

ABSTRACT
Cell migration frequently involves the formation of lamellipodial protrusions, the initiation of which requires Rac GTPases signalling to heteropentameric WAVE regulatory complex (WRC). While Rac-related RhoG and Cdc42 can potently stimulate lamellipodium formation, so far presumed to occur by upstream signalling to Rac activation, we show here that the latter can be bypassed by RhoG and Cdc42 given that WRC has been artificially activated. This evidence arises from generation of B16-F1 cells simultaneously lacking both Rac GTPases and WRC, followed by reconstitution of lamellipodia formation with specific Rho-GTPase and differentially active WRC variant combinations. We conclude that formation of canonical lamellipodia requires WRC activation through Rac, but can possibly be tuned, in addition, by WRC interactions with RhoG and Cdc42.
KEYWORDS: Arp2/3 complex, CRISPR/Cas9, Rho-GTPase, filopodium, lamellipodium, blebbing, stress fibre, migration, protrusion
Introduction
Cell migration is essential for many physiological and pathological processes, such as embryonic development, immunity and metastasis [1–3]. Protrusion of the plasma membrane to enable cell migration is commonly achieved by the formation of thin branched arrays of actin filaments, called lamellipodia [4,5]. Small GTPases of the Rac family (i.e. Rac1, Rac2, Rac3 in mammals) are essential for lamellipodia initiation and maintenance, at least in part, by activation and continuous interaction through two binding sites on Sra-1 (or its orthologue PIR121) embedded into heteropentameric WAVE regulatory complex (WRC) [6,7]. The closest relatives of Rac GTPases are RhoG and Cdc42. Both fail to initiate lamellipodia in fibroblasts lacking Rac expression [6], but can clearly promote Rac-dependent lamellipodia formation. This activity is thought to derive from crosstalk involving distinct Rac-GEF complexes [8–11]. The inability of RhoG and Cdc42 to initiate lamellipodia has hitherto been thought to be due to a lack of sufficient interaction with WRC. This is because both RhoG and Cdc42 show at best a weak interaction with Sra-1/PIR121 [6,12], and Cdc42, as opposed to Rac1, fails to activate native WRC in vitro [13].
Aside from Sra-1 (or PIR121), WRC is composed of four additional subunits – WAVE2 (or its paralogues WAVE1/WAVE3), the Sra-1/PIR121 interactor Nap1 (or Hem1 in the hematopoietic system), Abi1 (or Abi2/Abi3) and HSPC300 [14–17]. The Sra-1 subunit is ‘transinhibiting’ the Arp2/3 complex-activating, so called WCA domain located on the C-terminal end of WAVE proteins, and Rac binding to Sra-1 outcompetes this inhibitory interaction to release the WCA domain, making it accessible for actin and Arp2/3 complex binding [18]. We have recently shown that the two aforementioned Rac binding sites on Sra-1/PIR121 [18,19] are essential for allosteric activation of WRC in cells [7]. However, in spite of the previously proposed safe box model requiring two keys to allow for WRC activation to occur [19], we have found surprisingly specific physiological functions for the two sites in live cells. Whereas the low affinity A site is crucial for activation in vivo, the high affinity D site is contributing to the efficiency of lamellipodial protrusion, but by no means as important for WRC activation as the A site. Aside from the apparent critical function of Rac in WRC activation, it is less well established if or if so to what extent Rac-WRC interactions also drive WRC recruitment to and accumulation in the lamellipodium. For instance, we have also found that lamellipodia formation can be initiated, in principle, without direct WRC-Rac interactions once WRC is rendered active, assuming at least that introduced, respective mutations of both A and D sites into active WRC abolished its interaction with Rac entirely [7,19]. This is consistent with the fact that deleting the CAAX-box in Rac1, which is crucial for plasma membrane association, does impair, but not abolish lamellipodia formation in Rac1 knockout (KO) fibroblasts [6]. In spite of the absence of an unequivocal, alternative mechanism of WRC recruitment, these data suggest that activation and lamellipodial targeting of WRC might potentially be separable. An example of such a separation clearly constitutes the related GTPase Cdc42, which mediates activation of its downstream effectors FMNL2 and −3 as prerequisite of their lamellipodial targeting [20,21], which however can fully occur with active variants completely lacking GTPase interaction surfaces ([22] and unpublished data). Therefore, effector recruitment to lamellipodia is possible, in principle, without engagement of a given GTPase in spite of its established relevance in effector activation. Here, we have developed novel cell lines to compare the capability of lamellipodia formation by activated WRC in the absence versus presence of endogenous Rac GTPases.
Results
Rac – WRC interactions at the plasma membrane are dispensable for lamellipodia formation
We sought to test if plasma membrane insertion of the prenyl group of Rac1 is required for lamellipodia induction in B16-F1 cells. For this, we expressed constitutively active (Q61L), myc-tagged Rac1 or an identical construct lacking the CAAX-box essential for C-terminal prenylation in B16-F1 cells lacking Rac1/2/3 (clone#1 [7]). In analogy to our previously published experiments employing fibroblasts genetically deleted for Rac1 [6], deletion of the CAAX-box reduced, but did not abolish lamellipodia formation in these conditions. More specifically, these structures were induced with overall slightly reduced frequency, and the majority of them (roughly 60%) appeared to be immature, according to the previously established categorization of underdeveloped lamellipodia [7]. In contrast, the majority of cells expressing full length, constitutively active (Q61L) Rac1 harboured fully developed lamellipodia (Figure S1(a,b)). These data suggest that the same effects seen in Rac1−/− fibroblasts [6] were not cell-type specific, and could not potentially be explained by remnants of Rac2 or −3 protein expressed perhaps at undetectable levels from respective genes not targeted in these fibroblasts.
In analogy, we assembled WRCs in Sra-1/PIR121 KO cells (clone #3) harbouring a Sra-1 variant mediating constitutive WRC activation, but lacking functional Rac binding sites (A + D site WCA* [7], and Figure S1(e)). In this construct, specific point mutations abolished the binding of Rac to the A site (C179R/R190D) and D site (Y967A) [7,18,19], whereas the WCA* mutation (L697D/Y704D/L841A/F844A/W845A) in the WH2- and C-region (W and C) contact sites of Sra-1 prevented the ‘transinhibitory’ binding of Sra-1 to the WCA domain of WAVE, which is then released for activating Arp2/3 complex [7,18]. We found that Sra-1/PIR121 KO cells (clone #3) harbouring A + D site WCA*-mutated WRC can still rescue lamellipodia formation, albeit at strongly compromised frequency [7]. Once formed though, and although compromised, these lamellipodia can still accumulate WRC at their tips (Figure S1(c), see also [7]), and mediate continuous protrusion that is less smooth though than with lamellipodia driven by WRCs harbouring WT Sra-1 (Figure S1(d)). Not surprisingly, average protrusion velocities of these rare examples of compromised lamellipodia were reduced as compared to those mediated by wildtype Sra-1 (Figure S1(f)). Together, all these datasets thus suggest that although helpful, continuous Rac-WRC interactions at the plasma are not absolutely obligatory for lamellipodium protrusion.
Generation of a cell line allowing further dissection of the Rac-WRC signalling module
Next we asked whether Rac is essential for WRC-mediated lamellipodia formation solely because of its essential function in WRC activation or because of serving additional functions. To test this, we had to develop cell systems in which endogenous WRC or Rac proteins could be replaced by active variants of each or functional deficiency mutants in a combinatorial fashion. In previous work, we had established cell lines lacking either Rac1/2/3 or functional WRC (Sra-1/PIR121-null), in which off-target effects potentially caused by CRISPR/Cas9-mediated genome editing were excluded by a combination of analysing multiple, independently generated clones as well as by rescue of phenotypes with exogenous Rac and Sra-1, respectively [7]. In the current work, we extended this approach to generate a novel cell line disrupted for all five genes (Sra-1/PIR121+ Rac1/2/3 KO#3/11; Figure 1(a)). The latter now allows deciphering Rac/WRC signalling in more detail. In these cells, lamellipodia formation is strikingly dependent on exogenous expression of both Sra-1 and Rac1. While neither expression of EGFP as control, EGFP-Sra-1 nor myc-Rac1L61 alone facilitated lamellipodia formation in these cells, co-transfection of EGFP-Sra-1 and myc-Rac1L61 potently restored lamellipodia, indicating the presence of lamellipodia in these cells to strictly require both Rac and Sra-1 (Figure 1(b,c)). Notably, Rac1L61 expression in these WRC-deficient cells also caused plasma membrane blebbing (Figure 1(b) for representative image), reminiscent of our previous observations upon Rac microinjection upon WRC subunit knockdown [15]. This phenotype was robust and occurred at high frequency (45 ± 13% in Rac1L61 expressing cells vs. 13 ± 12% in EGFP-expressing controls), although a precise, mechanistic understanding of the phenomenon is currently lacking. To test if constitutive activation of WRC would be sufficient to trigger lamellipodia formation in the absence of Rac, we expressed EGFP-tagged Sra-1 WCA*. Surprisingly, however, EGFP-tagged Sra-1 WCA*, lacking the need for Rac-mediated activation of WRC was still incapable of driving lamellipodia formation in this cell line and conditions. If comparing this result with constitutively active WRCs compromised in Rac binding, but giving rise to inefficient lamellipodia formation in Sra-1/PIR121-KO cells ([7] and Figure S1(c,d)), two theoretical explanations for this discrepancy are thinkable: Firstly, the A + D site-mutated, active WRC used in Figure S1 can still inefficiently bind to endogenous Rac proteins present in these cells. Alternatively, Rac possesses additional, essential functions absent in cells harbouring active WRC but lacking endogenous Rac proteins (as in Figure 1(b,c)). Future experiments will have to distinguish between these two possibilities.
Figure 1.
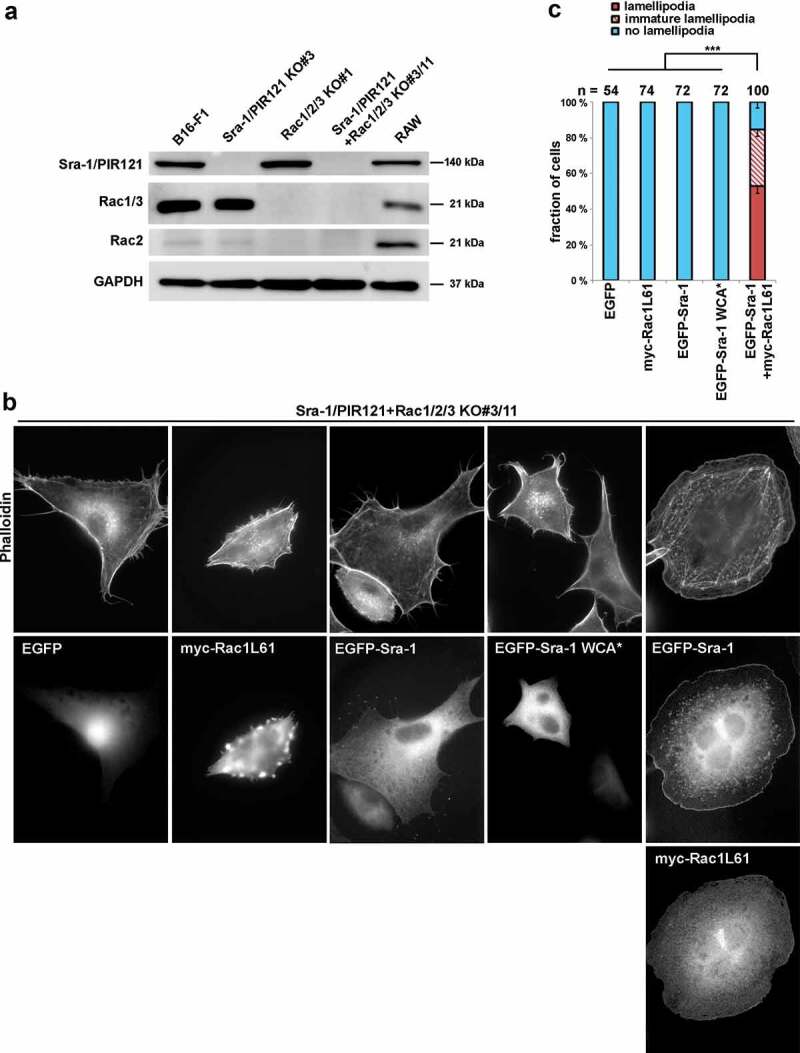
Generation of Sra-1/PIR121+ Rac1/2/3 KO cells
(a) Western blotting of distinct cell lines to probe for expression levels of endogenous Sra-1/PIR121 and/or Rac GTPases. (b) Cell morphologies of Sra-1/PIR121+ Rac1/2/3 KO cells (clone #3/11) expressing respective constructs, as indicated. Panels in top row show stainings of the actin cytoskeleton with phalloidin, and middle and/or bottom row images show fluorescence of the same cells derived from either EGFP or anti-myc antibody stainings, as indicated. (c) Quantification of lamellipodial phenotypes. EGFP-Sra-1 WCA* denotes a construct rendering WRC constitutively active due to mutations relieving the autoinhibitory interaction of Sra-1 with the C-terminal WCA-domain (hence WCA* or active WCA) of WAVE [7,18]. Lamellipodial actin networks that were generally small, narrow, irregular or displayed multiple ruffles were defined as ‘“immature lamellipodia”’, as opposed to regular, fully developed lamellipodia. n gives number of cells analysed, data correspond to arithmetic means ± SEM from at least three independent experiments. Statistical significance was assessed for differences between percentages of cells with ‘no lamellipodia’ phenotype. ***p < 0.001 (two-sample, two-sided t-test).
Rac-related Rho GTPases fail to activate WRC, but can substitute for Rac in the presence of activated WRC
Up to this date, the literature contains conflicting results concerning the relevance of the closest relatives of Rac GTPases in mammals, in particular RhoG, but to a certain extent also Cdc42. In spite of prominent studies establishing functions for RhoG in signalling complexes operating upstream of Rac [10–12], RhoG has also already been concluded to contribute to fibroblast migration independent of Rac activation [23], although it has remained unclear how that might occur mechanistically [24]. Of note, and again in full accordance with our previously published fibroblast data [6], our Rac1/2/3-deficient B16-F1 melanoma line failed entirely to form lamellipodia even upon expression of constitutively active RhoG (Figure 2(a,b)). Identical results were obtained with overexpressed, constitutively active Cdc42 but not Rac1, which robustly restored lamellipodia formation (Figure 2(a,b)), as expected [7]. Aside from the incapability of Cdc42 to induce lamellipodia, we found a prominent induction of stress fibres in these conditions (Figure 2(a); 89 ± 2% of transfected cells), which was not seen with RhoG (2 ± 3%). Such an activity is traditionally still mostly attributed to RhoA/B/C activity in the literature, and thus not followed up further in the context of the current study. However, since our previous efforts allowed us to experimentally separate Rac-mediated WRC activation from other potential functions ([7] and see above), we wondered whether we might – in analogy to Rac – be able to establish connections between RhoG or Cdc42 activities and WRC function independent of Rac-mediated WRC activation. For this, we explored RhoG- or Cdc42-driven actin cytoskeleton remodelling in cells exclusively harbouring active WRC but lacking endogenous Rac GTPases (see Figure 1). And indeed, co-transfection of active (Q61L), myc-tagged RhoG or Cdc42 together with Sra-1 WCA* led to partial rescue of lamellipodia formation in Sra-1/PIR121+ Rac1/2/3 KO cells (clone #3/11) (Figure 3(a,c) for representative images and quantitations, respectively). While RhoG was able to induce immature lamellipodia in more than 40% of transfected cells and even occasional, fully developed lamellipodia, Cdc42 caused lamellipodia formation only in a small subfraction of cells (see Figure 3(a), asterisks, and Figure 3(c) for quantification). Although the low frequency of Cdc42-mediated induction of (at least immature) lamellipodia in cells co-expressing Sra WCA* could not be ignored, it was not significantly different statistically from cells co-expressing constitutively active Cdc42 with wildtype Sra-1 (not shown). This result was obtained with two-sided, two-sample t-test, but when using Fisher’s exact test also appropriate for comparing two experimental groups (Cdc42 co-expression with Sra-1 WT versus WCA*) falling into two categories (with or without lamellipodia), the difference between them turned out to be statistically significant (p = 0.023). Moreover, both RhoG and Cdc42 were capable, in principle, of driving accumulation of Sra-1 WCA* at protrusion sites given that lamellipodia were formed (Figure 3(b)). Specific Sra-1 WCA* enrichment upon co-expression of both RhoG and Cdc42 was also confirmed by linescan analyses of respective images (Figure 3(b)). In contrast, co-tranfection of Sra-1 WCA* with myc-tagged, constitutively active RhoA (G14V) failed to stimulate lamellipodia or lamellipodia-like structures in all cells analysed (96 cells from 3 independent experiments; Figure 3(a,c) for quantification). This was consistent with the lack of any accumulation of EGFP-tagged Sra-1 WCA* at the periphery of these cells (Figure 3(b), right panel), in spite of constitutively active RhoA clearly being functional in this case, as evidenced by the expected, prominent stimulation of stress fibres (Figure 3(a), right panels).
Figure 2.

Rac-related GTPases RhoG and Cdc42 fail to induce lamellipodia in the absence of Rac expression
(a) Cell morphologies of Rac1/2/3 KO cells (clone #1) expressing EGFP-tagged GTPases, as indicated. (b) Quantification of lamellipodial phenotypes was performed as described for Figure 1(c). n gives number of cells analysed, data correspond to arithmetic means ± SEM from at least three independent experiments. Statistical significance was assessed for differences between percentages of cells with ‘no lamellipodia’ phenotype. ***p < 0.001 (two-sample, two-sided t-test).
Figure 3.
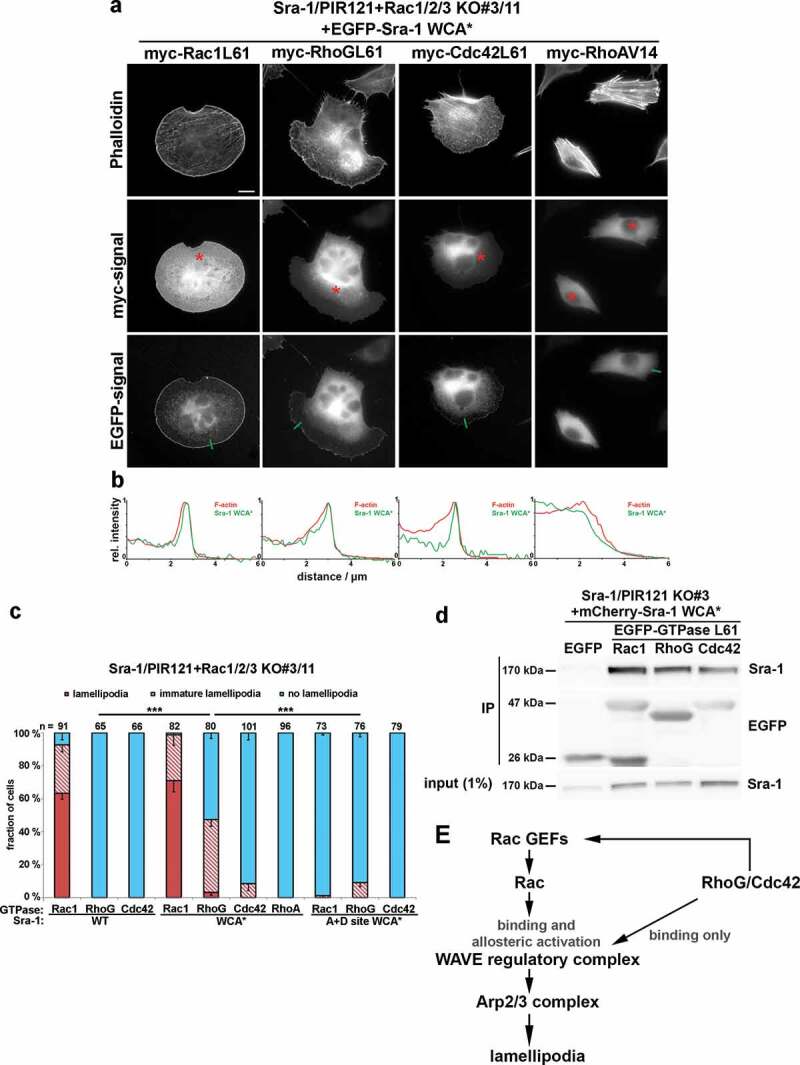
Induction of lamellipodia by RhoG or Cdc42 in conjunction with activated WRC, but in the absence of Rac expression
(a) Representative images of Sra-1/PIR121+ Rac1/2/3 KO cells (clone #3/11) expressing EGFP-Sra-1 WCA* together with myc-tagged, small GTPases, as indicated. Transfected cells identified by myc-staining (asterisks) were analysed concerning their cell morphologies (top row, actin filament staining with phalloidin) and the capability to accumulate active WRC (Sra-1 WCA*) at peripheral lamellipodia (b). Measurements shown in b were performed along line scans as shown in the images provided in a (green lines). (c) Quantification of lamellipodial phenotypes, done as described for Figure 1(c). n gives number of cells analysed, and differentially coloured columns are arithmetic means ± SEM from at least three independent experiments. Statistical significance was assessed for differences between the percentages of cells with ‘no lamellipodia’ phenotype. ***p < 0.001 (two-sample, two-sided t-test). (d) Sra-1/PIR121 KO cells (clone # 3) were co-transfected with mCherry-Sra-1 WCA* and either EGFP or constitutively active (Q61L), EGFP-tagged small GTPases, lysed and subjected to immunoprecipitation against EGFP. Note the complete absence of immunoprecipitation of Sra-1 upon co-expression of EGFP alone (negative control), and significant interactions of Sra-1 WCA* with Rac-1, RhoG and Cdc42. (e) Model for how RhoG and Cdc42 might regulate WRC and lamellipodia formation.
As already mentioned in the context of statistical analysis, the ability of RhoG and Cdc42 to induce lamellipodia in these experiments was strictly dependent on constitutive WRC activity in this cell line (through using Sra-1 WCA*), as both RhoG and Cdc42 failed to induce lamellipodia when co-expressed with WT-Sra-1 (Figure 3(c)). Finally, additional mutation of the two Rac binding sites on constitutively active WRC completely prevented (in case of Cdc42) or at least strongly inhibited (in case of RhoG) the induction of lamellipodia in these conditions (Figure 3(a,c)), indicating that the action of RhoG/Cdc42 may be explained by direct, (Rho-GTPase binding surface-dependent) interaction with WRC. In line with this, immunoprecipitation experiments showed clearly detectable interactions of RhoG and Cdc42 with Sra-1 WCA*, albeit somewhat weaker perhaps than observed for Rac1 (Figure 3(d)). Assuming that the described, potential RhoG/Cdc42 -WRC interactions may be direct, we also wondered whether these GTPases might preferentially target the A or the D site previously established as interaction surfaces on WRC with Rac1 [19]. Due to the low frequency of Cdc42-mediated lamellipodia formation in this assay (Figure 3), we focused on RhoG in these experiments compared to Rac1 used as control. We again co-transfected our cell line lacking endogenous WRC plus all Rac-GTPases with the individual A and D site mutants of WCA* Sra-1 and constitutively active Rac1 (as control) versus RhoG. While mutating either the A or D site caused significant impairment of lamellipodia formation induced by active Rac1, albeit for distinct reasons, as expected (for comparison of analogous, but not identical experiments, see ref [7].), mutating the A site did not cause a statistically significant reduction of RhoG-mediated lamellipodia formation in this assay. In contrast, abrogation of D site function caused a severe reduction of lamellipodia (Figure S2(a)), virtually identical to the level observed upon simultaneous mutation of both sites (Figure 3(c)). From this, we concluded that the potential, perhaps direct WRC-RhoG interaction is largely mediated by the D site of Sra-1.
All these results prompted us to compare by additional in silico analyses the putative binding surfaces of RhoG and Cdc42 with the D site of Sra-1, which also constitutes the high affinity binding site for Rac GTPases [19]. Sequence alignments revealed that two (in case of RhoG) or three (in case of Cdc42) amino acids differed from Rac1 in the putative binding interface (Figure S2(b)). However, neither of these residues caused significant changes in electrostatic surface potentials (Figure S2(c)). RhoA, on the contrary, showed obvious differences in surface electrostatic potential in the putative binding interface, particularly caused by the glutamine to valine substitution at position 33 in RhoA, the analogous substitution of which in Rac1 (E31V) apparently interfered with proper lamellipodia formation [7,25]. Together, these data suggest that both RhoG and Cdc42 can specifically interact with WRC, in principle, and in a physiologically relevant manner once WRC has been activated by Rac1.
Discussion
Although puzzling initially, we have previously found and confirmed here using distinct experimental systems that Rac may not have to associate with the plasma membrane in order to activate or recruit WRC during lamellipodia initiation and maintenance. Yet, and not inconsistent with this view, Rac GTPases remain to be obligatory for both WRC activation and lamellipodia formation, as cells lacking both endogenous Rac GTPases and WRC can only form lamellipodia upon additional, specific manipulation. To our surprise, we establish for the first time here that lamellipodia formation is possible, in principle, without Rac GTPases, given that cells lack the need for WRC activation (because it is already activated or does not need to be activated), and that they over-express constitutively active variants of either RhoG or Cdc42. In other words, overexpression of RhoG, and to a lesser extent Cdc42 can cause the accumulation of constitutively active WRC even in the complete absence of endogenous Rac GTPases, presumably causing WRC-dependent Arp2/3 complex activation. This suggests that aside from previously established signalling crosstalk between Rac GTPases and RhoG/Cdc42 [8–10], the latter may directly contribute to the maintenance and/or activity of WRC at protruding lamellipodia edges once Rac has managed to activate individual WRCs. It is thus thinkable that RhoG and Cdc42 may directly support Rac signalling by partially taking over Rac functions concerning WRC positioning irrespective of WRC activation.
Consistent with this view, we have previously found average turnover times for WRC subunits at the protruding plasma membrane that certainly fit the hypothesis of continuous activation events of Arp2/3 complexes, mediated by individual WRC units in a Rho GTPase binding-dependent fashion [7]. Slow turnover and molecular crowding of individual WRCs at the membrane likely contributes to efficient Arp2/3 activation at these sites (see also [26,27]), last, not least because efficient Arp2/3 complex activation was previously demonstrated to involve simultaneous engagement of two WCA domains [28–30]. Therefore, it seems plausible that efficient Arp2/3 complex activation at the lamellipodium tip coincides with WRC clustering, with the latter being affected, at least in part, by GTPase signalling.
The described, direct contribution of RhoG and Cdc42 to WRC-mediated actin remodelling is also found to occur in spite of at best weak or entirely non-specific interactions of RhoG and Cdc42 with wildtype WRC [6,12], as we show here that the situation changes dramatically if constitutively active WRC is used, which can be precipitated quite efficiently by both Rac-related GTPases. Moreover, we also found that at least the interaction of RhoG with WRC is largely mediated through the D site of Sra-1 in WRC, and not the A site (Figure S2(a)), established previously to be crucial for WRC activation by Rac1 [7]. This then could explain the complete failure of RhoG (and Cdc42) to activate WRC in the absence of endogenous Rac GTPases (Figures 2 and Figure 3(c)). All these considerations are also confirmed by sequence alignments and structural considerations concerning effector interaction surfaces present on Rac1, RhoG and Cdc42 versus RhoA (Figure S2). Future structural studies will be needed to solidify the hypothesis of direct interactions and explain why – if confirmed to be direct – the comparably robust interaction of RhoG and Cdc42 with Sra-1 observed here cannot occur with inactive WRC and/or translate into WRC activation.
Although the binding efficiency of RhoG and Cdc42 to active WRC appeared comparable in immunoprecipitation experiments, there was clearly measurable differences between the efficiency of the output response (lamellipodia in this case), the precise reasons for which remain to be determined. Yet, the vast majority of both RhoG and Cdc42-dependent lamellipodia formed in these conditions (absence of endogenous Rac GTPases) were still immature, clearly illustrating the relevance of Rac in formation and turnover of these structures beyond its established, essential function in WRC activation.
The novel cell system lacking endogenous Rac and WRC proteins (B16-F1 Sra-1/PIR121+ Rac1/2/3 KO clone #3/11) also harbours the potential of emphasizing previously mentioned, but less well studied phenotypes caused by active, small GTPases, indicative for the commonly established, but chronically underestimated complexity arising from Rho-GTPase crosstalk [31,32]. For instance, without WRC, Rac failed to induce lamellipodia, but not infrequently caused plasma membrane blebbing much less common to B16-F1 cells expressing endogenous WRC and thus capable of lamellipodia formation. Similar observations were previously reported for overexpression of the Rac effector loop mutant F37A [33], which we now consider to be impaired in driving lamellipodia formation as a result of compromised WRC interaction [7]. Whether this blebbing activity arises from intrinsic, Rac-specific features (i.e. requiring direct Rac-effector binding) or is a more indirect result of crosstalk to RhoA/B/C signalling remains unclear. Moreover, our preliminary observations also indicated that Cdc42 activities can prominently funnel into contractile stress fibre formation if Rac signalling (or lamellipodia formation) is missing. This could likely be mediated through signalling to MRCK kinases, previously shown to cooperate with Rho-ROCK signalling [34–36], which hitherto appeared less prevalent in our cell systems without compromised lamellipodia formation (see [37,38]). As the lamellipodia response seen with the Cdc42 – (active) WRC combination was much less prominent than seen for the RhoG – (active) WRC couple, it is tempting to speculate that the stress fibre induction phenotype mentioned above might interfere with more robust lamellipodia formation. This might be explained perhaps by the widely accepted and long-standing antagonism between protrusion- (lamellipodia) versus contractility-dependent (stress fibres) processes [39,40]. Future experiments will have to reveal whether this assumption is correct.
Whatever the case, our data add to the view that systematic generation and side-by-side comparison of Rho GTPase and effector knockouts in the same parental cell line will continue to unfold mechanistic insights into the intricacies of Rho signalling and crosstalk relevant for actin remodelling processes.
Materials and methods
Cell culture
B16-F1 cell line was purchased from ATCC (CRL-6323, sex:male). B16-F1 derived Sra-1/PIR121 KO cells (clone #3), as well as Rac1/2/3 KO cells (clone #1) were as described [7]. B16-F1 cells and derivatives were cultured in DMEM (4.5 g/l glucose; Invitrogen), supplemented with 10% FCS (Gibco), 2 mM glutamine (Thermo Fisher Scientific) and penicillin (50 Units/ml)/streptomycin (50 µg/ml) (Thermo Fisher Scientific). B16-F1 cells were routinely transfected in 35 mm dishes (Sarstedt), using 0.5 µg DNA in total and 1 µl JetPrime for controls, and 1 µg DNA in total and 2 µl JetPrime for B16-F1-derived knockout cells. After overnight transfection, cells were plated onto acid-washed, laminin-coated (25 µg/ml) coverslips and allowed to adhere for at least 5 hours prior to analysis.
DNA constructs
pEGFP-C1 and – C2 vectors were purchased from Clontech Inc. (Mountain View, CA, USA). pEGFP-C2-Sra-1, and derived mutant constructs (i.e. WCA*, A site WCA*, D site WCA*, A + D site WCA*) were described previously [7] and correspond to the splice variant CYFIP1a, sequence AJ567911. mCherry-tagged Sra-1 WCA* was generated by swapping EGFP with mCherry, kindly provided by Dr. Roger Tsien (University of California at San Diego, La Jolla, California, USA) using NheI/BsrGI restriction sites. pRK5-myc-Rac1L61 and pRK5-myc-RhoAV14 were kindly provided by Alan Hall and Laura Machesky (CRUK Beatson Institute, Glashow, UK). Cdc42L61 (placental isoform) was synthesized by Eurofins Genomics and cloned into pRK5-myc and pEGFP-C1 vectors. pEGFP-C1-Rac1L61 and pRK5-myc-RhoGL61 were as described [6,7]. For generation of pEGFP-C1-RhoGL61, the corresponding DNA fragment immobilized from pRK5-myc-RhoGL61 with BamHI/EcoRI was ligated into pEGFP-C1 vector digested with BglII/EcoRI. pRK5-myc-Rac1L61-∆CAAX was generated by site directed mutagenesis using 5ʹ-GAGGAAGAGAAAATGACTGCTGTTGTAAGTC-3ʹ as forward primer. The fidelity of all constructs was verified by sequencing.
CRISPR/Cas9-mediated genome editing
B16-F1 cells lacking functional CYFIP1 and CYFIP2 genes, as well as Rac1, Rac2 and Rac3 genes were generated by treating Sra-1/PIR121 KO cells (clone #3) with pSpCas9(BB)-2A-Puro (PX459) vectors targeting Rac1, Rac2 and Rac3 genes. Specifically, cells were co-transfected with plasmids targeting ATGCAGGCCATCAAGTGTG (Rac1/2) and ATGCAGGCCATCAAGTGCG (Rac3) genomic regions as described [7]. After puromycin selection of transfected cells (3 days), cells were extensively diluted and a few days later, macroscopically visible colonies picked, to obtain single cell-derived clones. Derived cell clones already lacking Sra-1/PIR121 were screened for the additional absence of Rac expression by Western Blotting (see Figure 1).
Western blotting
Preparation of whole cell lysates was performed essentially as described [7]. Western blotting was carried out using standard techniques. Primary antibodies used were Sra-1/PIR121 [15], Rac1/3 (23A8, Merck), Rac2 [6], GAPDH (6C5, Calbiochem) and GFP (clones 7.1 and 13.1, Roche). HRP-conjugated secondary antibodies were purchased from Invitrogen. Chemiluminescence signals were obtained upon incubation with ECL™ Prime Western Blotting Detection Reagent (GE Healthcare), and were recorded with ECL Chemocam imager (Intas, Goettingen, Germany).
Immunoprecipitation
For EGFP-immunoprecipitation experiments, Sra-1/PIR121 KO cells (clone #3) co-expressing EGFP alone or EGFP-tagged variants of constitutively active (Q61L) GTPases together with mCherry-tagged Sra-1 WCA* were lysed with lysis buffer (1% Triton X-100, 140 mM KCl, 50 mM Tris/HCl pH 7.4/50 mM NaF, 10 mM Na4P2O7, 2 mM MgCl2 and Complete Mini, EDTA-free protease inhibitor [Roche]). Lysates were cleared and incubated with GFP-Trap agarose beads for 60 min. Subsequently, beads were washed three times with lysis buffer lacking Triton X-100 and protease inhibitor, mixed with Laemmli buffer, boiled for 5 min and subjected to Western Blotting.
Fluorescence microscopy, phalloidin and antibody stainings and quantification
B16-F1-derived cell lines were seeded onto laminin-coated (25 µg/ml), 15 mm-diameter glass coverslips and allowed to adhere for at least 5 hours. Cells were fixed with pre-warmed, 4% paraformaldehyde (PFA) in PBS for 20 min, and permeabilized with 0.05% Triton-X100 in PBS for 30 sec.
PFA-fixed cell samples following transfections with plasmids mediating expression of EGFP-tagged proteins were counterstained with ATTO-594-conjugated phalloidin (1:200). For stainings with myc antibodies, permeabilized cells were blocked with 5% horse serum and 1% BSA in PBS, followed by staining with monoclonal anti-myc antibody (9E10; undiluted, home-made hybridoma supernatant). Primary antibodies were visualized with Alexa Fluor 350-coupled anti-mouse IgG. Linescans were generated using MetaMorph software by drawing lines (width of 15 pixels) from inside the cell and across the lamellipodium. For quantitation of stress fibers or blebs, cells derived from three independent experiments and stained for the actin cytoskeleton with phalloidin were acquired using digital imaging as described above, followed by manual categorization as positive or negative for these structures.
Time-lapse microscopy
Live cell imaging shown in Figure S1(c) was done with Sra-1/PIR121 KO #3 cells transfected with respective EGFP-tagged Sra-1 variants and migrating on laminin-coated glass (25 µg/ml). Cells were observed in µ-Slide 4 well (Ibidi), and maintained in microscopy medium (F12 HAM HEPES-buffered medium, Sigma), including 10% FCS (Gibco), 2 mM glutamine (Thermo Fisher Scientific) and penicillin (50 Units/ml)/streptomycin (50 µg/ml) (Thermo Fisher Scientific). Conventional video microscopy was performed on an inverted microscope (Axiovert 100TV, Zeiss) equipped with an HXP 120 lamp for epifluorescence illumination, a halogen lamp for phase-contrast imaging, a Coolsnap-HQ2 camera (Photometrics) and electronic shutters driven by MetaMorph software (Molecular Devices). Live cell images were obtained using a 100 x/1.4 NA Plan apochromatic oil objective at a frame rate of 12/min. Kymographs were generated using MetaMorph software by drawing lines from inside the cell and across the lamellipodium, and the protrusion velocity determined by measuring the advancement of lamellipodia tips over time.
In silico-comparison of binding interfaces of small GTPases to the D site of Sra-1
Sequence alignments were carried out using http://www.uniprot.org. The structure of RhoG was predicted with Phyre2 [41]. For comparison of surface electrostatic potentials of different small GTPases, indicated structures were superimposed with Rac1 occupying the D site of Sra-1 [7,19] using CCP4MG.
Statistical analysis
To assess statistical significance, two-sided, two-sample t-test was applied when data passed normality and equal variance tests (i.e. in Figure 1(c), Figure 2(b), Figure 3(c), S1(b) and S2(a)), otherwise nonparametric Mann-Whitney-Rank-Sum test was performed (Figure S1(f)). Fisher’s exact test was conducted for comparing cells with or without lamellipodia in Sra-1/PIR121+ Rac1/2/3 KO#3/11 clone, co-expressing Cdc42 with Sra-1 WT versus WCA* (Figure 3(c)). Statistical analyses were performed using Sigma plot 12.0 (Systat Software). Observed differences between groups were considered to be statistically significant if the error probability (p-value) of this assumption was below 5% (p < 0.05).
Supplementary Material
Acknowledgments
This work was supported by grants within the framework of a graduate programme supported by the Deutsche Forschungsgemeinschaft (DFG), called GRK2223/1 (to K.R. and W.B). We also thank Brigitte Denker for excellent technical assistance, Prof. Laura Machesky (CRUK Beatson Institute, Glasgow, UK) for Rho-GTPase expression constructs and Prof. Baoyu (Stone) Chen (Iowa State University, U.S.A.) for fruitful discussions.
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft [GRK2223/1].
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
The supplemental data for this article can be accessed here.
References
- [1].Lambert AW, Pattabiraman DR, Weinberg RA.. Emerging biological principles of metastasis. Cell. 2017;168:670–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Worbs T, Hammerschmidt SI, Förster R. Dendritic cell migration in health and disease. Nat Rev Immunol. 2017;17:30–48. [DOI] [PubMed] [Google Scholar]
- [3].Kurosaka S, Kashina A. Cell biology of embryonic migration. Birth Defects Res C Embryo Today. 2008;84:102–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rottner K, Faix J, Bogdan S, et al. Actin assembly mechanisms at a glance. J Cell Sci. 2017;130:3427–3435. [DOI] [PubMed] [Google Scholar]
- [5].Rottner K, Schaks M. Assembling actin filaments for protrusion. Curr Opin Cell Biol. 2018;56:53–63. [DOI] [PubMed] [Google Scholar]
- [6].Steffen A, Ladwein M, Dimchev GA, et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J Cell Sci. 2013;126:4572–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Schaks M, Singh SP, Kage F, et al. Distinct interaction sites of Rac GTPase with WAVE regulatory complex have non-redundant functions in vivo. Curr Biol. 2018;28:3674–3684.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Baird D, Feng Q, Cerione RA. The cool-2/α-pix protein mediates a Cdc42-Rac signaling cascade. Curr Biol. 2005;15:1–10. [DOI] [PubMed] [Google Scholar]
- [9].Nishimura T, Yamaguchi T, Kato K, et al. PAR-6–PAR-3 mediates Cdc42-induced Rac activation through the Rac GEFs STEF/Tiam1. Nat Cell Biol. 2005;7:270–277. [DOI] [PubMed] [Google Scholar]
- [10].Katoh H, Negishi M. RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature. 2003;424:461–464. [DOI] [PubMed] [Google Scholar]
- [11].Katoh H, Hiramoto K, Negishi M. Activation of Rac1 by RhoG regulates cell migration. J Cell Sci. 2006;119:56–65. [DOI] [PubMed] [Google Scholar]
- [12].Meller J, Vidali L, Schwartz MA. Endogenous RhoG is dispensable for integrin-mediated cell spreading but contributes to Rac-independent migration. J Cell Sci. 2008;121:1981–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Lebensohn AM, Kirschner MW. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009;36:512–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Kunda P, Craig G, Dominguez V, et al. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Curr Biol. 2003;13:1867–1875. [DOI] [PubMed] [Google Scholar]
- [15].Steffen A, Rottner K, Ehinger J, et al. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. Embo J. 2004;23:749–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Innocenti M, Zucconi A, Disanza A, et al. Abi1 is essential for the formation and activation of a WAVE2 signalling complex. Nat Cell Biol. 2004;6:319–327. [DOI] [PubMed] [Google Scholar]
- [17].Eden S, Rohatgi R, Podtelejnikov AV, et al. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature. 2002;418:790–793. [DOI] [PubMed] [Google Scholar]
- [18].Chen Z, Borek D, Padrick SB, et al. Structure and control of the actin regulatory WAVE complex. Nature. 2010;468:533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chen B, Chou H-T, Brautigam CA, et al. Rac1 GTPase activates the WAVE regulatory complex through two distinct binding sites. Elife. 2017;6:29795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Block J, Breitsprecher D, Kühn S, et al. FMNL2 drives actin-based protrusion and migration downstream of Cdc42. Curr Biol. 2012;22:1005–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kage F, Winterhoff M, Dimchev V, et al. FMNL formins boost lamellipodial force generation. Nat Commun. 2017;8:14832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Dimchev G, Steffen A, Kage F, et al. Efficiency of lamellipodia protrusion is determined by the extent of cytosolic actin assembly. MBoC. 2017;28:1311–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Monypenny J, Zicha D, Higashida C, et al. Cdc42 and Rac family gtpases regulate mode and speed but not direction of primary fibroblast migration during platelet-derived growth factor-dependent chemotaxis. Mol Cell Biol. 2009;29:2730–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Steffen A, Koestler SA, Rottner K. Requirements for and consequences of Rac-dependent protrusion. Eur J Cell Biol. 2014;93:184–193. [DOI] [PubMed] [Google Scholar]
- [25].Westwick JK, Lambert QT, Clark GJ, et al. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol. 1997;17:1324–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lai FP, Szczodrak M, Block J, et al. Arp2/3 complex interactions and actin network turnover in lamellipodia. Embo J. 2008;27:982–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Iwasa JH, Mullins RD. Spatial and Temporal Relationships between actin-filament nucleation, capping, and disassembly. Curr Biol. 2007;17:395–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Padrick SB, Doolittle LK, Brautigam CA, et al. Arp2/3 complex is bound and activated by two WASP proteins. PNAS. 2011;108:E472–E479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Padrick SB, Cheng H-C, Ismail AM, et al. Hierarchical regulation of WASP/WAVE proteins. Mol Cell. 2008;32:426–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Boczkowska M, Rebowski G, Kast DJ, et al. Structural analysis of the transitional state of Arp2/3 complex activation by two actin-bound WCAs. Nat Commun. 2014;5:3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lawson CD, Burridge K. The on-off relationship of Rho and Rac during integrin-mediated adhesion and cell migration. Small GTPases. 2014;5:e27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Boulter E, Garcia-Mata R, Guilluy C, et al. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol. 2010;12:477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Schwartz MA, Meredith JE, Kiosses WB. An activated Rac mutant functions as a dominant negative for membrane ruffling. Oncogene. 1998;17:625–629. [DOI] [PubMed] [Google Scholar]
- [34].Wilkinson S, Paterson HF, Marshall CJ. Cdc42–MRCK and Rho–ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat Cell Biol. 2005;7:255–261. [DOI] [PubMed] [Google Scholar]
- [35].Unbekandt M, Lilla S, Zanivan S, et al. The CDC42 effector protein MRCKβ autophosphorylates on Threonine 1108. Small GTPases. 2019; DOI: 10.1080/21541248.2018.1564472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zhao Z, Manser E. Myotonic dystrophy kinase-related Cdc42-binding kinases (MRCK), the ROCK-like effectors of Cdc42 and Rac1. Small GTPases. 2015;6:81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Rottner K, Stradal TE. Actin dynamics and turnover in cell motility. Curr Opin Cell Biol. 2011;23:569–578. [DOI] [PubMed] [Google Scholar]
- [38].Kage F, Steffen A, Ellinger A, et al. FMNL2 and −3 regulate Golgi architecture and anterograde transport downstream of Cdc42. Sci Rep. 2017;7:9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Sanz-Moreno V, Gadea G, Ahn J, et al. Rac activation and inactivation control plasticity of tumor cell movement. Cell. 2008;135:510–523. [DOI] [PubMed] [Google Scholar]
- [40].Rottner K, Hall A, Small JV. Interplay between Rac and Rho in the control of substrate contact dynamics. Curr Biol. 1999;9:640–S1. [DOI] [PubMed] [Google Scholar]
- [41].Kelley LA, Mezulis S, Yates CM, et al. The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.