

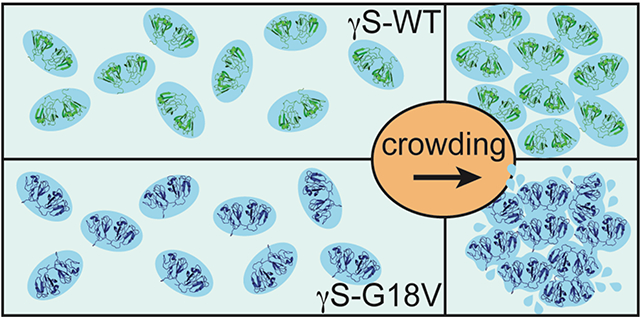
Abstract
We demonstrate that the effect of protein crowding is critically dependent on the stability of the protein’s hydration shell, which can dramatically vary between different proteins. In the human eye lens, γS-crystallin (γS-WT) forms a densely packed transparent hydrogel with a high refractive index, making it an ideal system for studying the effects of protein crowding. A single point mutation generates the cataract-related variant γS-G18V, dramatically altering the optical properties of the eye lens. This system offers an opportunity to explore fundamental questions regarding the effect of protein crowding, using γS-WT and γS-G18V: (i) how do the diffusion dynamics of hydration water change as a function of protein crowding?; and (ii) upon hydrogel formation of γS-WT, has a dynamic transition occurred generating a single population of hydration water, or do populations of bulk and hydration water coexist? Using localized spin probes, we separately probe the local translational diffusivity of both surface hydration and interstitial water of γS-WT and γS-G18V in solution. Surprisingly, we find that under the influence of hydrogel formation at highly crowded γS-WT concentrations up to 500 mg/mL, the protein hydration shell remains remarkably dynamic, slowing by less than a factor of two, if at all, compared to that in dilute protein solutions of ~5 mg/mL. Upon self-crowding, the population of this robust surface hydration water increases, while a significant bulk-like water population coexists even at ~500 mg/mL protein concentrations. In contrast, surface water of γS-G18V irreversibly dehydrates with moderate concentration increases or subtle alterations to the solution conditions, demonstrating that the effect of protein crowding is highly dependent on the stability of the protein-specific hydration shell. The core function of γS-crystallin in the eye lens may be precisely its capacity to preserve a robust hydration shell, whose stability is abolished by a single G18V mutation.
Graphical Abstract
Introduction
Protein crowding not only exerts protein intermolecular forces when reducing protein intermolecular distances by way of entropic, excluded volume, effects1,2, but also by amplifying complex interactions when hydration water layers overlap within several nanometer distances off the protein surface.3–6 Thus, under crowding conditions, weak attractive interactions among the proteins or other crowding partners can play a role,5 and even quinary protein structures exerting protein-matrix contacts may become highly relevant.7,8 For decades, it was thought that increased protein stability upon macromolecular crowding was due mainly to an entropic effect, in that simply the decrease in available volume for the protein to inhabit increasingly favors compact protein conformations.1,9,10 Recent findings, however, show that increased protein stabilization upon crowding can also be highly dependent on the chemistry of the crowder involved.11–13 Due to crowding agents’ preferential exclusion from the protein surface, it was hypothesized that their effect on protein stabilization is indirect, most likely due to changes to the hydration network near the protein surface.11 In stark contrast, others have reported the opposite, in which decreased protein stability accompanies macromolecular crowding.3,4,6,14,15 The destabilization may lead to protein unfolding, and in some cases, protein misfolding followed by amyloid fibril formation.16 While excluded volume effects have been the traditional explanation, an alternative or additional factor for the decreased protein stability with increased crowding forces is that confinement may suppress the dielectric response of the solvent.14 These effects reduce the favorability of hydrophobic interactions and enhance both the electrostatic attraction and hydrogen bonding of proteins, affecting the stability of the native state.14 However, it is likely that many more factors and interactions play into the effects of crowding;7 one such factor is the nature of the to-be-crowded protein surface itself and its hydration shell—the subject of this study, and a factor that has not been the subject of focus to date. The core hypothesis that this paper aims to test is that as crowding is increased, an increased overlap, and thus squeezing of the hydration water lining the protein surface will result in different aggregation properties, depending on whether its hydration water is strongly bound and stable vs. less favorably bound and fragile. In the first case, strongly bound surface water is costly to remove and constitutes a repulsive hydration barrier, and thus may inhibit direct inter-protein contacts, while in the second case the less strongly bound water is more prone to dehydration from the protein surface, enabling direct inter-protein contacts that may lead to aggregation. Such properties or effects are not easily discernable or predictable for proteins in dilute solution. The occurrence of a dynamic transition above a certain crowding threshold (explained as the sharp convergence of hydration and bulk water into one population of hydration water above a threshold protein concentration) has been reported;17 though the question remains whether this is a general phenomenon, and if so whether this phenomenon occurs under accessible or relevant sample conditions for most proteins or not. Due to water’s promise as a key factor relating protein stability and protein crowding, we seek to compare water dynamics at the protein surface with those in the interstitial volume to observe the effects of crowding forces from a dual perspective. The model system we chose to investigate the role of protein-specific surface properties on the effect of protein self-crowding is the wild-type human γS-crystallin (γS-WT), which naturally thrives in the crowded eye lens, and its cataract-related G18V variant, (γS-G18V), which demonstrates reduced stability and aggregation-prone behavior.
The transparency and refractive index gradient of the eye lens result from the unique properties of its structural proteins, the crystallins, which are distributed across the concentration gradient of the lens. The α-crystallins are small heat shock proteins, while the purely structural crystallins of the βγ-superfamily are evolutionarily selected for their high solubility and high refractive indices. Together, they constitute one of the most crowded populations of soluble, non-renewing proteins in the human body. Their aggregation, whether induced by mutation, age, oxidation, UV irradiation, or a combination of these and other factors, is a primary cause of cataract formation. In the water-rich cortex—the outermost part of the lens, γS-crystallin is the most abundant structural protein, although it is also found among the even more crowded γ-crystallins of the lens nucleus.18 It experiences the full concentration gradient of the lens fibers from approximately 200 mg/mL total protein concentration in the outer cortical fibers to approximately 400 mg/mL in the inner cortical fibers, and slightly higher still in the compressed and dehydrated lens nucleus.19 Long-term lens function requires the population of crystallins to remain soluble and stable at these concentrations for the lifespan of the organism. In this context, the specifics of γS-crystallin assembly and aggregation—though less comprehensively explored than the chaperone functions of the α-crystallins in their binding of intermediate aggregatory species—are essential to the critical questions of long-term crystallin stability and solubility. The age-related breakdown of γS-crystallin into protein fragments can lead to stable and soluble association with β-crystallin fragments—a pathway hypothesized to reduce or delay their respective tendencies toward aggregation.20 γS-crystallin has also been shown to suppress the phase-separation process that occurs during formation of cold cataract.21 However, γS-crystallin can induce disruptive aggregation, opacification, and cataract formation when mutated, as exemplified by γS-G18V. This clinically-discovered variant presents reduced structural stability and solubility relative to γS-WT,22 and is associated with hereditary childhood-onset cataracts.23 Atomistic MD simulations and biophysical measurements of γS-G18V indicate that thermodynamic explanations for the difference in aggregation propensity between γS-crystallin variants are by themselves insufficient to explain the dramatically different aggregation propensities,24 while structural biology studies reveal only subtle changes in the backbone arrangement of the N-terminal domain of γS-G18V compared to γS-WT. Given that what leads to an increased aggregation of γS-G18V and enhanced binding of γS-G18V by αB-crystallin are subtle changes to the surface of γS-G18V facing the solvent,25 it is plausible that the nature of protein surface-hydration water interaction is the key difference between γS-G18V and γS-WT. Although it is generally assumed that dehydration accompanies opacification, surface hydration water properties within nanometer lengthscales of protein surfaces have not been experimentally and directly characterized. We seek to resolve three questions related to the gelation or aggregation propensity of wild-type γS-crystallin and its variant: (i) At what stage of inter-protein approach, as the protein concentration is increased, are γS-WT and γS-G18V differentiable in their surface hydration dynamics in vitro, if at all? (ii) What are the properties of the hydration shell of the stable γS-WT protein? (iii) Under high self-crowding conditions, do bulk and hydration water populations coexist, or has a dynamic transition occurred with a single population of water hydrating the γS-WT hydrogel?
Previous studies of structural crystallins have suggested that the quantity of bound hydration water differs from the quantity of water around an average globular protein, but conflicting experimental measurements of bound water quantity abound.26,27 Rather, the relevant property for the extraordinary solubility of γS-crystallin may be the stability of the surrounding hydration water, not its quantity. Answering this question requires a dynamics-based experimental approach to hydration water under ambient solution conditions. Equally critical is the ability to study hydration dynamics under crowded conditions, analogous to the situation in the eye lens under physiological conditions, in both the healthy and disease states. Both of these capabilities have only recently become available with Overhauser dynamic nuclear polarization (ODNP) relaxometry to measure surface hydration water dynamics28–30—the technique of choice in this study. By exploiting ODNP, concurrently with continuous wave electron paramagnetic resonance (CWEPR) lineshape analysis, the dynamics of the surface hydration water and of the interstitial water were explored with increasing protein concentration of γS-WT and γS-G18V, across 100-fold variation between 5 and ~500 mg/mL. This concentration range includes the threshold concentration where hydrogel formation of γS-crystallin occurs and encompasses protein concentrations found in the cortical fibers of the eye. CWEPR lineshape analysis can extract changes in the local protein packing and tumbling from the rotational mobility of a paramagnetic nitroxide radical-based spin label tethered to the protein surface. ODNP at 0.35 Tesla probes the translational diffusive dynamics of freely moving water within ~10–15 Å, i.e. of 2–4 layers of water, near the stable nitroxide radical based-spin probes, by transferring polarization of the electron spin to local water protons upon saturation of the nitroxide’s CWEPR line.28,31–34 Thus, the experimental access to hydration water was enabled by covalently tethering a stable nitroxide radical-based spin label to a solvent-exposed cysteine (either endogenous or added by substituting a cysteine for another surface residue) on the surfaces of S-WT or γS-G18V, while the access to interstitial water was enabled by imbibing unbound stable nitroxide radical spin probes in the interstitial volume of the crowded protein solution. In other words, the hydration water discussed here is referring to the surface water that the ODNP technique detects when employing a protein surface-tethered spin label, i.e. of 2–4 layers of water near the protein surface. In contrast, what we refer to here as bulk water is the water that ODNP detects when employing freely dissolved nitroxide spin probes in bulk solution. Both the dynamics of hydration and interstitial water are compared to and referenced against that of bulk water (measured in the absence of protein). Local translational water diffusion necessarily involves hydrogen bond breaking and reforming of the diffusing water molecules, and as such directly reflects on the strength of the hydrogen bond network of mobile hydration water near the spin probe. The strength of the local hydrogen bond network probed by the spin label tethered to the protein surface is exerted collectively by the adhesion between water and the protein surface and the cohesion within the surface hydration water network.35 Thus, the local protein surface water diffusivity can be viewed as an inverse local surface water viscosity—an appropriate analogy given that the cooperative behavior of water within the protein hydration shell is thought to be key to the modulation of its diffusion dynamics. If γS-crystallin’s function is to simply sustain its hydration water network under gel-forming conditions, this would be reflected in an unaltered hydration water dynamics and viscosity as protein concentration is increased. If γS-crystallin’s function is to corral a more robust surface water network under gel-forming conditions, hydration water dynamics will slow as protein concentration is increased and its population will rapidly increase at the expense of bulk water. These are among the key questions we seek to answer.
Accompanying the ODNP measurements, high-resolution solution NMR spectra of the protein amide N-H spins were obtained to characterize the protein-solvent interaction from the protein structural perspective. Backbone N-H residues that are intramolecularly hydrogen-bonded within the protein can be distinguished from those that are hydrogen-bonded to the solvent by the temperature-dependent change in the chemical shift of the N-H proton resonances. The amide N-H temperature coefficient (Δδ/ΔT) has been used in other studies to determine which protons are involved in intramolecular hydrogen bonds in folding intermediates of an aggregation-prone form of human lysozyme36 and in a collagen-mimetic peptide variant engineered to mimic osteogenesis imperfecta.37 Here we compare the landscape of Δδ/ΔT values of γS-WT and γS-G18V, which have previously been shown to exhibit only minor structural differences25, but significant differences in solubility, solvent accessibility, and interaction with binding partners.22,25,38 When directly comparing their surface and interstitial water dynamics by ODNP, we learn that the stability of the surface hydration water is the key functional difference, consistent with discernable differences in their amide N-H temperature coefficients that reflect differences in protein-solvent interactions. This combination of experiments allows us to correlate the water-centric results from ODNP with the site-specific protein-centric hydration information afforded by the N-H temperature coefficients.
Results
For the purpose of carrying out CWEPR and ODNP measurements, the spin probe is either tethered to the protein surface or dissolved into the interstitial water volume of the protein solution or gel. To examine the surface of γS-WT and γS-G18V, a nitroxide moiety was tethered to one or more solvent-exposed cysteines. As the goal was to evaluate and compare the average properties of the protein surface water layer (2–4 layers off the protein surface) of γS-WT and γS-G18V, three different spin-labeling schemes were chosen in order to map out the protein-water interactions near the mutation site. The relevant residues are highlighted in the full structures presented in the Supplemental Information (SI) (Figure S1,S2). First, we spin-labeled the endogenous cysteines in loop 2, as highlighted in Figure 1A and Figure S2. In the unmodified proteins, the exposed residues where the spin label could bind are C25 for γS-WT and one or more of three solvent-accessible cysteines, C23, C25, or C27 for γS-G18V (Figure 1A). The spin labeled constructs of the unmodified γS-WT and γS-G18V in the context of ODNP and CWEPR will be referred to simply as γS-WT and γS-G18V. In order to account for potential artifacts resulting from comparing WT and γS-G18V hydration at different surface sites and/or proteins having different numbers of exposed cysteine residues, four additional protein constructs were made in which all three endogenous cysteines in Loop 2 were replaced with serines. Then, an additional single cysteine was substituted in to enable the spin labeling of γS-WT and γS-G18V at the same single site, either at position 28 (Figure 1B) or 32 (Figure 1C). These constructs, C23S/C25S/C27S/A28C, G18V/C23S/C25S/C27S/A28C, C23S/C25S/C27S/T32C, and G18V/C23S/C25S/C27S/T32C, will henceforth be referred to as S3-A28C, S3-G18V/A28C, S3-T32C, and S3-G18V/T32C, respectively, for the sake of brevity. CD spectra indicated that all four S3 constructs were folded with primarily β-sheet secondary structure similar to γS-WT, as described in the SI (Figure S3). In all cases, the spin-labeled sites are placed in the N-terminal domain near the G18V mutation site located in loop 1 (Figure 1A,B,C), within a ~20 Å radius of G18/V18 (backbone N to N), well within the region of structural disruption caused by the G18V mutation. In order to investigate the interstitial water volume of the protein solution or gel, the hydrophilic small molecule probe 4-hydroxy TEMPO, was dissolved in the protein solution.
Figure 1.

The loop 2 region where the spin labels are placed is shown for γS-WT (light green) and γS-G18V (light blue) where cysteine residues are highlighted in yellow (A) Endogenous cysteine residues. (B) Spin-labeling Cys substitution at position 28 (C) Spin-labeling Cys substitution at position 32.
CWEPR spectra reveal soluble γS-WT and aggregation-prone γS-G18V.
CWEPR spectra were taken of γS-WT and γS-G18V to determine differences between their local protein dynamics near the spin-labeled site in dilute solution, as well as with increasing protein concentration to probe concentration-dependent changes in the local protein dynamics and/or the onset of aggregation. First, we focus on the CWEPR lineshape in dilute solution. Here, the CWEPR spectrum of γS-WT (spin labeled, otherwise unmodified WT) is comparable to that of γS-G18V (Figure 2A). Both are highly dynamic, revealing no signature of protein-protein interaction, and with line-shapes characteristic of a surface-exposed and mobile nitroxide label with a rotational correlation time of order 1–3 ns. In fact, the CWEPR line-shape of γS-G18V (spin labeled, unmodified G18V) is slightly narrower than that of γS-WT, most likely because the spin-labeled site (C23, C25, C27) on the solvent exposed loop 2 is less ordered and slightly extended, making particularly sites C23 and C25 more solvent-exposed than in γS-WT—this can be gleaned from examining the loop 2 region from the solution-state NMR structure,25 as shown in Figure 1A with the relevant cysteine residues highlighted in yellow. The CWEPR spectra of S3-A28C and S3-G18V/A28C under dilute conditions (5–25 mg/ml) exhibit similar CWEPR line-shapes as γS-WT and γS-G18V (Figure S4A). Based on the spin label rotational correlation time (~1–3 ns) reflected in the CWEPR spectra and the protein hydrodynamic diameter (~5 nm) as measured by DLS,24 we conclude that both γS-WT and γS-G18V, as well as the S3-A28C and S3-G18V/A28C variants exist as monomers in freshly prepared samples and under dilute conditions. The CWEPR spectrum of the S3-T32C variant has a slightly broadened, but single component, line-shape, while that of S3-G18V/T32C exhibits two components, similar to those that will be seen in the CWEPR spectra of γS-G18V at higher concentrations (Figure 2B and S4A,B)—in this case, we cannot rule out some degree of aggregation of the S3-G18V/T32C construct even at dilute concentrations.
Figure 2.

A. CWEPR spectra of γS-WT (green) and γS-G18V (blue) at low concentration (~10mg/ml). B. Representative CWEPR spectra of γS-G18V in the monomeric state (I.), with onset of aggregation (II.), and upon aggregation (III.). C Representative CWEPR spectra of γS-WT stored below ~100 mg/ml prior to spin labeling show mobile spin labels at 5 and 270 mg/ml, respectively (IV,V). A broader line (VI.) is observed when a threshold concentration (>100 mg/ml) for γS-WT is reached prior to spin labeling, as observed for protein concentrations between 5–550 mg/ml (see Section 2 in SI).
Next, we study the CWEPR line-shape with increasing protein concentration. We observe high variability in the CWEPR line-shape of γS-G18V, exemplified in the difference between spectra I and III presented in Figure 2B. This is more comprehensively shown in Figure 2D, where the distance between the outermost hyperfine peaks, 2A’zz, is displayed as a function of protein concentration for all variants studied here. Changes in the 2A’zz value empirically and qualitatively reflect on changes in the local protein environment. For γS-G18V, 2A’zz is split into two distinct groupings, which is attributed to the appearance of a secondary broad feature—indicating an immobile spectral component due to possible tertiary contact between the spin label and a protein interface (marked as i in Figure 2B (II.)). This immobile component appears when γS-G18V is concentrated above a certain concentration (e.g. 130 mg/mL). The intensity of this immobile signature drastically increases and the overall lineshape dramatically broadens as the sample is further concentrated (Figure 2B (III)), signifying the local burial and global slowing of the spin label (the rotational correlation time slows about 3 times), suggestive of progressing aggregation. Once γS-G18V is subject to aggregating conditions, the broad component does not vanish nor diminish upon subsequent dilution to concentrations where monomeric states have been previously observed, (e.g. dilution of γS-G18V from 150 mg/mL to 30 mg/mL still yield a spectrum as shown in Figure 2B (III)), indicating that the protein aggregation state is essentially irreversible. Consistently, once this type of EPR line-shape is observed, the samples tend to become unusable due to protein precipitation. This type of immobile spectral component is also seen in both the S3-G18V/A28C and S3-G18V/T32C variants. In S3-G18V/A28C, the immobile component appears at higher concentrations (70 mg/mL) (Figure S4B) and with similar magnitude as observed with γS-G18V at elevated concentrations (130 mg/mL, Figure 2B (II)). The S3-G18V/T32C variant presents a more significant immobile component even at low concentrations (Figure S4A), which continued to increase in intensity as the protein is further concentrated (Figure S4B). The CWEPR spectra show that all γS-G18V variants have a tendency to aggregate at elevated protein concentrations.
The distinct immobile spectral component (indicated with i in Figure. 2B, Figures. S4A,B) seen in the γS-G18V variants is not present in any of the γS-WT CWEPR spectra (see Figure 2C, Figures S4A,B), as reflected in an unaltered 2A’zz (distance between the two outermost hyperfine peaks) across the entire concentration range of 5 and 550 mg/ml for the given γS-WT protein batch (Fig. 2D). This suggests that spontaneous aggregation does not occur with γS-WT. The relatively mobile CWEPR spectra IV and V of Figure 2B are representative of γS-WT that was stored below ~100 mg/ml prior to spin labeling and concentrated to the desired protein concentration. An overall broader line is observed when a threshold concentration (>100 mg/mL) for γS-WT is reached prior to spin labeling, but still no secondary component is observed for protein concentrations between 5–550 mg/mL. The difference in line broadening, depending on the threshold concentration prior to spin labeling is attributed to the formation of an intramolecular disulfide bond between C23 and C27 within loop 2, and further discussed in Section 1 of SI (Figure S5). This is reflected in two distinct 2A’zz values for γS-WT—3.5 mT for γS-WT stored below 100 mg/mL and ~5 mT for γS-WT stored below 100 mg/ml prior to spin labeling (Figure 2D). However importantly, the distinct immobile spectral component is missing, as reflected in an unaltered 2A’zz (distance between the two outermost hyperfine peaks) across the entire concentration range of 5 and 550 mg/ml for the given γS-WT protein batch (Figure 2D). The 2A’zz values for the S3-A28C and S3-T32C constructs fall into the 3.5 mT or 5 mT category as observed with γS-WT (Figure 2D). While the 2A’zz value increases slightly between low (~25 mg/ml) and high concentrations (~450 mg/ml) for S3-A28C, the 2A’zz value for S3-T32C shows an increase from ~3.5 mT to 5 mT between low and high concentrations, but neither show the same magnitude of changes as seen in γS-G18V (Figure 2D, Figure S5B). This firmly validates that γS-WT is not aggregated and shows robust solubility across a 100-fold change in protein concentration. In general, the EPR line-shape broadens with increasing protein concentration, starting above ~350 mg/mL, which we attribute to the global increase in solution viscosity (similar line-shape broadening was observed when systematically adding sucrose to a dilute protein solution, as discussed in Section 2 and as shown in Figure S7). In other words, the 2A’zz values for γS-WT qualitatively demonstrate an unchanging local protein environment with increasing γS-WT protein concentration, while the overall broadening in line-width reflects a changing global protein tumbling due to increased solution viscosity with increasing γS-WT protein concentration.
In summary, under monomeric conditions, γS-WT and γS-G18V present only subtle conformational differences near the site of spin labeling. Differences in the properties of these two variants only become apparent at increased protein concentration or when exposed to adverse conditions, in which CWEPR was found to be sensitive at detecting the onset of irreversible aggregation of γS-G18V. The observation of discernable differences of the CWEPR features between γS-WT and γS-G18V with increasing concentration provide motivation to further explore the stability versus fragility of the protein surface hydration water through the direct characterization of surface water diffusivity.
ODNP reveal stable hydration shell for γS-WT and fragile hydration shell for γS-G18V.
We performed ODNP on the same spin labeled samples as characterized by CWEPR to determine the diffusivity of surface hydration water accessible by ODNP, in the local vicinity of the spin-labeled side chain on a protein surface. Although we are only probing hydration water around loop 2 in the N-terminal domain of the protein (residues marked in yellow in Figure 1A–C and shown in Figure S1 in the SI), its proximity to the site of the G18V point mutation makes it an ideal region to probe both the inherent differences in hydration between the protein variants, as well as any global changes in the surface hydration landscape that may occur as the protein concentration is increased. The ODNP-derived translational correlation time (τ, time needed for water to diffuse across ~10 Å distance) values found on γS-WT surfaces at concentrations up to 550 mg/mL are presented in Figure 3. For γS-WT protein concentration of 20 mg/mL τ was found to be ~ 150–200 ps, which falls within the range of expected surface water diffusion dynamics measured on other globular proteins by ODNP (Figure 3A).29 Crucially, this value increases by less than a factor of 2 from τ ~ 150–200 ps at 20 mg/mL to τ = ~ 200–300 ps at >100 to ~550 mg/mL (Figure 3A and B, green), across two orders of magnitudes of protein concentration (5 to ~550 mg/mL). This is different from the solution behavior of most proteins, which would aggregate at such high concentrations.39 Importantly, we verified (shown in Supplemental Figure S6) that the measured hydration dynamics of γS-WT is insensitive to small protein conformational changes, such as the intramolecular disulfide bond formation that was suggested to yield the slightly broadened CWEPR spectrum, as reflected in different 2A’zz values (~3.5 vs. ~5) for γS-WT when spin labeled below or above ~100 mg/mL (Figure 2D). The ODNP results for S3-A28C (left flag, orange, Figure 3) and S3-T32C (right flag, pink, Figure 3) yield τ values that consistently fall within 250–325 ps over the entire protein concentration range studied (5–550 mg/mL). For these two variants, the τ values with 250–300 ps fall within error between ~20 and ~450 mg/mL protein concentrations, not even displaying the factor of 2 seen with γS-WT. Together, these results indicate that the apparent robustness of the γS-WT hydration shell is not dependent on a particular local property of the labeled sites, such as endogenous C25 vs. the introduced C28 or C32 sites, but is reflective of protein-intrinsic properties of the γS-WT surface.
Figure 3.

A. ODNP-measured translational correlation time of surface hydration water around γS-WT (green open square), S3-A28C (orange open circle), S3-T32C (pink open triangle), and interstitial hydration water (black open square), along with G18V surface hydration (sample 1v: blue square, 2v: blue circle, 3v: blue triangle, 4v: blue upside-down triangle, and 5v: blue diamond), S3-G18V/A28C (orange left flag), and S3-G18V/T32C (pink right flag). B. Region from 0 to 350 ps zoomed in to show γS-WT variation. The surface hydration dynamics of γS-WT remain relatively invariant up to 550 mg/mL. The diffusivity of interstitial water was similar to that of bulk water at γS-WT < 200 mg/mL (reference value for bulk water given at 0 mg/mL), and slowed about two fold at > 400 mg/mL. The translational correlation time of γS-G18V in the monomeric form (blue square) was indistinguishable from that of γS-WT (green open square) in dilute solution, though the results for γS-G18V were highly sample preparation dependent, with slower water correlation times observed for samples stored at high concentrations, even after subsequent dilution.
The G18V point mutation was previously found to result in significantly reduced solubility and increased aggregation propensity, leading us to expect the surface hydration properties of γS-G18V to differ significantly from the wild type. Counter to the original hypothesis, the translational correlation time of hydration water of monomeric γS-G18V was found to be indistinguishable from γS-WT (τ = ~ 200 ps, see Figure 3A, blue), consistent with the comparable and mobile CWEPR spectra found for the monomers (Figure 2A). The difference, again, was found in the dramatic variability of the translation correlation time of γS-G18V’s hydration water, whose dynamics slow down significantly when the γS-G18V concentration passes a threshold value or is stored for a prolonged duration. The hydration dynamics of γS-G18V is sensitive to even subtle changes in sample preparation; the mere storage at 150 mg/mL yielded τ ~800 ps (see Figure 3A (2v), orange), with the very slow hydration water dynamics remaining invariant after dilution to 30 mg/mL, implying that irreversible aggregation has occurred. Note that protein surface water diffusion at a binding/aggregation interface will dramatically slow, either due to molecular confinement of local water, or more likely, due to longer distance of closest approach between the spin label and surface water upon its expulsion from the interface. Generally, a large range for τ was found for γS-G18V samples with nominally equal protein concentrations, but prepared under different sample preparation conditions (Figure 3A). This dramatic variability in hydration water translation correlation time was also found in S3-G18V/A28C and S3-G18V/T32C with τ values ranging from 200–800 ps. Taken together, ODNP using protein surface-tethered spin labels verify that the G18V variant reveals concentration- and aging- dependent changes in its hydration shell, consistent with its propensity for aggregation.
ODNP of free spin probes reveal bulk-like interstitial water to persist in highly concentration γS-WT “gel”.
Next, we perform ODNP measurements using a freely dissolved hydrophilic nitroxide spin probe, 4-hydroxy TEMPO, in the γS-WT solution at protein concentrations ranging from 0 to 550 mg/mL, with 0 referring to the bulk solvent (which is the protein-free buffered solution, not pure water). The purpose was to see whether the interstitial water in the protein solution is perturbed, and to what degree, especially above a threshold concentration of ~150 mg/mL where the γS-WT solution appears to form a gel. Given that the interstitial dimensions necessarily decrease with increasing protein concentrations under crowded conditions, leading to an overlap between the hydration and bulk water, the interstitial water dynamics could reasonably be expected to exhibit dynamic properties between that of surface hydration and bulk water. The translational correlation time of the interstitial water at protein concentrations up to 130 mg/mL was found to be indistinguishable from that of bulk water, as shown in Figure 3 (black open square, compare 130 and 0 mg/ml). The interstitial water dynamics begin to slow above a concentration of 200 mg/mL, and by a total of about 2-fold, from ~50 ps for the protein-free solution to ~100 ps for the protein solution containing ~550 mg/ml γS-crystallin (Figure 3B). Crucially, the latter τ ~ 100 ps value for the interstitial water in the concentrated protein solution is significantly smaller compared to the τ ~250 ps value of the protein surface hydration water measured with surface-tethered spin labels (Note that this is not merely a geometric effect, as surface hydration dynamics of order τ ~100 ps has been measured before with surface-tethered spin labels40). This shows that at ~200 mg/mL, the hydration shell begins to constitute a detectable population of interstitial water, but does not entirely overlap nor dominate even at protein concentrations of ~550 mg/mL. Notably, the bulk viscosity increases by more than 10 times from 50 to 500 mg/mL, while the interstitial water diffusivity slows only 2 times at protein concentrations where the inter-protein (center-to-center) separation is on the order of ~5–6 nm. This demonstrates the coexistence of at least two populations of water in this highly crowded protein gel of the eye lens—bulk and surface hydration water, and that the properties of the protein surface hydration water and interstitial water are distinct, not inter-dependent.
Differences in hydrogen bonding between γS-WT and γS-G18V.
In order to understand whether the differences in the stability/fragility of the hydration shell of γS-WT and γS-G18V are reflected in microscopic differences in the interaction between specific protein residues with the solvent, the amide proton temperature coefficient (Δδ/ΔT), which is the change in backbone amide N-H chemical shift as a function of temperature, was measured. Thermal expansion of hydrogen bonds gives rise to the change in chemical shift as a function of temperature, which is more pronounced for N-H protons forming hydrogen bonds to the solvent.41 For globular proteins, the temperature coefficients of the backbone resonances are usually linear over a temperature range up to about 15 K below the thermal denaturation temperature, with nonlinear dependences indicating more than one conformer in solution. Typical values range from −16 to +2 ppb/K.42 Below the denaturation temperature, increasing the temperature increases the magnitudes of local fluctuations in the protein structure, causing lengthening of each H-bond and hence movement of the chemical shift toward its random coil value. Based on previous studies,43 residues with temperature coefficients less than −4.6 ppb/K (Figure 4A, dotted line) are taken to be involved in intermolecular hydrogen bonds with the solvent, while more positive values are interpreted as intramolecular hydrogen bonds within the protein. Although Δδ/ΔT is an imperfect measure of hydrogen-bonding status,44 this heuristic was found to correctly classify 90% of the hydrogen bonds in β-sheets.45 Selected amide proton temperature coefficients are plotted in Figure S9 of the SI, while all the available data are summarized in Figure 4 and tabulated in the Supplementary Table S1.
Figure 4.

A. Comparison of the NH temperature coefficient, Δδ/ΔT, for γS-WT (green) and γS-G18V (blue). Most Δδ/ΔT values for residues in the C-terminal domain are unchanged, while, many Δδ/ΔT values for residues in the N-terminal domain differ between γS-G18V and γS-WT. Values above the dotted line at −4.6 ppb are indicative of residues involved in intramolecular hydrogen bonds, and those below ones that are hydrogen bond to solvent. B. NH temperature coefficients, Δδ/ΔT, for γS-WT (x-axis) vs. γS-G18V (y-axis). The largest deviations in the NH temperature coefficient are observed in the N-terminal domain closest to the mutation site (dark teal), with significant deviations also occurring in the latter part of the N-terminal domain (light blue). Few differences are seen in the C-terminal domain (salmon), mostly corresponding to residues in the interdomain interface. The dashed line has a slope of 1 as a guide to identify the residues for which γS-WT and γS-G18V have the same Δδ/ΔT values.
Four tryptophan residues, W47, W73, W137, and W163, are common to all mammalian βγ-crystallins and are thought to be important for stabilizing the fold and for protecting both the crystallin proteins and the retina against UV-photodamage via fluorescence quenching. The Δδ/ΔT for the tryptophan residues reveal that in both proteins W47 (−7.76 and −6.40 ppb/K for γS-WT and γS-G18V, respectively) is hydrogen bonded to the solvent, while W73 and W163 have values in the range of ±1 ppb /K, consistent with intramolecular hydrogen bonding, as expected for W73 and W163 buried in the hydrophobic core of the protein with well-defined hydrogen bonds to crystallographically observed waters46,47. Most importantly, the characteristic Δδ/ΔT signatures of the four tryptophan residues are preserved between the γS-WT and γS-G18V variant, implying that the core structural properties are similar.
Extending the comparison of the NH temperature coefficients (Figure 4) to each amino acid of γS-WT and γS-G18V, most temperature coefficients in the C-terminal domain are found to be comparable (CTD 90–178), while modest differences are apparent throughout the N-terminal domain, particularly near the mutation site and in the interdomain interface, consistent with the previous finding that most structural differences between γS-WT and γS-G18V are found in this region25. In Figure 4A, the values of Δδ/ΔT are plotted for both γS-WT and γS-G18V as a function of sequence position. The threshold value of −4.6 ppb/K is indicated by a dotted line. Figure 4B shows the same data, plotted as Δδ/ΔT (γS-WT) vs. Δδ/ΔT (γS-G18V), color-coded according to sequence position (darker to lighter moving from N-terminal to C-terminal). The dotted line has a slope of 1; points that fall along this line are residues for which the values of Δδ/ΔT are the same for both proteins. At the mutation site (residue 18), a large chemical shift difference is observed, however the difference in Δδ/ΔT for G18 (−4.98 ppb/K) of γS-WT vs. V18 (−3.00 ppb /K) of γS-G18V is small, indicating that the local H-bonding is similar. Still, overall more residues in γS-G18V show Δδ/ΔT less than −4.6 ppb/K, i.e. tend to form intermolecular hydrogen bonds with the solvent, whose corresponding residues in γS-WT have Δδ/ΔT greater than 1 ppb/K. These include residues T4, F10, R19, D26, E43, G44, Y70, and H87. In contrast, residues of γS-G18V that present larger Δδ/ΔT than in γS-WT, whose Δδ/ΔT in γS-WT was less than −4.6 ppb/K, include S35, C37, and L88. Thus on balance, the cataract-related variant more strongly interfaces the solvent than γS-WT.
These observations are consistent with molecular dynamics simulations that predicted greater exposure of residues in the N-terminal domain and the interdomain interface of γS-G18V to the solvent.24 The exposure of more residues to solvent in γS-G18V does not imply a more strongly or stably hydrated protein. In fact, likely the additional solvent-exposed residues in γS-G18V expose more hydrophobic residues to the protein surface. Whether this is the case is difficult to predict from microscopic measurements of the amide hydration landscape or from the counting of individual amino acids that are exposed to or hidden from the solvent, as the effective local surface hydrophobicity/hydrophilicity even of a given amino acid as part of a protein surface is determined by a collective behavior involving neighboring amino acid residues and their interactions with water48. Rather, we verify the increase in the effective surface hydrophobicity of γS-G18V by the increased fluorescence of bis-1-anilinonaphthalene-8-sulfonate (bis-ANS) bound to γS-G18V as compared to γS-WT (Figure 5). Because the fluorescence emission of bis-ANS fluorescence increases upon binding to hydrophobic protein regions,49 this assay is commonly used to detect differences in exposed hydrophobic surface area between different proteins.50. Here, the observed fluorescence intensity of bound bis-ANS is greater for γS-G18V compared to γS-WT, implying that the surface hydrophobicity of γS-G18V is higher compared to γS-WT, although the particular residues involved cannot be identified from this data.
Figure 5.

γS-WT and γS-G18V present subtle differences in intramolecular/solvent hydrogen bonding and surface hydrophobicity. Increased fluorescence of bis-ANS at 500 nm was observed upon binding to γS-G18V (blue) compared to γS-WT (green), consistent with an increase in exposed hydrophobic surface area for γS-G18V.
Discussion
ODNP and CWEPR methods permit measurements of protein surface hydration and interaction under highly crowded conditions—representing inaccessible experimental conditions for high-resolution NMR spectroscopy. Note that the natural concentration of γS-crystallin in the eye lens outer cortex is as low as ~200 mg/mL, while the inner cortex can reach ~400 mg/mL, making our chosen experimental conditions highly relevant. The observation for γS-WT that the surface hydration water dynamics slow by less than a factor of 2 across such a broad protein concentration range, including the highly crowded regime of 550 mg/mL, is remarkable. This suggests that the surface water diffusivity measured by ODNP is not a function of molecular confinement, but an intrinsic property defined by the protein surface. Crucially, the hydration layer of γS-WT is particularly robust, and thus highly resistant to strong intermolecular interactions typically found with other proteins at high concentrations. Even at protein concentrations that lead to the formation of a transparent hydrogel at > 150 mg/mL, the hydration shell is sufficiently stable, so that the crowding force exerted by closely approaching crystallin molecules does not overcome the repulsive hydration force by the protein surface hydration layer. Similar results are also found with S3-A28C and S3-T32C, in which all endogenous cysteine residues in Loop 2 have been mutated to serine, and a single cysteine engineered in for singly spin-labeling of the γS-crystallin. The comparable correlation times for the surface hydration water found for γS-WT and its corresponding S3 variants indicate that the robustness of the γS-WT hydration shell extends beyond the immediate vicinity of C25, and appears to be a general feature of this protein. Our hypothesis is that γS-crystallin is an effective lens protein precisely because its hydration shell remains intact for several layers, even under the most crowded conditions. This property helps the protein maintain its exceptionally high solubility, as the stable hydration shell acts as a barrier to direct inter-protein contact and binding.
Our suggestion that the stable protein surface hydration shell is an intrinsic surface property of γS-WT is further supported by our finding that the interstitial water in the γS-WT solution displays significantly faster dynamics compared to the protein surface hydration water, and constitutes a distinct population. The interstitial water dynamics remains bulk-like between 0 and 130 mg/mL protein concentration with unaltered τ ~ 50 ps, and only slows at concentrations exceeding 200 mg/mL. The maximal retardation for the interstitial water is a factor of ~2 from τ ~ 50 ps to τ ~ 100 ps when the γS-crystallin concentration increases from 0 to ~550 mg/mL. Notably, the bulk viscosity slows by well over a factor of 10 over this concentration regime. This demonstrates the coexistence of at least two populations of water in this highly crowded protein gel of the eye lens—bulk-like and surface hydration water, and that the coalescing of bulk-like and surface hydration water does not occur with γS-WT crowding. This reveals that the surface water dynamics, and thus the water network stability in the environs of γS-WT, is determined by the intrinsic properties of the protein surface, not by the crowding effect alone. A recent computational study showed that intermolecular crowding of protein G and protein G/villin systems can cause a significant slowing of diffusion of the interstitial hydration (as much as 3-times slower than bulk) at intermolecular distances that span more than 10 layers of water across ~4–6 nm inter-protein distances—affecting even the protein’s first hydration shell.14 Furthermore, an ultrafast 2D IR study found the occurrence of what they refer to as a dynamic transition upon macromolecular crowding of the protein lysozyme, postulated to be due to a dramatic extension of hydration layers upon reducing inter-protein distances below a threshold.17 This is in contrast to our study conducted at similarly close inter-protein distances and concentrations, in which we find the hydration shell of γS-WT to largely resist changes in surface water diffusivity that coexists with and connects to a distinct bulk-like interstitial water population, ensuring high protein solubility. These seemingly contrary findings can be reconciled when assuming that the extent, stability and deformability of a protein hydration shell is protein-specific—this is a major finding of this study.
Based on the CWEPR line-shape and long correlation times observed for hydration water under crowded conditions and in aged samples, we conclude that γS-G18V harbors a fragile hydration shell. Here, we use the terminology fragile because this hydration water tends to dry up under crowded conditions, allowing direct contact between the protein molecules leading to aggregation. Irreversible aggregation is accompanied by the eviction of the hydration water shell that is shielding the protein surface, and the overwhelming of water-surface bonds by intermolecular bonds. This water eviction occurs quite easily and irreversibly in the γS-G18V variant as protein concentration is increased, even to modest concentrations. However, if a sample of monomeric γS-G18V is prepared at low enough concentrations, and handled carefully, it is possible to measure similar rates of diffusion to that of γS-WT monomers in dilute solutions. It becomes clear that the monomeric form of γS-G18V that presents indistinguishable surface protein hydration from that of the γS-WT monomer is a meta-stable state.
The structurally subtle, yet functionally consequential, differences between γS-G18V and γS-WT are confirmed to be based on microscopic alterations to protein-solvent interactions. These differences were identified by the temperature dependence of the amide proton chemical shifts for several residues in the N terminal region, which is also the region thought to form the protein interaction interface. In general, γS-G18V has more residues interacting with the solvent than γS-WT, in particular near (less than 15 Å from) the G18V mutation site, while fluorescence of bound bis-ANS verified a higher effective surface hydrophobicity of γS-G18V compared to γS-WT, all caused by a single G18V point mutation. Such an increase in the exposed hydrophobic surface area has also been found by Pande et al. for an aggregation-prone variant of human γD-crystallin, suggesting that this might be a common feature in the aggregation of crystallin variants.51 This is consistent with the current understanding that hydrophobic protein surfaces more easily dewet than hydrophilic ones, possibly because the hydration waters in such regions are in a state resembling that a liquid-vapor interface.52 Even so, the effective hydrophobicity of a protein cannot be reliably predicted by accounting for the surface hydropathy of a given protein made up of the hydropathy of the solvent-exposed amino acids, i.e. “sum of the parts”.53 The main reason that such predictions are extremely difficult is that a stable hydration shell, leading to a hydrophilic protein surface requires cooperativity within the water network, whose nature and stability is determined by the protein surface topology and/or the folded protein structure itself.54 The sustained and exceptionally stable hydration shell across a wide range of protein concentrations is a specific property of γS-WT, as determined by the specific topology of the γS-WT protein surface, which however cannot be easily predicted a priori from known structural or surface properties. We have empirically determined, by direct measurement of protein surface and interstitial water dynamics, that γS-WT harbors an exceptionally stable hydration shell. The core function of γS-crystallin in the eye lens may be precisely its capacity to preserve a robust and protein-intrinsic, hydration shell, whose stability is abolished by a single G18V mutation.
Conclusion
In summary, the complementary information provided by the solvent-interaction measurements by solution-state NMR and ODNP alongside CWEPR provide empirical views at different timescales and properties of the solvation shells of γS-WT and γS-G18V proteins, which exhibit highly disparate behaviors in the water-rich cortical fibers, yet present strikingly similar structural characteristics in the monomeric state. ODNP finds a highly robust hydration shell of γS-WT that does not respond to systematic crowding from 5 to ~550 mg/mL, and reveals decoupling between surface hydration and bulk water, in the sense that these represent different populations of water with distinctly different dynamic properties. By comparing disease-related variants with their wild-type counterparts, we gained potential insight into the molecular basis of function/malfunction—the core function of γS-crystallin in the eye lens may be precisely its capacity to simply preserve the stable hydration shell intrinsic to the protein. An outcome of broad interest of this study is that the effect of protein crowding depends on the stability of the protein-specific and -intrinsic hydration shell.
Experimental Methods
CWEPR/ODNP Sample Preparation.
Natural abundance γS-WT and γS-G18V were expressed and purified as previously described24,25. Both γS-WT and γS-G18V were diluted to 1 mg/mL prior to spin labeling. The γS-WT protein was stored in 10 mM phosphate buffer (pH 6.9, 0.05% sodium azide), while the γS-G18V was stored in 10 mM acetate buffer (pH 4.3, 0.05% sodium azide). γS-G18V was stored in the more acidic acetate buffer in hopes to achieve higher concentrations without significant aggregation.
The natural spin labeled site C23, C25 and C27 in γS-WT and γS-G18V were mutated to serine creating label free backgrounds. The introduction of a new spin labeling site was chosen by ranking the proximity of small uncharged residues to position 18, resulting in A28C and T32C mutations being chosen. Mutations were introduced to each label free background, resulting in two pairs of proteins. The mutations were introduced using standard mutagenesis and cloning procedures using the following forward primers and their reverse compliment:
C23S,C25S,C27S,
5’- CGCTATGACTCTGATAGCGACTCTGCAGATTTC-3’;
A28C, 5’- GATAGCGACTCTTGCGATTTCCACAC-3’;
T32C, 5’- GCAGATTTCCACTGCTACCTAAGTCG-3’.
Mutants were expressed at 20 °C in autoinduction media,55 then purified as previously reported. 24 The fold of each mutant was verified by CD spectra.
Five sample conditions were utilized for ONDP measurement on γS-G18V variant: Sample 1v was stored at 0.5 – 1.5 mg/mL prior to spin-labeling, Sample 2v from a different growth was stored at ~150 mg/mL prior to dilution for spin-labeling, Sample 3v consisted of sample 1v, but that had been stored for 3–4 months at 4 ˚C, Sample 4v was protein from another growth stored >3 months at 0.5 – 1.5 mg/mL at 4 ˚C, Sample 5v was protein from another subsequent growth stored at 0.5–1.5 mg/mL and measured soon after the protein was purified. Two sample conditions were utilized for ODNP measurements on the γS-WT: Sample 1w was stored at 100–150 mg/mL prior to dilution for spin-labeling, while Sample 2w was stored at ~30 mg/mL prior to spin-labeling. For all of the S3 variants, the protein was measured soon after protein was purified and stored at dilute conditions (0.5–1.5 mg/ml). All protein samples were spin-labeled with a 2-fold excess of MTS for 2 hours at 4 ˚C. The samples were then washed of excess MTSL using a PD-10 desalting column.
For both CWEPR and ODNP measurements, a 3.5 μL spin labeled sample was loaded into a quartz round capillary tube of 0.60 mm inner diameter and 0.84 mm outer diameter as previously described.29 For ODNP enhancement and T1 relaxation measurements a combination of spin labeled protein and non spin-labeled protein was utilized to reach high protein concentrations. For T10 measurements, strictly non-spin labeled protein was used under the same experimental conditions as performed for the T1 measurements. Interstitial water dynamics were measured via ODNP using non-spin labeled proteins along with free 4-hydroxy Tempo at 20 mM.
CWEPR Procedure.
For CWEPR, the sample was continuously illuminated with microwave radiation at a fixed frequency (9.8 GHz) at 25 dB, while the magnetic field was swept (modulation frequency =100.0 KHz, modulation amplitude = 0.50 Gauss).
ODNP Procedure.
1H ODNP experiments were performed at 0.35 T at a 14.8 MHz 1H Larmor frequency and at 9.8 GHz electron spin Larmor frequency using a homebuilt U-shaped NMR coil inside an ER4123D dielectric resonator. Sample temperature under microwave irradiation was calibrated by the 1H T1(p), as a function of microwave power, p. During ODNP measurements, the center field of the nitroxide hyperfine transitions lines were pumped continuously by microwave irradiation, while the 1H NMR signal was measured, as previously described29.
Solution-state NMR.
1H-15N labeled γS-WT and γS-G18V were expressed and purified as previously described41,56, and dialyzed against 10 mM sodium phosphate, pH 6.9, containing 0.05% sodium azide, 2 mM TMSP, and 10% D2O. The protein concentrations were 1.5 mM for both γS-WT and γS-G18V.
2D 1H-15N HSQCs of γS-WT and γS-G18V were acquired on an 800 MHz Varian UnityINOVA spectrometer (Agilent, Inc.) equipped with a 1H-13C-15N 5 mm tri-axis PFG triple resonance probe every 5 °C as the samples were heated between 22–47 °C. Decoupling of 15N nuclei was performed using the GARP sequence57. The samples were allowed to equilibrate at each temperature for several minutes before data collection. Sample precipitation and loss of signal occurred at temperatures above 47 °C. 1H shifts were referenced to TMSP and 15N shifts were referenced indirectly to TMSP. NMR data were processed using NMRPipe58 and analyzed using Sparky.59
Bis-ANS fluorescence assay.
Fluorescence spectra were collected as a function of bis-ANS binding for γS-WT and γS-G18V crystallins with a F4500 Hitachi fluorescence spectrophotometer. The excitation and emission wavelengths were 390 nm and 500 nm, respectively. Protein concentrations for both γS-WT and γS-G18V were ~1 mg/mL in 10 mM sodium phosphate buffer (pH 6.9, 0.05 % sodium azide). Stock solutions of bis-ANS were prepared in methanol with final concentrations maintained below 5% (v/v) varying from 5 to 750 μM when mixed with the crystallins. Bis-ANS concentrations were measured using ε = 16,790 M−1 cm−1 at 385 nm.
Circular Dichroism (CD).
Cysteine variant samples were concentrated to 0.10 mg/mL in 10 mM sodium phosphate (pH 6.9, 0.05 % sodium azide). CD measurements were preformed using a Jasco J-810 CD spectropolarimeter to access secondary structure. Spectra were measured from 260 nm to 195 nm and repeated in triplicate. Measurement parameters were 50 nm/sec continuous scanning with a 2 nm band width and 4 sec response. Raw spectra data was processed using 5 nm convolution Savitzkey-Golay smoothing.60 Variant spectra match native wild-type γS-crystallin and variant G18V previously in Brubaker et al. 24
Supplementary Material
ACKNOWLEDGMENT
S.H., K.H., and R.S. made us of the MRL Facilities supported by the NSF through the Materials Research Science and Engineering Centers under Grant DMR 1121053. S.H., K.H., and R.S. acknowledge the support of the IRG1 group of the MRL. Brendan Allison is acknowledged for his aid in researching the background literature relevant to our study, as well as significant contribution to composing and editing this article. S.H., K.H., and R.S. acknowledge support from the 2011 NIH Director New Innovator Award and the Cluster of Excellence RESOLV (EXC 1069) funded by the Deutsche Forschungsgemeinschaft. R.W.M, C.N.K., J.C.B, K.W.R., and D.K. acknowledge support from NIH R01EY021514 to R.W.M.
Funding Sources
Any funds used to support the research of the manuscript should be placed here (per journal style).
ABBREVIATIONS
- NMR
Nuclear magnetic resonance
- ODNP
Overhauser dynamic nuclear polarization
- CWEPR
continuous wave electron paramagnetic resonance
- CD
circular dichroism
Footnotes
Any additional relevant notes should be placed here.
ASSOCIATED CONTENT
Supporting Information Available: CWEPR spectra of DTT treated γS-WT samples, translational correlation time (ODNP) vs. ΔH0 (CWEPR) plot of corresponding γS-WT samples, CWEPR spectra of γS-WT with various concentrations of added viscogens, ODNP theory, δΔ /ΔΤ data for all residues tested. This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- (1).Minton AP Biopolymers 1981, 20, 2093. [Google Scholar]
- (2).Zhou H-X; Rivas G; Minton AP Annual review of biophysics 2008, 37, 375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Miklos AC; Sarkar M; Wang Y; Pielak GJ Journal of the American Chemical Society 2011, 133, 7116. [DOI] [PubMed] [Google Scholar]
- (4).Predeus AV; Gul S; Gopal SM; Feig M The Journal of Physical Chemistry. B 2012, 116, 8610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Gnutt D; Gao M; Brylski O; Heyden M; Ebbinghaus S Angewandte Chemie 2015, 54, 2548–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Inomata K; Ohno A; Tochio H; Isogai S; Tenno T; Nakase I; Takeuchi T; Futaki S; Ito Y; Hiroaki H; Shirakawa M Nature 2009, 458, 106. [DOI] [PubMed] [Google Scholar]
- (7).Wirth AJ; Gruebele M BioEssays 2013, 35, 984. [DOI] [PubMed] [Google Scholar]
- (8).Monteith WB; Cohen RD; Smith AE; Guzman-Cisneros E; Pielak GJ Proceedings of the National Academy of Sciences 2015, 112, 1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dhar A; Samiotakis A; Ebbinghaus S; Nienhaus L; Homouz D; Gruebele M; Cheung MS Proceedings of the National Academy of Sciences 2010, 107, 17586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Dong H; Qin S; Zhou H-X PLoS Computational Biology 2010, 6, e1000833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Senske M; Törk L; Born B; Havenith M; Herrmann C; Ebbinghaus S J Am Chem Soc 2014, 136, 9036. [DOI] [PubMed] [Google Scholar]
- (12).Benton LA; Smith AE; Young GB; Pielak GJ Biochemistry 2012, 51, 9773. [DOI] [PubMed] [Google Scholar]
- (13).Wang Y; Sarkar M; Smith AE; Krois AS; Pielak GJ J Am Chem Soc 2012, 134, 16614. [DOI] [PubMed] [Google Scholar]
- (14).Harada R; Sugita Y; Feig MJ Am. Chem. Soc. 2012, 134, 4842–4849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hatters DM; Minton AP; Howlett GJ The Journal of Biological Chemistry 2002, 277, 7824–7830. [DOI] [PubMed] [Google Scholar]
- (16).Batra J; Xu K; Qin S; Zhou H-X Biophysical Journal 2009, 97, 906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).King JT; Arthur EJ; Brooks CL; Kubarych KJ J Am Chem Soc 2014, 136, 188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bloemendal H; de Jong W; Jaenicke R; Lubsen NH; Slingsby C; Tardieu A Progress in Biophysics and Molecular Biology 2004, 86, 407. [DOI] [PubMed] [Google Scholar]
- (19).Augusteyn RC Experimental eye research 2010, 90, 643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Su SP; McArthur JD; Truscott RJW; Aquilina JA Bba-Proteins Proteom 2011, 1814, 647. [DOI] [PubMed] [Google Scholar]
- (21).Liu C; Asherie N; Lomakin A; Pande J; Ogun O; Benedek GB Proceedings of the National Academy of Sciences of the United States of America 1996, 93, 377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ma Z; Piszczek G; Wingfield PT; Sergeev YV; Hejtmancik JF Biochemistry 2009, 48, 7334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sun H; Ma Z; Li Y; Liu B; Li Z; Ding X; Gao Y; Ma W; Tang X; Li X; Shen Y Journal of Medical Genetics 2005, 42, 706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Brubaker WD; Freites JA; Golchert KJ; Shapiro RA; Morikis V; Tobias DJ; Martin RW Biophysical Journal 2011, 100 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kingsley CN; Brubaker WD; Markovic S; Diehl A; Brindley AJ; Oschkinat H; Martin RW Structure 2013, 21, 2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Petitt P; Edwards ME; Forciniti D Eur J Biochem 1997, 243, 415. [DOI] [PubMed] [Google Scholar]
- (27).Zhao HY; Chen YW; Rezabkova L; Wu ZR; Wistow G; Schuck P Protein Sci 2014, 23, 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Armstrong BD; Han SG J Am Chem Soc 2009, 131, 4641. [DOI] [PubMed] [Google Scholar]
- (29).Franck JM; Pavlova A; Scott JA; Han S Progress in Nuclear Magnetic Resonance Spectroscopy 2013, 74, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Cheng C-Y; Varkey J; Ambroso MR; Langen R; Han S Proceedings of the National Academy of Sciences 2013, 110, 16838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Armstrong BD; Han S The Journal of Chemical Physics 2007, 127, 104508. [DOI] [PubMed] [Google Scholar]
- (32).Hausser KH; Stehlik D In Advances in Magnetic and Optical Resonance; John S,W, Ed.; Academic Press: 1968; Vol. Volume 3, p 79. [Google Scholar]
- (33).Höfer P; Parigi G; Luchinat C; Carl P; Guthausen G; Reese M; Carlomagno T; Griesinger C; Bennati M J Am Chem Soc 2008, 130, 3254. [DOI] [PubMed] [Google Scholar]
- (34).Sezer D; Prandolini MJ; Prisner TF Physical Chemistry Chemical Physics 2009, 11, 6626. [DOI] [PubMed] [Google Scholar]
- (35).Song J; Allison B; Han S MRS Bulletin 2014, 39, 1082. [Google Scholar]
- (36).Dhulesia A; Cremades N; Kumita JR; Hsu S-TD; Mossuto MF; Dumoulin M; Nietlispach D; Akke M; Salvatella X; Dobson CM Journal of the American Chemical Society 2010, 132, 15580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Li Y; Brodsky B; Baum J Journal of Biological Chemistry 2009, 284, 20660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Khago D; Wong E; Kingsley CN; Freites JA; Tobias DJ; Martin RW Biochimica et Biophysica Acta 2016, 1860, 325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kramer Ryan M.; Shende Varad R.; Motl N; Pace CN; Scholtz JM Biophysical Journal 2012, 102, 1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Franck J; Ding Y; Stone K; Qin P; Han S Journal of the American Chemical Society 2015, 137, 12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Hong J; Jing Q; Yao L Journal of Biomolecular Nmr 2013, 55, 71. [DOI] [PubMed] [Google Scholar]
- (42).Baxter NJ; Williamson MP Journal of Biomolecular NMR 1997, 9, 359. [DOI] [PubMed] [Google Scholar]
- (43).Cierpicki T; Zhukov I; Byrd RA; Otlewski J Journal of Magnetic Resonance 2002, 157, 178. [DOI] [PubMed] [Google Scholar]
- (44).Tomlinson JH; Williamson MP Journal of Biomolecular NMR 2012, 52, 57. [DOI] [PubMed] [Google Scholar]
- (45).Cierpicki T; Otlewski J Journal of Biomolecular NMR 2001, 21, 249. [DOI] [PubMed] [Google Scholar]
- (46).Purkiss AG; Bateman OA; Goodfellow JM; Lubsen NH; Slingsby C Journal of Biological Chemistry 2002, 277, 4199. [DOI] [PubMed] [Google Scholar]
- (47).Basak A; Bateman O; Slingsby C; Pande A; Asherie N; Ogun O; Benedek GB; Pande J Journal of Molecular Biology 2003, 328, 1137. [DOI] [PubMed] [Google Scholar]
- (48).Chong SH; Ham S Angew Chem Int Edit 2014, 53, 3961. [DOI] [PubMed] [Google Scholar]
- (49).Bothra A; Bhattacharyya A; Mukhopadhyay C; Bhattacharyya K; Roy S Journal of Biomolecular Structure & Dynamics 1998, 15, 959. [DOI] [PubMed] [Google Scholar]
- (50).Hawe A; Sutter M; Jiskoot W Pharmaceutical Research 2008, 25, 1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Pande A; Zhang J; Banerjee PR; Puttamadappa SS; Shekhtman A; Pande J Biochemical and Biophysical Research Communications 2009, 382, 196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Berne BJ; Weeks JD; Zhou RH Annual Review of Physical Chemistry 2009, 60, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Jamadagni SN; Godawat R; Garde S In Annual Review of Chemical and Biomolecular Engineering; Prausnitz JM, Ed.; Annual Reviews: Palo Alto, 2011; Vol. 2, p 147. [DOI] [PubMed] [Google Scholar]
- (54).Patel AJ; Varilly P; Jamadagni SN; Hagan MF; Chandler D; Garde S Journal of Physical Chemistry B 2012, 116, 2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Studier FW Protein Expression and Purification 2005, 41, 207. [DOI] [PubMed] [Google Scholar]
- (56).Brubaker WD; Martin RW Biomolecular NMR Assignments 2012, 6, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Shaka AJ; Barker PB; Freeman R Journal of Magnetic Resonance 1985, 64, 547. [Google Scholar]
- (58).Delaglio F; Grzesiek S; Vuister GW; Zhu G; Pfiefer J; Bax A J. Biomol. NMR. 1995, 6, 277–293. [DOI] [PubMed] [Google Scholar]
- (59).Goddard TD; Kneller DG; University of California, San Francisco. [Google Scholar]
- (60).Savitzky A; Golay M Anal. Chem. 1964, 36, 1627–1639. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.