

ABSTRACT
R-loops are intermediate structures of transcription that can accumulate when transcriptional elongation is blocked by inhibiting BRD4. In normal cells, R-loop persistence suppresses firing of adjacent replication origins. This control is lost in a subset of cancer cells, where BRD4 inhibition results in R-loop accumulation, leading to transcription-replication collisions and DNA double-strand breaks during S-phase, followed by cell death. This finding sheds new light on the mechanisms by which BRD4 inhibitors function as cancer therapies, and indicates that targeting other cellular events to cause R-loop accumulation may be useful for cancer treatment.
KEYWORDS: BRD4, bromodomain proteins, R-loops, DNA damage, replication stress, transcription-replication conflicts
Genome stability in eukaryotic cells depends on the coordination of transcription and replication machinery as they move along a common DNA template. Discordance between these events leads to transcription-replication conflicts (TRCs), increased replication fork stress, DNA damage, and chromosomal instability.1 One potential source of TRCs are R-loops, DNA:RNA hybrids that occur during gene transcription. R-loops occupy up to 5% of the human genome, and normally occur within the promoter and termination regions of RNA Polymerase II (RNAPII)-dependent, actively transcribing genes.2 R-loops are involved in diverse cellular processes including Ig class switching in B cells, mitochondrial DNA replication, and telomere homeostasis; hence, their presence or absence on chromatin can have dynamic effects on genome maintenance and disease.3
Dysregulated R-loop homeostasis can have deleterious effects, the most concerning of which is genomic instability caused by collisions with replication forks leading to increased fork stress and DNA damage.1,4 Not surprisingly, there exists a symbiosis between specific DNA damage signaling and repair pathways and R-loop maintenance proteins to assist in R-loop suppression, resolution, and tolerance. Proteins involved in homologous recombination such as BRCA1 and BRCA2 recruit the DNA helicase Senataxin (SETX) to replication forks to assist in the resolution of R-loops at transcription termination sites.4 The Cimprich lab showed that the ATR/Chk1 DNA damage signaling pathway is particularly responsive to head-on TRCs, which increase R-loop accumulation and replication fork stress, while co-directional collisions have minimal effects on RNAPII dynamics, R-loop homeostasis, and fork stalling, but can trigger the ATM/Chk2 DNA damage checkpoint (Figure 1a).5 Similarly, decreased expression of the 9-1-1 complex, ATR, or Chk1 causes R-loop-dependent replication fork stalling, while decreased expression of ATM, Chk2, or the post-replicative repair factors UBE2B and RAD18 causes increased R-loop accumulation with unrepaired double-strand breaks and post-replicative single-stranded DNA gaps.6 Importantly, trans-expression of RNase H can reduce the DNA damage observed following depletion of the 9-1-1 complex, ATR, or Chk1, further incriminating R-loops as a major source of TRCs and replication-associated DNA damage.6
Figure 1.
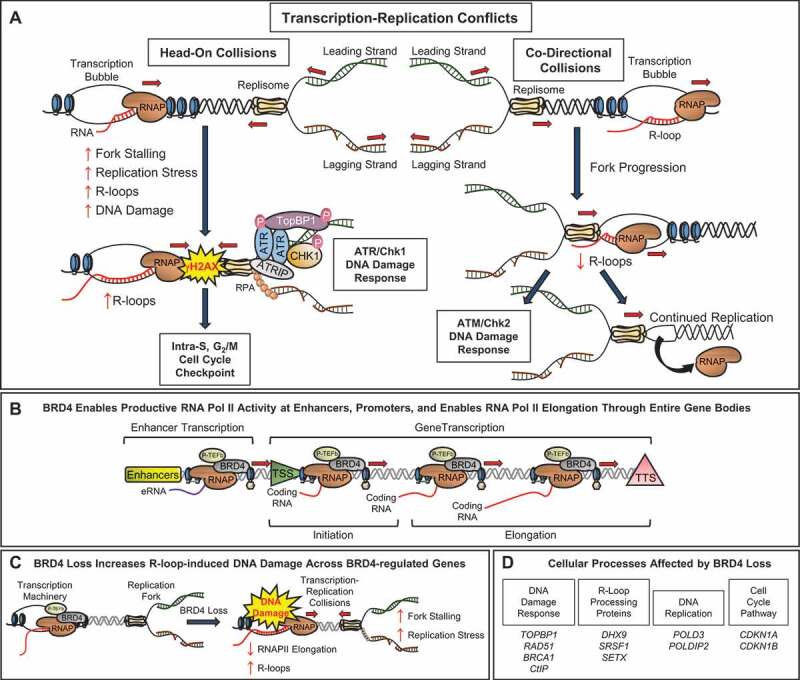
Mechanisms through which BRD4 prevents transcription-replication conflicts in oncogenic cells. (a) DNA damage response signaling pathways associated with head-on vs co-directional transcription-replication collisions. (b) BRD4 facilitates RNA polymerase (RNAP) II activity at upstream enhancer regions, at sites of transcription initiation, and also facilitates productive RNAPII elongation through entire gene bodies. (c) BRD4 loss decreases RNAPII elongation, increases accumulation of R-loops, replication fork stress and stalling, and DNA damage at sites of BRD4-regulated genes. (d) Lists of genes corresponding to cellular processes negatively affected by BRD4 loss
We and others recently reported that the epigenetic reader BRD4, plays a critical role in maintaining R-loop homeostasis and preventing TRCs in a subset of cancer cells.7–9 BRD4 and other members of the bromodomain-containing family of proteins, including BRD2 and BRD3, are transcriptional co-activators that recognize acetylated lysine residues on histones directly via their conserved tandem bromodomains. BRD4 in particular, is enriched at enhancer regions of the MYC oncogene, leading to the development of bromodomain inhibitors as a treatment for Myc-driven cancers. Curiously, BRD4 inhibitors also show potent anti-tumor effects in some tumors that are not Myc-driven, an effect that we postulate results from TRC-induced DNA breaks independent of any effect on MYC transcription. BRD4 normally regulates RNAPII activity in multiple ways, including recruiting PTEF-b to enhancers and proximal-promoter pause sites to enable RNAPII initiation and elongation, and then driving productive RNAPII elongation along entire bodies of genes via direct interactions with hyperacetylated lysine residues on histones (Figure 1b). We propose a model where loss of BRD4 function causes stalling of RNAPII elongation throughout entire gene bodies of a subset of BRD4-regulated genes.9 This results in accumulation of R-loops, TRCs, and replication fork stress during S-phase, eventually leading to DNA damage and apoptosis as a consequence of mitotic catastrophe.9
One might expect R-loop-induced TRCs and replication fork stress to cause cell cycle arrest rather than S-phase progression, double-strand DNA breaks, and mitotic cell death. Indeed, our findings revealed multiple mechanisms through which loss of BRD4 drives cell death in oncogenic cell lines. First, we found that reduced RNAPII elongation following BRD4 loss leads to increased R-loop-driven DNA damage throughout the bodies of BRD4-, JMJD6-, and CHD4-co-regulated genes (Figure 1c). Second, we noted that loss of BRD4 function suppressed expression of the ATR/Ck1 replication stress DNA damage response checkpoint mediator TopBP1, causing a paradoxical inactivation of the G1/S and G2/M checkpoints, and slippage of DNA-damaged cells into mitosis, where they died by mitotic catastrophe.9 Third, BRD4 loss caused R-loop-induced DNA damage throughout the gene bodies of key R-loop processing genes, including DHX9, SRSF1, and SETX, steadily reducing the cellular stores of these proteins over time, thereby contributing to the inability of cells to resolve R-loops at 24–48 hours after bromodomain inhibition. Finally, we and others found that loss of BRD4 impairs transcription of several key genes – BRCA1, RAD51, and CtIP – involved in DNA repair by homologous recombination (HR, Figure 1d). This effect on HR repair has been shown to result in synergy between BRD4 and PARP inhibitors in multiple preclinical models of cancer.10
Interestingly, we also found DNA damage throughout the gene body of DNMT1 following BRD4 loss.9 As R-loops are key regulators of DNMT targeting to chromatin with important roles in cancer progression and treatment,4 our findings hint that combining DNMT inhibitors, or other novel therapies that exacerbate TRCs and replication stress-induced DNA damage, with bromodomain inhibitors, could increase their therapeutic potential for cancer treatment.
These findings that TRCs, DNA damage, and cell death result from R-loop accumulation following BRD4 inhibition suggests that therapeutic induction of R-loop persistence could be a generalizable approach for inducing oncogenic cell death. In particular, one could imagine inhibiting other molecules involved in suppressing R-loop accumulation, such as RNA-binding proteins, particularly when combined with inhibition of DNA damage-induced cell cycle checkpoints by ATR or Chk1 inhibitors, could prove highly effective as new anti-cancer therapies.
Funding Statement
This work was supported by the National Cancer Institute [P30-CA14051]; National Institutes of Health [R01-CA226898]; National Institutes of Health [R35-ES028374]; National Institutes of Health [R01-ES015339]; Bridge Project, a partnership between the Koch Institute for Integrative Cancer Research at MIT and the Dana-Farber/Harvard Cancer Center; KI Quinquennial Cancer Research Fellowship; MIT Center for Precision Cancer Medicine; Koch Institute Frontier Research Program; STARR Consortium grant.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hamperl S, Cimprich KA.. Conflict resolution in the genome: how transcription and replication make it work. Cell. 2016;167:1–3. doi: 10.1016/j.cell.2016.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allison DF, Wang GG.. R-loops: formation, function, and relevance to cell stress. Cell Stress. 2019;3:38–46. doi: 10.15698/cst2019.02.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Muse T, Aguilera A, Loops: R. From physiological to pathological roles. Cell. 2019;179:604–618. doi: 10.1016/j.cell.2019.08.055. [DOI] [PubMed] [Google Scholar]
- 4.Wells JP, White J, Stirling PC, Loops R. Their composite cancer connections. Trends Cancer. 2019;5:619–631. doi: 10.1016/j.trecan.2019.08.006. [DOI] [PubMed] [Google Scholar]
- 5.Hamperl S, Bocek MJ, Saldivar JC, Swigut T, Cimprich KA. Transcription-replication conflict orientation modulates R-loop levels and activates distinct DNA damage responses. Cell. 2017;170:774–786. doi: 10.1016/j.cell.2017.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barroso S, Herrara-Moyano E, Munoz S, Garcia-Rubio M, Gomez-Gonzalez B, Aguilera A. The DNA damage response acts as a safeguard against harmful DNA-RNA hybrids of different origins. EMBO Rep. 2019;20:e47250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kim JJ, Lee SY, Gong F, Battenhouse AM, Boutz DR, Bashyal A, Refvik ST, Chiang C-M, Xhemalce B, Paull TT, et al. Systematic bromodomain protein screens identify homologous recombination and R-loop suppression pathways involved in genome integrity. Genes Dev. 2019;33:1751–1774. doi: 10.1101/gad.331231.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edwards DS, Maganti R, Tanksley JP, Luo J, Park JJH, Balkanska-Sinclair E, Ling J, Floyd SR. BRD4 prevents R-loop formation and transcription-replication conflicts by ensuring efficient transcription elongation. Cell Rep. 2020;32:108166. doi: 10.1016/j.celrep.2020.108166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam FC, Kong YW, Huang Q, Vu Han T-L, Maffa AD, Kasper EM, Yaffe MB. BRD4 prevents the accumulation of R-loops and protects against transcription-replication collision events and DNA damage. Nat Commun. 2020;11:4083. doi: 10.1038/s41467-020-17503-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, Vellano CP, Ju Z, Zhao W, Zhang D, et al. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33(401–416):e408. doi: 10.1016/j.ccell.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]