

ABSTRACT
CRL and APC/C belong to the RING-finger-type E3 ligases, and both play important roles in cell cycle regulation. Recently, we found that SAG, a RING component of CRL, acts as an endogenous inhibitor of APC/C by competing with APC2 for E2s binding; while APC/CCDH1 targets SAG for ubiquitylation and degradation at G1 phase. The negative crosstalk between these two E3s ensures the orderly cell cycle progression.
KEYWORDS: SAG, APC/C, UBE2C, UBE2S, cell cycle
The RING-finger-type E3 ligases are the largest family of E3 ubiquitin ligases, and mainly include the CRL (Cullin-RING ligase) and APC/C (Anaphase-Promoting Complex). The CRL complex is composed of four components: the scaffold protein Cullin, the substrate recognition protein F-box, the adaptor protein, and the RING finger protein with only two family members, RBX1/ROC1 and RBX2/ROC2/SAG.1 SAG (sensitive to apoptosis gene) was originally cloned as a redox-inducible gene, which encodes a protein of 113 amino acids with 12 cysteine residues. By forming intra- and inter-molecule disulfide bonds in response to oxidant exposure, SAG protein possesses the non-enzymatic antioxidant activity.2 SAG was subsequently found to be the second member of a RING finger component in CRLs with E3 ligase activity.3
APC/C is another RING-finger-type E3 complex containing at least 15 different subunits, including the cullin-related protein APC2 and the RING finger protein APC11. APC/C ligase is mainly activated during the cell cycle between metaphase and the end of the next G1 depending on the association with two activator proteins CDC20 and CDH1. Specifically, APC/CCDC20 triggers the progression of metaphase to anaphase, while APC/CCDH1 is activated to initiate mitotic exit and maintain a stable G1 state.4
The negative crosstalk between the CRL, especially RBX1/E3, and APC/C has been revealed by many previous studies, mostly focusing on mutual degradation of the two E3 components.4,5 Whether and how the crosstalk also exists between SAG/E3 and APC/C was completely unknown. Our recent studies showed that unlike RBX1, which solely complexes with E2 ubiquitin-conjugating enzymes CDC34 or UBCH2C to promote substrate ubiquitylation via the K48 linkage, SAG prefers to complex with UBE2C and UBE2S E2 to promote substrate ubiquitylation via the K11 linkage.6,7 Given that APC/C E3 also couples with UBE2C and UBE2S for substrate ubiquitylation, and the levels of UBE2C and UBE2S fluctuates during the cell cycle,8 we hypothesized that SAG and APC/C may negatively cross-talk with each other by competitively binding for the same E2s to ensure proper cell cycle progression.
In our most recent study,9 we reported that SAG knockdown significantly enhanced the binding between APC2 and UBE2C and UBE2S, and vice versa for enhanced SAG-UBE2C/UBE2S binding upon APC2 knockdown. Significantly, enhanced binding of APC2-UBE2C/2S triggered by SAG-knockdown biochemically activates APC/C ligase activity, leading to increased degradation of APC/C substrates, and biologically accelerated mitotic progression in both unsynchronized and synchronized cells.
The APC/C ligase activity was shown to confer drug resistance by triggering mitotic slippage via progressively degrading cyclin B1 in response to continuous anti-microtubule drugs.10 Indeed, we found that SAG depletion promoted mitotic slippage and apoptosis protection under continuous exposure to anti-microtubule drugs in a manner dependent of APC/C activity, eventually leading to drug resistance in both in vitro cell culture and in vivo xenograft tumor models. Under this condition, SAG targeting may trigger the unfavorable results when used in combination with antimitotic drugs.
On the other hand, we found interestingly that the SAG protein levels were also fluctuated during cell cycle progression, with the highest levels at the M phase and lowest levels at the G1 phase. While the high levels of SAG at the M phase likely keep the APC/C activity under the control by competitive E2 binding to ensure proper mitotic progression, the low levels of SAG at the G1 phase, where CDH1 is expressed at the highest level, were the result of targeted degradation by APC/CCDH1. Specifically, we identified an evolutionarily conserved D-box in the SAG protein sequence, which can be recognized by CDH1. As such, APC/CCDH1 promoted the ubiquitylation and degradation of SAG through a D-box-dependent manner at the G1 phase. Moreover, degradation of SAG by APC/CCDH1 to maintain the low level of SAG at the G1 phase was critical for the orderly G1-to-S transition, since ectopic expression of D-box mutant SAG, a degradation resistant version of SAG, impaired the APCCDH1 activity by competing for UBE2C/2S binding, thus promoting the G1-to-S transition, and conferring tumor cell survival.
Based on these results, we proposed a working model that the competition between SAG and APC/C for UBE2C/S binding is a dynamic process. In normal physiological conditions, APC/CCDH1, mainly activated at the G1 phase, triggers SAG ubiquitylation and degradation to maintain the activity of APC/C by reducing the SAG-E2s competition, which is like a feed-forward loop to ensure the APC/C activity at the G1 phase. When in a high SAG condition (e.g. overexpression in cancers), SAG will “win” the competition for UBE2C/2S binding with CDH1, which, to some extent, impairs the ligase activity of APC/CCDH1, therefore leading to accelerated G1-to-S transition. Under the low SAG expression conditions (e.g. SAG knockdown by siRNAs or SAG inactivation by drugs), more free UBE2C/2S then couple with APC/C to enhance its ligase activity, thereby promoting the mitotic progression and slippage, eventually leading to antimitotic drug resistance (Figure 1).
Figure 1.
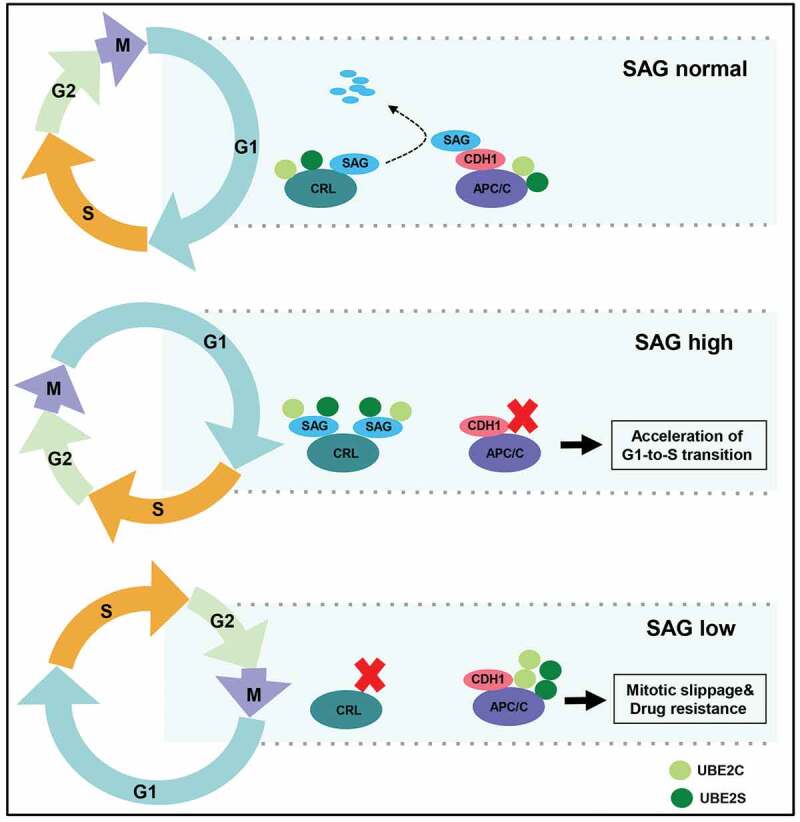
The competition between SAG and APC/C for UBE2C/S binding during cell cycle
Under physiological conditions, activated APC/CCDH1 at the G1 phase targets SAG for ubiquitylation and degradation to maintain the stable G1 status. Under high SAG conditions (e.g. SAG overexpression in cancers), SAG “sucks” UBE2C/2S from CDH1, thus impairing the APC/CCDH1 ligase activity to accelerate the G1-to-S transition. Under low SAG conditions (e.g. SAG knockdown by siRNAs or SAG inactivation by drugs), more UBE2C/2S are freed up to couple with APC/C, thus prematurely activating its ligase activity to promote the mitotic slippage, eventually leading to resistance to antimitotic drugs. SAG: sensitive to apoptosis gene, APC/C: Anaphase-Promoting Complex.
In summary, the two RING-finger-type E3 ubiquitin ligases, SAG/E3 and APC/C, negatively regulate each other to ensure the fidelity of cell cycle progression. Disrupting this fine balance will lead to unfavorable cell proliferation and drug resistance.
Funding Statement
This work is supported in part by the National Key R&D Program of China [2016YFA0501800] (YS).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Sun Y. Introduction. In: Sun Y, Wei W, Jin J, editors. Cullin-RING ligases and protein neddylation. Advances in Experimental Medicine and Biology. Singapore: Springer Singapore; 2020;1217:1–3. 10.1007/978-981-15-1025-0_1 [DOI] [Google Scholar]
- 2.Duan H, Wang Y, Aviram M, Swaroop M, Loo JA, Bian J, Tian Y, Mueller T, Bisgaier CL, Sun Y.. SAG, a novel zinc RING finger protein that protects cells from apoptosis induced by redox agents. Mol Cell Biol. 1999;19:3145–3155. doi: 10.1128/MCB.19.4.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Swaroop M, Wang Y, Miller P, Duan H, Jatkoe T, Madore S, Sun Y. Yeast homolog of human SAG/ROC2/Rbx2/Hrt2 is essential for cell growth, but not for germination: chip profiling implicates its role in cell cycle regulation. Oncogene. 2000;19:2855–2866. doi: 10.1038/sj.onc.1203635. [DOI] [PubMed] [Google Scholar]
- 4.Bassermann F, Eichner R, Pagano M. The ubiquitin proteasome system - implications for cell cycle control and the targeted treatment of cancer. Biochim Biophys Acta. 2014;1843:150–162. doi: 10.1016/j.bbamcr.2013.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jang SM, Redon CE, Thakur BL, Bahta MK, Aladjem MI. Regulation of cell cycle drivers by Cullin-RING ubiquitin ligases. Exp Mol Med. 2020. doi: 10.1038/s12276-020-00508-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuang P, Tan M, Zhou W, Zhang Q, Sun Y. SAG/RBX2 E3 ligase complexes with UBCH10 and UBE2S E2s to ubiquitylate beta-TrCP1 via K11-linkage for degradation. Sci Rep. 2016;6:37441. doi: 10.1038/srep37441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou W, Xu J, Li H, Xu M, Chen ZJ, Wei W, Pan ZQ, Sun Y. Neddylation E2 UBE2F promotes the survival of lung cancer cells by activating CRL5 to degrade NOXA via the K11 linkage. Clin Cancer Res. 2017;23:1104–1116. doi: 10.1158/1078-0432.CCR-16-1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wickliffe KE, Williamson A, Meyer HJ, Kelly A, Rape M. K11-linked ubiquitin chains as novel regulators of cell division. Trends Cell Biol. 2011;21:656–663. doi: 10.1016/j.tcb.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang S, Shen Y, Li H, Bi C, Sun Y, Xiong X, Wei W, Sun Y. The negative cross-talk between SAG/RBX2/ROC2 and APC/C E3 ligases in regulation of cell cycle progression and drug resistance. Cell Rep. 2020;32:108102. doi: 10.1016/j.celrep.2020.108102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Topham CH, Taylor SS. Mitosis and apoptosis: how is the balance set? Curr Opin Cell Biol. 2013;25:780–785. doi: 10.1016/j.ceb.2013.07.003. [DOI] [PubMed] [Google Scholar]