

ABSTRACT
Multiple Myeloma (MM) is a malignant disorder of plasma cells which, despite significant advances in treatment, remains incurable. Daratumumab, the first CD38 directed monoclonal antibody, has shown promising activity alone and in combination with other agents for MM treatment. Daratumumab is thought to have pleiotropic mechanisms of activity including natural killer (NK) cell-mediated antibody-dependent cellular cytotoxicity (ADCC). With the knowledge that CD38-expressing NK cells are depleted by daratumumab, we sought to investigate a potential mechanism of enhancing macrophage-mediated antibody-dependent cellular phagocytosis (ADCP) by combining daratumumab with cyclophosphamide (CTX). Cyclophosphamide’s immunomodulatory function was investigated by conditioning macrophages with tumor cell secretome collected from cyclophosphamide treated MM cell lines (CTX-TCS). Flow cytometry analysis revealed that CTX-TCS conditioning augmented the migratory capacity of macrophages and increased CD32 and CD64 Fcγ receptor expression on their cell surface. Daratumumab-specific tumor clearance was increased by conditioning macrophages with CTX-TCS in a dose-dependent manner. This effect was impeded by pre-incubating macrophages with Cytochalasin D (CytoD), an inhibitor of actin polymerization, indicating macrophage-mediated ADCP as the mechanism of clearance. CD64 expression on macrophages directly correlated with MM cell clearance and was essential to the observed synergy between cyclophosphamide and daratumumab, as tumor clearance was attenuated in the presence of a FcγRI/CD64 blocking agent.
Cyclophosphamide independently enhances daratumumab-mediated killing of MM cells by altering the tumor microenvironment to promote macrophage recruitment, polarization to a pro-inflammatory phenotype, and directing ADCP. These findings support the addition of cyclophosphamide to existing or novel monoclonal antibody-containing MM regimens.
KEYWORDS: Multiple myeloma, daratumumab, cyclophosphamide, macrophages, ADCP
Introduction
Multiple Myeloma (MM) is characterized by clonal expansion of malignant plasma cells in the bone marrow (BM). MM remains an incurable disease, however, with treatment regimens evolving, this dogma is being challenged.1,2 The difficulty in treating MM can in part be attributed to the supportive role of the BM microenvironment to malignant plasma cell differentiation, migration, clonal expansion, survival and resistance to therapies.3,4 It is thought that transformation to MM requires the development of a permissive tumor microenvironment (TME), which facilitates “immune escape”.5,6 Current MM therapies include proteasome inhibitors (e.g. bortezomib), immunomodulatory agents (e.g. lenalidomide), and monoclonal antibodies, particularly the IgG1 kappa (IgG1κ) CD38 monoclonal antibody daratumumab.7,8 The anti-MM activity of these therapies relies upon the presence of an intact immune system.9 Thus, an improved understanding of the mechanisms underlying the immune-escape observed in MM could provide new insights into disease pathogenesis and opportunities for therapeutic intervention.
With progression to MM, there are an increasing number of tumor-associated macrophages (TAMs) detectable in the BM.10 These are predominantly of an anti-inflammatory phenotype and promote tumor survival and immune suppression, which enables disease progression.11 A high number of anti-inflammatory TAMs in the BM has been associated with inferior survival in MM.12 TAMs likely accumulate in the BM under the influence of chemokines such as CCL2 (MCP-1), CCL3 (MIP-1α) and CCL5 (RANTES) secreted by the myeloma cells.13 CCL5, in conjunction with other chemokines including CCL2, promotes macrophage recruitment and survival and may act as a pro-survival factor.14
Circulating monocytes are attracted into the BM along this chemokine gradient where they are polarized toward an anti-inflammatory macrophage phenotype under the influence of factors such as prostaglandin E2 (PGE2) and interleukin (IL)-10 (reviewed in15). Indeed, many of these chemoattractants and polarizing factors are known to be produced by MM cells, and have been reported to be associated with adverse outcomes.16 The presence of a large number of TAMs is generally considered to be undesirable.17 Some therapeutic approaches, such as antibodies targeting colony stimulating factor (CSF)-1 receptor, have been designed to eliminate macrophages from the TME, although this approach has had limited success.18,19 An alternative approach is to activate or reprogram the cells to harness their anti-tumor potential. Repolarization of TAMs to an anti-tumor phenotype has been achieved by reprogramming TAMs using, for example, anti-CD47 antibodies,20 histone deacetylase inhibitors21 and Toll-like receptor agonists.22 In the tumor microenvironment, anti-tumor macrophages have the capacity to clear tumor cells by several mechanisms of cytotoxicity including: direct cytotoxicity by releasing cytotoxic agents e.g. reactive oxygen species, antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP).23 Macrophages are thought to be critical effectors of monoclonal antibody (mAb) therapy, with reduced therapeutic efficacy recorded when macrophage levels were depleted in the TME .24 Monoclonal antibodies bind to effector macrophages via Fc gamma (Fcγ) receptors, a family of glycoproteins which bind to the Fc portion of IgG antibodies.25 These receptors can be activating or inhibitory. The activating Fcγ receptors in humans are; FcγRI/CD64, FcγRIIa/CD32a, FcγRIIc/CD32c and FcγRIIIa/CD16a.26 Unlike NK cells which predominantly only express FcγRIIIa/CD16a, macrophages express all types of Fc receptors.26 Their depletion has been associated with reduced in vivo efficacy of antibodies and improved outcome has been seen in conjunction with high affinity polymorphisms of FcγRIIa/CD32a, which are not expressed on NK cells.27 This may be particularly important in the context of treatment with daratumumab. Originally, it was thought that ADCC mediated by NK cells would constitute one of the most important mechanisms of action of daratumumab.28 However, with the benefit of careful correlative studies from clinical trials, we now know that treatment with daratumumab leads to rapid depletion of NK cells, which are strongly CD38 positive, and that this can last up to 6 months following cessation of treatment.29 Therefore, to maximize the clinical efficacy of daratumumab, it may be necessary to have a sufficient number of activated TAMs.30 This has recently been demonstrated by Viola et al. who analyzed bone marrow samples for the presence of CD14 positive macrophages, and reported significantly higher numbers in patients responding to daratumumab compared with relapsed/refractory MM (RRMM) or patients with daratumumab-resistant disease. They also observed increased macrophage surface expression of CD80/CD86, T-cell co-stimulatory antigens promoting T-cell activation and expansion.31
It has been known for some time that cyclophosphamide has important immunomodulatory effects with low doses able to selectively eliminate regulatory T cells (Tregs) leading to immune activation.32,33 However, the effects on innate immune cells, such as macrophages, is less well understood. In a study focused on B cell leukemia, Pallasch and colleagues have shown that cyclophosphamide enhanced the anti-tumor effects of monoclonal antibody therapy through enhancing macrophage-mediated anti-tumor activity.34 We therefore sought to assess the effects of cyclophosphamide on the anti-tumor activity of macrophages in combination with daratumumab in the context of MM. Ex vivo data from a clinical trial, set up to assess the addition of cyclophosphamide to a daratumumab-containing regimen (CyBorD-DARA), uncovered observations suggesting that this treatment combination increased the vulnerability of MM cells to phagocytosis by macrophages.30 Our study investigates the specific mechanism of action of cyclophosphamide in the induction of ADCP in vitro.
Here, we show that low dose cyclophosphamide, in addition to its ability to induce MM cell death, alters the tumor microenvironment and promotes macrophage recruitment and a pro-inflammatory phenotype. Conditioning macrophages with the secretome from cyclophosphamide-treated MM cells (CTX-TCS) potentiated daratumumab-mediated killing in a dose-dependent manner. The synergy identified here directly correlated with FcγRI/CD64 surface expression and function on macrophages, indicating FcγRI/CD64 as a key player in daratumumab-mediated ADCP of MM cells by CTX-TCS conditioned macrophages. We believe these data support the continued development of cyclophosphamide-daratumumab combinations in the treatment of MM.
Materials and methods
Cell culture
The MM (MM1.S and RPMI-8226) cell lines and monocyte/macrophage mononuclear (THP-1)35,36 cell line, purchased from the American Type Culture Collection (ATCC, Virginia, USA), were sub-cultured in RPMI-1640 media (Thermofisher Scientific, MA, USA) supplemented with 10% fetal bovine serum (FBS) (Sigma, MO, USA), 1% L-glutamine (Thermofisher Scientific) and 1% penicillin/streptomycin (Thermofisher Scientific). Plating densities for MM1.S, RPMI-8226 and THP-1 cell lines were 5 × 105 cells/ml, 3 × 105 cells/ml and 6 × 105 cells/ml, respectively.
Flow cytometric analysis of cell surface proteins
Following treatment, the MM cells or macrophages were washed in PBS and multi-parameter flow cytometry was carried out as previously described.37,38 Briefly, single cell suspensions were plated in duplicate at 1 × 105 cells/well of a V-bottomed 96-well plate in FACS buffer. Cells were washed three times with 150 µl FACS buffer. After the final wash, cell pellets were re-suspended in the diluted primary antibody and incubated at 4°C for 15 minutes in the dark. All antibodies and associated reagents used here (Sytox, CD38, CD32/FcγRIIA, CD64/FCγRI, CD47, SIRP-α, PD-L1 and PD-1), along with their respective suppliers and catalog numbers, are provided in Supplementary Table 1. Cells were washed three times with 150 µl FACS buffer per wash and re-suspended in 100 µl FACS buffer, transferred to appropriate 5 ml tubes and left on ice until ready to analyze on the BD FACSCanto II flow cytometer (BD Biosciences, NJ, USA). Output from the BD FACS DIVA software (BD Biosciences) was analyzed using FlowJo V10.6..2 software. The BD Biosciences, NJ, USA fluorescence intensity (MFI) was calculated by expressing the fluorescence intensity of each sample relative to the cell size. The relative fluorescence intensity (RFI) was calculated by expressing the MFI of each treatment relative to the control group. For imaging flow cytometry, samples were prepared in the same manner as for conventional flow cytometry and were run on the ImageStream® X Mark II imaging flow cytometer (Merck Millipore, Cork, Ireland). Imaging flow data was analyzed using IDEAS® software (Amnis, WA, USA).
Immunoassays
Inflammatory chemokines, tumor-modifying factors and cytokine levels were quantified in MM cell secretomes using the high performance Human Magnetic Luminex Performance Assay Base Kit A (R&D Systems) and was carried out in accordance with the manufacturer’s recommended protocol. The Prostaglandin E2 (PGE2) Parameter Assay Kit (R&D Systems) was used to quantify PGE2 levels in the MM cell secretomes.39 This is a competitive enzyme immunoassay that was carried out in accordance with the manufacturer’s recommended protocol.
Generation of the cyclophosphamide-tumor cell secretome (CTX-TCS)
MM1.S or RPMI-8226 MM cells were plated in 6-well plates 48 hours in advance of treatment. Cells were treated with the reduced form of cyclophosphamide (CTX) (Santa Cruz Biotechnology, TX, USA) (0 µM, 2.5 μM, 5 μM, 10 μM and 20 µM) for 24 and 48 hours. Following treatment, media was discarded and cells were washed three times using warm Dulbecco’s phosphate buffered saline solution (PBS) (Thermofisher Scientific). Fresh media was replenished and left for a further 24 hours. The CTX-TCS was collected at 24 hours and used for subsequent experiments (as depicted in the Figure 2(d) schematic).
Figure 2.
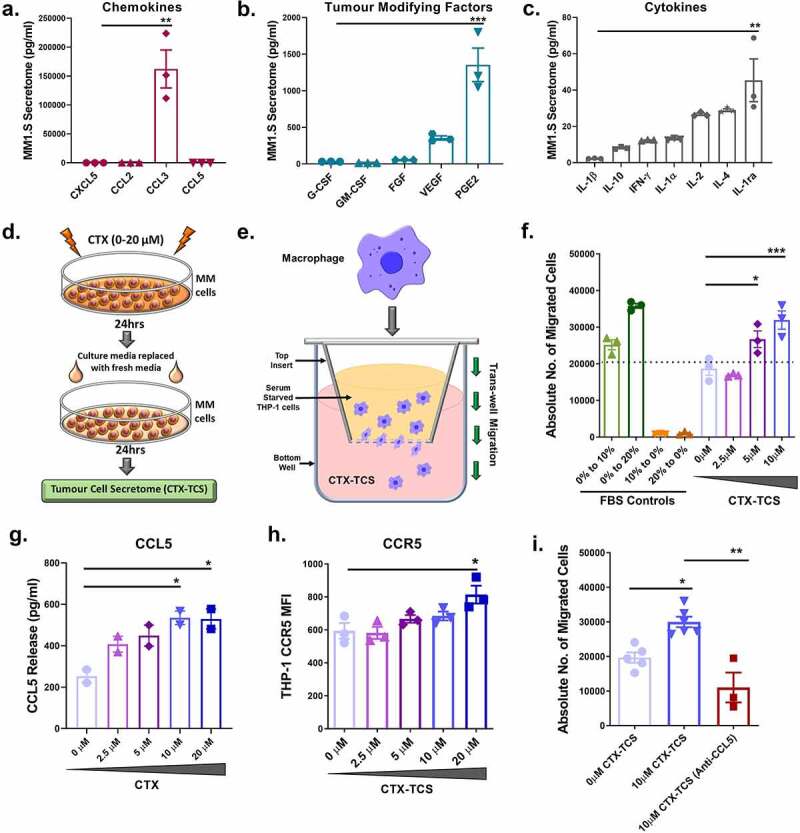
Low dose cyclophosphamide enhances macrophages migration to the tumor microenvironment. (a-c) Multiplex immunoassay of macrophage specific chemokine, tumor promoting factor and cytokine release from MM1.S cells (pg/ml). [Note: PGE2 data was generated using a competitive ELISA]. Significance was investigated relative to the factor with the lowest secreted concentration (Chemokine – CXCL5; Tumor modifying factor – G-CSF; Cytokine- IL-1β). (d) Schematic of the generation and collection of the cyclophosphamide tumor cell secretome (CTX-TCS). (e) Schematic of the transwell migration assay setup, with serum deprived THP-1 suspension in the well insert and FBS containing media or CTX-TCS in the underlying well. (f) The total number of THP-1 cells migrating toward 0–10 µM CTX-TCS, quantified by flow cytometry. Absolute cell counts are reported here. FBS gradients were used as transwell migration controls (0% to 10% and 0 to 20% FBS serving as positive controls; 10% to 0% and 20% to 0% FBS serving as negative controls). (g) CCL5 release (pg/ml) from MM1.S cells in the presence of increasing concentrations of cyclophosphamide, quantified using a multiplex immunoassay (h) Expression of CCR5 on THP-1 cell surface following treatment with 0–20 µM CTX-TCS, quantified by flow cytometry and presented as median fluorescent intensity (MFI). (i) The total number of THP-1 cells migrating toward 10 µM CTX-TCS, in the absence or presence of 0.1 µg/ml anti-CCL5, quantified by flow cytometry and reported as absolute cell counts. Data from two -three independent experiments (n = 2-3) are presented here as scatter plots with bars (mean ± S.E.M). One-way ANOVA statistical analysis carried out, followed by Tukey’s post-hoc test. *p < 0.05. **p < 0.01. ***p < 0.001
Macrophage migration assay
THP-1 cells were serum-starved for 24 hours. THP-1 cell suspensions (1x105 cells) were added into the top wells of 8 μm transwell inserts which were placed into a 24-well plate. Culture media was placed into the bottom of the 24-well plate (as depicted in the Figure 2(e) schematic). 10% FBS-containing culture media and 20% FBS-containing culture media were used as positive migration controls. TCS released from cyclophosphamide treated MM cells (CTX-TCS), as described above, was placed into the wells of the 24-well plate in the absence or presence of 0.1 µg/ml anti-CCL5 (R&D Systems, MN, USA). The plate was returned to the incubator at 37°C for 4 hours, after which the media containing migrated THP-1 cells in the 24-well plates was collected. The media was centrifuged (400 g for 5 minutes), the cells were collected and counted using an Accuri™ C6 flow cytometer (BD Biosciences). Cell number was reported as absolute counts.
Human peripheral blood mononuclear cell isolation
Freshly drawn peripheral blood (PB) was collected in 10 ml ethylene diamine tetraacetic acid (EDTA) Vacutainer® tubes (BD Medical Supplies, Crawley, UK). Mononuclear cells (MC) were isolated from PB in 15 ml falcon tubes (Sarstedt, Wexford, Ireland) by layering 3 ml EDTA-anti-coagulated sample over 3 ml endotoxin-free Ficoll®-Paque density-gradient medium (GE Healthcare, Little Chalfont, United Kingdom). Samples were centrifuged for 22 minutes at 420 g at 4°C without brake. Using a plastic Pasteur pipette (Sarstedt), the visible “buffy coat” layer of mononuclear cells was removed. PBMCs were transferred into a fresh falcon tube (Sarsedt) and subsequently washed with 10 ml fluorescence-activated cell sorting (FACS buffer) [phosphate buffered saline (Thermofisher Scientific), 2% FBS (Sigma), and 0.05% sodium azide (Sigma)]. This suspension was centrifuged at 300 g for 10 minutes at 4°C. The pelleted mononuclear cells were re-suspended in 1 ml FACS buffer after the supernatant was discarded. Isolated PBMCs were treated with the reduced form of cyclophosphamide (CTX) (Santa Cruz Biotechnology) (0 µM, 2.5 μM, 5 μM, 10 μM and 20 µM) for 24 hours and subsequently stained for phenotypic characterization by flow cytometry. This research was approved by the local hospital ethics committee according to the requirements of Irish regulations, and it was conducted in accordance with the International Conference on Harmonization of Good Clinical Practice Guidelines and the principles of the Declaration of Helsinki.
Acquisition of samples from the translational CyBorD-DARA (16-BCNI-001/CTRIAL-IE 16-02) study
Patient samples were obtained from Phase 1b open-label, single-arm, dose escalation study (CyBorD-DARA 16-BCNI-001/CTRIAL-IE 16–02 study), as previously described.30 This trial is registered at www.clinicaltrials.gov as NCT02955810. All patients provided written informed consent. The local hospital ethics committee according to the requirements of Irish regulations approved this research, and it was conducted in accordance with the International Conference on Harmonization of Good Clinical Practice Guidelines and the principles of the Declaration of Helsinki. Mononuclear cells from PB and BM aspirates were isolated from samples collected from patients pre-treatment and post-treatment with CyBorD and DARA, as previously described.30 The mononuclear cell aliquots were incubated with optimized concentrations of fluorochrome-conjugated antibodies (mouse anti-human CD45-V500, CD16-FITC, CD14-PerCP, CX3CR1-Pe-Cy7, HLA-DR-APC, CD33-Vio770, CD56-V450, CD32-PE, CD163-APC) for 20 min at 4°C to allow identification of monocytes/macrophages, as well as monocyte/macrophage subsets. Fluorescence compensation was set using single-stained controls, and matching median compensation algorithms were applied. Fluorescence minus one controls were used to set analysis gate controls using a published gating strategy.30 The Median fluorescence intensity (MFI) was calculated by expressing the fluorescence intensity of each sample relative to the cell size. Data were analyzed using Diva v8.0.1 acquisition software (BD Biosciences) or FlowJo V10.6..2 software.
Antibody-dependent cellular phagocytosis (ADCP) assays
MM cell lines were fluorescently labeled using the CellTrace™ CFSE Cell Proliferation Kit (Thermofisher Scientific). Cells were stained as outlined in the manufacturer’s instructions. Stained cells were co-cultured with control or CTX-TCS conditioned macrophages at an effector to target (E:T) ratio of 2:1 for 18 hours. MM cells were pre-incubated in the presence of 1 μg/ml daratumumab (Janssen, PA, USA), an IgG1 anti-CD38 antibody, or the relevant isotype control and then added to cultures. After 18 h incubation, cells were imaged by fluorescent microscopy and then detached by pipetting to single cell suspensions. MM cell clearance by ADCP was assessed by flow cytometry. Percentage daratumumab-specific cell clearance was calculated as follows; [100–100*(% CFSE labeled MM cells (daratumumab treated)/% CFSE labeled MM cells (isotype control))]. To confirm that clearance was due to macrophage-mediated phagocytosis, experimental wells containing macrophages were pre-incubated with 1 μg/ml Cytochalasin D (Sigma), which inhibits actin polymerization and thus inhibits phagocytosis. When investigating the role played by FcγRI/CD64 in daratumumab-specific killing, macrophages were pre-incubated with anti-CD64 blocking antibody (eBiosciences, CA, USA).
Statistical analysis
Statistical analysis was carried out using GraphPad® Prism Version 8 (GraphPad Software, CA, USA) and Microsoft® Excel (Microsoft Corporation, WA, USA). Data were assessed for normal distribution using Shapiro Wilks normality test. Parametric data were analyzed using independent t-tests, one-way ANOVA and two-way ANOVA, followed by Tukey’s multiple comparison post hoc testing. For analysis of patient samples from CyBorD-Dara translational study, Wilcoxon matched pairs signed rank test and paired two-tailed t-tests were used. Statistical significance was considered at p < 0.05.
Results
Low dose cyclophosphamide induces low levels of MM cell death, an effect that is not potentiated by either lenalidomide or bortezomib
Findings from the CyBorD-DARA Phase 1b clinical trial suggested that combining cyclophosphamide with daratumumab increased MM cell vulnerability to ADCP.30 Firstly, we assessed the direct effects of cyclophosphamide alone or in combination with typical frontline MM therapies lenalidomide and bortezomib on MM cell death. MM1.S cells were treated with low dose concentrations (0–20 µM) of cyclophosphamide alone or in combination with 1 µM lenalidomide or 0.8 nM bortezomib over a 48 hour time course, after which in vitro cell death was quantified by flow cytometry (Figure 1(a)). A representative gating strategy for excluding debris, identifying viable cells and eliminating doublets is provided in Figure 1(b). Low-dose cyclophosphamide treatment increased MM cell death in a dose-dependent manner, inducing moderate levels of cell death at 20 µM cyclophosphamide after 24 hours (Figure 1(c)) and from 10 µM cyclophosphamide after 48 hours (Figure 1(d)). Immunomodulatory drugs (IMiD) and proteasome inhibitors (PI) are ubiquitous MM drugs that contribute to the majority of MM treatment regimens. The effects of low dose (10 µM) cyclophosphamide in combination with these standard frontline therapies were investigated by flow cytometry. The immunomodulatory agent (lenalidomide) and proteasome inhibitor (bortezomib) did not increase MM cell death effects when administered alone or in combination with cyclophosphamide over the 48 hour time course (Figure 1(e–f)). These results show that low dose cyclophosphamide induces low levels of cell death, an effect that is not potentiated by either lenalidomide or bortezomib. Considering the potent effects of cyclophosphamide previously identified, it suggests that functional effects other than those induced by cell death may be important in cyclophosphamide-mediated anti-tumor mechanisms.34 Therefore, we next assessed the immunomodulatory function of cyclophosphamide using immunoassays.
Figure 1.
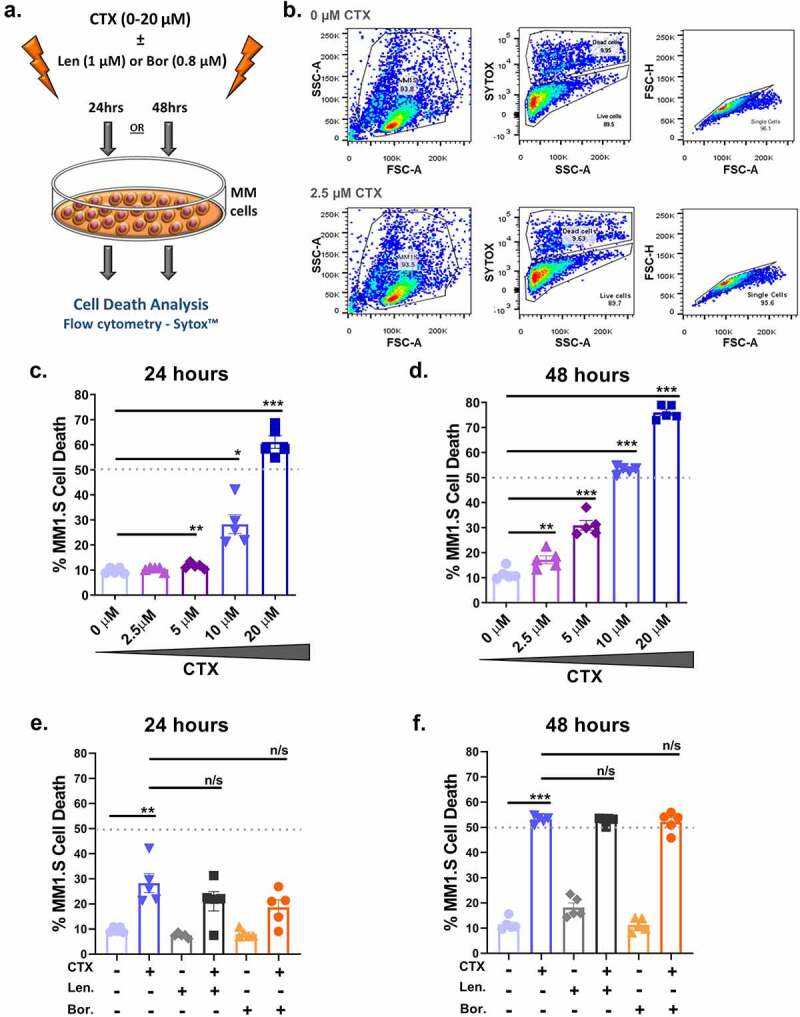
Low dose cyclophosphamide induces low level cell death in MM cell lines, which is not potentiated by either lenalidomide or bortezomib. (a) Schematic of MM1.S cells directly treated with 0–20 µM cyclophosphamide (CTX), 1 µM lenalidomide (Len) and 0.8 nM bortezomib (Bor) alone or in combination for 24 or 48 hours. (b) Sample gating strategy for excluding debris, identifying viable cell populations with Sytox™ and eliminating doublets. (c-d) Flow cytometry analysis (Sytox™) of MM1.S cell death following 0–20 µM cyclophosphamide for 24 or 48 hours. (e-f) Flow cytometry analysis (Sytox™) of MM1.S cell death in response to 10 µM cyclophosphamide, 1 µM lenalidomide or 0.8 nM bortezomib alone or in combination for 24 or 48 hours. Data from four independent experiments (n = 4) are presented here as scatter plots with bars (mean ± S.E.M). One-way ANOVA statistical analysis carried out, followed by Tukey’s post-hoc test. *p< 0.05. **p < 0.01. ***p < 0.001. n/s p > 0.05
Cyclophosphamide alters the tumor microenvironment and enhances macrophage migration
Cyclophosphamide has previously been shown to have immunomodulatory effects in the TME, diminishing Treg levels32 as well as increasing macrophage infiltration and function in leukemia in vivo.34 Next we sought to assess the effects of cyclophosphamide on macrophage phenotype and function in response to the MM TME. Immune effector cells, including macrophages and NK cells, are attracted to tumors by chemokines released from the target cells. Here, we assessed the effects of low dose cyclophosphamide treatment in vitro on the MM cell secretome and subsequent macrophage recruitment. MM cell secretome was collected and analyzed by a multiplex immunoassay to quantify baseline release of macrophage-specific chemokines (CXCL5, CCL2, CCL3 and CCL5), tumor-modifying factors (G-CSF, GM-CSF, FGF, VEGF and PGE2) and cytokines (IL-1β, IL-10, IFN-γ, IL-1α, IL-2, IL-4 and IL-1ra) from tumor cells. The levels of chemokines and tumor-modifying factors in TCS exceeded cytokine levels, by a factor of >1000-fold and >10-fold, respectively (Figure 2(a–c))
Similar patterns of release were identified in a second MM cell line (RPMI-8226), with macrophage-specific chemokines and tumor-modifying factors representing the greatest portion of the secretome (Supplementary Figure 1a-c). Together, these data indicate that macrophage-specific chemokines are most highly released by MM cells. Next, we investigated the effects of cyclophosphamide treatment on the MM cell secretome and subsequent immune effector (macrophage) cell recruitment and functional responses. The TCS released from MM cells treated with low dose (0–20 µM) cyclophosphamide (CTX-TCS) was collected for subsequent experimental setup, as depicted in Figure 2(d).
To assess macrophage recruitment to the tumor microenvironment, THP-1 cell migration toward CTX-TCS was analyzed, as depicted in Figure 2(e). The number of macrophages that migrated from a serum-starved suspension toward 0–10 µM CTX-TCS was quantified by flow cytometry. THP-1 macrophage migration toward the MM1.S secretome was enhanced to the levels of the positive controls (10% and 20% FBS) when the MM cells had been treated with 5 μM and 10 μM cyclophosphamide (Figure 2(f)). This indicates that MM cell TCS are altered by cyclophosphamide, resulting in enhanced secretion of chemokines that subsequently recruit macrophages to the tumor microenvironment.
Next we investigated potential mediators of this enhanced immune effector cell recruitment. Macrophage chemokine response is mediated via MM cell surface ligands binding receptors on macrophages. Treatment of MM1.S cells with cyclophosphamide did not increase the release of CXCL5, CCL2, CCL3 (data not shown), but did significantly increase CCL5 release in a dose-dependent manner (Figure 2(g)). A similar trend in CCL5 release from cyclophosphamide treated RPMI-8226 cells was also observed (Supplementary Figure 1d). Although some studies have published pro-tumourigenic effects of CCL5,14 more recent studies have identified potent anti-tumor activity of CCL5.40 CCL5 has previously been shown to sustain leukocyte activation and inflammatory responses.41 To investigate whether these infiltrating macrophages have pro- or anti-tumor effects, we investigated their cellular function. Informed by the increase in CCL5 release, we assessed the expression of the corresponding receptor on macrophages after 0–20 µM CTX-TCS conditioning by flow cytometry. Conditioning of THP-1 cells with CTX-TCS caused a significant upregulation in CCR5 (CCL3 and CCL5 receptor) protein expression at 20 μM cyclophosphamide dose (Figure 2(h)). These results indicate that low dose cyclophosphamide treatment of MM cells enhances macrophage migration to the tumor microenvironment potentially by increasing expression of chemokine receptors on the inflammatory macrophage cell surface. To determine if the enhanced macrophage migration toward CTX-TCS was mediated primarily via CCL5, we measured THP-1 migration toward MM1.S CTX-TCS in the absence or presence of anti-CCL5 (Figure 2(i)). There was a statistically significant reduction in migration of THP-1 cells toward 10 µM CTX-TCS in the presence of anti-CCL5. These results suggest CCL5 mediated migration is enhanced by cyclophosphamide.
Cyclophosphamide enhances the expression of proteins that positively regulate ADCP
ADCC by NK cells42 and ADCP by macrophages43 in the TME are two of the major mechanisms underlying monoclonal antibody anti-tumor activity. Cell death induced by daratumumab requires binding of the antibody to both CD38, highly expressed on multiple myeloma cells, and Fcγ receptors, found on both macrophages and NK cells (Figure 3(a)). As we previously showed, NK cells are depleted in patients treated with daratumumab, therefore, in this study, we focused on monocyte/macrophages as the macrophage number is not altered by daratumumab treatment.30 First, we assessed the effects of cyclophosphamide on the expression of the daratumumab target CD38 on MM cells. Importantly, the expression of CD38 on MM1.S (Figure 3(b)) and RPMI-8226 (Supplementary Figure 2b) cell surfaces was maintained following treatment with cyclophosphamide at 24 and 48 hours. In fact, a significant increase was observed in CD38 expression following cyclophosphamide treatment at 24 hours (Figure 3(b)).
Figure 3.
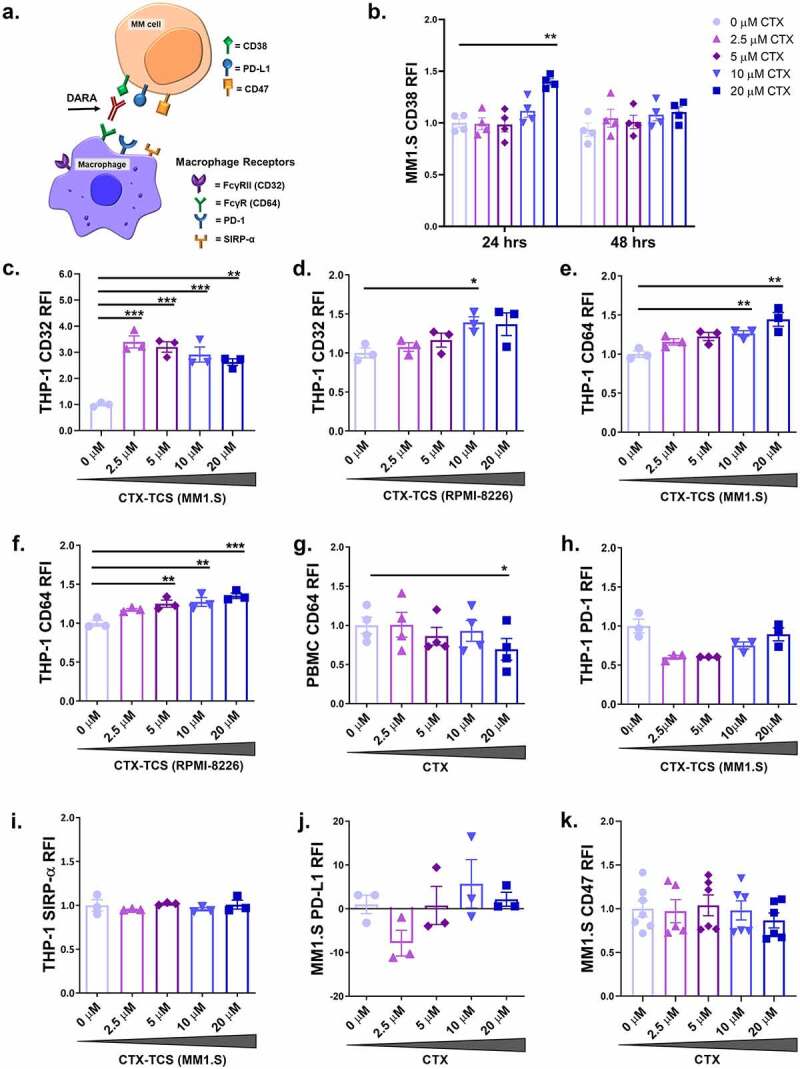
Low dose cyclophosphamide enhances expression of ADCP promoting proteins on MM cells and macrophages. (a) Schematic of receptors on macrophages and their corresponding ligands on MM cells that regulate ADCP. FcγRI/CD64 binds CD38, PD-1 binds PD-L1 and SIRP-α binds CD47. Daratumumab targets FcγRI/CD64 on macrophages and CD38 on MM cells. (b) CD38 expression on MM1.S cells following treatment with 0–20 µM cyclophosphamide for 24 or 48 hours. (c-d) Expression of FcγRII/CD32 on THP-1 cells following treatment with 0–20 µM CTX-TCS from MM1.S or RPMI-8226 cells, respectively, for 24 hours. (e-f) Expression of FcγRI/CD64 on THP-1 cells following treatment with 0–20 µM CTX-TCS from MM1.S or RPMI-8226 cells, respectively, for 24 hours. (g) FcγRI/CD64 expression on healthy volunteer PBMCs following direct treatment with 0–20 µM cyclophosphamide for 24 hours. (h-i) PD-1 and SIRP-α expression on THP-1 cells conditioned with 0–20 µM CTX-TCS for 24 hours. (j-k) PD-L1 and CD47 expression on MM1.S cells that received 0–20 µM cyclophosphamide for 24 hours. Expression was quantified by flow cytometry and presented as RFI (expression relative to corresponding untreated (0 µM) control group). Data from three or more independent experiments (n ≥ 3) are presented here as scatter plots with bars (mean ± S.E.M). One-way ANOVA statistical analysis carried out, followed by Tukey’s post-hoc test. *p < 0.05. **p < 0.01. ***p< 0.001
ADCP is regulated by macrophage cell surface receptors, including pro-phagocytic Fcγ receptors (FcγRII/CD32, FcγRI/CD64 and FcγRIII/CD16)26 and anti-phagocytic PD-1 and SIRP-α44,45 (Figure 3(a)). Conditioning THP-1 macrophages with CTX-TCS generated from MM1.S cells increased FcγRII/CD32 expression at each of the concentrations assessed relative to the 0 µM CTX-TCS control (Figure 3(c)). An increase in FcγRII/CD32 was also recorded when THP-1 cells were conditioned with 10 µM CTX- TCS from RPMI-8226 cells relative to the 0 µM CTX-TCS control (Figure 3(d)). However, the FcγRII/CD32 receptor has two subunits, one activating and one inhibitory (CD32a and CD32b respectively), and since the antibody used does not discriminate between these subunits we cannot determine whether FcγRII/CD32 increases are indicative of more active or inactive macrophages. The ratio of activating versus inhibitory subunit is a key factor in determining the function of CD32 binding.46,47 As CD64 has the highest affinity for IgG1 antibodies, we focussed on the expression of CD64 and its role in regulation of daratumumab-mediated ADCP. Data published from the CyBorD-DARA clinical trial reported increased FcγRI/CD64 expression on the surface of macrophages isolated from the bone marrow and PBMCs of MM patients who received cyclophosphamide treatment.30 Our in vitro studies support this finding, with CTX-TCS from MM1.S cells increasing FcγRI/CD64 expression on THP-1 cells in a dose-dependent manner, reaching statistical significance at 10 μM and 20 µM CTX-TCS (Figure 3(e)). We also observed a dose-dependent increase in FcγRI/CD64 on THP-1 cells conditioned with CTX-TCS from RPMI-8226 cells (Figure 3(f)). Ex vivo analysis of patient BM and peripheral blood samples from the CyBorD-DARA clinical trial mirrored this finding, with significantly enhanced FcγRI/CD64 expression observed after 24 hours of treatment.30 FcγRIII/CD16 expression in patient PBMC and bone marrow samples was analyzed by flow cytometry but was not found to increase following cyclophosphamide treatment (data not shown). Together, the significant increase in CD38 on MM1.S cells and FcγRI/CD64 on macrophages indicates a potential functional role for FcγRI/CD64 in ADCP.
To elucidate whether cyclophosphamide can alter macrophage activity directly or whether it relies on the tumor microenvironment to elicit its effects, THP-1 cells or healthy PBMCs were treated directly with 0–20 µM cyclophosphamide for 24 hours, after which surface expression of macrophage-activating Fcγ receptors was analyzed by flow cytometry. Cyclophosphamide did not alter FcγRII/CD32 or FcγRI/CD64 expression when applied directly to the THP-1 cells (Supplementary Figure 3). This lack of activation was also recorded with healthy PBMCs. In fact, 20 µM cyclophosphamide was found to reduce FcγRI/CD64 surface expression on human PBMCs (Figure 3(g)). Together, these findings indicate that enhanced anti-tumourigenic effects of cyclophosphamide are dependent on responses of the tumor microenvironment that enhance recruitment and activation of macrophages.
Evasion of phagocytosis by myeloma cells can be achieved when PD-1 and SIRP-α anti-phagocytic receptors on the macrophage cell surface bind PD-L1 and CD47 ligands present on the MM cells, respectively44,45 (Figure 3(a)). The effect of cyclophosphamide on these regulatory receptors and ligands was analyzed by flow cytometry. Conditioning THP-1 macrophages with CTX-TCS did not alter the expression of PD-1 (Figure 3(h)) or SIRP-α (Figure 3(i)) receptors at any dose assessed (0–20 µM). Previous ex vivo analysis of bone marrow and peripheral blood from cyclophosphamide treated myeloma patients also recorded no change in SIRP-α expression.30 Cyclophosphamide did not alter the expression of PD-L1 (Figure 3(j)) or CD47 (Figure 3(k)) ligands on MM1.S cells at any dose assessed (0–20 µM) in vitro. However, it was previously shown that cyclophosphamide reduced CD47 expression on tumor cells in the bone marrow of MM patients participating in the CyBorD-DARA clinical trial.30 This suggests that the complexity of the TME may not be fully recapitulated ex vivo in these assays. Our results suggest that cyclophosphamide induces pro-phagocytic markers on the macrophages. These data propose that the mechanism by which cyclophosphamide potentiates daratumumab anti-myeloma activity is by enhancing pro-phagocytic FcγRI/CD64 expression on the macrophage cell surface.
Low dose cyclophosphamide potentiates the anti-myeloma activity of daratumumab
Macrophage-mediated ADCP can be activated and enhanced by inclusion of functional Fc- containing antibodies. We next assessed the level of macrophage-mediated ADCP in combination with daratumumab. Using fluorescent microscopy and flow cytometry, we assessed the effect of daratumumab on CTX-TCS conditioned macrophages’ ability to phagocytose MM cells, that have been fluorescently labeled with CFSE, in the presence or absence of a phagocytosis inhibitor; Cytochalasin D (CytoD) (Figure 4(a)).
Figure 4.
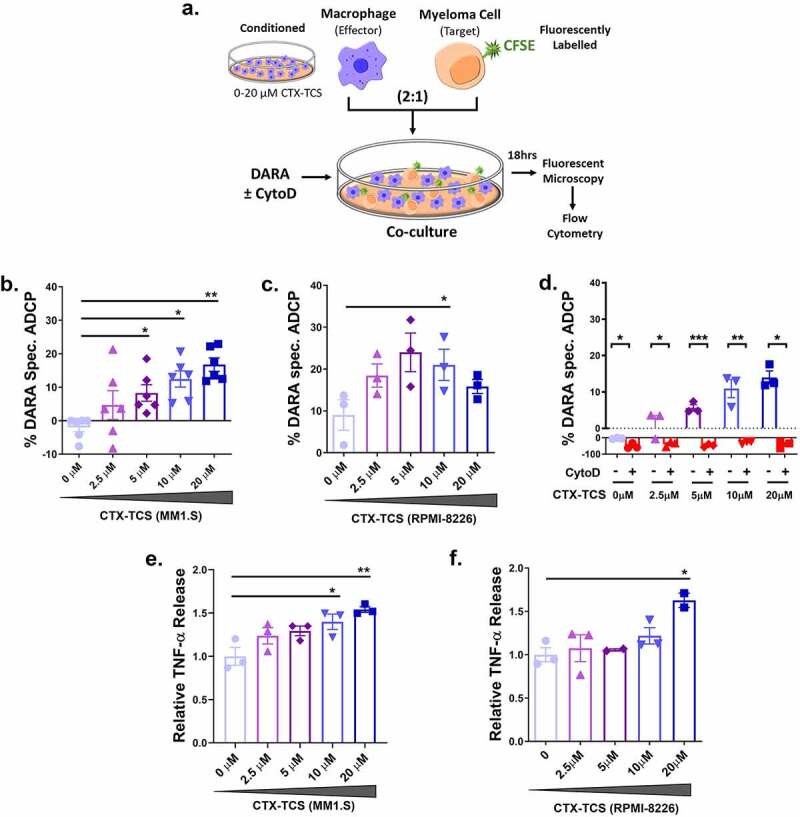
Low dose cyclophosphamide potentiates daratumumab mediated macrophage anti-myeloma activity. (a) Schematic of experimental setup. CFSE-labeled multiple myeloma cells were co-cultured with THP-1 macrophages pre-conditioned in 0–20 µM CTX-TCS. Following 18 hr incubation, fluorescent cells were analyzed by fluorescent microscopy and flow cytometry. (b) Daratumumab-mediated antibody-dependent cellular phagocytosis (ADCP) of MM1.S cells by 0–20 µM CTX-TCS conditioned THP-1 macrophages. (c) Daratumumab-mediated ADCP of RPMI-8226 cells by 0–20 µM CTX-TCS conditioned THP-1 macrophages. (d) Daratumumab-mediated ADCP of MM1.S cells by 0–20 µM CTX-TCS conditioned THP-1 macrophages in the absence or presence a phagocytosis inhibitor, 1 μg/ml CytoD. (e-f) Relative TNF-α release (fold change relative to control) from MM1.S and RPMI-8226 cells in the presence of CTX-TCS. Data from at least three independent experiments (n ≥ 3) are presented here as scatter plots with bars (mean ± S.E.M). Independent t-tests and one-way ANOVA statistical analysis carried out, followed by Tukey’s post-hoc test. *p < 0.05. **p < 0.01. ***p < 0.001
The effect of conditioning with CTX-TCS on the efficacy of daratumumab anti-myeloma activity was investigated by flow cytometric analysis. The percentage increase in the number of CFSE-stained MM cells encapsulated by THP-1 macrophage cells was calculated from MFI values. Low dose CTX-TCS was found to increase daratumumab-specific killing of MM1.S cells in a dose-dependent manner, with statistical significance observed from 5 µM – 20 µM cyclophosphamide (Figure 4(b)). A similar increase in ADCP was observed for RPMI-8226 cells (Figure 4(c)).
ADCP depends on cellular cytoskeletal re-arrangement; therefore, we assessed the effects of an inhibitor on the level of ADCP to confirm that the mechanism of action was in fact ADCP and not direct cytotoxicity (as macrophages have the capacity to directly kill myeloma cells by releasing high levels of IFN-γ and nitric oxide). THP-1 macrophages were treated with 1 μg/ml CytoD, which inhibits actin polymerization and therefore blocks phagocytosis. Flow cytometric analysis revealed the synergistic increase in MM cell clearance observed following cyclophosphamide and daratumumab treatment was diminished in the presence of CytoD. Dose-dependent increases in daratumumab-directed MM1.S cell phagocytosis by CTX-TCS conditioned THP-1 macrophages was found to be completely abrogated in the presence of CytoD, thereby suggesting phagocytosis as the primary mechanism of action of daratumumab (Figure 4(d)). A similar trend was also observed when quantifying RPMI-8226 MM cell phagocytosis (Supplementary Figure 4). Pro-inflammatory macrophages release numerous factors, including TNF-α, which can induce cancer cell death.48 Therefore, we investigated TNF-α levels following CTX-TCS treatment to investigate the functional consequences of conditioning with CTX-TCS. Cyclophosphamide was found to increase TNF-α release from both MM1.S and RPMI-8226 myeloma cell lines in a dose-dependent manner (Figure 4(e–f)). Taken together, these data provide evidence for cyclophosphamide increasing daratumumab-dependent MM cell phagocytosis by enhancing macrophage anti-tumor activity.
Daratumumab’s mechanism of anti-myeloma activity is partially dependent on FcγRI/CD64 expression on tumor-associated macrophages
It has been shown that FcyRI/CD64 expression can positively regulate ADCP, as it mediates IgG1 specific binding with the highest affinity.26 To investigate the potential role of CD64 in the context of daratumumab-induced ADCP, we used imaging flow cytometry in the presence and absence of a CD64 blocking antibody. Here, we visualized ADCP by flow cytometry of fluorescently labeled target cells that were co-cultured with effector cells (Figure 5(a)). Daratumumab-dependent cellular phagocytosis of MM cells was analyzed by CFSE-labeling MM1.S MM cells (green) and labeling CD45+ (purple) and CD64+ (blue) THP-1 macrophages. Imaging flow cytometry showed macrophages phagocytosing myeloma cells. The ability of macrophages to encapsulate MM1.S cells was dependent on FcγRI/CD64 expression. CD64 high (+) and very high (++) expressing macrophages were primarily responsible for the observed phagocytosis, whereas no or very low levels of phagocytosis were observed with CD64 low macrophages (Figure 5(b)).
Figure 5.
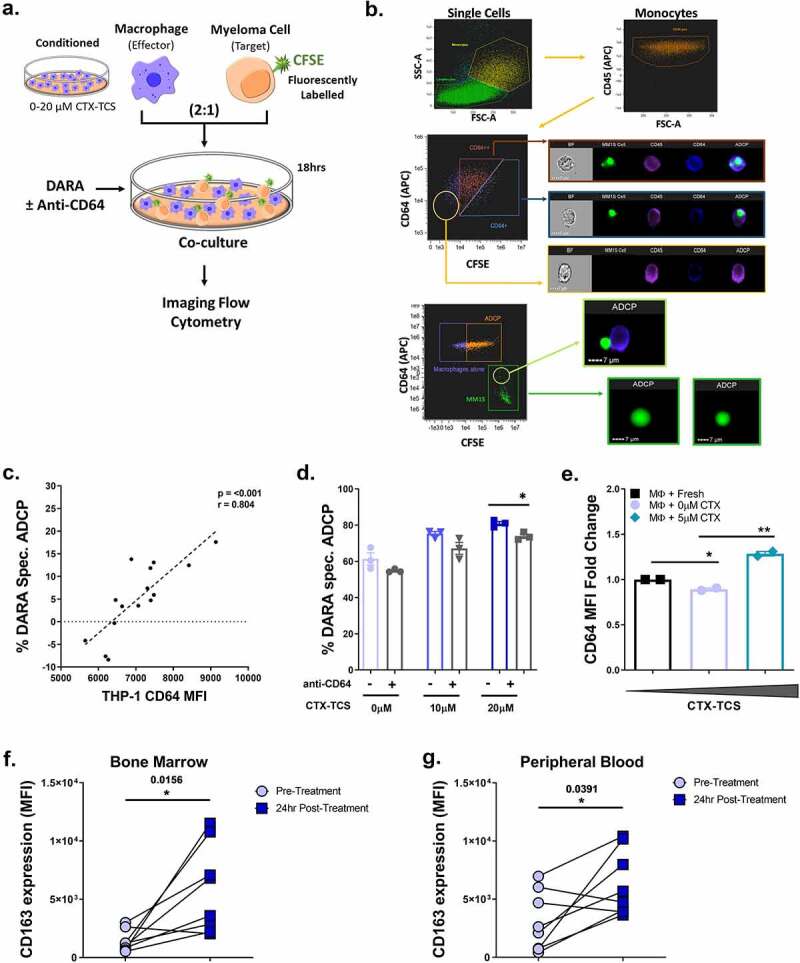
Daratumumab’s mechanism of anti-myeloma activity is partially dependent on FcγRI/CD64 expression on tumor associated macrophages. (a) Schematic of experimental setup. CFSE-labeled multiple myeloma cells were co-cultured with THP-1 macrophages pre-conditioned in 0–20 µM CTX-TCS. Following 18 hr incubation, fluorescent cells were quantified by imaging flow cytometry. (b) Imaging flow cytometry showing the encapsulation of MM1.S cells (CFSE-labeled, green) in macrophage cells (CD45+ cells, purple; CD64+ cells, blue), thereby demonstrating ADCP (green MM cells inside blue/purple macrophages). (c) Correlation analysis of THP-1 CD64 expression and corresponding percentage daratumumab -specific MM1.S cell clearance. Data from three independent experiments (n = 3) are presented here. (d) Daratumumab-specific ADCP of CTX-TCS conditioned MM1.S cells in the absence or presence of a CD64 blocking antibody. Data from three independent experiments (n = 3) are presented here as scatter plots with bars (mean ± S.E.M). (e) Flow cytometry data showing CD64 expression on macrophages from active multiple myeloma patients that were conditioned with fresh media, 0 µM or 5 µM CTX-TCS ex vivo. Data from two clinical samples (n = 2) are presented here as scatter plots with bars (mean ± S.E.M). (f-g) Dot plots indicate the Median Fluorescent intensity of CD163 expression on CD33+ CX3CR1+ CD56− CD14+ macrophages from active multiple myeloma patient bone marrow or PBMCs, respectively. Data from eight separate clinical samples collected before (Pre-Treatment) and 24 hours after treatment with cyclophosphamide (24 hr Post-Treatment) are presented here. Lines between dots indicate paired samples. Statistical analysis was performed by independent t-tests or the Wilcoxon matched pairs signed rank test. *p < 0.05. **p < 0.01. ***p < 0.001
We next investigated the relationship between FcγRI/CD64 expression on macrophages and their capacity to induce phagocytosis in the presence of daratumumab. Plotting THP-1 FcγRI/CD64 expression level versus percentage clearance of MM1.S cells by daratumumab revealed a direct correlation between FcγRI/CD64 expression and macrophage function (Figure 5(c)).
To functionally assess the impact of FcγRI/CD64, we repeated the experiment in the presence of an anti-CD64 blocking antibody. As seen in our previous experiments, low dose CTX-TCS (20 µM) significantly increased daratumumab-specific killing of MM1.S cells. This enhanced clearance of MM cells was abrogated in the presence of the FcγRI/CD64 blocking antibody (Figure 5(d)). Although not reaching statistical significance, a similar trend was recorded in daratumumab-specific killing of RPMI-8226 cells in the presence of an FCγRI CD64 blocking agent (Supplementary Figure 5). Together, these data indicate FcγRI/CD64 as a key player in daratumumab-specific ADCP of MM cells by CTX-TCS conditioned macrophages. We next investigated the effect of cyclophosphamide on macrophages from patients with active MM. FcγRI/CD64 was increased following conditioning macrophages from MM patients with 5 µM CTX-TCS ex vivo (Figure 5(e)). Previous data from our lab indicates that patients treated with CyBorD-DARA induces expression of FCγRI/CD64 on patient derived blood and bone marrow monocyte/macrophage cells. In addition to this, CyBorD was found to increase CD163, a marker of phagocytic activity, expression on macrophages isolated from both the bone marrow (Figure 5(f)) and PBMCs (Figure 5(g)). Although only a small sample set, this data would suggest that tumor-associated macrophages from MM patients are responsive to cyclophosphamide, showing increased FCγRI/CD64 surface expression and enhanced phagocytic activity, thereby supporting a potential synergistic increase in daratumumab-specific phagocytosis.
Discussion
Despite increasing use of antibody-based therapies clinically, the mechanisms underlying their therapeutic efficacy is only partly understood. Daratumumab is the first CD38-directed monoclonal antibody approved for the treatment of MM. At the time of commencement of the Phase 1b CyBorD-DARA trial, daratumumab IV was only approved for use in RRMM as a single agent. It is now approved in RRMM in combination with dexamethasone and the IMiD lenalidomide or the PI bortezomib, based on results of two large phase 3 studies which showed synergism between these agents.8,49 Approval as initial therapy was subsequently granted for non-transplant eligible patients with lenalidomide/dexamethasone or bortezomib, melphalan and prednisolone (VMP), 50,51 and most recently, for transplant-eligible patients in combination with thalidomide, bortezomib and dexamethasone (VTD) .52
Daratumumab is thought to have pleiotropic mechanisms of activity, killing myeloma cells through induction of complement-dependent cytotoxicity (CDC), ADCC, apoptosis via cross-linking and ADCP.23 Given the rapid, marked, and prolonged depletion of NK cells seen in daratumumab-treated patients,29 we focused our attention on identifying possible means of enhancing macrophage-mediated ADCP, by harnessing the anti-tumor potential of TAMs. A previous study reported that resistance to macrophage-mediated killing in lymphoid malignancies is overcome by combining monoclonal antibodies with cyclophosphamide .34 Cyclophosphamide was shown to induce an acute secretory activating phenotype (ASAP) in treated tumor cells, which led to macrophage infiltration and enhanced phagocytic activity in the bone marrow. Given this information, we sought to establish the existence of this phenomenon in MM in the context of other drugs likely to be used in combination with daratumumab, i.e. lenalidomide or bortezomib. These in vitro results accompany ex vivo data from the Phase 1b CyBorD-DARA trial, reported previously.30
Our in vitro data show that low dose cyclophosphamide induces MM cell death via effects on the TME rather than direct cellular toxicity, and that these effects are neither produced nor potentiated by the IMiD lenalidomide or the PI bortezomib. Cyclophosphamide alters the TME, promoting macrophage migration to the site of the tumor as shown in migration studies, and by upregulation of THP-1 expression of the chemoattractant receptors CCR2 and CCR5. This is supported by ex vivo data showing a trend toward increased circulating levels of CCL5/RANTES alongside small but significant increases in the levels of pro-inflammatory TNF-α and IFN-γ, known to skew macrophages toward a pro-inflammatory/anti-tumor phenotype.11 Considering the hypothesis that cyclophosphamide potentiates daratumumab-induced macrophage-mediated ADCP, we assessed factors relevant to this process. Critically, our data show that low dose cyclophosphamide upregulates both the target antigen CD38 on MM cells and also FcγRI/CD64 on macrophages. In addition, the phagocytosis inhibitory ligands on myeloma cells, CD47 and PD-L1, and their corresponding receptors on macrophages, SIRP-α and PD-1 respectively, remained unchanged by cyclophosphamide in vitro. However, CD47 was decreased on bone marrow MM cells analyzed from CyBorD-DARA clinical trial patients.30 This contrasting in vitro and in vivo data may be a result of the tumor microenvironment not being fully recapitulated in vitro, resulting in an underestimation of cyclophosphamide’s impact on CD47 expression. Daratumumab-specific killing assays confirmed enhanced killing in the presence of low dose cyclophosphamide. Imaging flow cytometry analyses provided visual evidence that this killing is mediated in part by phagocytosis, supported by the partial inhibition seen by blocking actin polymerization. Furthermore, this killing was shown to be ADCP-mediated, demonstrated by its dependence upon intact FcγRI/CD64 function.
In combination with our previous observations in the Blood Advances paper30 supporting the role of CD64, this new data shows the enhanced expression of CD163 on monocytes/macrophages after CyBorD treatment. Although CD163 has been shown to have both pro- and anti- tumor effects, data exists to support its role in the phagocytic activity of macrophages in multiple myeloma patients.53 These data highlight the complexity and plasticity of macrophage phenotypes in various tumor microenvironments .54 A recent study indicated that CD163 expression identifying M2 macrophages, in combination with targeting CD47, increased anti-tumor phagocytosis. In the associated published clinical trial,30 we observed a decrease in CD47 on multiple myeloma cells following cyclophosphamide treatment. This data, alongside the favorable outcomes observed in the clinical trial, indicates an enhanced CD64/CD163 macrophage expression in combination with a reduction in MM cell CD47 with increased anti-tumor activity.30
In the clinical setting, cyclophosphamide has been demonstrated to synergize with several other myeloma therapies enabling responses to be attained in patients with resistant disease.55 For example, in the phase 1/2 REPEAT study, patients with documented lenalidomide-resistant disease were treated with lenalidomide, dexamethasone and low dose daily cyclophosphamide, with an overall response of 67% achieved.55 Low dose cyclophosphamide has recently been combined with daratumumab, the IMiD pomalidomide and dexamethasone in RRMM patients previously treated with lenalidomide and a PI. The regimen was efficacious and well tolerated in a heavily pre-treated group.56 Additionally, the ANDROMEDA study, a randomized controlled phase 3 study comparing CyBorD-DARA with CyBorD in patients with light chain amyloidosis, a related plasma cell dyscrasia to MM is ongoing, with a positive outcome reported.57
Our results show that cyclophosphamide enhances daratumumab-mediated killing of MM cells via alterations in the TME, which promote macrophage recruitment, polarization to a pro-inflammatory phenotype, and ADCP. These effects may complement the activity of immunomodulatory agents and proteasome inhibitors. Cyclophosphamide is a widely accessible, orally available, well-tolerated and relatively inexpensive treatment for multiple myeloma. It has numerous other immunomodulatory activities including the ability to promote cytotoxic T cell function, downregulate anti-inflammatory Tregs, and improve presentation of tumor antigens by dendritic cells.32 Our findings support the addition of cyclophosphamide to existing or novel immunotherapy regimens for the treatment of MM patients and those with related plasma cell disorders, and shed light on its mechanism of action in the context of monoclonal antibody therapy.
Supplementary Material
Acknowledgements
This study was supported by research funding from Janssen Research & Development. Grant support to M.O.D. was provided by Science Foundation Ireland and the Irish Cancer Society (14/ICS/B3042 Blood Cancer Network Ireland (BCNI)). A.E.R. research was supported by translational funding from Janssen (ICD827536), the Irish Cancer Society (CRF12RYA) and Science Foundation Ireland (15/SIRG/3456 and 19/FFP6446) and Galway University Foundation (a foundation research lectureship to A.E.R.). T.R. research was supported by a Science Foundation Ireland Investigator Award (grant number 12/IA/1624) and by the Health Research Board of Ireland (grant number HRA_POR/2013/341).
All flow cytometry experiments were performed in the NUI Galway Flow Cytometry Core Facility which is supported by funds from NUI Galway, Science Foundation Ireland, the Irish Government’s Programme for Research in Third Level Institutions, Cycle 5 and the European Regional Development Fund. Technical and consultative support for flow cytometry experiments was provided by Dr Shirley Hanley of the NUI Galway Flow Cytometry Core Facility. Funding in support of imaging cytometry was received from Science Foundation Ireland under research infrastructure grant no. 16/RI/3760 and from the European Regional Development Fund. This research was supported by the HRB-Clinical Research Facility Galway, a unit of NUI Galway and Saolta University Health Care Group.
Funding Statement
This work was supported by the Janssen Research and Development (Grant Ref -ICD827536) .
Declaration of interest statement
M.O.D. and A.E.R. received research support for this study from Janssen Pharmaceuticals. M.O.D. holds equity in Onk Therapeutics and Carrick Therapeutics, has been a member of the Advisory Board for Janssen, Celgene, Adaptive Biotechnologies and has performed consultancy for Janssen and Abbvie. S.D.N. and K.L. received travel bursaries from Janssen Pharmaceuticals to present this research at the American Society of Hematology. The remaining authors declare no competing financial interests.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Barlogie B, Mitchell A, van Rhee F, Epstein J, Morgan GJ, Crowley J.. Curing myeloma at last: defining criteria and providing the evidence. Blood. 2014;124(20):3043–14. doi: 10.1182/blood-2014-07-552059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ravi P, Kumar SK, Cerhan JR, Maurer MJ, Dingli D, Ansell SM, Rajkumar SV. Defining cure in multiple myeloma: a comparative study of outcomes of young individuals with myeloma and curable hematologic malignancies. Blood Cancer J. 2018. February 28;8(3):26. doi: 10.1038/s41408-018-0065-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghobrial IM. Myeloma as a model for the process of metastasis: implications for therapy. Blood. 2012;120(1):20–30. doi: 10.1182/blood-2012-01-379024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kawano Y, Moschetta M, Manier S, Glavey S, Görgün GT, Roccaro AM, Anderson KC, Ghobrial IM. Targeting the bone marrow microenvironment in multiple myeloma. Immunol Rev. 2015;263(1):160–172. [DOI] [PubMed] [Google Scholar]
- 5.Noonan K, Borrello I. The immune microenvironment of myeloma. Cancer Microenviron. 2011;4(3):313–323. doi: 10.1007/s12307-011-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.de Haart SJ, van de Donk NW, Minnema MC, Huang JH, Aarts-Riemens T, Bovenschen N, Yuan H, Groen RWJ, McMillin DW, Jakubikova J, et al. Accessory cells of the microenvironment protect multiple myeloma from T-cell cytotoxicity through cell adhesion-mediated immune resistance. Clin Cancer Res Offj Am Assoc Cancer Res. 2013. October 15;19(20):5591–5601. doi: 10.1158/1078-0432.CCR-12-3676. [DOI] [PubMed] [Google Scholar]
- 7.Richardson PG, Weller E, Lonial S, Jakubowiak AJ, Jagannath S, Raje NS, Avigan DE, Xie W, Ghobrial IM, Schlossman RL, et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood. 2010. August 5;116(5):679–686. doi: 10.1182/blood-2010-02-268862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dimopoulos MA, Oriol A, Nahi H, San-Miguel J, Bahlis NJ, Usmani SZ, Rabin N, Orlowski RZ, Komarnicki M, Suzuki K, et al. Daratumumab, lenalidomide, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(14):1319–1331. doi: 10.1056/NEJMoa1607751. [DOI] [PubMed] [Google Scholar]
- 9.Rodríguez-Otero P, Paiva B, Engelhardt M, Prósper F, San Miguel JF. Is immunotherapy here to stay in multiple myeloma? Haematologica. 2017;102(3):423–432. doi: 10.3324/haematol.2016.152504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng Y, Cai Z, Wang S, Zhang X, Qian J, Hong S, Li H, Wang M, Yang J, Yi Q, et al. Macrophages are an abundant component of myeloma microenvironment and protect myeloma cells from chemotherapy drug-induced apoptosis. Blood. 2009;114(17):3625–3628. doi: 10.1182/blood-2009-05-220285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019. June 01;19(6):369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu Y, Chen X, Zheng Y. The role of tumor associated macrophages in multiple myeloma and its pathophysiological effect on myeloma cells survival, apopotosis and angiogenesis. Blood. 2015;126(23):4204. doi: 10.1182/blood.V126.23.4204.4204. [DOI] [Google Scholar]
- 13.Li Y, Zheng Y, Li T, Wang Q, Qian J, Lu Y, Zhang M, Bi E, Yang M, Reu F, et al. Chemokines CCL2, 3, 14 stimulate macrophage bone marrow homing, proliferation, and polarization in multiple myeloma. Oncotarget. 2015. September 15;6(27):24218–24229. doi: 10.18632/oncotarget.4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lapteva N, Huang XF. CCL5 as an adjuvant for cancer immunotherapy. Expert Opin Biol Ther. 2010. May;10(5):725–733. doi: 10.1517/14712591003657128. [DOI] [PubMed] [Google Scholar]
- 15.Ruffell B, Affara NI, Coussens LM. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012;33(3):119–126. doi: 10.1016/j.it.2011.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang H, Wang L, Chi P-D, Wang WD, Chen X-Q, Geng Q-R, Xia Z-J, Lu Y. High level of interleukin-10 in serum predicts poor prognosis in multiple myeloma. Br J Cancer. 2016;114(4):463–468. doi: 10.1038/bjc.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006. January 15;66(2):605–612. doi: 10.1158/0008-5472.CAN-05-4005. [DOI] [PubMed] [Google Scholar]
- 18.Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018. December 01;17(12):887–904. [DOI] [PubMed] [Google Scholar]
- 19.Ries Carola H, Cannarile Michael A, Hoves S, Benz J, Wartha K, Runza V, Rey-Giraud F, Pradel L, Feuerhake F, Klaman I, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014. June 16;25(6):846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 20.Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, Wang J, Contreras-Trujillo H, Martin R, Cohen JD, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012. April 24;109(17):6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, Johnson SF, Carrasco RD, Lazo S, Bronson RT, et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature. 2017. March 01;543(7645):428–432. doi: 10.1038/nature21409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Mercier I, Poujol D, Sanlaville A, Sisirak V, Gobert M, Durand I, Dubois B, Treilleux I, Marvel J, Vlach J, et al. Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res. 2013. August 1;73(15):4629–4640. doi: 10.1158/0008-5472.CAN-12-3058. [DOI] [PubMed] [Google Scholar]
- 23.Gül N, Babes L, Siegmund K, Korthouwer R, Bögels M, Braster R, Vidarsson G, Ten Hagen TLM, Kubes P, van Egmond M, et al. Macrophages eliminate circulating tumor cells after monoclonal antibody therapy. J Clin Invest. 2014. February 03;124(2):812–823. doi: 10.1172/JCI66776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Minard-Colin V, Xiu Y, Poe JC, Horikawa M, Magro CM, Hamaguchi Y, Haas KM, Tedder TF. Lymphoma depletion during CD20 immunotherapy in mice is mediated by macrophage FcγRI, FcγRIII, and FcγRIV. Blood. 2008;112(4):1205–1213. doi: 10.1182/blood-2008-01-135160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stewart R, Hammond SA, Oberst M, Wilkinson RW. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J ImmunoTher Cancer. 2014. August 19;2(1):29. doi: 10.1186/s40425-014-0029-x. [DOI] [Google Scholar]
- 26.Rosales C. Fcγ receptor heterogeneity in leukocyte functional responses. Front Immunol. 2017. March 20;8:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mellor JD, Brown MP, Irving HR, Zalcberg JR, Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J Hematol Oncol. 2013. January 4;6:1. doi: 10.1186/1756-8722-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Weers M, Tai Y-T, van der Veer MS, Bakker JM, Vink T, Jacobs DCH, Oomen LA, Peipp M, Valerius T, Slootstra JW, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol (Baltimore, Md: 1950). 2011. February;186(3):1840–1848. doi: 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- 29.Casneuf T, Xu XS, Adams HC 3rd, Axel AE, Chiu C, Khan I, Ahmadi T, Yan X, Lonial S, Plesner T, et al. Effects of daratumumab on natural killer cells and impact on clinical outcomes in relapsed or refractory multiple myeloma. Blood Adv. 2017;1(23):2105–2114. doi: 10.1182/bloodadvances.2017006866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O’Dwyer M, Henderson R, Naicker SD, Cahill MR, Murphy P, Mykytiv V, Quinn J, McEllistrim C, Krawczyk J, Walsh J, et al. CyBorD-DARA is potent initial induction for MM and enhances ADCP: initial results of the 16-BCNI-001/CTRIAL-IE 16-02 study. Blood Adv. 2019. June 25;3(12):1815–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Viola D, Dona A, Caserta E, Troadec E, Besi F, McDonald T, Ghoda L, Gunes EG, Sanchez JF, Khalife J, et al. Daratumumab induces mechanisms of immune activation through CD38+ NK cell targeting. Leukemia. 2020. April 16. doi: 10.1038/s41375-020-0810-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ghiringhelli F, Menard C, Puig PE, Ladoire S, Roux S, Martin F, Solary E, Le Cesne A, Zitvogel L, Chauffert B, et al. Metronomic cyclophosphamide regimen selectively depletes CD4+CD25+ regulatory T cells and restores T and NK effector functions in end stage cancer patients. Cancer Immunol Immunother. 2007. May 01;56(5):641–648. doi: 10.1007/s00262-006-0225-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Swan D, Gurney M, Krawczyk J, Ryan AE, O’Dwyer M. Beyond DNA damage: exploring the immunomodulatory effects of cyclophosphamide in multiple myeloma. Hemasphere. 2020. April;4(2):e350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pallasch CP, Leskov I, Braun CJ, Vorholt D, Drake A, Soto-Feliciano YM, Bent E, Schwamb J, Iliopoulou B, Kutsch N, et al. Sensitizing protective tumor microenvironments to antibody-mediated therapy. Cell. 2014. January 30;156(3):590–602. doi: 10.1016/j.cell.2013.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qin Z. The use of THP-1 cells as a model for mimicking the function and regulation of monocytes and macrophages in the vasculature. Atherosclerosis. 2012. March 01;221(1):2–11. doi: 10.1016/j.atherosclerosis.2011.09.003. [DOI] [PubMed] [Google Scholar]
- 36.Chanput W, Mes JJ, Wichers HJ. THP-1 cell line: an in vitro cell model for immune modulation approach. Int Immunopharmacol. 2014. November 01;23(1):37–45. doi: 10.1016/j.intimp.2014.08.002. [DOI] [PubMed] [Google Scholar]
- 37.Ryan AE, Colleran A, O’Gorman A, O’Flynn L, Pindjacova J, Lohan P, O’Malley G, Nosov M, Mureau C, Egan LJ, et al. Targeting colon cancer cell NF-κB promotes an anti-tumour M1-like macrophage phenotype and inhibits peritoneal metastasis. Oncogene. 2015. March 19;34(12):1563–1574. doi: 10.1038/onc.2014.86. [DOI] [PubMed] [Google Scholar]
- 38.O’Malley G, Treacy O, Lynch K, Naicker SD, Leonard NA, Lohan P, Dunne PD, Ritter T, Egan LJ, Ryan AE, et al. Stromal cell PD-L1 inhibits CD8+T-cell antitumor immune responses and promotes colon cancer. Cancer Immunol Res. 2018. November;6(11):1426–1441. doi: 10.1158/2326-6066.CIR-17-0443. [DOI] [PubMed] [Google Scholar]
- 39.Lynch K, Treacy O, Chen X, Murphy N, Lohan P, Islam MN, Donohoe E, Griffin MD, Watson L, McLoughlin S, et al. TGF-β1-licensed murine MSCs show superior therapeutic efficacy in modulating corneal allograft immune rejection in vivo. Mol Ther. 2020. May 30;28:2023–2043. doi: 10.1016/j.ymthe.2020.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Huffman AP, Lin JH, Kim SI, Byrne KT, Vonderheide RH. CCL5 mediates CD40-driven CD4+ T cell tumor infiltration and immunity. JCI Insight. 2020. May 21;5(10). doi: 10.1172/jci.insight.137263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee C-M, Peng -H-H, Yang P, Liou J-T, Liao -C-C, Day Y-J. C-C Chemokine Ligand-5 is critical for facilitating macrophage infiltration in the early phase of liver ischemia/reperfusion injury. Sci Rep. 2017. June 16;7(1):3698. doi: 10.1038/s41598-017-03956-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hatjiharissi E, Xu L, Santos DD, Hunter ZR, Ciccarelli BT, Verselis S, Modica M, Cao Y, Manning RJ, Leleu X, et al. Increased natural killer cell expression of CD16, augmented binding and ADCC activity to rituximab among individuals expressing the FcγRIIIa-158 V/V and V/F polymorphism. Blood. 2007;110(7):2561–2564. doi: 10.1182/blood-2007-01-070656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, Jan M, Cha AC, Chan CK, Tan BT, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-hodgkin lymphoma. Cell. 2010. September 03;142(5):699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Okazaki T, Honjo T. The PD-1–PD-L pathway in immunological tolerance. Trends Immunol. 2006. April 01;27(4):195–201. doi: 10.1016/j.it.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 45.Veillette A, Chen J. SIRPα–CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. 2018. March 01;39(3):173–184. doi: 10.1016/j.it.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 46.Holgado MP, Sananez I, Raiden S, Geffner JR, Arruvito L. CD32 ligation promotes the activation of CD4+ T cells. Front Immunol. 2018. November 30;9(2814). doi: 10.3389/fimmu.2018.02814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Anania JC, Chenoweth AM, Wines BD, Hogarth PM. The human FcγRII (CD32) family of leukocyte FcR in health and disease. Front Immunol. 2019. March 19;10(464). doi: 10.3389/fimmu.2019.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491. doi: 10.3389/fimmu.2014.00491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palumbo A, Chanan-Khan A, Weisel K, Nooka AK, Masszi T, Beksac M, Spicka I, Hungria V, Munder M, Mateos MV, et al. Daratumumab, bortezomib, and dexamethasone for multiple myeloma. N Engl J Med. 2016;375(8):754–766. doi: 10.1056/NEJMoa1606038. [DOI] [PubMed] [Google Scholar]
- 50.Facon T, Kumar S, Plesner T, Orlowski RZ, Moreau P, Bahlis N, Basu S, Nahi H, Hulin C, Quach H, et al. Daratumumab plus lenalidomide and dexamethasone for untreated myeloma. N Engl J Med. 2019;380(22):2104–2115. doi: 10.1056/NEJMoa1817249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mateos M-V, Dimopoulos MA, Cavo M, Suzuki K, Jakubowiak A, Knop S, Doyen C, Lucio P, Nagy Z, Kaplan P, et al. Daratumumab plus bortezomib, melphalan, and prednisone for untreated myeloma. N Engl J Med. 2017;378(6):518–528. doi: 10.1056/NEJMoa1714678. [DOI] [PubMed] [Google Scholar]
- 52.Moreau P, Attal M, Hulin C, Arnulf B, Belhadj K, Benboubker L, Béné MC, Broijl A, Caillon H, Caillot D, et al. Bortezomib, thalidomide, and dexamethasone with or without daratumumab before and after autologous stem-cell transplantation for newly diagnosed multiple myeloma (CASSIOPEIA): a randomised, open-label, phase 3 study. Lancet. 2019. July 6;394(10192):29–38. doi: 10.1016/S0140-6736(19)31240-1. [DOI] [PubMed] [Google Scholar]
- 53.Pinto ML, Rios E, Durães C, Ribeiro R, Machado JC, Mantovani A, Barbosa MA, Carneiro F, Oliveira MJ. The two faces of tumor-associated macrophages and their clinical significance in colorectal cancer. Front Immunol. 2019. August 20;10(1875). doi: 10.3389/fimmu.2019.01875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Skytthe MK, Graversen JH, Moestrup SK. Targeting of CD163(+) macrophages in inflammatory and malignant diseases. Int J Mol Sci. 2020. July 31;21(15):5497. doi: 10.3390/ijms21155497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nijhof IS, Franssen LE, Levin M-D, Bos GMJ, Broijl A, Klein SK, Koene HR, Bloem AC, Beeker A, Faber LM, et al. Phase 1/2 study of lenalidomide combined with low-dose cyclophosphamide and prednisone in lenalidomide-refractory multiple myeloma. Blood. 2016;128(19):2297–2306. doi: 10.1182/blood-2016-07-729236. [DOI] [PubMed] [Google Scholar]
- 56.Sebag M, Bahlis N, Venner CP, McCurdy A, Kouroukis CT, Shustik J, White DJ, Kotb R, Stakiw J, Laferriere NB, et al. A randomized Phase II, open label, study of daratumumab, weekly low-dose oral dexamethasone and cyclophosphamide with or without pomalidomide in patients with relapsed and refractory multiple myeloma. Blood. 2019;134(Supplement_1):3121. doi: 10.1182/blood-2019-124457. [DOI] [Google Scholar]
- 57.Palladini G, Kastritis E, Maurer MS, Zonder JA, Minnema MC, Wechalekar AD, Jaccard A, Lee HC, Bumma N, Kaufman JL, et al. Daratumumab plus CyBorD for patients with newly diagnosed AL amyloidosis: safety run-in results of ANDROMEDA. Blood. 2020. April 3;136:71–80. doi: 10.1182/blood.2019004460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.