

Abstract
Chitotriosidase activity and CCL18 concentration are interchangeably used for monitoring Gaucher disease (GD) activity, together with clinical assessment. However, comparative studies of these two biomarkers are scarce and of limited sample size. The aim of this systematic review with meta-analysis of individual participant data (IPD) was to compare the accuracy of chitotriosidase activity and CCL18 concentration for assessing type I GD severity. We identified cross-sectional and prospective cohort studies by searching Medline, EMBASE, and CENTRAL from January 1995 to June 2017, and by contacting research groups. The primary outcome was a composite of liver volume >1.25 multiple of normal (MN), spleen volume >5 MN, hemoglobin concentration <11 g/dL, and platelet count <100x109/L. Overall, IPD included 1,109 observations from 334 patients enrolled in nine primary studies, after excluding 111 patients with undocumented values and 18 patients with deficient chitotriosidase activity. IPD were unavailable for 14 eligible primary studies. The primary outcome was associated with a 5.3-fold (95% Confidence Interval [CI]: 4.2-6.6) and 3.0-fold (95% CI: 2.6-3.6) increase of the geometric mean for chitotriosidase activity and CCL18 concentration, respectively. The corresponding areas under the receiver operating characteristics curves were 0.82 and 0.84 (summary difference, 0.02, 95% CI: -0.02 to 0.05). The addition of chitotriosidase activity did not improve the accuracy of the CCL18 concentration. Estimates remained robust in the sensitivity analysis and consistent across subgroups. Neither the chitotriosidase activity nor the CCL18 concentration varied significantly according to a recent history of bone events among 97 patients. In conclusion, the CCL18 concentration is as accurate as chitotriosidase activity in assessing hematological and visceral parameters of GD severity and can be measured in all GD patients. This meta-analysis supports the use of CCL18 rather than chitotriosidase activity for monito-ring GD activity in routine practice.
Introduction
Gaucher disease (GD; OMIM#230800) is a recessively inherited lysosomal sto-rage disorder caused by biallelic mutations in the GBA1 gene that encodes the lysosomal acid -glucosidase.1 The metabolic defect in GD results in bone marrow and visceral organ infiltration by Gaucher cells (i.e., glucosylceramide-laden macrophages), leading to anemia, thrombocytopenia, hepatosplenomegaly, and skeletal manifestations.2-4 Three main GD types (I, II and III) have been described, on the basis of the clinical features and age of onset. Type I (i.e., non-neuropathic) GD is predominant (85-94%).5
GD natural course is rather unpredictable, even within subgroups of patients with the same GBA1 mutation.6 Therefore, evaluating the disease severity and prognosis is challenging for clinicians. In this context, a surrogate blood biochemical marker is highly desirable for assessing GD severity and helping decision-making for specific therapy initiation or adjustment.7 Several biomarkers have been identified, including chitotriosidase, CCL18, and glucosylsphingosine. 8-10
Chitotriosidase, the human analogue of the non-vertebrate chitinase, is directly secreted by Gaucher cells and is considered an indicator of the overall Gaucher cell burden.11 In patients with GD, chitotriosidase activity is about 1,000-fold higher than the normal values, and its level correlates with liver and spleen volume, hemoglobin concentration, platelet count, and some bone manifestations. 12,13 Plasma chitotriosidase activity decreases dramatically after initiation of enzyme replacement therapy (ERT) and rises again when the treatment is stopped.14 However, plasma chitotriosidase activity has major limitations for monitoring GD activity. Indeed, measuring plasma chitotriosidase activity is technically complex and not standardized across laboratories.15 Moreover, 6% of the general population is homozygous for a chitotriosidase variant harboring a 24-base pair duplication in the CHIT1 gene and is deficient in chitotriosidase activity.16 In addition, 35% of the general population is heterozygous for this chitotriosidase variant, and displays about half of the activity observed in people with wild-type CHIT1. Finally, other CHIT1 gene polymorphisms have been reported that slightly impair the enzyme activity.11
CC chemokine ligand 18 (CCL18), originally named pulmonary and activation-regulated chemokine (PARC), is also directly secreted by Gaucher cells and is considered an indicator of the overall Gaucher cell burden.8 Plasma CCL18 concentration in GD patient is 10- to 50-fold higher than in healthy subjects.8 CCL18 concentration correlates with the liver and spleen volume, platelet count, history of osteonecrosis, and number of anatomical sites of osteonecrosis.12,17 Importantly, the CCL18 concentration is not subject to genetic variation and can be measured in all patients, including those with deficient chitotriosidase activity.8
Hitherto, both chitotriosidase activity and CCL18 concentration are used for monitoring GD activity and assessing response to treatment, based on the findings from single-center studies of relatively limited sample size. The lack of large scale, multicenter, head-to-head comparison studies have hindered full validation of these biomarkers and formulation of context of use in the clinical practice. We hypothesized that evidence on the comparative accuracy of these two biomarkers could be strengthened by the secondary analysis of individual participant data (IPD) from primary studies on patients with GD. Therefore, our primary objective was to compare the accuracy of CCL18 concentration and chitotriosidase activity for assessing hematological and visceral parameters of type I GD severity. The secondary objective was to compare the accuracy of these two biomarkers for discriminating type I GD patients with symptomatic bone events.
Methods
This systematic review with IPD meta-analysis was performed according to current guidelines18,19 and complied with the Preferred Reporting Items for Systematic review and Meta- Analysis (PRISMA)-IPD statement.20 The rationale and methods were pre-specified and reported in a protocol21 registered at PROSPERO (CRD42015027243). This meta-analysis was carried out on data from primary studies for which ethical approval had been obtained by the investigators. The Comité de Protection des Personnes Sud Est 6, Clermont-Ferrand, France (IRB 00008526) reviewed the protocol and considered that it did not qualify for biomedical research requiring patient informed consent, provided that no supplementary data would be collected from the participants enrolled in primary studies.21
Eligibility criteria
Eligible studies included cross-sectional and cohort studies that measured both chitotriosidase activity and CCL18 concentration at baseline and/or at follow-up in consecutive patients with type I GD (Online Supplementary Materials and Methods). Studies with fewer than 10 participants were excluded from this systematic review.
Information sources
Studies were identified by searching Medline, EMBASE, and Cochrane Central Register of Controlled Trials (CENTRAL) from January 1995 to June 2017. Our electronic search was supplemented by scanning the reference lists of the retrieved articles and by contacting research groups.
Search strategy
The search concepts included chitotriosidase activity, CCL18, biological markers, ERT, and GD (Online Supplementary Appendices S1-3). No restriction of document type and language was applied, and no methodology filter was used.
Study selection
Two review authors (TR and JL) assessed potentially relevant full-text articles against pre-specified eligibility criteria.
Data collection
Information on primary studies were collected using a standardized data extraction form. Where possible, IPD were extracted from published articles. Otherwise, the corresponding authors or principal investigators were invited to collaborate in this systematic review project by supplying de-identified IPD.21 The requested IPD included baseline characteristics and pre-specified outcomes.21 Organ volumes were expressed as multiples of normal (MN) adjusted for body weight.
Outcomes
The primary outcome was a composite of hemoglobin concentration <11 g/dL (<10 g/dL for patients aged 12-59 months of age), platelet count <100x109/L, spleen volume >5 MN, and liver volume >1.25 MN. The secondary outcomes included symptomatic bone events, a composite of hemoglobin concentration <8 g/dL (<7 g/dL for patients 12-59 months of age), platelet count <50x109/L, spleen volume >15 MN, and liver volume >2.5 MN, and individual components of the primary and secondary composite outcomes. Bone events included skeletal fracture, osteonecrosis, or avascular necrosis that occurred within the previous 12 months of biomarker analysis.21 All outcomes and cut-off values for continuous parameters were set according to published guidelines or previous studies,22,23 and were pre-specified.21
Statistical analysis
We performed logarithm transformation for chitotriosidase activity and CCL18 concentration and derived geometric mean ratios along with 95% Confidence Intervals (CI), in order to account for skewed distributions.24,25 The comparative accuracy of CCL18 concentration relative to chitotriosidase activity in discriminating patients with the outcomes of interest was quantified by the difference in the areas under the receiver operating characteristic (AUC-ROC) curves. Data synthesis was performed with one- and two-stage approaches.26,27
Figure 1.

Identification and selection of studies for the meta-analysis of individual participant data. *Seventy three additional participants (222 observations) had splenectomy at baseline. IDP: individual participant data.
Results
Study selection and obtained IPD
Overall, 2,636 records were identified by database searching (Figure 1). Two additional records were found by contacting field experts and searching clinical trial registries, respectively. After removing 13 duplicates, the titles and abstracts of 2,625 records were screened for eligibility. Of these, 318 records were identified as being potentially relevant and full-text articles were retrieved for a more thorough review. After excluding 295 records based on the full-text article, our systematic review included 23 primary studies from which IPD were sought.
IPD were available only for nine of these studies totaling 463 patients with type I GD (Figure 1). The sponsor of four trials of ERT with velaglucerase28-31 and the principal investigators of three observational studies17,32,33 supplied computerized IPD upon request. The IPD of the other two randomized controlled trials on ERT with taliglucerase were extracted from the published articles34,35 (Online Supplementary Appendix S4). Our meta-analytical sample consisted of 1,109 observations from 334 patients with type I GD, after excluding 111 patients with undocumented values for chitotriosidase activity and/or serum CCL18 concentration, and 18 patients with deficient chitotriosidase activity (Figure 1). The median number of observations per patient was 2 (range: 1-14).
IPD were not available for the other 14 eligible primary studies that included 11 academic observational studies8,9,14,36-43 and three industry-sponsored clinical trials of ERT or substrate reduction therapy44-46 (Online Supplementary Appendix S5). Overall, these studies enrolled 565 patients with GD, although overlap in study populations could not be excluded for five studies carried out in the Netherlands8,9,14,37,42 (201 patients) and four studies performed in Spain36,39,40,41 (203 patients).
Study characteristics
Among the nine studies that supplied IPD, six were international industry-funded clinical trials of ERT28-31,34,35 and three were observational in design17,32,33 (Online Supplementary Appendix S4). The study enrollment periods extended from 2003 to 2015, and the length of the followup ranged from 12 to 132 months for longitudinal studies. The median number of primary study participants and observations contributing to the meta-analysis was 32 (range: 9-98) and 117 (range: 20-224), respectively. All but one study recruited adult or mixed populations. The exception was a clinical trial performed in a pediatric setting. 35 Children younger than 16 years of age accounted for 13% of all participants.
Chitotriosidase activity was measured using the 4MUdeoxy- chitobiose28-31,33,35 or 4MU-chitotriose17,32 fluorogenic substrates, in compliance with the published methods. Serum CCL18 concentration was assayed using DELFIA or ELISA in five and three studies, respectively (Online Supplementary Appendix S6). Information on chitotriosidase activity and serum CCL18 assays was not available for one study.34 The median values ranged from 1,340-14,809 nmol/mL/h for chitotriosidase activity, and from 237-1,113 ng/mL for serum CCL18 concentration. In five clinical trials, 28-31,35 chitotriosidase activity and serum CCL18 concentration were both measured at a single central core laboratory (i.e., the Academic Medical Center in Amsterdam, the Netherlands). Liver and spleen volumes were quantified using magnetic resonance imaging in eight studies, assessed by independent blinded reviewers in five studies, and undocumented in one study. The median values in primary studies ranged from 0.8-1.7 MN for liver volume, 2.7-14.1 MN for spleen volume, 11.7-14.0 g/dL for hemoglobin concentration, and 82-260 x109/L for platelet count (Online Supplementary Appendix S6).
Study quality assessment
Six of the eight studies that evaluated the primary composite outcome fulfilled five or more QUADAS-2 tool criteria (Online Supplementary Appendix S7). The other two studies met four QUADAS-2 tool criteria. As data were collected by retrospective chart review, the Yale’s National Gaucher Disease Treatment Center study33 was considered at high risk of bias for index tests and pre-specified surrogate outcome assessment. This study was also considered at high risk of flow bias due to undocumented chitotriosidase activity or serum CCL18 concentration in 68% (113 of 167) of participants. In a randomized controlled trial that enrolled only treatment-naive patients, the applicability concern was high and it was not possible to formally determine whether biomaker assessment was blinded to the pre-specified outcomes and which analytical methods were used.34
Chitotriosidase activity and serum CCL18 concentration according to the outcomes
In one-stage meta-analysis involving 492 observations nested within 177 participants from eight primary studies (Figure 1), the primary composite outcome was associated with increased geometric mean of chitotriosidase activity (7,623 vs. 1,478 nmol/mL/h; geometric mean ratio: 5.29, 95% CI: 4.24-6.61, P<0.001) and CCL18 concentration (679 vs. 198 ng/mL; geometric mean ratio: 3.04, 95% CI: 2.57-3.58, P<0.001) compared with the absence of outcome (Table 1). Despite substantial between-study heterogeneity, the two-stage meta-analysis yielded comparable results, using random-effect models (Online Supplementary Appendices S8-9).
The effect size was quite homogeneous among the individual components of the primary composite outcome, with geometric mean ratio point estimates ranging from 2.96-5.43 for chitotriosidase activity and from 2.28-3.22 for serum CCL18 concentration (Table 1). Similar results were obtained for the secondary composite outcome and its individual components (Table 1), except for serum CCL18 concentration, which did not differ according to severe anemia (445 vs. 666 ng/mL, geometric mean ratio, 1.39, 95% CI: 0.82-2.37, P=0.22).
The geometric means of chitotriosidase activity (3,556 vs. 1,618 nmol/mL/h; geometric mean ratio: 1.47, 95% CI: 0.78-2.79, P=0.24) and serum CCL18 concentration (786 vs. 449 ng/mL; geometric mean ratio: 1.22, 95% CI: 0.81-1.81, P=0.34) did not differ according to symptomatic bone events with imaging confirmation, among 218 observations nested within 97 participants from a single primary study (Table 1). The decrease in CCL18 concentration was paralleled by a similar trend in chitotriosidase activity over the 24 months of follow-up, among participants enrolled in four industry-sponsored clinical trials evaluating enzyme replacement therapy (ERT) (Online Supplementary Appendix S10).
Comparative accuracy of chitotriosidase activity and CCL18 concentration for assessing GD severity
In one-stage meta-analysis, area under the curve receiver operating characteristics (AUC-ROC) curve point estimates were 0.82 for chitotriosidase activity and 0.84 for serum CCL18 concentration for discriminating patients with GD according to the primary composite outcome (summary difference: 0.02, 95% CI: -0.02 to 0.05, P=0.32, Table 2). Adding chitotriosidase activity did not improve serum CCL18 concentration accuracy, as indicated by the AUC-ROC curve estimates (0.85 with and 0.84 without chitotriosidase activity, P=0.18) (Figure 2).
The summary estimates of the difference in the AUCROC curves were consistent in the two-stage meta-analysis (Online Supplementary Appendix S11). No evidence of selective reporting for the primary composite outcome was found graphically (Online Supplementary Appendix 12) or statistically (P=0.20). However, this result should be interpreted with caution, given the limited number of primary studies.
Table 1.
One-stage unpaired comparisons of chitotriosidase activity and CCL18 concentration according to pre-specified outcomes among type I Gaucher disease patients.

Comparable effect sizes were observed for the secondary composite outcome and most of the individual components (Table 2). However, the AUC-ROC curve of serum CCL18 concentration for predicting severe anemia was lower than that of chitotriosidase activity (0.64 vs. 0.81, summary difference, -0.17, 95% CI: -0.33 to -0.08, P=0.008).
Subgroup analysis
No evidence of heterogeneity in the biomarker levels associated with the primary composite outcome was observed across the age groups and provision of ERT (Online Supplementary Appendix S13). Conversely, lowerquality studies (<5 QUADAS-2 criteria) displayed higher geometric mean ratios for chitotriosidase activity, according to the primary outcome (Online Supplementary Appendix 13).
Differences in the AUC-ROC curves for the primary composite outcome were homogeneous across the age groups, QUADAS-2 criteria, fluorogenic substrate, and CCL18 assay type (Online Supplementary Appendix S14). However, serum CCL18 concentration was more accurate than chitotriosidase activity in discriminating patients with the primary composite outcome among those receiving ERT (Online Supplementary Appendix S14). No evidence of heterogeneity in serum CCL18 concentration accuracy as a function of chitotriosidase activity deficiency was found (Online Supplementary Appendices S15-16).
Sensitivity analysis
In leave-one-out sensitivity analysis that iteratively removed one study at a time, geometric mean ratios (Online Supplementary Appendix 17) and differences in the AUCROC curves (Online Supplementary Appendix 18) for the two biomarkers remained unchanged relative to the primary outcome, confirming that our results were not driven by any single study. In the additional sensitivity analysis, coding splenectomy as splenomegaly did not modify the associations of the primary and secondary composite outcomes with the two biomarkers (Online Supplementary Appendix S19), although the AUC-ROC curve for chitotriosidase activity decreased significantly (Online Supplementary Appendix S20).
Discussion
Most published studies reporting on chitotriosidase activity and CCL18 concentration accuracy in assessing GD severity are of relatively limited sample size. In this context, our meta-analysis summarizes evidence from 1,109 observations nested within 334 participants enrolled in nine primary studies, with a broad range of patient age and disease severity.
Our analysis indicates that CCL18 concentration is as accurate as chitotriosidase activity in discriminating patients on the basis of our primary composite outcome. This finding was robust in the sensitivity analysis, and we did not find evidence of between-study heterogeneity (I²=0%, P=0.68) in two-stage meta-analysis. Additionally, we did not detect any significant heterogeneity among studies and patient subgroups, although CCL18 concentration may be more accurate in patients receiving ERT. Our primary result is also supported by consistency in the effect sizes for most of the individual outcome components and the secondary composite outcomes.
Figure 2.
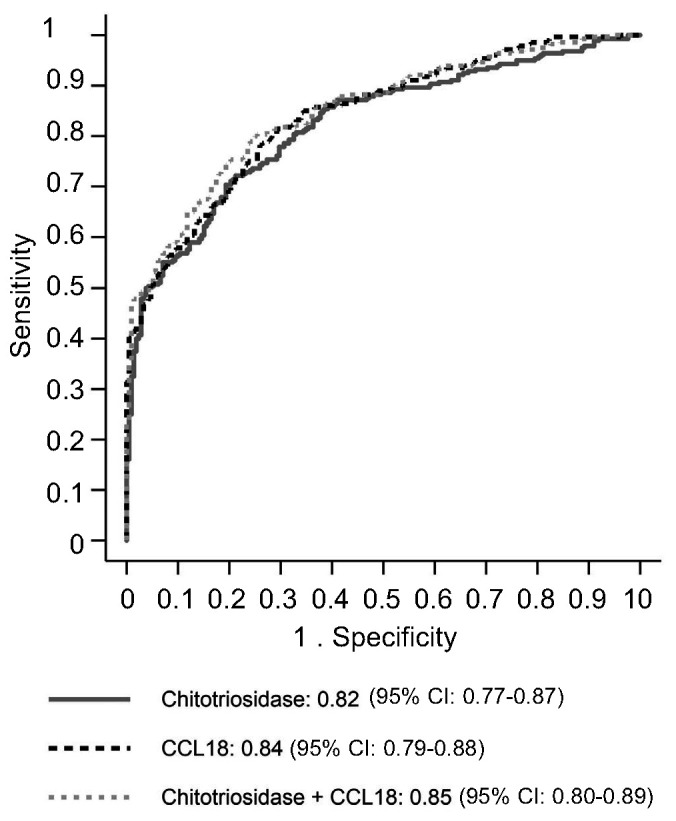
The AUC-ROC curves receiver operating characteristics curves for chitotriosidase activity, CCL18 concentration, and the combination of these two biomarkers. CI: Confidence Interval. AUC: area under the curve; ROC: receiver operating characterists.
Our meta-analysis corroborates and extends evidence from previous studies showing that chitotriosidase activity and CCL18 concentration relate to visceral and hematological parameter abnormalities in treated and untreated patients.8,12,13,15 A noteworthy exception was severe anemia (< 8 g/dL), for which CCL18 showed a lower AUC-ROC curve than chitotriosidase activity. This observation is consistent with our failure to show a significant difference in the CCL18 level for patients with and without severe anemia. Interestingly, weak or variable associations were previously reported between hemoglobin concentration and chitotriosidase activity or CCL18 concentration.12,13,15 A potential explanation might relate to the failure of physicians to correctly diagnose concurrent causes of severe anemia in GD patients. Yet, this cannot explain why the accuracy differed between chitotriosidase activity and CCL18 concentration. Due to the limited number of severe anemia cases in our meta-analytical sample (i.e., 7 among the 1,036 observations from 309 participants), our findings should be interpreted with caution and deserve confirmation in independent samples. Consistent with previous studies,47,48 we found that splenectomy altered the accuracy of chitotriosidase activity in discriminating patients with the primary outcome.
Another striking result of our meta-analysis is that the addition of chitotriosidase activity did not improve the accuracy of serum CCL18 concentration in identifying patients with the primary outcome, as shown by the unchanged AUC-ROC curve. This negative finding suggests that combining chitotriosidase activity and serum CCL18 concentration would have a limited incremental value, probably because they convey redundant information on Gaucher cell burden.14,49-51
Table 2.
One-stage paired comparisons of AUC-ROC curves for chitotriosidase activity and CCL18 concentration for discriminating type I Gaucher disease patients with prespecified outcomes.

The recent glucosylsphingosine biomarker9 may provide complementary information about the pathophysiological process in GD.14,33 Indeed, plasma glucosylsphingosine relates to sphingolipid turnover while chitotriosidase activity and CCL18 concentration are indicative of overall Gaucher cell mass. Glucosylsphingosine appeared to be highly sensitive and specific for the primary diagnosis and severity assessment of GD in previous studies.33,52 It would be interesting to compare the accuracy of glucosylsphingosine and CCL18 concentration using the same approach as in our meta-analysis although insufficient data exist to perform this analysis to date. Meanwhile a prospective comparative study of the three biomarkers is warranted to evaluate their contribution in monitoring GD activity.
In contrast to CCL18, glucosylsphingosine can be measured in dried blood spot and has emerged as a second-tier biomarker for newborn screening for GD.53 Measuring biomarkers in dried blood spot samples might facilitate GD activity monitoring for clinics where the technology is not available.
A limitation of our primary composite outcome is that it did not capture bone manifestations, despite their major impact on the quality of life.2,24 Previous studies reported that chitotriosidase activity level correlates with bone complications, 13 history of osteonecrosis,17 and number of anatomical sites of osteonecrosis,17 although these associations were inconsistent.12,15 Similarly, CCL18 concentration has been inconsistently associated with history of osteonecrosis and number of anatomical sites of osteonecrosis.12,17 Unfortunately, information on symptomatic bone events was available in only one study included in our meta-analysis,17 and we could not reach a definitive conclusion for this outcome.
There are several potential explanations for the see-mingly inconsistent findings between our analysis of bone events and the study by Deegan et al.17 First, we used a stricter definition of bone manifestations, which included symptomatic major events (i.e., skeletal fracture, osteonecrosis, or avascular necrosis) that could be dated and excluded progressive alterations (i.e., osteoporosis, Erlenmeyer flask femur deformity). Second, we took into account the data temporality by including bone events that occurred within the previous 12 months of biomarker analysis while Deegan et al.17 analyzed the previous history of bone events that occurred during the lifetime. Third, the unit of analysis was different between our analysis (218 observations nested within 97 patients) and the study by Deegan et al.17 (100 patients).
Two other clinical trials included in our meta-analysis recorded bone mineral density,30,34 but they did not report clinically relevant endpoints for bone disease (i.e., skeletal fracture, osteonecrosis, or avascular necrosis). Thus, the imprecise definition of diagnostic terms, the lack of standardized criteria for bone assessment, and the slow improvement in bone outcomes after specific treatment probably hamper the identification of a relationship between biomarkers and this important clinical outcome.54-57 Another approach in assessing skeletal disease is to measure residual biomarker levels after remission of visceral disease (reversal of hepatomegaly in splenectomized patients or reversal of hepatosplenomegaly in intact spleen patients).
Our findings have clinical implications for GD monitoring. Due to the comparable accuracy of the two biomarkers, the impact of common genetic polymorphism on chitotriosidase activity and the analytical robustness of CCL18 assays, it should be logical to recommend measuring CCL18 concentration rather than chitotriosidase activity as a biomarker of Gaucher cell burden and visceral and hematological damage.
Measuring chitotriosidase activity in addition to CCL18 concentration does not appear useful in the routine practice or in clinical research. Indeed, the two biomarkers are strongly correlated and convey redundant information on GD severity. Additionally, chitotriosidase activity has limited incremental value in improving CCL18 concentration accuracy, as reflected by the unchanged AUC-ROC curve estimate.
Our meta-analysis demonstrates that chitotriosidase activity and CCL18 concentration relate to visceral and hematological parameters that can be easily monitored in the routine practice. Hence, additional studies are needed to further investigate the usefulness of these two biomar-kers. In the current practice, they are used to monitor an early response to treatment although their value for initiating specific treatment or adjusting treatment dosage remains questioned. Only prospective patient management studies or randomized controlled trials can establish the effectiveness of biomarker-guided therapy. The lack of robust data and measurement heterogeneity may also explain why some of the GD severity scores do not include biomarkers.58 Lastly, the relationships between GD biomarkers and patient-centered outcomes, including quality of life, fatigue, chronic pain, and restriction of daily activities should be evaluated.55,59
This meta-analysis analyzed IPD from nine primary studies that were completed since 2004, summarizing the most recent available evidence on the accuracy of chitotriosidase activity and CCL18 concentration in assessing type 1 GD severity. All studies included in this meta-analysis evaluated both biomarkers in the same patients, providing unconfounded comparative accuracy estimates.60 Moreover, our findings have strong generali-zability because we combined primary studies that enrolled various populations of patients worldwide.60
However, our meta-analysis also has a few caveats that must be considered. First, the finding interpretation is inevitably limited by the unavailability of IPD from the other 14 potentially eligible primary studies. Second, there was substantial between-study heterogeneity in chitotriosidase activity and, to a lesser extent, in CCL18 concentration. The lack of assay standardization may have contributed to this heterogeneity, although chitotriosidase activity and CCL18 concentration were measured at a single central core laboratory in five clinical trials. Therefore, we used mixed-effect regressions to account for this heterogeneity and performed sensitivity and subgroup analyses that supported the robustness of the summary estimates.
In conclusion, CCL18 concentration is as accurate as chitotriosidase activity in assessing hematological and visceral parameters of GD activity and can be measured in patients who are homozygous for null chitotriosidase variants. However, the observed between-study heterogeneity and the limitations of this meta-analysis should encourage the international community to i) implement a strategy of homogenization and standardization of dosage techniques, more than 15 years after their implementation in clinical practice and ii) set up a prospective large-scale study to evaluate GD biomarkers, including glucosyl-sphingosine. This latter biomarker seems interesting from a pathophysiological perspective, but its superiority over chitotriosidase activity and CCL18 concentration remains to be documented.
Supplementary Material
Acknowledgment
The authors are indebted to the investigators of all primary studies for their contributions that allowed carrying out this secondary analysis. They are also grateful to Zoya Panahloo, Farid Merazi, Hak-Myung Lee, Bjorn Mellgard, Michael Craig, and Hartmann Wellhoefer from Shire, Lexington, MA, for sharing individual participant data from clinical trials and facilitating the pooled analysis. Statistical analysis was performed within the framework of the Grenoble Alpes Data Institute (ANR-15-IDEX-02).
References
- 1.Zimran A, Elstein D. Chapter 72: Gaucher disease and related lysosomal storage diseases. Kaushansky K, Lichtman MA, Prchal JT, et al., eds. Williams Hematology. New York: McGraw-Hill Education; 2016:1121-1131. [Google Scholar]
- 2.Charrow J, Andersson HC, Kaplan P, et al. The Gaucher registry: demographics and disease characteristics of 1698 patients with Gaucher disease. Arch Intern Med. 2000; 160(18):2835-2843. [DOI] [PubMed] [Google Scholar]
- 3.Grabowski GA, Zimran A, Ida H. Gaucher disease types 1 and 3: phenotypic characterization of large populations from the ICGG Gaucher Registry. Am J Hematol. 2015; 90(Suppl 1):S12-18. [DOI] [PubMed] [Google Scholar]
- 4.Zimran A, Belmatoug N, Bembi B, et al. Demographics and patient characteristics of 1209 patients with Gaucher disease: descriptive analysis from the Gaucher Outcome Survey (GOS). Am J Hematol. 2018;93(2): 205-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nalysnyk L, Rotella P, Simeone JC, et al. Gaucher disease epidemiology and natural history: a comprehensive review of the literature. Hematology. 2017;22(2):65-73. [DOI] [PubMed] [Google Scholar]
- 6.Mistry PK, Belmatoug N, Vom Dahl S, et al. Understanding the natural history of Gaucher disease. Am J Hematol. 2015; 90(Suppl 1):S6-S11. [DOI] [PubMed] [Google Scholar]
- 7.Weinreb NJ, Aggio MC, Andersson HC, et al. Gaucher disease type 1: revised recommendations on evaluations and monitoring for adult patients. Semin Hematol. 2004; 41(4 Suppl 5):S15-22. [DOI] [PubMed] [Google Scholar]
- 8.Boot RG, Verhoek M, de Fost M, et al. Marked elevation of the chemokine CCL18/PARC in Gaucher disease: a novel surrogate marker for assessing therapeutic intervention. Blood. 2004;103(1):33-39. [DOI] [PubMed] [Google Scholar]
- 9.Dekker N, van Dussen L, Hollak CE, et al. Elevated plasma glucosylsphingosine in Gaucher disease: relation to phenotype, storage cell markers, and therapeutic response. Blood. 2011;118(16):e118-127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hollak CE, van Weely S, van Oers MH, et al. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest. 1994;93(3):1288-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Aerts JM, Kallemeijn WW, Wegdam W, et al. Biomarkers in the diagnosis of lysosomal storage disorders: proteins, lipids, and inhibodies. J Inherit Metab Dis. 2011;34(3):605-619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deegan PB, Moran MT, McFarlane I, et al. Clinical evaluation of chemokine and enzymatic biomarkers of Gaucher disease. Blood Cells Mol Dis. 2005;35(2):259-267. [DOI] [PubMed] [Google Scholar]
- 13.van Dussen L, Hendriks EJ, Groener JE, et al. Value of plasma chitotriosidase to assess nonneuronopathic Gaucher disease severity and progression in the era of enzyme replacement therapy. J Inherit Metab Dis. 2014;37(6):991-1001. [DOI] [PubMed] [Google Scholar]
- 14.Smid BE, Ferraz MJ, Verhoek M, et al. Biochemical response to substrate reduction therapy versus enzyme replacement therapy in Gaucher disease type 1 patients. Orphanet J Rare Dis. 2016;11:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schoonhoven A, Rudensky B, Elstein D, et al. Monitoring of Gaucher patients with a novel chitotriosidase assay. Clin Chim Acta. 2007;381(2):136-139. [DOI] [PubMed] [Google Scholar]
- 16.Boot RG, Renkema GH, Verhoek M, et al. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J Biol Chem. 1998;273(40):25680-25685. [DOI] [PubMed] [Google Scholar]
- 17.Deegan PB, Pavlova E, Tindall J, et al. Osseous manifestations of adult Gaucher disease in the era of enzyme replacement therapy. Medicine (Baltimore). 2011; 90(1):52-60. [DOI] [PubMed] [Google Scholar]
- 18.Riley RD, Lambert PC, Abo-Zaid G. Metaanalysis of individual participant data: rationale, conduct, and reporting. BMJ. 2010; 340:c221. [DOI] [PubMed] [Google Scholar]
- 19.Stewart LA, Clarke MJ. Practical methodology of meta-analyses (overviews) using updated individual patient data. Cochrane Working Group. Stat Med. 1995;14(19): 2057-2079. [DOI] [PubMed] [Google Scholar]
- 20.Stewart LA, Clarke M, Rovers M, et al. Preferred reporting items for systematic review and meta-analyses of individual participant data: the PRISMA-IPD Statement. JAMA. 2015;313(16):1657-1665. [DOI] [PubMed] [Google Scholar]
- 21.Raskovalova T, Deegan PB, Yang R, et al. Plasma chitotriosidase activity versus CCL18 level for assessing type I Gaucher disease severity: protocol for a systematic review with meta-analysis of individual participant data. Syst Rev. 2017;6(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hollak CE, Belmatoug N, Cole JA, et al. Characteristics of type I Gaucher disease associated with persistent thrombocytopenia after treatment with imiglucerase for 4-5 years. Br J Haematol. 2012;158(4):528-538. [DOI] [PubMed] [Google Scholar]
- 23.WHO. Haemoglobin concentrations for the diagnosis of anaemia and assessment of severity. Vitamin and Mineral Nutrition Information System. Geneva: World Health Organization; 2011. [Google Scholar]
- 24.Bland JM, Altman DG. Transformations, means, and confidence intervals. BMJ. 1996;312(7038):1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bland JM, Altman DG. The use of transformation when comparing two means. BMJ. 1996;312(7039):1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burke DL, Ensor J, Riley RD. Meta-analysis using individual participant data: one-stage and two-stage approaches, and why they may differ. Stat Med. 2017;36(5):855-875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stewart GB, Altman DG, Askie LM, et al. Statistical analysis of individual participant data meta-analyses: a comparison of methods and recommendations for practice. PLoS One. 2012;7(10):e46042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ben Turkia H, Gonzalez DE, Barton NW, et al. Velaglucerase alfa enzyme replacement therapy compared with imiglucerase in patients with Gaucher disease. Am J Hematol. 2013;88(3):179-184. [DOI] [PubMed] [Google Scholar]
- 29.Elstein D, Mehta A, Hughes DA, et al. Safety and efficacy results of switch from imiglucerase to velaglucerase alfa treatment in patients with type 1 Gaucher disease. Am J Hematol. 2015;90(7):592-597. [DOI] [PubMed] [Google Scholar]
- 30.Gonzalez DE, Turkia HB, Lukina EA, et al. Enzyme replacement therapy with velaglucerase alfa in Gaucher disease: results from a randomized, double-blind, multinational, Phase 3 study. Am J Hematol. 2013; 88(3):166-171. [DOI] [PubMed] [Google Scholar]
- 31.Zimran A, Altarescu G, Philips M, et al. Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood. 2010;115(23): 4651-4656. [DOI] [PubMed] [Google Scholar]
- 32.Berger J, Vigan M, Pereira B, et al. Intra-monocyte pharmacokinetics of imiglucerase supports a possible personalized management of Gaucher disease type 1. Clin Pharmacokinet. 2019;58(4):469-482. [DOI] [PubMed] [Google Scholar]
- 33.Murugesan V, Chuang WL, Liu J, et al. Glucosylsphingosine is a key biomarker of Gaucher disease. Am J Hematol. 2016; 91(11):1082-1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zimran A, Brill-Almon E, Chertkoff R, et al. Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood. 2011;118(22):5767-5773. [DOI] [PubMed] [Google Scholar]
- 35.Zimran A, Gonzalez-Rodriguez DE, Abrahamov A, et al. Safety and efficacy of two dose levels of taliglucerase alfa in pediatric patients with Gaucher disease. Blood Cells Mol Dis. 2015;54(1):9-16. [DOI] [PubMed] [Google Scholar]
- 36.Andrade-Campos MM, Garcia-Sobreviela MP, Medrano-Engay B, et al. Assessing underlying chronic inflammation status in type 1 Gaucher disease patients (GD1): Involvement of lipocaline (LCN2) and other monocyte cytokines. Mol Genet Metab. 2017;120:S22. [Google Scholar]
- 37.Boot RG, Verhoek M, Langeveld M, et al. CCL18: a urinary marker of Gaucher cell burden in Gaucher patients. J Inherit Metab Dis. 2006;29(4):564-571. [DOI] [PubMed] [Google Scholar]
- 38.Di Rocco M, Giona F, Carubbi F, et al. A new severity score index for phenotypic classification and evaluation of responses to treatment in type I Gaucher disease. Haematologica. 2008;93(8):1211-1218. [DOI] [PubMed] [Google Scholar]
- 39.Giraldo P, Alfonso P, Atutxa K, et al. Realworld clinical experience with long-term miglustat maintenance therapy in type 1 Gaucher disease: the ZAGAL project. Haematologica. 2009;94(12):1771-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giraldo P, Irun P, Alfonso P, et al. Evaluation of Spanish Gaucher disease patients after a 6-month imiglucerase shortage. Blood Cells Mol Dis. 2011;46(1):115-118. [DOI] [PubMed] [Google Scholar]
- 41.Giraldo P, Pérez-López J, Núñez R, et al. Patients with type 1 Gaucher disease in Spain: a cross-sectional evaluation of health status. Blood Cells Mol Dis. 2016;56(1):23-30. [DOI] [PubMed] [Google Scholar]
- 42.Groener JE, Poorthuis BJ, Kuiper S, et al. Plasma glucosylceramide and ceramide in type 1 Gaucher disease patients: correlations with disease severity and response to therapeutic intervention. Biochim Biophys Acta. 2008;1781(1-2):72-78. [DOI] [PubMed] [Google Scholar]
- 43.Limgala RP, Ioanou C, Plassmeyer M, et al. Time of initiating enzyme replacement therapy affects immune abnormalities and disease severity in patients with Gaucher disease. PLoS One. 2016;11(12):e0168135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lukina E, Watman N, Dragosky M, et al. Eliglustat, an investigational oral therapy for Gaucher disease type 1: phase 2 trial results after 4 years of treatment. Blood Cells Mol Dis. 2014;53(4):274-276. [DOI] [PubMed] [Google Scholar]
- 45.Mistry PK, Lukina E, Ben Turkia H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA. 2015;313(7):695-706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pastores GM, Petakov M, Giraldo P, et al. A phase 3, multicenter, open-label, switchover trial to assess the safety and efficacy of taliglucerase alfa, a plant cell-expressed recombinant human glucocerebrosidase, in adult and pediatric patients with Gaucher disease previously treated with imiglucerase. Blood Cells Mol Dis. 2014;53(4):253-260. [DOI] [PubMed] [Google Scholar]
- 47.Drugan C, Drugan TC, Grigorescu-Sido P, et al. Modelling long-term evolution of chitotriosidase in non-neuronopathic Gaucher disease. Scand J Clin Lab Invest. 2017; 77(4):275-282. [DOI] [PubMed] [Google Scholar]
- 48.Stirnemann J, Belmatoug N, Vincent C, et al. Bone events and evolution of biologic markers in Gaucher disease before and during treatment. Arthritis Res Ther. 2010;12(4): R156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boven LA, van Meurs M, Boot RG, et al. Gaucher cells demonstrate a distinct macrophage phenotype and resemble alternatively activated macrophages. Am J Clin Pathol. 2004;122(3):359-369. [DOI] [PubMed] [Google Scholar]
- 50.Gordon S. Alternative activation of macrophages. Nat Rev Immunol 2003;3(1): 23-35. [DOI] [PubMed] [Google Scholar]
- 51.Mantovani A, Sica A, Sozzani S, et al. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677-686. [DOI] [PubMed] [Google Scholar]
- 52.Rolfs A, Giese AK, Grittner U, et al. Glucosylsphingosine is a highly sensitive and specific biomarker for primary diagnostic and follow-up monitoring in Gaucher disease in a non-Jewish, Caucasian cohort of Gaucher disease patients. PLoS One. 2013;8(11): e79732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gelb MH. Newborn screening for lysosomal storage diseases: methodologies, screen positive rates, normalization of datasets, second- tier tests, and post-analysis tools. Int J Neonatal Screen. 2018;4(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Charrow J, Scott CR. Long-term treatment outcomes in Gaucher disease. Am J Hematol. 2015;90(Suppl 1):S19-24. [DOI] [PubMed] [Google Scholar]
- 55.Hollak CE, Maas M, Aerts JM. Clinically relevant therapeutic endpoints in type I Gaucher disease. J Inherit Metab Dis. 24;24(Suppl 2):S97-105. [DOI] [PubMed] [Google Scholar]
- 56.Shemesh E, Deroma L, Bembi B, et al. Enzyme replacement and substrate reduction therapy for Gaucher disease. Cochrane Database Syst Rev. 2015;3:CD010324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sims KB, Pastores GM, Weinreb NJ, et al. Improvement of bone disease by imiglucerase (Cerezyme) therapy in patients with skeletal manifestations of type 1 Gaucher disease: results of a 48-month longitudinal cohort study. Clin Genet. 2008; 73(5):430-440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weinreb NJ, Cappellini MD, Cox TM, et al. A validated disease severity scoring system for adults with type 1 Gaucher disease. Genet Med. 2010;12(1):44-51. [DOI] [PubMed] [Google Scholar]
- 59.Johnston BC, Miller PA, Agarwal A, et al. Limited responsiveness related to the minimal important difference of patient-reported outcomes in rare diseases. J Clin Epidemiol. 2016;79(11):10-21. [DOI] [PubMed] [Google Scholar]
- 60.Leeflang MM, Deeks JJ, Gatsonis C, et al. Systematic reviews of diagnostic test accuracy. Ann Intern Med. 2008;149(12):889-897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.