

Abstract
Children with rare genetic diseases that cause respiratory dysregulation are at particularly high mortality risk due to development of respiratory failure. The tectonin β-propeller–containing protein 2 (TECPR2) mutations are proposed to cause autophagy defect affecting axonal integrity and development of progressive neurodegenerative and neuromuscular disease. Published TECPR2 mutation cases have described a high prevalence of respiratory failure. We review respiratory pathology in previously published cases and a new case of a 5-year-old girl with previously undescribed TECPR2 mutation demonstrating progressive central apnea due to respiratory cycle dysregulation. This is the first TECPR2 mutation case to demonstrate an ataxic (Biot’s) breathing pattern with consistently inconsistent inspiratory and expiratory times and with relatively intact chemoreception during sleep. Therefore, we propose that the central apnea index alone may not be the appropriate marker for mortality risk. Rather, the morbidity and mortality associated with TECPR2 mutations are multisystem in nature and this burden complicates the ultimate needs for ventilation support and prognosis.
Citation:
Patwari PP, Wolfe LF, Sharma GD, Berry-Kravis E. TECPR2 mutation–associated respiratory dysregulation: more than central apnea. J Clin Sleep Med. 2020;16(6):977–982.
Keywords: central apnea, circadian rhythm disturbance, Biot’s breathing, ataxic breathing, respiratory and autonomic dysregulation
INTRODUCTION
Children affected by rare genetic diseases that cause respiratory and autonomic dysregulation are at particularly high mortality risk due to development of respiratory failure. Therefore, close attention to the respiratory phenotype is essential. Rare genetic disorders with altered control of breathing include Rett syndrome, Joubert syndrome, familial dysautonomia (aka hereditary sensory and autonomic neuropathy type 3),1 central congenital hypoventilation syndrome, rapid-onset obesity with hypothalamic dysfunction, hypoventilation and autonomic dysregulation,2 and Prader-Willi syndrome. Hereditary sensory autonomic neuropathies (HSANs), which also have partial overlap with hereditary spastic paraplegia (HSP), have a broad phenotypic spectrum and have generally not included respiratory pathology as a primary concern. One relatively recently proposed subtype of HSAN involves mutation of tectonin β-propeller–containing protein 2 (TECPR2).3 The TECPR2 mutations have also been included as a subtype 49 of familial spastic paraplegia (SPG49).4 While the respiratory phenotype has been described as progressive central apnea (CA), our case is the first to demonstrate an ataxic (Biot’s) breathing pattern with relatively intact chemoreception during sleep.
Control of breathing is a complex process that involves input and output from central and peripheral signals (Figure 1). Chemoreception based on partial pressures of oxygen and carbon dioxide values will send signals to the controller/integration area (central pattern generator) of the brainstem that will then send signals to affect motor response to adjust minute ventilation.5 The differences in respiratory pattern based on state can be attributed to absence or presence of other neuromodulatory factors. Dissection of control of breathing networks becomes more difficult as there are layers of redundancy as fail-safes to ensure life-sustaining gas exchange, such that respiratory load meets physiologic demand. In pathologic conditions, respiratory insufficiency may only become apparent with a change of state (exercise, resting, awake, versus sleep) or with acute challenge (pneumonia, sepsis, sedation). This failure of respiratory regulation often overlaps with other components of autonomic nervous system dysregulation.
Figure 1. Simplified pathway for respiratory control.
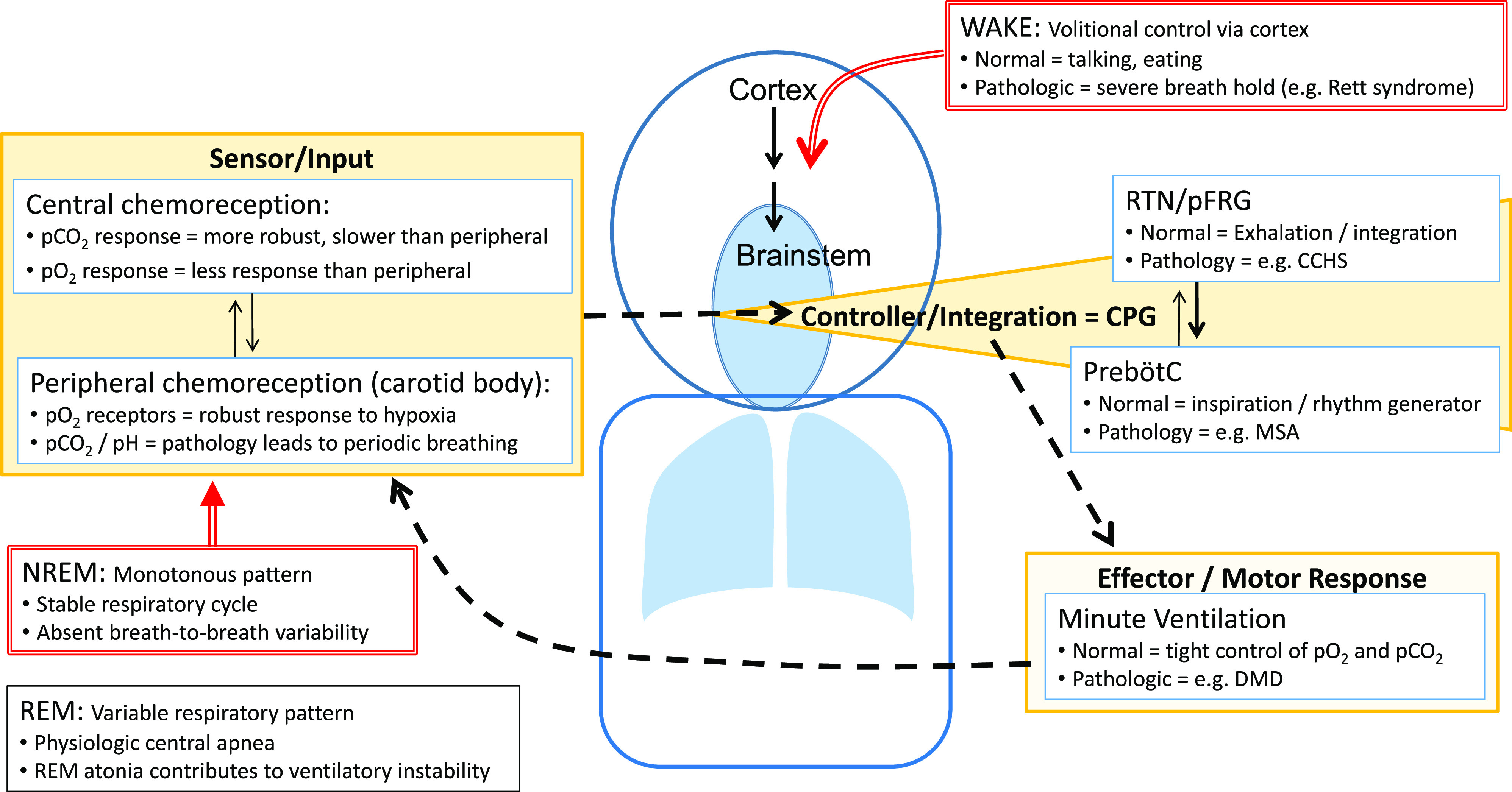
In the midbrain, the pre-Bötzinger complex is considered the pacemaker responsible for rhythm generation (setting breathing frequency) and a proposed pattern generator is responsible for the respiratory cycle (duration of breath and tidal volume); together, this is referred to as the CPG. Input to the CPG comes from central chemoreceptors (in response to hypercarbia), peripheral chemoreceptors (in response to hypoxia), and other peripheral sensory receptors. The integration of this input also occurs at multiple levels. The output then drives the motor response that manifests as minute ventilation (alveolar ventilation). To further fine-tune control of breathing, multiple feedback loops are simultaneously in play, of which some are well established (chemoreflex or loop gain) and others are yet to be defined. During wakefulness, simplistically, volitional control of breathing can override CPG to allow for speaking and eating (although, in normal conditions, also maintaining appropriate gas exchange). During NREM sleep, breathing drive is predominantly influenced by chemoreception and absent behavioral influences, resulting in a stable (monotonous) respiratory pattern. CCHS = congenital central hypoventilation syndrome; CPG = central pattern generator; DMD = Duchenne muscular dystrophy; MSA = multiple system atrophy; NREM = non–rapid eye movement; pCO2 = partial pressure of carbon dioxide; pFRG = parafacial respiratory group; pO2 = partial pressure of oxygen; PreBötzC = pre-Bötzinger complex; REM = rapid eye movement; RTN = retrotrapezoid nucleus.
Limited information exists regarding the respiratory phenotype for individuals affected by TECPR2 mutations (Table 1). The majority of cases are affected by hypotonia, progressive CA, and recurrent pulmonary infections with mortality due to respiratory failure.3,6–8 Therefore, attention to respiratory status and progression is paramount. We present the first detailed evaluation of respiratory dysregulation and progression in a child with TECPR2 mutations.
Table 1.
Summary of reported TECPR2 mutations and associated phenotype.
| Case | Mutation | Presentation | Neuro/Ophth | Autonomic | Behavior | Sleep | Respiratory |
|---|---|---|---|---|---|---|---|
| 1–5 (Oz-Levi 2012)6 | c.3416delT (frameshift, p.Leu1139Argfs*75) | Hypotonia and developmental delay at 2 years old | Developmental delay, motor delay, hypotonia. In 4 of 5 who walked, later developed spastic rigid ataxic gait. Areflexia. Four of 5 with episodes of decreased alertness with exacerbation of hypotonia. One with transient severe encephalopathy. | — | Friendly disposition | — | Recurrent pulmonary infections. Severe CA initially during sleep, later evolving to wake state. Four of 5 required MV (3 with spontaneous remission, 1 with chronic MV at 20 years). PSG with CAs (>90/h) and hypoxia. |
| Homozygous | |||||||
| 4 of 5 with genetic testing | |||||||
| Premature truncation of protein | |||||||
| 6 (Heimer 2015)3 | p.Leu440/p.Thr1891le | Developmental delay, GERD | Hypotonia, decreased reflexes, global developmental delay, astigmatism, myopia | Decreased pain sensitivity, delayed gastric emptying | Cheerful, hyperactive | Frequent arousals at 9 months | Postoperative ARDS, breath holding, PSG at 4 years with AHI 12 events/h, SpO2 nadir 80%, ETCO2 peak 55 mmHg, pneumonia and shock at 6 years leading to death |
| 7 (Heimer 2015)3 | p.Leu440/p.Leu440Arfs*19 | Floppy infant, dysphagia, and GERD | Developmental delay, areflexia, abrupt somnolence with EEG encephalopathy, esotropia, ataxic gait | Decreased pain sensitivity, temperature dysregulation, episodes of bradycardia with hypertension, sodium/water dysregulation | Friendly, violent mood changes | Arousals on PSG | Tonsillectomy, recurrent aspiration and pneumonia, chronic lung disease, tracheostomy with mechanical ventilator, “apnea” with elevated CO2 (>100 mmHg). |
| 8 (Heimer 2015)3 | p.Leu440/p.Leu1139Arfs*75 | Dysphagia and GERD in infancy | Developmental delay, areflexia, axial hypotonia | Decreased pain sensitivity, temperature dysregulation, postural hypotension, sodium/water dysregulation | Apathy alternating with restlessness and hyperactivity | Recurrent awakenings | Aspiration, desaturation, recurrent intubation, CA |
| 9 (Covone 2016)8 | p.Leu684Val/p.Thr903Met | Gait instability at 4 years | Normal intelligence, brisk reflexes at knee and upper extremities, areflexia at ankles, muscle atrophy, strabismus, ocular apraxia | — | — | — | Respiratory failure at 13 years of age, tracheostomy and mechanical ventilator |
| Compound heterozygous missense mutation | |||||||
| 10 (Zhu 2015)7 | p.Leu440ArgfsTer19 | — | Developmental delay, areflexia, severe hypotonia | — | — | — | “Breathing abnormalities” |
| Homozygous | |||||||
| Frameshift mutation | |||||||
| 11 (CASE) | p.D259MfsX44c.774delA | Hypotonia at 6 months | Developmental delay, low axial tone, motor coordination immaturity, strabismus | Photophobia, decreased pain sensitivity, chronic constipation | Friendly, hyperactive | Circadian disturbance with advanced sleep phase | Progressive CA, Biot’s breathing |
| p.K343RfsX2c.1028_1032delAAGGA | |||||||
| Compound heterozygous mutation | |||||||
| Large truncation of the protein |
AHI = apnea-hypopnea index; ARDS = acute respiratory distress syndrome; CA = central apnea; EEG, electroencephalogram; ETCO2 = end-tidal carbon dioxide; GERD = gastroesophageal reflux disease; MV = mechanical ventilation; PSG = polysomnogram; SpO2 = oxygen saturation; TECPR2, tectonin β-propeller–containing protein 2.
REPORT OF CASE
Birth and family history
The patient was born full term after an unremarkable pregnancy and newborn course. The mother has hypothyroidism, the father has Crohn’s disease, and 2 younger siblings are healthy.
Presentation
The patient was noted to have hypotonia around 5–6 months of age and subsequent mild gross motor delays that included pulling to stand at 11 months, crawling at 16 months, and walking at 22 months of age. She was prescribed glasses by 16 months of age for strabismus. She received appropriate therapies for motor and speech/language delay. The initial neurology evaluation at 23 months of age included magnetic resonance imaging of the brain, which showed scattered areas of relative T2 hyperintensity predominantly in the peri-atrial and subcortical white matter, read as nonspecific gliosis with suspicion for myoneural disorder causing hypotonia. Dysphagia was found on a swallow study at 2.5 years of age with improvement with paced eating. The patient was also noted to have possible signs of autonomic dysregulation, such as photophobia (without an overt pupil abnormality), decreased pain sensitivity, and chronic constipation. She was also noted to have hand flapping without other rhythmic/repetitive behaviors. Video electroencephalography at 34 months of age demonstrated mild background slowing and excess fast activity diffusely, rare bursts of delta activity, and multifocal epileptiform discharges without clinical correlate. The patient showed slow progress of motor and language development. By 3.5 years of age, she was able to speak about 50 words but had articulation difficulties that made her difficult to understand. By 5 years of age, she was treated with intensive cognitive-behavioral therapy. She has had no clinical seizures.
Physical examination
At 4 years of age, height and weight were at the fifth percentile for age. The patient was very social, with appropriate eye contact, and engaging (excessively friendly). She was normocephalic without dysmorphic features. She had prominent strabismus with difficulty abducting either eye. Cardiac examination revealed normal heart sounds without murmurs and appropriate peripheral perfusion. Lungs were clear to auscultation with regular respiratory pattern (no breath-holding or hyperpneic episodes). The abdomen was soft without organomegaly. The neurologic examination including cranial nerves, motor tone and strength, deep tendon reflexes, sensory examination, and cerebellar functions was unremarkable with the exception of low axial tone and motor coordinational immaturity with a poorly coordinated gait and foot pronation and eversion. The patient showed near-constant activity requiring frequent redirection by her parents.
Genetic testing
At 3.5 years of age, she had whole-exome sequencing and was found to be a compound heterozygote for pathogenic mutations in TECPR2 anticipated to result in large protein truncation (Table 1).
Respiratory course
The patient had lingual frenulectomy at 6 weeks of age (reportedly to facilitate feeding). At 28 months, adenoidectomy was performed due to report of snoring without apnea and difficulty sleeping. She had subsequent improvement in snoring, restless sleep, and speech (although continued with advanced sleep phase and fragmented sleep). Due to the persistence of occasional snoring, frequent arousals, and increased irritability, her first polysomnogram was completed at 32 months of age to evaluate for residual obstructive sleep apnea. This was reported to show “a complex respiratory pattern consisting of expiratory hypopneas, central appearing apneas, and questionable apneustic breathing.” On repeat polysomnograms (Table 2), the obstructive component improved, CA progressed, and Biot’s pattern persisted (Figure 2). Furthermore, decreased heart rate variability was noted on review of a Poincaré plot indicating progression of autonomic dysregulation. Noninvasive ventilation (NIV) was well tolerated during the titration study and for at least 1 week at home; then, the patient developed significant resistance to continued use of NIV despite desensitization attempts, which has been attributed to behavioral and sensory issues. Her continued difficulty with tolerance has been attributed to behavior and sensory issues. Sleep endoscopy at 4.3 years of age demonstrated absence of airway obstruction. Notable CAs were observed during sedation by the pediatric anesthesiologist requiring prolonged postprocedure observation. By 5 years of age, the patient continues without NIV support.
Table 2.
Summary of polysomnographic findings by age.
| Age at Study | ||||
|---|---|---|---|---|
| 2 Years, 8 Months | 3 Years, 9 Months | 4 Years, 7 Months | 5 Years | |
| TST, minutes | 363.5 | 314.5 | 308.5 | 410.5 |
| Sleep efficiency, % | 70 | 65.9 | 64.7 | 78.2 |
| AHI, events/h | 9.3 | 36.4 | 55.2 | 45.7 |
| REM AHI, events/h | 23.6 | 9.1 | 44.4 | 30.5 |
| Obstructive AHI, events/h | ? | 9.9 | 1.4 | 3.2 |
| Obstructive apnea index, events/h | 0 | 0 | 0 | 0 |
| Central apnea index, events/h | ? | 13.7 | 47.1 | 28.9 |
| *Hypopneas (obstructive + central), count of events | 57 | 52 + 67 | 7 + 35 | 22 + 93 |
| SpO2 nadir, % | 85 | 83 | 89 | 89 |
| SpO2 <90%, minutes | 1.2 | 7.9 | 1.9 | 0.5 |
| ETCO2 average, peak, mmHg | 38, 46 | 36, 48 | 37, 48 | 41, 54 |
| Average RR, breaths/minute | 8–14 | 8–14 | 6–10 | 6–10 |
*Hypopnea events are listed as total count. In this case of central respiratory dysregulation and in the absence of scoring criteria for Biot’s breathing, the apnea-hypopnea indices are not necessarily meaningful markers of disease severity or progression. AHI = apnea-hypopnea index; ETCO2 = end-tidal carbon dioxide; REM = rapid eye movement; RR = repiratory rate; SpO2 = oxygen saturation; TST = total sleep time.
Figure 2. Polysomnogram demonstrating respiratory cycle variability in NREM sleep.
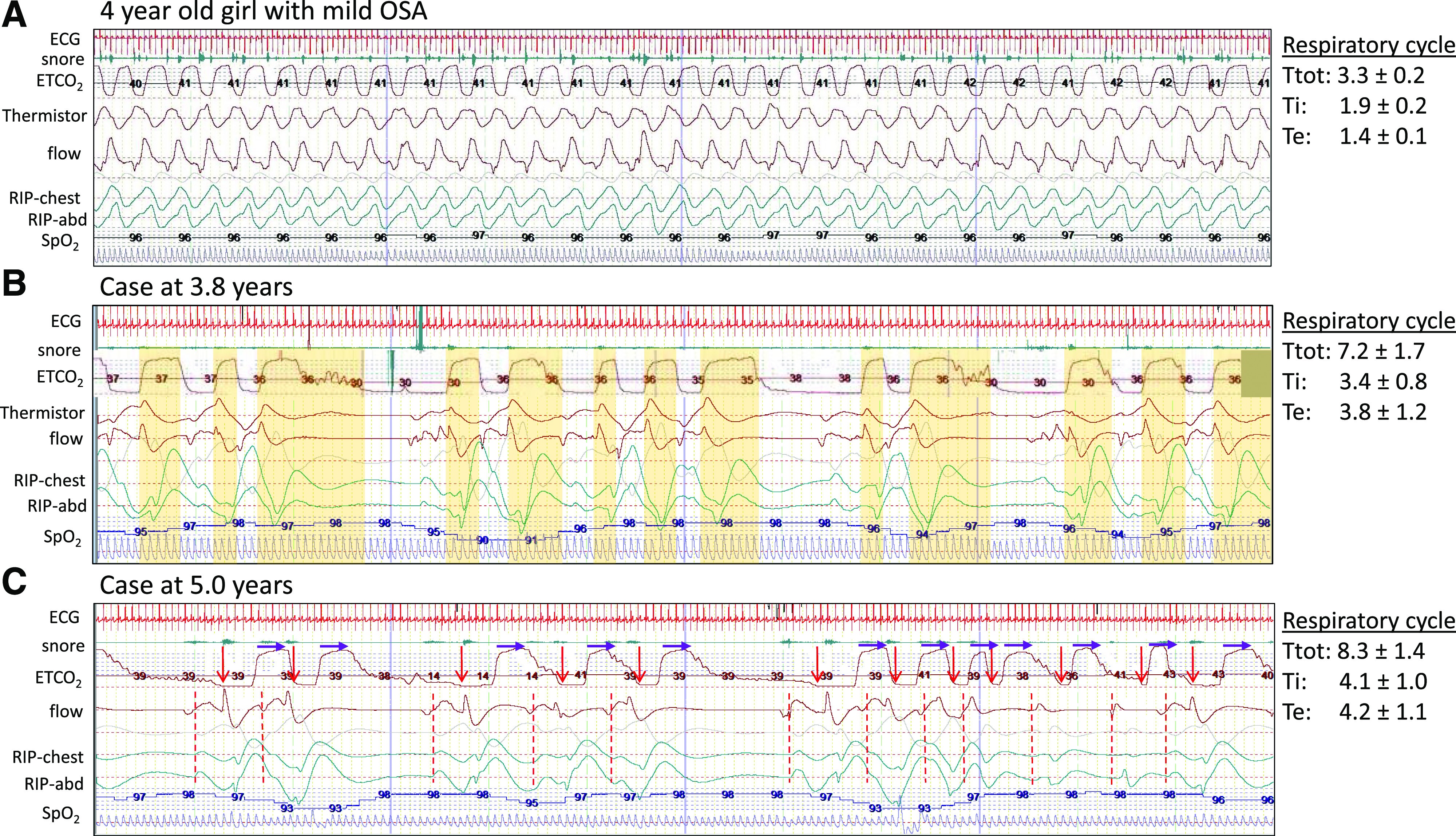
Images are of a 2-minute segment from baseline polysomnogram comparing an age-matched, relatively healthy child with the patient at 3.8 years and 5 years of age. The respiratory cycle values to the right note the average and standard deviation of total cycle time, inspiratory time, and expiratory time for the breaths in the first 30 seconds of the 2-minute segment. (A) A 4-year-old girl with mild pediatric OSA. Note the normal (monotonous) respiratory pattern and stable respiratory cycle times. (B) Our patient at 3.8 years of age. The ETCO2 waveform is manually shifted 3 seconds earlier to account for recording delay compared to flow and respiratory effort bands (RIP). The vertical yellow bars indicate the expiratory component of each respiratory cycle. Note the wide variability and absent pattern. (C) Our patient at 5 years of age. The start of inhalation is noted by a red arrow and dashed line. The purple arrows are of fixed length and indicate the start of exhalation. Again, note the absence of distinct pattern and wide variability in both inspiratory time and expiratory time with slower respiratory rate at 6–10 breaths per minute. ETCO2 = end-tidal carbon dioxide; NREM = non–rapid eye movement; OSA = obstructive sleep apnea; RIP = respiratory inductance plethysmography.
DISCUSSION
This is the first detailed report of the evolving respiratory changes in a patient with TECPR2 mutations including the unique pattern of breathing. Specifically, the “central sleep apnea” found with ataxic breathing is distinct from periodic breathing, Cheyne-Stokes breathing, postarousal respiratory events, and apneustic breathing and not necessarily associated with hypoventilation. Both periodic breathing and Cheyne-Stokes breathing pattern include alternating episodes of hyperpnea followed by apnea (Cheyne-Stokes with a characteristic crescendo-decrescendo pattern between apneic periods). Postarousal respiratory events are of questionable clinical relevance.9 The characteristic feature of apneustic breathing is a gasp with inspiratory hold that is classically found with injury to the pons. Another form of erratic breathing is Biot’s breathing, which is characterized by irregular cycles of breathing without distinct clusters of breathing,10 such as is found with this case and as demonstrated by waveforms recorded during baseline polysomnograms (Figure 2).
TECPR2 encodes a 1411–amino acid protein with 3 WD11 (tryptophan-aspartic acid) repeat domains located at the N-terminal region of the protein and 6 TECPR domains concentrated around amino acid 1000 and 1250. The p.Leu1139Argfs, which has already been demonstrated as pathogenic,12 would result in a loss of 4 out of 6 TECPR domains. The p.D259MfsX44 and TECPR2 p.K343RfsX2 mutations in this patient would be expected to cause a large truncation of the protein and loss of all 6 TECPR domains, leading to non-sense–mediated decay, as would be expected for multiple other mutations described in previous patients with TECPR2 mutations (Table 1), and these are expected to function as null mutations. The null mutations in TECPR2 have been linked to abnormalities of autophagy with decreased accumulation of the autophagy initiation protein MAPILCII/LC3II and attenuation of activity of both LC3II and the cargo recruiting protein SQSTM1/p62 in patient fibroblasts.13 An autophagy defect was proposed to affect axonal integrity and function in the nervous system through secondary effects on endoplasmic reticulum shaping, endosomal trafficking and microtubule stability,3 although definitive identification of specific mechanisms involved in TECPR2 deficiency has not been clarified.
In general, a disruption in cellular autophagy is particularly devastating to neurons and has been shown to cause progressive neurodegenerative and neuromuscular disease (lysosomal storage diseases, Huntington disease, amyotrophic lateral sclerosis) with a wide phenotypic spectrum. With TECPR2 mutations, neuromuscular dysfunction appears to be a key feature in all reported cases based on hypotonia and recurrent pneumonia. In this case, there is altered respiratory cycle timing (Biot’s breathing pattern) while maintaining sufficient ventilatory response to maintain normal oxygen saturation and carbon dioxide values (intact chemoreception). Hypoventilation appears to be a component of previously reported cases based on development of hypoxia and need for chronic mechanical ventilation.
The respiratory management of this patient is in line with standard of care for patients with neuromuscular weakness, requiring aggressive airway clearance, screening for dysphagia and aspiration, repeated polysomnograms (with end-tidal carbon dioxide monitoring to provide breath-to-breath waveform and values to identify prolonged exhalations and hypoventilation) to track progression, and institution of supported ventilation at the first signs of hypoventilation. In this case, the CA index alone may not be the appropriate marker for mortality risk. Rather, mortality is more likely due to the multisystem nature of the disease, which complicates the ultimate needs for ventilation support. Therefore, the CA index and total apnea-hypopnea index do not predict the need for supported ventilation.
Future studies should examine genotype–phenotype relationships in patients with TECPR2 mutations with respect to respiratory functioning, so that the potential effects of these patients’ mutations can be used to identify high-risk patients. In addition to implementation of appropriate treatment strategies due to respiratory dysfunction, further clarification of TECPR2 cellular pathways is needed to design mechanism-based treatments.
DISCLOSURE STATEMENT
All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work. The patient’s parents provided written consent to publication of this de-identified case report. The authors report no conflicts of interest.
ACKNOWLEDGMENTS
Author contributions: Drs. Patwari and Berry-Kravis conceptualized and drafted the initial manuscript, contributed to data collection and data analysis, and revised the manuscript. Drs. Wolfe and Sharma contributed to data interpretation, critically reviewed the manuscript for important intellectual content, and reviewed and revised the manuscript.
ABBREVIATIONS
- CA
central apnea
- HSAN
hereditary sensory autonomic neuropathy
- HSP
hereditary spastic paraplegia
- TECPR2
tectonin β-propeller–containing protein 2
REFERENCES
- 1.Palma J-A, Gileles-Hillel A, Norcliffe-Kaufmann L, Kaufmann H. Chemoreflex failure and sleep-disorderd breathing in familial dysautonomia: implications for sudden death during sleep. Auton Neurosci. 2019;218:10–15. 10.1016/j.autneu.2019.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Patwari PP, Wolfe LF. Rapid-onset obesity with hypothalamic dysfunction, hypoventilation, and autonomic dysregulation: review and update. Curr Opin Pediatr. 2014;26(4):487–492. 10.1097/MOP.0000000000000118 [DOI] [PubMed] [Google Scholar]
- 3.Heimer G, Oz-Levi D, Eyal E, et al. TECPR2 mutations cause a new subtype of familial dysautonomia like hereditary sensory autonomic neuropathy with intellectual disability. Eur J Paediatr Neurol. 2016;20(1):69–79. 10.1016/j.ejpn.2015.10.003 [DOI] [PubMed] [Google Scholar]
- 4. Spastic Paraplegia 49, Autosomal Recessive; SPG49, OMIM®. Johns Hopkins University, Baltimore, MD. MIM Number: 615031. https://omim.org/. Updated February 9, 2018. Accessed November 27, 2019.
- 5.Ramirez JM, Baertsch N. Defining the rhythmogenic elements of mammalian breathing. Physiology. 2018;33:302–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oz-Levi D, Ben-Seev B, Ruzzo EK, et al. Mutation in TECPR2 reveals a role for autophagy in hereditary spastic paraparesis. Am J Hum Genet. 2012;91(6):1065–1072. 10.1016/j.ajhg.2012.09.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu X, Petrovski S, Xie P, et al. Whole-exome sequencing in undiagnosed genetic disease: interpreting 119 trios. Genet Med. 2015;17(10):774–781. 10.1038/gim.2014.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Covone AE, Fiorillo C, Acquaviva M, et al. WES in a family trio suggests involvement of TECPR2 in a complex form of progressive motor neuron disease. Clin Genet. 2016;90(2):182–185. 10.1111/cge.12730 [DOI] [PubMed] [Google Scholar]
- 9.Haupt ME, Goodman DM, Sheldon SH. Sleep related expiratory obstructive apnea in children. J Clin Sleep Med. 2012;8(6):673–679. 10.5664/jcsm.2262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wijdicks EFM. Biot’s breathing. J Neurol Neurosurg Psychiatry. 2007;78(5):512–513. 10.1136/jnnp.2006.104919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. WD Repeats. In: Schwab M, ed. Encyclopedia of Cancer. Berlin, Heidelberg: Springer; 2011.
- 12.Oz-Levi D, Gelman A, Elazar Z, Lancet D. TECPR2: a new autophagy link for neurodegeneration. Autophagy. 2013;9(5):801–802. 10.4161/auto.23961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stadel D, Millarte V, Tillmann KD, et al. TECPR2 cooperates with LC3C to regulate COPII-Dependent ER export. Mol Cell. 2015;60(1):89–104. 10.1016/j.molcel.2015.09.010 [DOI] [PubMed] [Google Scholar]