, Assum G, Galonska C, Siebert R, et al. 2019. Analysis of short tandem repeat expansions and their methylation state with nanopore sequencing. Nat Biotechnol 37: 1478–1481. 10.1038/s41587-019-0293-x [DOI] [PubMed] [Google Scholar]
- Gilpatrick T, Lee I, Graham JE, Raimondeau E, Bowen R, Heron A, Downs B, Sukumar S, Sedlazeck FJ, Timp W. 2020. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol 38: 433–438. 10.1038/s41587-020-0407-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Globisch D, Münzel M, Müller M, Michalakis S, Wagner M, Koch S, Brückl T, Biel M, Carell T. 2010. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS One 5: e15367 10.1371/journal.pone.0015367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunau C, Clark SJ, Rosenthal A. 2001. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res 29: 65e 10.1093/nar/29.13.e65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JU, Su Y, Shin JH, Shin J, Li H, Xie B, Zhong C, Hu S, Le T, Fan G, et al. 2014. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci 17: 215–222. 10.1038/nn.3607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafford-Tear NJ, Tsai YC, Sadan AN, Sanchez-Pintado B, Zarouchlioti C, Maher GJ, Liskova P, Tuft SJ, Hardcastle AJ, Clark TA, et al. 2019. CRISPR/Cas9-targeted enrichment and long-read sequencing of the Fuchs endothelial corneal dystrophy–associated TCF4 triplet repeat. Genet Med 21: 2092–2102. 10.1038/s41436-019-0453-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg Ja, He C, Zhang Y. 2011. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science 333: 1300–1303. 10.1126/science.1210597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Olsen HE, Turner DJ, Stoddart D, Bulazel KV, Paten B, Haussler D, Willard HF, Akeson M, Miga KH. 2018. Linear assembly of a human centromere on the Y chromosome. Nat Biotechnol 36: 321–323. 10.1038/nbt.4109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney SM, Chin HG, Vaisvila R, Bitinaite J, Zheng Y, Estève PO, Feng S, Stroud H, Jacobsen SE, Pradhan S. 2011. Tissue-specific distribution and dynamic changes of 5-hydroxymethylcytosine in mammalian genomes. J Biol Chem 286: 24685–24693. 10.1074/jbc.M110.217083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27: 1571–1572. 10.1093/bioinformatics/btr167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdyukov S, Bullock M. 2016. DNA methylation analysis: choosing the right method. Biology (Basel) 5: 3 10.3390/biology5010003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9: 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q-M, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322. 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND, Lucero J, Huang Y, Dwork AJ, Schultz MD, et al. 2013. Global epigenomic reconfiguration during mammalian brain development. Science 341: 1237905 10.1126/science.1237905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Siejka-Zielińska P, Velikova G, Bi Y, Yuan F, Tomkova M, Bai C, Chen L, Schuster-Böckler B, Song C-X. 2019. Bisulfite-free direct detection of 5-methylcytosine and 5-hydroxymethylcytosine at base resolution. Nat Biotechnol 37: 424.–. 10.1038/s41587-019-0041-2 [DOI] [PubMed] [Google Scholar]
- Liu Y, Cheng J, Siejka-Zielińska P, Weldon C, Roberts H, Lopopolo M, Magri A, D'Arienzo V, Harris JM, McKeating JA, et al. 2020. Accurate targeted long-read DNA methylation and hydroxymethylation sequencing with TAPS. Genome Biol 21: 54 10.1186/s13059-020-01969-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard MO, Gurdasani D, Mentzer AJ, Porter T, Sandhu MS. 2018. Long reads: their purpose and place. Hum Mol Genet 27: R234–R241. 10.1093/hmg/ddy177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand AC, Jain M, Eizenga JM, Musselman-Brown A, Olsen HE, Akeson M, Paten B. 2017. Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods 14: 411–413. 10.1038/nmeth.4189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna: https://www.R-project.org/. [Google Scholar]
- Salter JD, Bennett RP, Smith HC. 2016. The APOBEC protein family: united by structure, divergent in function. Trends Biochem Sci 41: 578–594. 10.1016/j.tibs.2016.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook HC. 1989. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY: https://ci.nii.ac.jp/naid/10003540621/ [accessed September 10, 2020]. [Google Scholar]
- Schutsky EK, DeNizio JE, Hu P, Liu MY, Nabel CS, Fabyanic EB, Hwang Y, Bushman FD, Wu H, Kohli RM. 2018. Nondestructive, base-resolution sequencing of 5-hydroxymethylcytosine using a DNA deaminase. Nat Biotechnol 36: 1083–1090. 10.1038/nbt.4204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson JT, Workman RE, Zuzarte PC, David M, Dursi LJ, Timp W. 2017. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods 14: 407–410. 10.1038/nmeth.4184 [DOI] [PubMed] [Google Scholar]
- Sun Z, Dai N, Borgaro JG, Quimby A, Sun D, Corrêa IR, Zheng Y, Zhu Z, Guan S. 2015. A sensitive approach to map genome-wide 5-hydroxymethylcytosine and 5-formylcytosine at single-base resolution. Mol Cell 57: 750–761. 10.1016/j.molcel.2014.12.035 [DOI] [PubMed] [Google Scholar]
- Xie W, Barr CL, Kim A, Yue F, Lee AY, Eubanks J, Dempster EL, Ren B. 2012. Base-resolution analyses of sequence and parent-of-origin dependent DNA methylation in the mouse genome. Cell 148: 816–831. 10.1016/j.cell.2011.12.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Sebra R, Pullman BS, Qiao W, Peter I, Desnick RJRJ, Geyer CR, DeCoteau JF, Scott SASA. 2015. Quantitative and multiplexed DNA methylation analysis using long-read single-molecule real-time bisulfite sequencing (SMRT-BS). BMC Genomics 16: 350 10.1186/s12864-015-1572-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Hon GCC, Szulwach KEE, Song C-X, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. 2012. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 149: 1368–1380. 10.1016/j.cell.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.