

Abstract
Objectives:
Secondhand smoke (SHS) is a major risk factor for lung cancer in nonsmokers. DNA damage-derived mutagenicity is a well-established mechanism of SHS-carcinogenicity; however very little is known about the impact of SHS exposure on the epigenome.
Materials and methods:
We have investigated whether exposure to SHS can modulate the expression of key epigenetic regulators responsible for the establishment and/or maintenance of DNA methylation and histone modification patterns in vivo. We have sub-chronically exposed mice to a mutagenic but non-tumorigenic dose of SHS, and subsequently determined the expression levels of major epigenetic modifiers in the lungs of SHS-exposed mice, immediately after termination of exposure and following 7-month recovery in clean air.
Results and conclusion:
Quantification of the expression of genes encoding DNA methyltransferases (Dnmt1, Dnmt3a, Dnmt3b and Dnmt3l), methyl binding domain proteins (Mecp2, Mbd2 and Mbd3) and histone deacetylases (Hdac1 and Hdac2) by quantitative reverse-transcription polymerase chain reaction analysis showed modest but not statistically significant differences in the relative transcription of these key epigenetic regulators between SHS-exposed mice and age-matched controls. The non-significant changes in the expression of main epigenetic modifiers in SHS-exposed mice imply that SHS may predominantly induce genotoxic effects, particularly at non-tumorigenic doses, whereas epigenetic effects may only be secondary and manifest en route to tumor formation.
Keywords: DNA methyltransferases (DNMTs), Epigenome, Histone deacetylases (HDACs), Lung cancer, Methyl-CpG binding domain proteins (MBDs), Nonsmokers
1. Introduction
Secondhand smoke (SHS), also known as passive smoke, involuntary smoke, or environmental tobacco smoke, is a significant public health concern, and a major risk factor for lung cancer and other diseases [1–4]. Recent estimates show that in 2004, SHS has caused a total of 603,000 deaths, equivalent to 1.0% of all deaths worldwide [3]. In the United States alone, about 10,000–15,000 never smokers die annually from lung cancer [5]. One third of these deaths are ascribed to exposure to SHS [5]. Despite the extensive epidemiologic data supporting the link between SHS exposure and lung cancer, the underlying molecular mechanisms remain largely unknown [2,4,6,7]. A better understanding of the mechanism(s) of action of SHS in the genesis and development of lung cancer in non-smokers can help devise future strategies for prevention, early detection, treatment and prognosis of this malignancy [6–8].
Recent work in our laboratory has established a genotoxic mode of action for SHS based on the ability of this carcinogen to induce DNA damage and mutation that may lead to deregulation of cancer-related genes [9–12]. Most recently, we have also identified epigenetic modulation of gene expression as an alternative mechanism of chemical carcinogenesis in vivo [11]. Whether SHS can impact the epigenetic machinery in the lungs of nonsmokers, however, remains to be determined [7,13]. Investigating the epigenetic regulation of gene expression by SHS is paramount to the identification of potential targets of cancer therapy because epigenetic alterations are reversible through pharmacologic interventions and/or genetic manipulations [14–17].
Genetic defects in the epigenetic enzymes, including those involved in the DNA methylation and histone modification pathways, are frequently found in human cancer [18–21]. Though DNA methylation and histone modifications require different sets of enzymes and chemical reactions for mark deposition, the two systems are known to mutually regulate each other during mammalian development and cancer [22–24]. In mammals, methylation at the 5-position of cytosine is catalyzed by DNA methyltransferases (DNMTs) [25,26]. DNMT1 is the most abundant methyltransferase; it binds preferentially to hemimethylated DNA and is involved in the maintenance of DNA methylation patterns in the daughter strands during DNA replication [27]. DNMT3A and DNMT3B related proteins bind mostly to unmethylated CpG dinucleotides and are responsible for de novo methylation during development [25,26]. Though lacking catalytic activity itself, DNMT3L enhances the binding activities of the de novo methyltransferases [28]. Expression levels of DNMTs, in particular those of DNMT1, DNMT3A, and DNMT3B, have been shown to be elevated in various types of human cancer, including lung cancer [20,26].
Methyl-CpG binding domain (MBD) proteins are a prominent class of epigenetic enzymes that play a crucial role in the transcriptional state of the epigenome [29]. MBD proteins bind to methylated DNA and coordinate the cross-talk between DNA methylation, histone modifications, and chromatin organization [30]. Disrupted gene expression of MBDs can affect the level of DNA methylation and has been detected in different types of human cancer, cancer cell lines and various mouse models of human cancer [30]. Another important family of epigenetic modifiers is histone deacetylases (HDACs) [31]. HDACs are known to play a key role in several physiological and pathological states, including cancer [32]. HDACs are recruited to chromatin remodeling complexes via the MBD proteins [33] and catalyze the removal of acetyl groups from lysine residues on histone tails, leading to chromatin condensation and gene transcriptional repression [31,32].To date, several classes of HDACs have been identified [31,34]. Aberrant expression of Class I subfamily of HDACs (i.e., HDAC1 and HDAC2) has been observed in a variety of human cancers, including lung cancer, and is often associated with higher stage and poor prognosis [31,34–36].
Accumulating evidence shows that genetic mutations in epigenetic regulators are associated with an altered epigenome in human cancer [18–21]. The re-shaping of the epigenome in human cancer has also been attributed to exposure to a variety of mutagens and carcinogens [37,38]. To determine whether epigenetic aberrancies in human cancer are indeed caused by exposure to mutagenic compounds, we have used the example of SHS-carcinogenicity and investigated whether exposure to SHS can influence the expression of key epigenetic modifiers in vivo. To this end, we have sub-chronically exposed mice, whole body, to SHS in exposure chambers of a microprocessor-controlled smoking machine, and subsequently determined the expression levels of major epigenetic regulators in the lungs of SHS-exposed mice. Total RNA was extracted from the lungs of SHS-exposed mice, immediately after discontinuation of exposure and following 7-month recovery in clean air to establish the short-term and long-term effects, respectively, of SHS. We have used standard quantitative reverse-transcription polymerase chain reaction (qRT-PCR) analysis to examine changes in the expression of genes encoding DNA methyltransferases (Dnmt1, Dnmt3a, Dnmt3b and Dnmt3l), methyl binding domain proteins (Mecp2, Mbd2 and Mbd3) and histone deacetylases (Hdac1 and Hdac2). Our results indicate that sub-chronic exposure of mice to SHS at a non-tumorigenic dose is insufficient to induce statistically significant changes in the transcription of key epigenetic modifiers.
2. Material and methods
2.1. Animal care and maintenance
Animals were bred and maintained in accordance with the recommendations of the National Institutes of Health provided in the Guide for the Care and Use of Laboratory Animals. The study was approved by the Institutional Animal Care and Use Committee (IACUC). All mice were kept in polypropylene cages in groups of 2–3 animals per cage, and housed in an air-conditioned animal room with ambient temperature of 21 ±1 °C and relative humidity of 55% with 12-h light/dark cycle. Throughout all experiments, including the exposure phase and recovery period, the mice had access to food (PicoLab Rodent Diet 20, PMI Nutrition International, LLC., Brentwood, MO) and water ad libitum.
2.2. SHS exposure
The protocol used for SHS treatment of mice has been previously described in Refs. [9,12]. The breeding colony of mice used in the present study as well as in our previous reports [9,12] was originally obtained from Stratagene (Stratagene, La Jolla, CA). Stratagene was later acquired by Agilent Technologies Inc. (Agilent Technologies Inc., Santa Clara, CA). Briefly, male adult mice (6–8 weeks old) on a C57BL/6 genetic background were divided into two treatment groups (n = 5 per group), including (I) four months exposure: T4 and (II) four months exposure + seven months recovery in clean air: T4+7. In our previously published studies, we have confirmed that this treatment protocol is well tolerated by mice, and can efficiently induce DNA damage and mutation as well as cause deregulation of gene expression [9,10,12,39]. We have also verified that short-term and long-term effects of SHS on the transcriptome can be established in this mouse model after a 4-month SHS exposure period and following seven months recovery in clean air, respectively [12]. SHS exposure was performed by burning 3R4F Reference Kentucky cigarettes (0.73 mg nicotine per cigarette, purchased from the Tobacco Research Institute, University of Kentucky, Lexington, KY) using a custom-made smoking machine (model TE-10, Teague Enterprises, Davis, CA) as previously described [9,12]. The TE-10 smoking machine was programmed to generate a mixture of sidestream smoke (89%) and mainstream smoke (11%), which is conventionally used to mimic SHS for experimental purposes [10,40].
All mice underwent an acclimatization period during which they were gradually exposed to incremental doses of SHS [9,12]. Following the acclimatization period, the animals were maintained on a SHS exposure regimen, which consisted of whole body exposure to SHS for 5 h per day, 5 days per week, for a duration of four months. Control mice were handled similarly to SHS-exposed animals, and underwent mock-exposure to filtered high-efficiency particulate-air (HEPA). At the end of SHS/mock-exposure and following the recovery period, the mice were euthanized by CO2 asphyxiation. Lungs were harvested, snap-frozen in liquid nitrogen, and kept at −80 °C until further analysis.
2.3. Quantitative reverse-transcription polymerase chain reaction
Standard quantitative reverse-transcription polymerase chain reaction (qRT-PCR) was used to measure the expression level of major DNA methyltransferases, including Dnmt1, Dnmt3a, Dnmt3b, and Dnmt3l; methyl-binding domain proteins, including Mecp2, Mbd2, Mbd3; and histone deacetylases, including Hdac1 and Hdac2 [41,42]. Total RNA was extracted from the lungs of experimental and control mice as previously described [12]. DNase-treated RNA (0.5 μg) from T4 and T4+7 mice was reverse transcribed into complementary DNA (cDNA) using SuperScript® VILO™ cDNA Synthesis kit (Invitrogen). The expression level of target genes was determined by qRT-PCR using the TaKaRa SYBR® Premix Ex TaqTM II (Tli RNaseH Plus) (Clontech Laboratories, Inc., Mountain View, CA, USA) and the CFX96 Touch™ Real-Time PCR detection system (Bio-Rad). All experiments were performed in triplicate and fold changes were determined using the 2−ΔΔCt method as previously described [41,42]. Data were normalized using the endogenous housekeeping gene, Gapdh, as reference. The primer sets used for qRT-PCR are available upon request.
3. Results and discussion
Exposure to SHS is a major risk factor for lung cancer in non-smokers [7]. However, the underlying molecular mechanism(s) of SHS-carcinogenicity is far from well-understood [7]. Recently, we have shown DNA damage-derived mutagenicity of SHS [9,10], and epigenetic deregulation of gene expression by a chemical constituent of SHS (i.e., benzo[a]pyrene) [11] in mice in vivo. The objective of this study was to determine whether SHS can indeed produce cancer-relevant changes in the epigenetic machinery in the same model system. Because aberrant DNA methylation and conformational changes in chromatin through post-translational modification of histones are the best-established mechanisms of carcinogenesis [17,43,44], we therefore investigated whether exposure to SHS can modulate the expression of key epigenetic regulators responsible for the establishment and/or maintenance of DNA methylation and histone modification patterns. We note that despite interspecies differences, the modulation of gene expression by cigarette smoke is mostly similar between humans and mice [45]. In addition, there is striking pathobiological resemblance between mouse and human lung cancer [46]. This validates the utility of mouse models for studying smoke-related carcinogenesis in humans [45].
Using genome-wide gene expression microarray analysis (Affymetrix Inc., Santa Clara, CA), we have previously characterized the lung transcriptome of mice exposed to sub-chronic doses of SHS [12]. Whereas we found a strong correlation between SHS exposure and aberrant gene expression (both reversible and irreversible) in cancer-related functional networks, we observed very subtle differences in the transcription of epigenetic modifiers between SHS-exposed mice and controls [12]. However, the microarray technology is known to suffer from inadequate sensitivity for detection of low-abundance transcripts and ratio compression that result in consistent underestimation of mRNA expression level ratios [47]. To circumvent this limitation of the microarray analysis, we herein used the ‘gold standard’ of gene expression analysis, i.e., quantitative real-time RT-PCR [48], to precisely measure the expression level of key epigenetic modifiers in the lungs of mice exposed to SHS using the same protocol as used in our previous report [12].
In the present study, we have exposed mice to a mutagenic, yet, non-tumorigenic dose of SHS for 5 h/day, 5 days/week for 4 months in exposure chambers of a microprocessor-controlled smoking machine. Subsequently, we have analyzed the pulmonary RNA of the SHS-exposed mice, immediately after termination of exposure (i.e., T4) and following seven months recovery in clean air (i.e., T4+7) to find the short-term and long-term effects, respectively, of SHS. Quantitative real-time RT-PCR was used to accurately quantify the mRNA levels of major DNA methyltransferases (DNMTs), methyl-CpG binding domain proteins (MBDs), and histone deacetylases (HDACs) in the lungs of SHS-exposed mice versus controls. Under the experimental conditions used in this study, we found no statistically significant differences in the transcription of Dnmt1, Dnmt3a, Dnmt3b and Dnmt3l genes between SHS-exposed mice and their age-matched controls (see, Fig. 1). Modest differences in gene expression were observed in T4+7 mice, with a 33–35% increase in de novo methyltransferases Dnmt3a and Dnmt3b, as compared to control. In T4+7 mice, relative transcription ofMbd2 and Mecp2 also increased to 35% and 30% respectively, while Mbd3 levels remained essentially unaffected (see, Fig. 2). These modest changes in the expression of major enzymes regulating CpG methylation patterns are consistent with our previous observations in which we found comparable patterns of DNA methylation between SHS-exposed mice and controls [39].
Fig. 1.
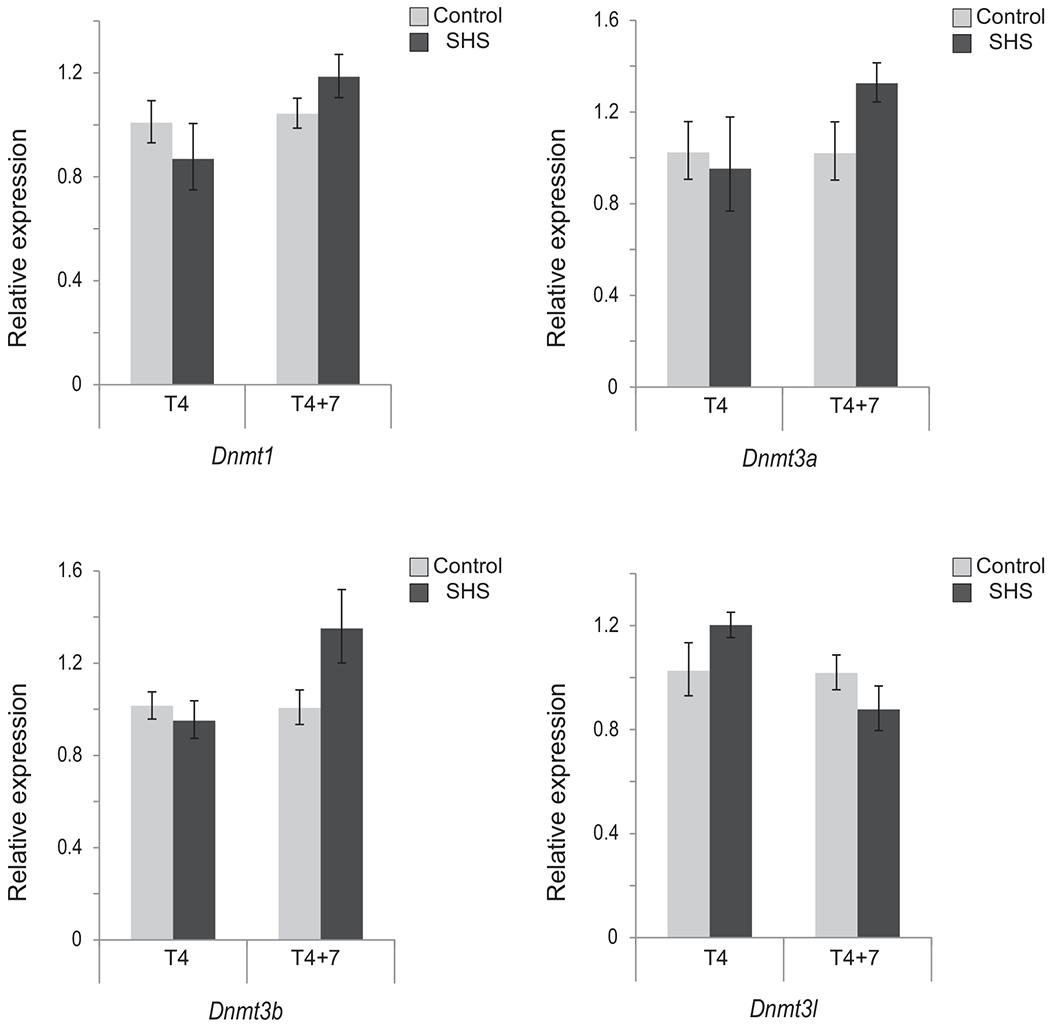
Expression of Dnmt1, Dnmt3a, Dnmt3b and Dnmt3l in SHS exposed mice versus control. Total RNA was extracted from the lungs of mice exposed to SHS for four months, immediately after discontinuation of exposure (T4) and following 7-month recovery in clean air (T4+7). The transcription level of genes encoding major DNMTs was examined by qRT-PCR using the 2−ΔΔCt method [41]. Bars represent the mean normalized expression (±SD) of triplicate samples in SHS-exposed mice and age-matched controls. Data were normalized using the endogenous housekeeping gene, Gapdh, as reference and controls as calibrators. Marginal differences in transcription levels between SHS-exposed and control mice were not statistically significant (Fisher’s exact test).
Fig. 2.
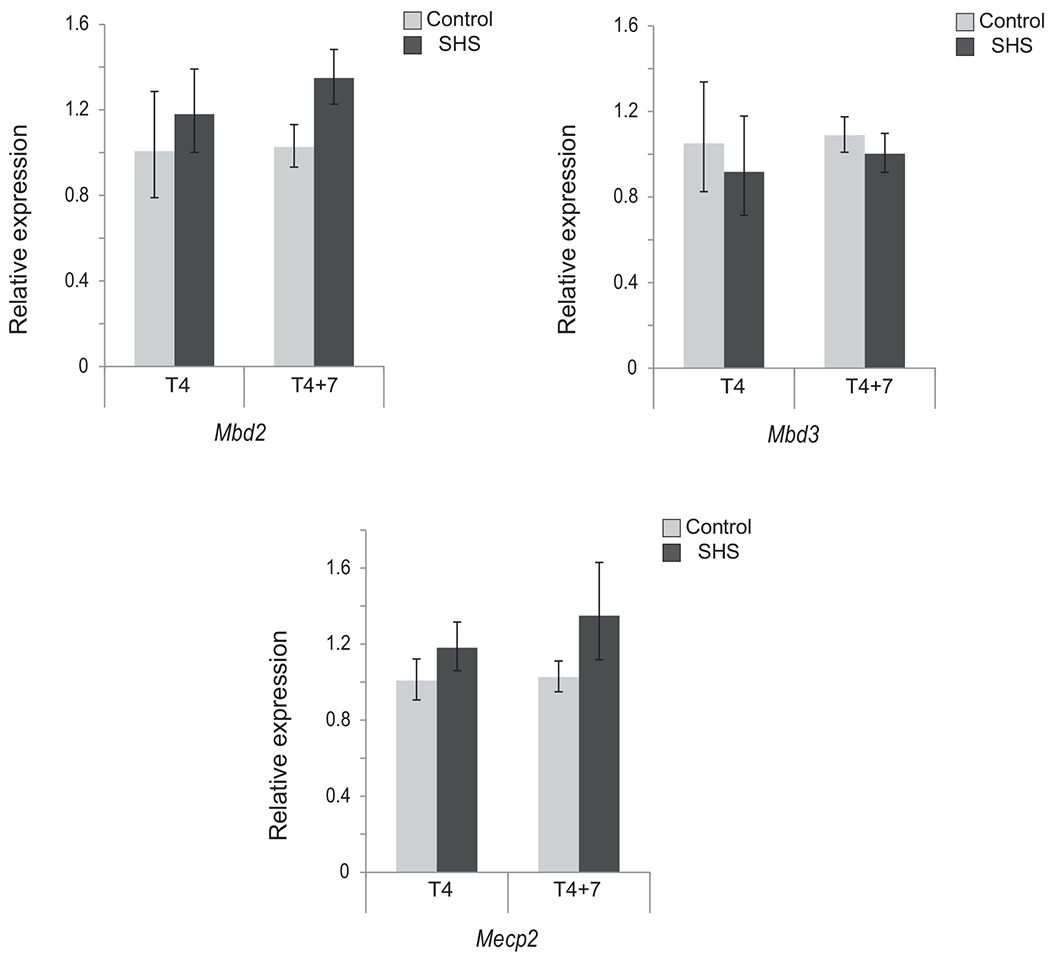
Expression of Mbd2, Mbd3, and Mecp2 in SHS exposed mice versus control. The transcription level of genes encoding major MBDs was examined in T4and T4+7 mice by qRT-PCR using the 2−ΔΔCt method [41]. Bars represent the mean normalized expression (±SD) of triplicate samples in SHS-exposed mice and age-matched controls. Data were normalized using the endogenous housekeeping gene, Gapdh, as reference and controls as calibrators. Marginal differences in transcription levels between SHS-exposed and control mice were not statistically significant (Fisher’s exact test).
Earlier reports have demonstrated that cigarette smoke causes hyperacetylation of histones and decreased histone deacetylase activity in the lungs of smokers and patients with chronic obstructive pulmonary disease (COPD), as well as in the lungs of rodents exposed to cigarette smoke [49,50].Thus, we examined the expression levels of two main HDACs, including Hdac1 and Hdac2. Our qRT-PCR analysis on the pulmonary RNA of SHS-exposed mice showed that Hdac1 and Hdac2 transcripts decreased only slightly in T4 mice (up to 26%), while returning to baseline levels in T4+7 mice (see, Fig. 3).
Fig. 3.
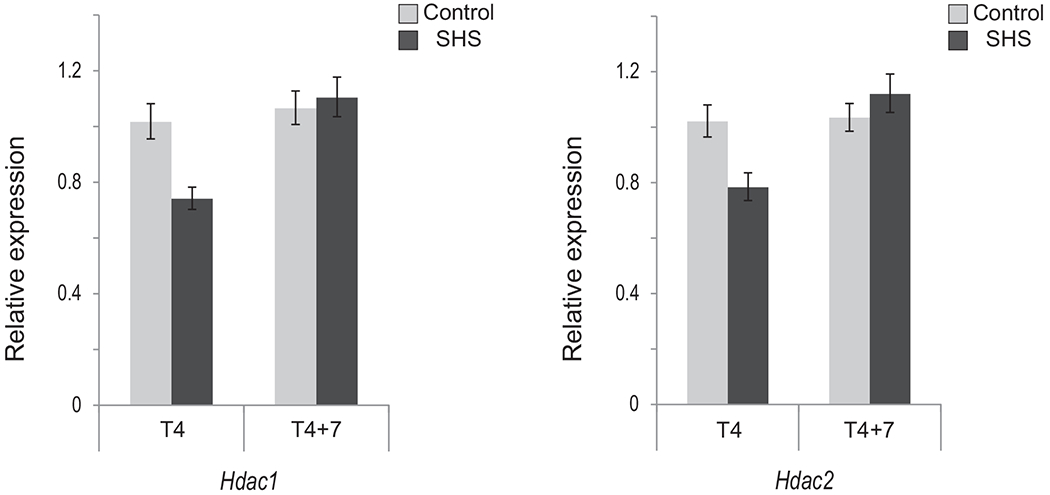
Expression of Hdac1 and Hdac2 in SHS exposed mice versus control. The transcription level of genes encoding Class I HDACs was examined in T4 and T4+7 mice by qRT–PCR using the 2−ΔΔCt method [41]. Bars represent the mean normalized expression (±SD) of triplicate samples in SHS-exposed mice and age-matched controls. Data were normalized using the endogenous housekeeping gene, Gapdh, as reference and controls as calibrators. Marginal differences in transcription levels between SHS-exposed and control mice were not statistically significant (Fisher’s exact test).
Recently, we and others have analyzed the transcriptional status of DNMTs, MBDs, and HDACs in relation to ectopic DNA methylation during multi-step chemically induced carcinogenesis in animal models [11,51–53]. In vivo treatment of mice with potent carcinogens, including urethane, ethylnitrosourea (ENU), diethylstilbestrol (DES), and benzo[a]pyrene (B[a]P), induced dynamic changes in the expression of major epigenetic regulators, including Dnmt1, Dnmt3a, Dnmt3b, Mbd1, Mbd2, Mecp2, Hdac1 and Hdac2 [11,51–53]. Deregulation of these epigenetic modifiers was associated with aberrant promoter CpG methylation of selected target genes [51–53] or found widespread in the genome [11]. Of importance, differences in DNA methylation patterns between carcinogen-treated animals and controls were readily detectable in apparently normal mice long before neoplastic lesion formation [11]. Unlike in the above reports, we have herein used SHS, which is a known weak mutagen and carcinogen [7,54,55]. To be consistent with our previous genetic [9,10], epigenetic [39], and transcriptomic [12] studies on SHS, we have also administered a mutagenic but non-tumorigenic dose of SHS since the focus of all our studies is on early biological effects of carcinogens. Comparing the findings of the present study with those of the above reports [11,51–53], one may ascribe the marginal changes in the expression levels of key epigenetic modifiers in our SHS-exposed mice, as opposed to those found in mice treated with powerful mutagens/carcinogens, to differences in test compounds and/or doses administered. Given the weak mutagenicity of SHS, it is also likely that this chemical is unable to sufficiently trigger active cell proliferation, a pre-requisite for establishment of epigenetic marks [27].
In addition to DNMTs, MBDs and HDACs, several other epigenetic regulators are emerging as key drivers, readers, editors or erasers of the epigenome [18–21]. Thus, one cannot rule out the possibility that SHS may modulate alternative epigenetic modifiers not analyzed in the present study. Potential candidates might include genes involved in miRNA regulation, chromatin remodeling, or other histone modifying enzymes [20]. Of relevance, our comprehensive analysis of the transcriptome in SHS-exposed mice indicated that several deregulated genes encode proteins that, alone or in combination with other remodeling factors, are involved in the deposition/reading of histone modifications, including ARID4B, KAT2B, and CHD4 [12]. Lastly, it is plausible that SHS may predominantly induce genotoxic effects, particularly at non-tumorigenic doses, whereas epigenetic effects may only be secondary and manifest en route to tumor formation.
In conclusion, we found no statistically significant changes in the expression of key epigenetic modifiers in the lungs of SHS-exposed mice as compared to age-matched controls (see, Figs. 1–3).
These findings are in agreement with those of our recent study in which we demonstrated comparable patterns of DNA methylation between SHS-exposed mice and controls [39]. In addition, the highly accurate single-gene RT-PCR analysis performed in this study reconfirms our previous genome-wide gene expression microarray analysis in which we showed fairly small changes in the transcription of epigenetic regulators in SHS-exposed mice [12]. Whether mutagenic SHS can cause genetic alterations in other master epigenetic regulators not analyzed in the present study and whether such alterations may result in reshaping of the epigenome in the lungs of nonsmokers is currently unknown and requires a thorough follow-up investigation.
Acknowledgments
All authors have directly participated in the planning, execution, and analysis of this study. They have read and approved the final version submitted. They had full access to all the data in the study, and had final responsibility for the decision to submit for publication. Work of the authors was supported by a grant from the American Cancer Society (RSG-11-083-01-CNE) to A.B. The sponsor of the study had no role in study design, data collection, data analysis, data interpretation, writing of the report, or in the decision to submit for publication.
Footnotes
Conflict of interest
All the authors declare no conflict of interest.
References
- [1].International Agency for Research on Cancer (IARC), Tobacco Smoke and Involuntary Smoking. World Health Organization (WHO), International Agency for Research on Cancer, Lyon, France, 2004, pp. 1191–1413. [Google Scholar]
- [2].The US. Surgeon General. The health consequences of involuntary exposure to tobacco smoke: A report of the surgeon general. 2006,12–136. [PubMed] [Google Scholar]
- [3].Oberg M, Jaakkola MS, Woodward A, Peruga A, Pruss-Ustun A, Worldwide burden ofdisease from exposure to second-hand smoke: a retrospective analysis ofdata from 192 countries, Lancet 377 (2011) 139–146. [DOI] [PubMed] [Google Scholar]
- [4].U.S. The Surgeon General, The Health Consequences of Smoking-50 Years of Progress: A Report of the Surgeon General, US Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health., Atlanta, GA, 2014. [Google Scholar]
- [5].Thun MJ, Henley SJ, Burns D, Jemal A, Shanks TG, Calle EE, Lung cancer death rates in lifelong nonsmokers,J. Natl. Cancer Inst. 98 (2006) 691–699. [DOI] [PubMed] [Google Scholar]
- [6].Sun S,Schiller JH, Gazdar AF, Lung cancer in never smokers–a different disease, Nat. Rev. Cancer 7 (2007) 778–790. [DOI] [PubMed] [Google Scholar]
- [7].Besaratinia A, Pfeifer GP, Second-hand smoke and human lung cancer, Lancet Oncol 9 (2008) 657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sato M, Shames DS, Gazdar AF,Minna JD,A translational view of the molecular pathogenesis of lung cancer, J. Thorac. Oncol 2 (2007) 327–343. [DOI] [PubMed] [Google Scholar]
- [9].Kim SI, Arlt VM,Yoon JI, Cole KJ, Pfeifer GP, Phillips DH, Besaratinia A, Whole body exposure of mice to secondhand smoke induces dose-dependent and persistent promutagenic DNA adducts inthe lung, Mutat. Res. 716(2011) 92–98. [DOI] [PubMed] [Google Scholar]
- [10].Kim SI,Yoon JI, Tommasi S, Besaratinia A, New experimental data linking secondhand smoke exposure to lung cancer in nonsmokers, FASEB J. 26 (2012)1845–1854. [DOI] [PubMed] [Google Scholar]
- [11].Tommasi S, Zheng A, Yoon JI, Besaratinia A, Epigenetic targeting of the Nanog pathway and signaling networks during chemical carcinogenesis, Carcinogenesis 35 (2014) 1726–1736. [DOI] [PubMed] [Google Scholar]
- [12].Tommasi S, Zheng A, Besaratinia A, Exposure of mice to secondhand smoke elicits both transient and long-lasting transcriptional changes in cancer-related functional networks, Int. J. Cancer 136 (2015) 2253–2263. [DOI] [PubMed] [Google Scholar]
- [13].Lee KW, Pausova Z, Cigarette smoking and DNA methylation, Front. Genet 4 (132) (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Rhee I, Bachman KE, Park BH, Jair ICW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, Baylin SB, Vogelstein B, DNMT1 and DNMT3b cooperate to silence genes in human cancer cells, Nature 416 (2002) 552–556. [DOI] [PubMed] [Google Scholar]
- [15].Gius D, Cui H, Bradbury CM,Cook J, Smart DK, Zhao S, Young L, Brandenburg SA, Hu Y, Bisht KS, Ho AS, Mattson D, Sun L, Munson PJ, Chuang EY,Mitchell JB, Feinberg AP, Distinct effects ongene expression of chemical and genetic manipulation of the cancer epigenome revealed by a multimodality approach, Cancer Cell 6 (2004) 361–371. [DOI] [PubMed] [Google Scholar]
- [16].Jacinto FV, Ballestar E, Esteller M, Impaired recruitment of the histone methyltransferase DOT1Lcontributes tothe incomplete reactivation oftumor suppressor genes upon DNA demethylation, Oncogene 28 (2009) 4212–4224. [DOI] [PubMed] [Google Scholar]
- [17].Rodriguez-Paredes M, Esteller M, Cancerepigenetics reaches mainstream oncology, Nat. Med. 17 (2011)330–339. [DOI] [PubMed] [Google Scholar]
- [18].You JS, Jones PA, Cancergenetics and epigenetics: two sides of the same coin? Cancer Cell 22 (2012) 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shen H, Laird PW, Interplay between the cancergenome and epigenome, Cell 153 (2013) 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Plass C, Pfister SM, Lindroth AM, Bogatyrova O, Claus R, Lichter P, Mutations in regulators of the epigenome and their connections to global chromatin patterns in cancer, Nat. Rev. Genet. 14 (2013) 765–780. [DOI] [PubMed] [Google Scholar]
- [21].Yang Z, Jones A, Widschwendter M, Teschendorff AE, An integrative pan-cancer-wide analysis of epigenetic enzymes reveals universal patterns of epigenomic deregulation in cancer, Genome Biol. 1640(1)(2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Cedar H, Bergman Y, D. N. A. Linking methylation and histone modification: patterns and paradigms, Nat. Rev. Genet. 10 (2009) 295–304. [DOI] [PubMed] [Google Scholar]
- [23].Jin B, Li Y, Robertson KD, DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes. Cancer 2(2011)607–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rose NR, Klose RJ, Understanding the relationship between DNA methylation and histone lysine methylation, Biochim. Biophys. Acta 1839 (2014)1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Jones PA, Liang G, Rethinking how DNA methylation patterns are maintained, Nat. Rev. Genet. 10 (2009) 805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Subramaniam D, Thombre R, Dhar A, Anant S, DNA methyltransferases: a novel target for prevention and therapy, Front. Oncol 4 (2014) 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Probst AV, Dunleavy E, Almouzni G, Epigenetic inheritance during the cell cycle, Nat. Rev. Mol. Cell Biol. 10 (2009) 192–206. [DOI] [PubMed] [Google Scholar]
- [28].Kareta MS, Botello ZM,Ennis JJ, Chou C, Chedin F, Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L, J. Biol. Chem. 281 (2006) 25893–25902. [DOI] [PubMed] [Google Scholar]
- [29].Hendrich B, Bird A, Identification and characterization of a family of mammalian methyl-CpG binding proteins, Mol. Cell Biol. 18 (1998) 6538–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Du Q, Luu PL, Stirzaker C, Clark SJ, Methyl-CpG-binding domain proteins: readers of the epigenome, Epigenomics (2015) 1–23. [DOI] [PubMed] [Google Scholar]
- [31].Barneda-Zahonero B, Parra M, Histone deacetylases and cancer, Mol. Oncol 6 (2012)579–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Falkenberg KJ, Johnstone RW, Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders, Nat. Rev. Drug Discov 13(2014) 673–691. [DOI] [PubMed] [Google Scholar]
- [33].Lopez-Serra L, Esteller M, Proteins that bind methylated DNA and human cancer: readingthe wrong words, Br. J. Cancer 98 (2008) 1881–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ropero S, Esteller M, The role of histone deacetylases (HDACs) in human cancer, Mol. Oncol 1 (2007) 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Minamiya Y, Ono T, Saito H, Takahashi N, Ito M, Mitsui M, Motoyama S, Ogawa J, Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung, Lung Cancer 74 (2011) 300–304. [DOI] [PubMed] [Google Scholar]
- [36].Sasaki H, Moriyama S, Nakashima Y, Kobayashi Y, Kiriyama M, Fukai I, Yamakawa Y, Fujii Y, Histone deacetylase 1 mRNA expression in lung cancer, Lung Cancer 46 (2004) 171–178. [DOI] [PubMed] [Google Scholar]
- [37].Cortessis VK, Thomas DC, Levine AJ, Breton CV, Mack TM, Siegmund KD, Haile RW, Laird PW, Environmental epigenetics: prospects for studying epigenetic mediation ofexposure-response relationships, Hum. Genet. 131 (2012)1565–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Herceg Z, Vaissiere T, Epigenetic mechanisms and cancer: an interface between the environment and the genome, Epigenetics 6 (2011) 804–819. [DOI] [PubMed] [Google Scholar]
- [39].Tommasi S, Zheng A, Yoon JI, Li AX, Wu X, Besaratinia A, Whole DNA methylome profiling in mice exposed to secondhand smoke, Epigenetics 7 (2012)1302–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].D’Agostini F, lzzotti A, Balansky R, Zanesi N, Croce CM, De Flora S, Early loss of Fhit inthe respiratory tract of rodents exposed to environmental cigarette smoke, Cancer Res. 66 (2006. 3936–3941. [DOI] [PubMed] [Google Scholar]
- [41].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods 25 (2001)402–408. [DOI] [PubMed] [Google Scholar]
- [42].Tommasi S, Zheng A, Weninger A, Bates SE, Li XA, Wu X, Hollstein M, Besaratinia A, Mammalian cells acquire epigenetic hallmarks of human cancer during immortalization, Nucleic Acids Res. 41 (2013) 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Portela A, Esteller M, Epigenetic modifications and human disease, Nat. Biotechnol. 28 (2010) 1057–1068. [DOI] [PubMed] [Google Scholar]
- [44].Baylin SB, Jones PA, A decade of exploring the cancer epigenome—biological and translational implications, Nat. Rev. Cancer 11 (2011) 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Morissette MC, Lamontagne M,Berube JC, Gaschler G, Williams A, Yauk C, Couture C Laviolette M,Hogg JC, Timens W, Halappanavar S, Stampfli MR, Bosse Y, lmpact of cigarette smoke on the human and mouse lungs: a gene-expression comparison study, PLoS One 9 (2014) e92498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Meuwissen R, Berns A, Mouse models for human lung cancer, Genes. Dev. 19 (2005) 643–664. [DOI] [PubMed] [Google Scholar]
- [47].Draghici S, Khatri P, Eklund AC, Szallasi Z, Reliability and reproducibility issues in DNA microarray measurements, Trends Genet. 22 (2006) 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Mocellin S, Rossi CR, Pilati P, Nitti D, Marincola FM, Quantitative real-time PCR: a powerful ally in cancer research,Trends Mol. Med. 9 (2003) 189–195. [DOI] [PubMed] [Google Scholar]
- [49].Barnes PJ, Adcock IM, Ito K, Histon acetylation and deacetylation: importance in inflammatory lung diseases, Eur. Respir. J. 25 (2005) 552–563. [DOI] [PubMed] [Google Scholar]
- [50].Sundar IK, Nevid MZ, Friedman AE, Rahman I, Cigarette smoke induces distinct histone modifications in lung cells: implications for the pathogenesis of COPD and lung cancer,J. Proteome Res. 13 (2014) 982–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Pandey M, Gupta KP, Epigenetics, an early event in the modulation ofgene expression by inositol hexaphosphate in ethylnitrosourea exposed mouse lungs, Nutr. Cancer 63 (2011) 89–99. [DOI] [PubMed] [Google Scholar]
- [52].Li Y, Hamilton KJ, Lai AY, Burns KA, Li L, Wade PA, Korach KS, Diethylstilbestrol (DES)-stimulated hormonal toxicity is mediated by ERalpha alteration of target gene methylation patterns and epigenetic modifiers (DNMT3A, MBD2, and HDAC2) in the mouse seminal vesicle, Environ. Health Perspect. 122 (2014) 262–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Pandey M, Sahay S, Tiwari P, Upadhyay DS, Sultana S, Gupta KP, Involvement of EZH2, SUV39H1, G9a and associated molecules in pathogenesis of urethane induced mouse lung tumors: potential targets for cancer control, Toxicol. Appl. Pharmacol. 280 (2014) 296–304. [DOI] [PubMed] [Google Scholar]
- [54].Hecht SS, Tobacco carcinogens, their biomarkers and tobacco-induced cancer, Nat. Rev. Cancer 3 (2003) 733–744. [DOI] [PubMed] [Google Scholar]
- [55].DeMarini DM, Genotoxicity of tobacco smoke and tobacco smoke condensate: a review, Mutat. Res. 567 (2004) 447–474. [DOI] [PubMed] [Google Scholar]