

ABSTRACT
The fork protection complex (FPC), comprising the TIMELESS (TIM)-TIPIN heterodimer, acts as a scaffold of the replisome to support seamless DNA replication. We recently showed that SDE2, a PCNA-associated DNA replication stress regulator, maintains the integrity of the FPC, and together with TIM, protects stalled replication forks from nucleolytic degradation.
KEYWORDS: Genome instability, DNA replication stress, SDE2, TIMELESS, fork protection complex, BRCA1/BRCA2
Maintaining DNA replication fork integrity is essential for genome stability and cellular survival. DNA replication stress impairs replication fork progression and contributes to the formation of aberrant DNA structures, leading to fork collapse and breakage, a major source of instability that drives tumorigenesis.1 Persistent DNA replication stress is a key feature of cancer, and exploiting the replication vulnerability of cancer cells has emerged as a new therapeutic strategy. For instance, poly (ADP-ribose) polymerase (PARP) inhibitors, including an FDA-approved compound olaparib, induce synthetic lethality in the context of BRCA1/BRCA2 deficiency by exacerbating the instability of stalled replication forks.2
DNA replication is coordinated by the replication machinery, or the replisome, in which the Cdc45-MCM-GINS (CMG) helicase unwinds duplex DNA, while replicative polymerases execute nascent strand synthesis with guidance from the processivity factor proliferating cell nuclear antigen (PCNA). PCNA also interacts with numerous genome surveillance factors to coordinate stress responses against DNA damage. A key component required for the replisome and replication fork stability is the evolutionarily conserved protein complex, the fork protection complex (FPC), which consists of the TIMELESS (TIM) and TIPIN core along with ancillary proteins AND-1 and CLASPIN.3 The TIM-TIPIN heterodimer couples the helicase-polymerase movement, thereby ensuring unperturbed fork progression, especially at repetitive DNA sequences where the replisome frequently pauses. A recent cryogenic electron microscopy (cryo-EM) study revealed that Tof1/Csm3 (TIM-TIPIN in S. cerevisiae) grips duplex DNA ahead of CMG, facilitating strand separation and replisome progression.4 The FPC also plays important roles in sister chromatid cohesion, polymerase α/primase chromatin loading, and ATR checkpoint by facilitating CHK1 phosphorylation, highlighting the multifaceted roles of the FPC in genome maintenance.5
Our recent study elucidated how the FPC is regulated during active replication and revealed a new role of the FPC in protecting hydroxyurea (HU) induced-stalled forks via its interaction with SDE2 (SDE2 telomere maintenance homolog; C1orf55).6 Human SDE2, originally identified as a replication stress response regulator at DNA replication forks, undergoes endolytic cleavage, which releases its N-terminal ubiquitin-like (UBL) domain via deubiquitinating enzyme activity and in a PCNA-dependent manner.7 We further showed that the remaining C-terminal (Ct) fragment, SDE2Ct, exposes a lysine at its N-terminus and thereby becomes a substrate of the Arg/N-end rule, a specialized proteolytic signaling that operates through the recognition of the first amino acid of a substrate.8 Chromatin-associated degradation of SDE2Ct by the Arg/N-end rule and ATP-driven segregase VCP/p97UFD1-NPL4 is required for activating the signaling of translesion DNA synthesis at ultraviolet C (UVC)-induced stalled forks, suggesting that cellular SDE2 levels may regulate dynamic changes of the replisome under replication stress.
Indeed, further analysis of SDE2-interacting proteins at replication forks led us to identify TIM as a new binding partner of SDE2.6 SDE2 interacts with the C-terminus of TIM via its conserved SDE2 domain, and this interaction is necessary for stabilizing TIM against proteasome-dependent degradation. Intriguingly, unlike TIPIN that forms an obligate heterodimer with TIM, SDE2 regulates TIM specifically engaged at replication forks, implicating to the replisome-associated SDE2 function toward TIM stability. Consequently, loss of SDE2 impairs localization of TIM at replication forks and causes uncoupling of helicase-polymerase activities, supporting SDE2 role in promoting the integrity of the FPC, as required for replisome progression.
We expected SDE2 deficiency to phenocopy TIM deficiency. Using single-molecule DNA combing analysis, we showed that SDE2 or TIM knockdown results in compromised fork progression, increase in replication fork asymmetry and new origin firing, and impaired stalled fork recovery upon HU treatment. Furthermore, SDE2 depletion abrogates CHK1 phosphorylation in a TIM- and CLASPIN-dependent manner.
One compelling phenotype that we observed upon SDE2 or TIM knockdown was a dramatic increase of RPA32 phosphorylation at Ser4/Ser8, indicative of DNA hyper-resection and break formation upon replication damage. DNA combing analysis revealed that stalled forks undergo excessive resection in knocked-down cells, indicating that SDE2 and TIM are required for stalled fork protection. Replication fork reversal, which involves the regression of a stalled fork to form a four-way junction by the action of RAD51 recombinase and several SWI/SNF family translocases, has emerged as a key protective mechanism for fork stabilization and recovery.9 Intriguingly, loss of SMARCAL1, a fork remodeler, or inhibition of MRE11 nuclease activity, rescues knocked-down cells from fork degradation, indicating that SDE2 and TIM protect reversed forks from nucleolytic degradation. These results establish a new role of TIM in engaging stalled fork protection besides its canonical role in active fork progression. Fork reversal is mediated by a disparate group of proteins involved in fork remodeling and processing, and dynamic changes of the FPC within the replisome, potentially prompted by SDE2, may therefore help engage new factors required for protecting remodeled forks.
In summary, our study provides new insights into how replication fork integrity is preserved by the SDE2-TIM interaction and defines the FPC as a new component in reversed fork protection (Figure 1). TIM is overexpressed in many cancers, suggesting that enhanced TIM activity may confer a survival advantage that mitigates elevated replication stress manifested in tumors.10 This may be particularly beneficial in a BRCA1/BRCA2-deficient background, thus influencing PARP inhibitor sensitivity and patient outcomes. Finding a way to modulate TIM-dependent fork protection may provide a new therapeutic strategy to exploit the replication vulnerability of cancer cells.
Figure 1.
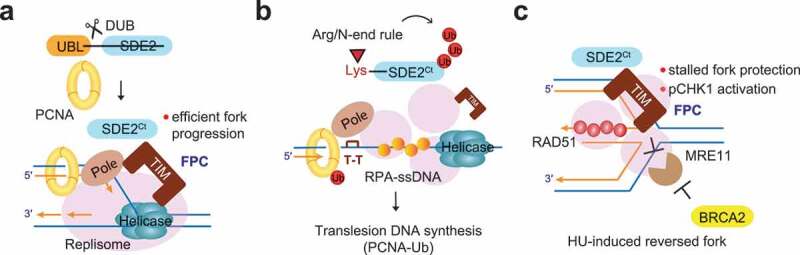
Dynamic regulation of replication forks by the fork protection complex (FPC). (a) Cleavage of SDE2 by a deubiquitinating enzyme (DUB) and upon PCNA interaction localizes the C-terminal (Ct) SDE2Ct at a replication fork. SDE2Ct interacts and stabilizes TIMELESS (TIM) in the FPC, which connects DNA unwinding-polymerization activities and ensures undisturbed fork progression. (b) In response to ultraviolet C (UVC)-induced damage (e.g. thymine dimer, T-T) that causes uncoupling of the helicase-polymerase ε function, SDE2Ct undergoes chromatin-associated degradation prompted by the Arg/N-end rule, which promotes RPA-single-stranded DNA (ssDNA) complex formation and activation of translesion DNA synthesis by PCNA ubiquitination. (c) SDE2Ct and TIM protect hydroxyurea (HU)-induced reversed forks from MRE11-dependent nucleolytic degradation together with BRCA2-dependent fork stabilization mechanism. The FPC also promotes ATR-dependent CHK1 phosphorylation. We propose that the replisome undergoes dynamic remodeling (schematically represented by three circles) to accommodate new DNA structures and protein entities necessary for stabilizing stalled forks and activating stress response signaling, which may be controlled by SDE2 and its interaction with the FPC. Ub: ubiquitin, UBL: ubiquitin-like
Funding Statement
This work was supported by National Institutes of Health [grant R01CA218132] and American Cancer Society [Grant 132235-RSG-18-037-DMC].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Branzei D, Foiani M.. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11(3):1–2. doi: 10.1038/nrm2852. [DOI] [PubMed] [Google Scholar]
- 2.Pommier Y, O’Connor MJ, de Bono J.. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8(362):362ps17. doi: 10.1126/scitranslmed.aaf9246. [DOI] [PubMed] [Google Scholar]
- 3.McFarlane RJ, Mian S, Dalgaard JZ.. The many facets of the Tim-Tipin protein families’ roles in chromosome biology. Cell Cycle. 2010;9(4):700–705. doi: 10.4161/cc.9.4.10676. [DOI] [PubMed] [Google Scholar]
- 4.Baretić D, Jenkyn-Bedford M, Aria V, Cannone G, Skehel M, Yeeles JTP. Cryo-EM structure of the fork protection complex bound to CMG at a replication fork. Mol Cell. 2020;78(5):926–40.e13. doi: 10.1016/j.molcel.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leman AR, Noguchi E. Local and global functions of Timeless and Tipin in replication fork protection. Cell Cycle. 2012;11(21):3945–3955. doi: 10.4161/cc.21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rageul J, Park JJ, Zeng PP, Lee E-A, Yang J, Hwang S, Lo N, Weinheimer AS, Schärer OD, Yeo J-E, et al. SDE2 integrates into the TIMELESS-TIPIN complex to protect stalled replication forks. Nat Commun. 2020;11(1):5495. doi: 10.1038/s41467-020-19162-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jo U, Cai W, Wang J, Kwon Y, D’Andrea AD, Kim H. PCNA-dependent cleavage and degradation of SDE2 regulates response to replication stress. PLoS Genet. 2016;12(12):e1006465. doi: 10.1371/journal.pgen.1006465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rageul J, Park JJ, Jo U, Weinheimer AS, Vu TTM, Kim H. Conditional degradation of SDE2 by the Arg/N-end rule pathway regulates stress response at replication forks. Nucleic Acids Res. 2019;47(8):3996–4010. doi: 10.1093/nar/gkz054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Quinet A, Lemacon D, Vindigni A. Replication fork reversal: players and guardians. Mol Cell. 2017;68(5):830–833. doi: 10.1016/j.molcel.2017.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bianco JN, Bergoglio V, Lin Y-L, Pillaire M-J, Schmitz A-L, Gilhodes J, Lusque A, Mazières J, Lacroix-Triki M, Roumeliotis TI, et al. Overexpression of Claspin and Timeless protects cancer cells from replication stress in a checkpoint-independent manner. Nat Commun. 2019;10(1):910. doi: 10.1038/s41467-019-08886-8. [DOI] [PMC free article] [PubMed] [Google Scholar]