

ABSTRACT
Palbociclib, a selective CDK4/6 kinase inhibitor, is approved in combination with endocrine therapies for the treatment of advanced estrogen receptor positive (ER+) breast cancer. In pre-clinical cancer models, CDK4/6 inhibitors act primarily as cytostatic agents. In two commonly studied ER+ breast cancer cell lines (MCF7 and T47D), CDK4/6 inhibition drives G1-phase arrest and the acquisition of a senescent-like phenotype, both of which are reversible upon palbociclib withdrawal (incomplete senescence). Here we identify an ER+ breast cancer cell line, CAMA1, in which palbociclib treatment induces irreversible cell cycle arrest and senescence (complete senescence). In stark contrast to T47D and MCF7 cells, mTORC1 activity is not stably suppressed in CAMA1 cells during palbociclib treatment. Importantly, inhibition of mTORC1 signaling either by the mTORC1 inhibitor rapamycin or by knockdown of Raptor, a unique component of mTORC1, during palbociclib treatment of CAMA1 cells blocks the induction of complete senescence. These results indicate that sustained mTORC1 activity promotes complete senescence in ER+ breast cancer cells during CDK4/6 inhibitor-induced cell cycle arrest. Consistent with this mechanism, genetic depletion of TSC2, a negative regulator of mTORC1, in MCF7 cells resulted in sustained mTORC1 activity during palbociclib treatment and evoked a complete senescence response. These findings demonstrate that persistent mTORC1 signaling during palbociclib-induced G1 arrest is a potential liability for ER+ breast cancer cells, and suggest a strategy for novel drug combinations with palbociclib.
KEYWORDS: Palbociclib, cdk4/6, mTORC1, er+ Breast Cancer, senescence
Introduction
Estrogen receptor positive (ER+) breast cancer accounts for nearly 84% of all breast cancer cases and is typically managed with endocrine therapy [1,2]. However, drug resistance remains a significant clinical concern [1,3] and requires rational-targeted therapies in combination with anti-estrogen treatments. ER+ breast cancers frequently coopt mechanisms to promote the action of cyclin-dependent kinases (CDKs) and their cognate cyclin proteins, overriding cell cycle checkpoints, and increasing cell proliferation [4]. Accordingly, cell cycle modulation represents an important therapeutic strategy for ER+ breast cancers.
Palbociclib, a potent and highly selective inhibitor of CDK4/CDK6 [5], was the first CDK inhibitor to be approved for the treatment of ER+/HER2- breast cancer in combination with endocrine therapy [6]. Palbociclib arrests cells in the G1-phase of the cell cycle by suppressing the phosphorylation of the retinoblastoma protein (Rb) [7]. The resulting hypo-phosphorylated Rb inhibits the transcriptional activity of E2F, thereby blocking the transcription of critical genes required for entry into S-phase. In principle, all cancers that retain Rb expression and functionally depend on the CDK4/6 pathway for proliferation should be sensitive to CDK4/6 inhibitors. However, the most significant clinical impact of CDK4/6 inhibitors has been with ER+/HER2- breast cancers, due in part to the fact that ER-dependent oncogenic signaling relies heavily on the activation of the CDK/Rb/E2F signaling axis [6,7]. In ER+ breast cancer cell lines, palbociclib induces phenotypic alterations associated with senescence [8]. Cellular senescence is a phenotypic transition marked by irreversible exit from the cell cycle and characterized by an increase in cell size, elevated senescence-associated-β-galactosidase (SA-β-Gal) activity, and expression of a senescence-associated secretory phenotype (SASP) [9]. However, the senescent phenotype and G1 arrest observed in palbociclib-treated ER+ breast cancer cell lines are not permanent but instead are reversible upon palbociclib removal [8].
The mechanistic target of rapamycin (mTOR) pathway is frequently upregulated in ER+ breast cancer [10]. Consequently, there are several clinical trials underway interrogating combinations of endocrine therapy/CDK4/6 inhibition with inhibitors of the mTOR pathway [11]. mTOR is a serine/threonine protein kinase that serves as a master regulator of metabolism, integrating mitogenic signals and nutrients to control cellular proliferation and growth based on nutrient availability [12]. The activation of mTOR complex 1 (mTORC1) by amino acids and glucose at the lysosomal surface promotes protein and lipid synthesis, and suppresses the catabolic macroautophagy pathway [12]. Crosstalk has been described between the cell cycle and mTOR via CDK4/6. Drosophila CDK4 and mammalian CDK4/6 phosphorylate and inhibit the tuberous sclerosis complex 2 (TSC2) protein, a negative regulator of mTORC1, leading to increased mTORC1 activity [13,14]. In triple-negative breast cancer (TNBC) cells, CDK4 phosphorylates the tumor suppressor folliculin to promote lysosome-dependent protein degradation, which increases the levels of free amino acids, and, in turn, mTORC1 activation [15]. Collectively, the cellular responses provoked by mTORC1 activation are associated with entry into senescence [16–21]. However, the consequences of altered mTORC1 activity during the senescent-like state resulting from CDK4/6 inhibition in ER+ breast cancer remain poorly understood.
Although preclinical studies have shown that CDK4/6 inhibition by palbociclib or abemaciclib decreases the activity of mTORC1 in most cancer cell lines [14,22–24], we have identified an atypical ER+ breast cancer cell line, CAMA1, that cannot sustain inhibition of mTORC1 activity during palbociclib treatment and acquires an irreversible senescent phenotype that we term complete senescence. Importantly, stabilizing mTORC1 activity in palbociclib-arrested MCF7 cells converts their response from reversible (incomplete) to complete senescence. The causal relationship between persistent mTORC1 activation and complete senescence suggests that a drug which stimulates mTORC1 activity in combination with palbociclib may drive breast cancer cells toward complete senescence with potential to improve response to CDK4/6 inhibition.
Materials and methods
Cell culture
CAMA1, T47D, and MCF7 cell lines were obtained from ATCC and genetic identities were confirmed by STR profiling (ATCC). The cell lines were screened for mycoplasma. The growth media in which the cell lines were maintained included RPMI-1640 with L-glutamine (Gibco 11,875–085) supplemented with 10% heat-inactivated fetal bovine serum (FBS-Gibco) at 37°C in a humidified incubator with 5% CO2. TSC1 wild-type (+/+) and TSC1 deficient (-/-) mouse embryonic fibroblast cell lines (MEFs) (kindly provided by David Kwiatkowski, Harvard University, Boston) were maintained in RPMI-1640 with L-glutamine supplemented with 10% FBS [25]. CAMA1 cells expressing inducible lentiviral shRNA were maintained in RPMI supplemented with 10% tetracycline (tet)-free FBS (Takara) + 2 ug ml−1 puromycin (Gibco). Preparation of stable shRNA cell lines (RNA interference and viral infection) and reagents used are described in Supplementary Materials and Methods.
Palbociclib treatment and recovery from palbociclib-arrest
Cells were seeded in 6-well culture plates in triplicates (Falcon) and allowed to attach overnight. Optimal seeding densities were established for each cell line to reach 70–80% confluence at the experimental endpoint. The next day, media were replaced with fresh growth media containing either drug vehicle only (50 mM lactic acid pH 4.0) or 500 nM palbociclib, unless mentioned otherwise, for various timepoints. Drug-containing media were replenished twice a week. At the end of the palbociclib treatment, cells were suspended from each culture by trypsinization and the cell densities for each suspension were measured using either an Invitrogen Countess Automated Cell Counter (Invitrogen) or Vi-Cell XR Cell Viability Analyzer (Beckman Coulter). To examine the recovery from palbociclib-induced arrest, cells were washed with fresh media three times and maintained in growth media without palbociclib until one of the treated groups recovered from growth arrest to 80–90% confluency. At the end of the recovery, cells were suspended from each culture by trypsinization and the cell densities were measured as described above.
Palbociclib was synthesized at Pfizer, and was dissolved in 50 mM lactic acid pH 4.0. Rapamycin was purchased from Sigma-Aldrich and dissolved in DMSO.
RNA sequencing
Cells were seeded at 100,000 cells per well into 6-well culture plates. After 24 h, cells were treated with 500 nM palbociclib for various duration. At the indicated timepoints, cells were lysed and processed for RNA-Seq. Detailed information regarding sample preparation and RNA-Seq analysis can be found in Supplementary Materials and Methods.
Senescence-associated-β-galactosidase (SA-β-Gal) activity
Qualitative SA-β-Gal staining was performed using Cellular Senescence Assay Kit (Cell BioLabs, CBA-230) according to the manufacturer’s recommendations. For the detection of senescent cells by flow cytometry, CellEvent Senescence Green Flow Cytometry Assay Kit (Invitrogen C10840) was used per manufacturer’s instructions. Detailed information can be found in Supplementary Materials and Methods.
Colony-forming assay
To examine the recovery from palbociclib-induced arrest when treated in combination with either rapamycin or with knockdown of Raptor or Rictor, the cells were washed once with 1x PBS, stained with 0.4% sulforhodamine B (SIGMA S1402) diluted in 1% acetic acid for 10 min at room temperature, rinsed three times with 1% acetic acid and air-dried. Images were scanned with Epson Perfection V600 Photo Scanner.
CRISPR-mediated genetic depletion of TSC2
Chemically modified synthetic single guide RNA (sgRNA) against TSC2 (sg1, sg2, and sg3) and non-targeting control sgRNA (negative sg) were obtained from Synthego and were nucleofected to T47D and MCF7 according to the manufacture’s protocol. For detailed information, see Supplementary Materials and Methods. The guide RNA sequences were:
TSC2_sg1: 5-UGAACUGGUGGAGAGAUGUG- 3
TSC2_sg2: 5-CAACAUGUGCCACCUCAUGG- 3
TSC2_sg3: 5-CGUCCAUGACCUGUUGACCA- 3
Proteomics and phospho-proteomics profiling
T47D and CAMA1 cells were treated in triplicate with 500 nM palbociclib, washed twice with ice-cold PBS containing protease and phosphatase inhibitors, and lysed in 6 M guanidinium chloride lysis buffer. The resulting proteins were digested with Lys-C/trypsin. The tryptic peptides were labeled with TMT-10 plex and analyzed with nano-LC coupled with Q Exactive™ Mass Spectrometer. For phospho-proteomic profiling, phosphopeptides were enriched with TiO2 prior to LC-MS analysis. Detailed information regarding sample preparation and analysis can be found in Supplementary Materials and Methods.
Western blotting
Detailed information regarding western blotting can be found in Supplementary Materials and Methods. Primary antibodies used were:
| Protein | Catalog | Dilution |
| Vinculin | Sigma V9131 | 1:2000 |
| p-Rb S780 | Cell Signaling 8180 | 1:1000 |
| Rb | Cell Signaling 9309 | 1:1000 |
| p-4EBP1 T37/46 | Cell Signaling 9459 | 1:1000 |
| 4EBP1 | Cell Signaling 9452 | 1:1000 |
| p-S6RP S235/36 | Cell Signaling 4856 | 1:1000 |
| S6RP | Cell Signaling 2217 | 1:1000 |
| p-S6K1 T389 | Cell Signaling 9205 | 1:1000 |
| S6K1 | Cell Signaling 2708 | 1:1000 |
| p-TSC2 S664 | Abcam ab133465 | 1:1000 |
| TSC2 | Cell Signaling 4308 | 1:1000 |
| p-p44/42 MAPK (ERK1/2) (T202/Y204) | Cell Signaling 4370 | 1:1000 |
| p44/42 MAPK (ERK1/2) | Cell Signaling 9107 | 1:1000 |
| p-AKT S473 | Cell Signaling 9271 | 1:1000 |
| p-AKT T308 | Cell Signaling 13,038 | 1:1000 |
| AKT | Cell Signaling 9272 | 1:1000 |
| Cyclin E1 | Abcam ab133266 | 1:1000 |
| Cyclin E2 | Abcam ab40890 | 1:1000 |
| TSC1 | Cell Signaling 6935 | 1:1000 |
Quantitative reverse transcription-PCR (qRT-PCR)
For details, see Supplementary Materials and Methods. Taqman Assay solutions used were:
| Primer name | Assay ID |
| IFIT3 | Hs01922752_s1 |
| CCL5 | Hs00982282_m1 |
| CXL10 | Hs00171042_m1 |
| CCL2 | Hs00234140_m1 |
| ICAM1 | Hs00164932_m1 |
| TNF | Hs00174128_m1 |
| IL6 | Hs00174131_m1 |
| RPTOR | Hs00375332_m1 |
| RICTOR | Hs00380903_m1 |
| ACTB | Hs01060665_g1 |
Results
Palbociclib induces complete senescence in CAMA1, but not in MCF7 and T47D cells
Proliferation of ER+ breast cancer cells has been shown to be much more sensitive to CDK4/6 inhibitors than normal breast epithelial cell lines [8,26]. Treatment of ER+ breast cancer cells with palbociclib results in G1 phase cell cycle arrest [8]. To more fully understand the differential effects of the CDK4/6 inhibitor palbociclib on cell cycle arrest of ER+ breast cancer cells, T47D, MCF7 and CAMA1 were treated with 500 nM palbociclib for 14 days, and molecular and morphological changes were monitored. We tested 500 nM palbociclib, since this drug concentration fully arrested proliferation in all 3 cell lines and has been shown to have on-target inhibitory effects on CDK4/6 in ER+ breast cancer cells [8]. Indeed, we did not observe an increase in cell number for palbociclib treated cell cultures over the entire 14 days of treatment (Figure 1A). Palbociclib treatment arrested CAMA1 cells in G1 phase similar to previous observations with T47D and MCF7 cells (Supplementary Figure 1A) [8]. CAMA1 cells also exhibited similar sensitivity to inhibition of cell proliferation by palbociclib as T47D cells (Supplementary Figure 1B). The proliferative arrest induced by 500 nM palbociclib in CAMA1 cells was reversed by knocking down retinoblastoma mRNA (RB1), consistent with CDK4/6 inhibition as the underlying mechanism and not the result of off-target drug activity (Supplementary Figure 1C). Palbociclib treatment also caused an increase in cell size and SA-β-Gal staining, which are features of cellular senescence, in all 3 cell lines (Figure 1B). Another key characteristic of cellular senescence is irreversible cell cycle arrest. To determine whether palbociclib treatment resulted in irreversible cell cycle arrest and if onset of irreversible arrest is dependent on the duration of treatment, we examined the ability of these cell lines to recover proliferation after removal of palbociclib following either a short (6 days) or prolonged (14 days) duration of treatment. At both time points, T47D and MCF7 cells reverted to their normal morphology, displayed reduced SA-β-Gal staining, and began to divide after palbociclib removal (Figure 1A). Thus, palbociclib-mediated cell cycle arrest and senescence phenotype is reversible or incomplete in these ER+ breast cancer cell lines. This palbociclib-mediated cell cycle arrest remained reversible after 28 days of palbociclib treatment (data not shown). Like T47D and MCF7 cells, CAMA1 cells resumed proliferation after release from 6 days of palbociclib treatment (Figure 1A). However, when palbociclib was removed from CAMA1 cells after 14 days of treatment, the cells remained stably enlarged, SA-β-Gal positive, and irreversibly arrested (Figure 1A). Moreover, this irreversible arrest eventually transitioned with time to cell death as indicated by a reduction in cell number (Figure 1A, note last two bars). Thus, prolonged palbociclib treatment drove CAMA1 cells into an irreversible, or complete, senescence. We also tested whether the complete senescence observed in CAMA1 cells occurred prior to the standard 14 days of palbociclib treatment. Indeed, we observed that palbociclib-induced growth arrest, flattened morphology, and increased SA-β-Gal staining became largely irreversible with 10 days of treatment (Supplementary Figure 1D). Consistent with the induction of a fully senescence phenotype, palbociclib treatment caused a robust upregulation of SASP genes in CAMA1 cells (Figure 1C; Supplementary Table 1). The SASP response was much weaker in T47D and MCF7 cells compared to CAMA1 (Figure 1C; Supplementary Table 1).
Figure 1.
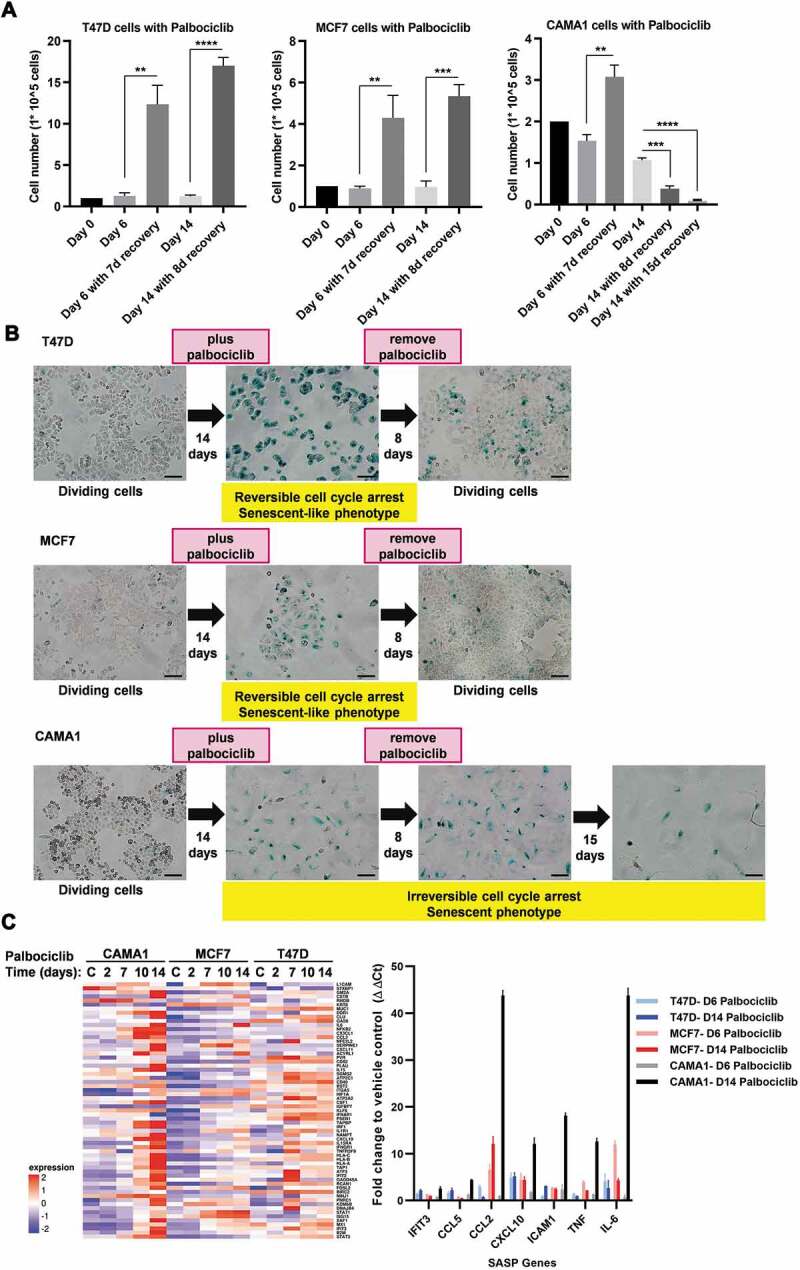
Palbociclib induces complete senescence in CAMA1, but not in MCF7 and T47D cells. (A) T47D (left), MCF7 (middle) or CAMA1 (right) were treated with palbociclib (500 nM) for 6 or 14 days and allowed to recover for indicated days (d) after palbociclib withdrawal. Cell number was measured at the end of the treatment and after recovery. Values are shown as mean ± standard deviation (s.d.) from three biological replicates. **P< 0.01; ***P< 0.001; ****P< 0.0001 (two-tailed unpaired t-test). (B) Cells were treated as in (A) for 14 days and allowed to recover for the indicated duration after palbociclib withdrawal. Cells were subjected to SA-β-Gal activity and representative images were captured with a 10× objective. Scale bars, 100 µm. The images shown are representative from at least two independent experiments. (C) (Left) Heatmap of relative transcript levels of SASP genes assessed by RNA-seq in the indicated cell lines treated with palbociclib (500 nM) at indicated times. “C” refers to drug vehicle -treated control cells. (Right) Mean ± s.d. of SASP gene expression measured by qRT-PCR in the indicated cell lines treated with palbociclib (500 nM) for 6 and 14 days. Values are normalized to Actb and are shown as fold change to vehicle control treatment
Phospho-proteomics profiling reveals differential regulation of mTORC1 signaling in T47D relative to CAMA1 cells in response to palbociclib treatment
In order to gain insights into the mechanism underlying the induction of complete versus incomplete senescence by palbociclib against ER+ breast cancer cells, we performed phospho-proteomic and total proteomic profiling of palbociclib-treated CAMA1 and T47D cells. Phospho-proteomic profiling revealed notably differential effects on the mTOR signaling pathway, with phosphorylation of mTORC1-related substrates sustained during palbociclib treatment in CAMA1, but reduced during palbociclib treatment of T47D cells (P-Cluster 3) (Figure 2A). Moreover, palbociclib exposure triggered an increase in phosphosites on proteins associated with activation of the MAPK signaling pathway in CAMA1, but not in T47D cells (P-cluster 2). Conversely, we also observed an increase of phosphosites on proteins involved in branched chain amino acids (valine, leucine and isoleucine) catabolism in palbociclib-treated T47D, but not in CAMA1 cells (P-cluster 1) (Figure 2A). Branched chain amino acids, particularly leucine, as well as MAPK signaling pathways are known to activate mTORC1 signaling [27,28]. In agreement with these results from phospho-proteomics, our total proteomic profiling indicated upregulation of proteins involved in lysosome-mediated catabolism by palbociclib in T47D and MCF7, but not in CAMA1 cells (Supplementary Figure 2A-D). mTORC1 signaling is known to suppress lysosomal gene expression [29], and the absence of induction of lysosomal proteins by palbociclib treatment of CAMA1 cells is consistent with the retention of high mTORC1 activity in the drug-treated cells. Overall, these changes suggest differential effects of palbociclib on mTORC1 signaling activity in CAMA1 compared with T47D and MCF7 cells.
Figure 2.
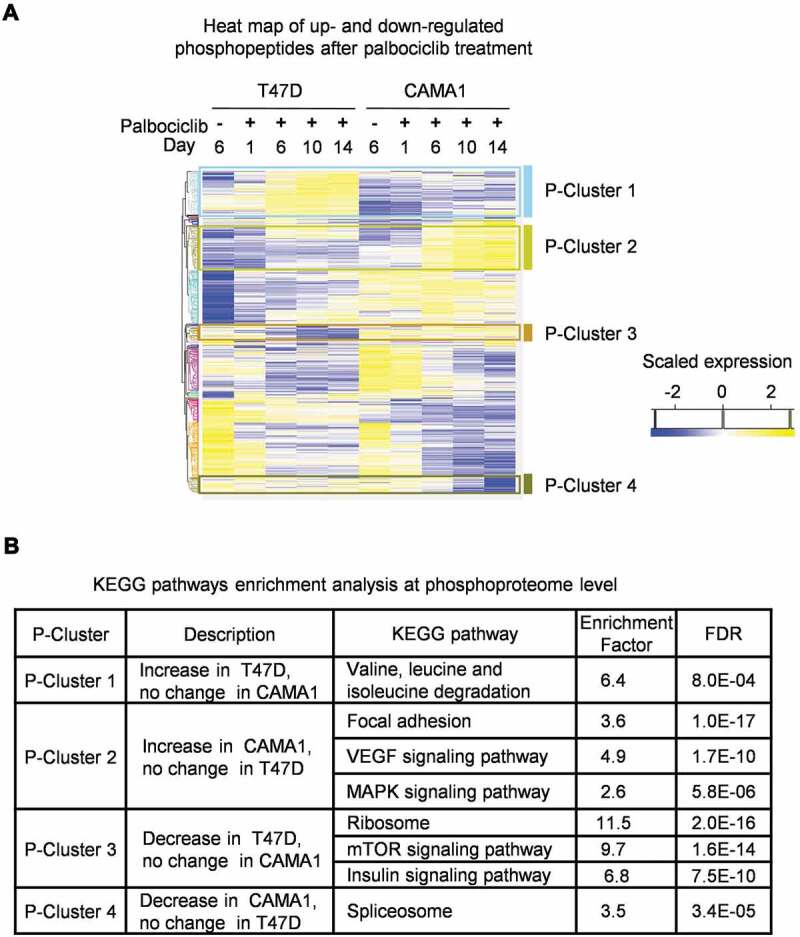
Phosphoproteomics profiling reveals differential regulation of mTORC1 signaling in T47D versus CAMA1 cells in response to palbociclib treatment. (A) Heatmap showing log2 transformed and scaled abundance of phosphopeptides with significant changes in T47D and CAMA1 cells treated with vehicle only (-) for 6 days (to ensure log phase growth) or palbociclib (500 nM) for the indicated time points from three independent samples per condition. Hierarchical clustering of phosphopeptides with significant changes with treatment. (B) Pathway enrichment analysis of phosphoproteome clusters using KEGG database with Fisher Exact Test (Benjamini-Hochberg FDR ≤ 0.02) is shown
Palbociclib treatment results in decreased mTORC1 signaling and TSC2 phosphorylation in T47D and MCF7, but not in CAMA1 cells
To confirm the above findings, we selected specific proteins and phosphoproteins for western blot analysis to monitor markers of CDK4/6 and mTORC1 activity in MCF7, T47D and CAMA1 cell lines treated with palbociclib for 6, 10 or 14 days. As expected, palbociclib treatment inhibited Rb phosphorylation, a cellular biomarker of palbociclib activity, in all 3 cell lines throughout the entire 14 days of treatment (Figure 3). Consistent with our phospho-proteomics data, palbociclib treatment of T47D and MCF7 cells resulted in decreased phosphorylation of downstream components of the mTORC1 pathway (4EBP1, S6K1 and S6RP) at all timepoints of drug exposure (Figure 3). These results confirm that palbociclib treatment reduces mTORC1 activity in T47D and MCF7 breast cancer cells. After 6 days of palbociclib exposure, CAMA1 cells displayed clear reductions in the phosphorylation of the two direct mTORC1 substrates, S6K1 and 4EBP1, whereas phosphorylation of the S6K1 substrate, S6RP was unchanged at this early timepoint (Figure 3; Supplementary Figure 3). In contrast to T47D and MCF7, this early decrease in the phosphorylation of mTORC1 direct substrates in CAMA1 cells was fully reversed after 10 and 14 days of palbociclib treatment (Figure 3; Supplementary Figure 3). These results suggest that the failure of CAMA1 cells to maintain suppression of mTORC1 activity during prolonged exposure to palbociclib might be linked to complete senescence phenotype observed in these cells.
Figure 3.

Palbociclib treatment inhibits TSC2 phosphorylation and mTORC1 activity in T47D and MCF7, but not in CAMA1 cells. Western blot analyses of mTORC1, TSC2, ERK and AKT signaling with the indicated antibodies. T47D, MCF7 and CAMA1 cells were treated with vehicle only (-) for 6 days or palbociclib (500 nM) for the indicated time points. Data shown are a representative of 2 or more independent experiments
We next investigated the effect of palbociclib on signaling pathways that regulate mTORC1 activity in the breast cancer cell lines. The TSC1 and TSC2 heterodimeric protein complex (TSC1/2) negatively regulates mTORC1 activity by acting as a GTPase-activating protein (GAP) for an obligate mTORC1 activator, GTP-bound Rheb. TSC1/2 stimulates the conversion of active, GTP-bound Rheb to inactive Rheb-GDP, which, in turn, suppresses mTORC1 activity [30,31]. The TSC2 subunit is targeted for phosphorylation by multiple protein kinases, including extracellular signal-regulated kinases, ERK1/2, that inhibit TSC2 activity via phosphorylation at serine 664 (S664) [32,33]. Although the phosphorylation of the S664 residue was inhibited after 6 days of palbociclib treatment in all 3 breast cancer cell lines, phosphorylation of this site on TSC2 increased uniquely in CAMA1 cells with longer palbociclib treatment (Figure 3). This increase in phospho-TSC2 corresponded with the increase in mTORC1 activity during prolonged palbociclib treatment of CAMA1 cells. Consistent with S664 being an ERK phosphorylation site on TSC2, we observed increased ERK1/2 phosphorylation, which is indicative of ERK reactivation [34] in palbociclib-treated CAMA1 cells compared to T47D and MCF7 cells. Moreover, this increase in phospho-ERK1/2 in CAMA1 occurred at the same timepoints of palbociclib treatment as the increase in phospho-TSC2 (i.e. ≥10 days) (Figure 3). In addition to increased ERK MAPK pathway activity, we also observed that palbociclib treatment increased AKT activation (phosphorylation on S473 and T308) uniquely in CAMA1 cells versus T47D and MCF7 cells (Figure 3). AKT is also known to regulate mTORC1 activity by inhibiting TSC2 function [35]. These results indicate that prolonged palbociclib treatment activates both the ERK and AKT signaling pathways uniquely in CAMA1 cells which may contribute to increased mTORC1 activity.
mTORC1 but not mTORC2 inhibition blocks complete senescence in CAMA1 cells by palbociclib
Active mTORC1 signaling during cell cycle arrest has been shown to induce irreversible proliferation arrest in melanoma, retinal pigment epithelium, and fibrosarcoma cell lines [19,20,36]. Our data suggest that differential responses of transiently versus persistently reduced mTORC1 activity during growth arrest of ER+ breast cancer cells underlies complete versus incomplete senescence provoked by palbociclib. Since palbociclib induces complete senescence in CAMA1 cells that have been arrested by drug treatment for 10 or more days, we hypothesized that concordant inhibition of mTORC1 activity in CAMA1 cells arrested with palbociclib for less than 10 days would block the cells from undergoing complete senescence. We treated CAMA1 cells with palbociclib for 7 days, then treated the cells with palbociclib with or without combination with the mTORC1 inhibitor rapamycin for an additional 7 days (Figure 4A). After the 14 total days of treatment, the drugs were removed, and the cells were scored for recovery from palbociclib-induced growth arrest. Consistent with the results presented in Figure 1B, CAMA1 cells that were treated with palbociclib alone for 14 days maintained an enlarged and flattened morphology and were irreversibly arrested (Figure 4B, left panel). In sharp contrast, these senescence-associated responses were abrogated by co-treatment of CAMA1 with rapamycin (Figure 4B, right panel). Thus, inhibition of mTORC1 signaling by rapamycin, as confirmed by western blot (Figure 4C), blocked palbociclib-induced complete senescence of CAMA1 cells.
Figure 4.
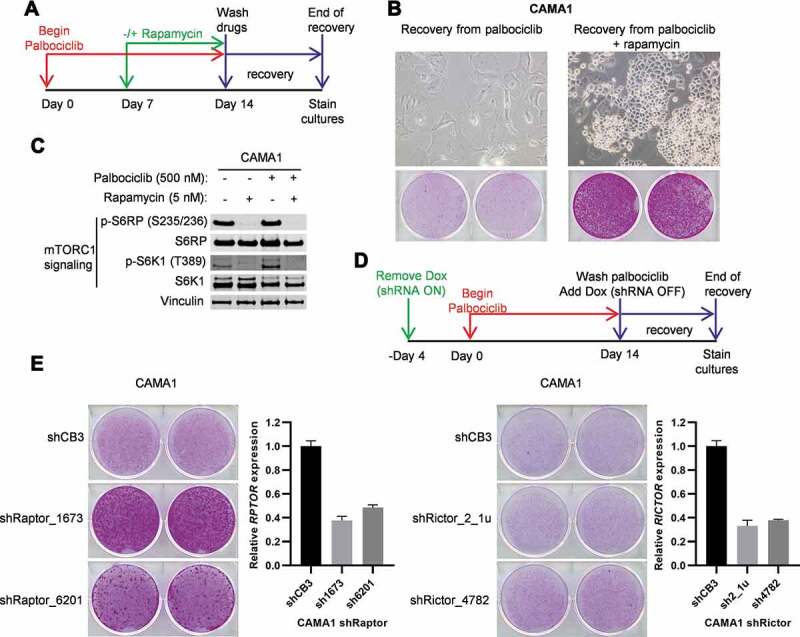
Inhibition of mTORC1 but not mTORC2 blocks complete senescence in CAMA1 cells by palbociclib. (A) Schematic of the treatment and recovery assay from palbociclib (500 nM) or palbociclib (500 nM) + rapamycin (5 nM) combination. (B) Colony forming assay of cells following treatment and recovery as indicated in (A). Representative images of cells captured with 10× objective without (upper) or with SRB staining (bottom) after the recovery from palbociclib-induced arrest (left) or palbociclib + rapamycin-induced arrest (right) are shown. (C) The cells were treated as in (A) with the indicated drugs and immunoblotted for mTORC1 activity. (D) Schematic of the recovery assays from palbociclib arrest for cells with Raptor or Rictor knockdown by Tet-OFF regulated shRNAs. “Dox” refers to doxycycline. (E) Colony forming assay for cells with Raptor or Rictor knockdown and treated with palbociclib and allowed to recover as indicated in (D). Data shown are a representative of 2 independent experiments. Bar graphs from qPCR analyses of RPTOR (left) and RICTOR (right) mRNA levels in CAMA1 cells after 7 days of shRNAs induction. Values are normalized to Actb and presented relative to shCB3 (non-targeting control)
Next, we performed genetic experiments to determine whether the observed effects of rapamycin were specifically due to inhibition of the mTORC1, as opposed to the alternative mTOR complex 2 (mTORC2). We selectively inhibited mTORC1 or mTORC2 activity with short hairpin RNAs controlled by a tetracycline-repressible promoter (Tet-OFF-shRNAs). Doxycycline, a derivative of tetracycline, was removed to induce shRNAs that specifically depleted Raptor (RPTOR) or Rictor (RICTOR), which are essential components of the mTORC1 and mTORC2 complexes, respectively [37–39]. We induced Raptor or Rictor knockdown for 4 days and then treated the CAMA1 cells with palbociclib for 14 days (Figure 4D). Palbociclib was then removed and expression of the shRNAs was silenced by re-addition of doxycycline to allow re-expression of Raptor or Rictor. Re-expression of Raptor or Rictor was important during the measurement of recovery because mTOR is essential for cell proliferation and Raptor or Rictor knockdown would interfere with the recovery from palbociclib-induced growth arrest. Whereas cells that expressed the non-targeting shRNA (shCB3) during palbociclib treatment underwent complete senescence, cells in which Raptor was knocked down during palbociclib treatment resumed proliferation after drug removal (Figure 4E, left panel). Recovery from the anti-proliferative effect of palbociclib correlated with the expression level of Raptor, which further suggested a tight correlation between mTORC1 activity and the reversibility of the drug-induced G1 arrest. In contrast to Raptor knockdown, Rictor knockdown in CAMA1 cells during palbociclib treatment failed to enable resumption of proliferation after palbociclib washout (Figure 4E, right panel). These results indicate that sustained mTORC1, but not mTORC2-dependent signaling, may underly complete senescence resulting from prolonged palbociclib-mediated cell cycle arrest of CAMA1 cells.
Palbociclib triggers complete senescence in TSC2-null MCF7 cells
To further investigate the role of sustained mTORC1 activity in the complete senescence response to prolonged palbociclib treatment, we investigated whether deregulated mTORC1 activity due to loss of TSC2 function would enable prolonged palbociclib treatment to induce complete senescence in a breast cancer cell line that normally undergoes incomplete senescence. We utilized CRISPR-Cas9 and tested 3 different single guide RNAs (sg1, sg2, and sg3) to cause deletions in the TSC2 gene in MCF7 cells. As a negative control, we transfected MCF7 cells with a non-targeting single guide RNA (negative sg). All 3 single guide RNAs (sgRNAs) targeting TSC2 significantly reduced TSC2 protein levels in unselected MCF7 cell populations, with the sg2- and sg3-treated populations demonstrating the lowest levels of TSC2 protein (Figure 5A). We refer to these cells as the MCF7 TSC2 KO cell lines. Palbociclib treatment inhibited phosphorylation of Rb and decreased total Rb protein levels equivalently in the MCF7 TSC2 KO cell lines compared to the MCF7 negative sg cell line (Figure 5A). This result indicated that loss of TSC2 did not alter the sensitivity of the MCF7 cells to palbociclib. Consistent with our previous results (Figure 3), we observed inhibition of mTORC1-dependent phosphorylation of S6K1, S6RP, and 4EBP1 in palbociclib-treated MCF7 negative sg cells (Figure 5A). However, in the TSC2 KO cell lines, palbociclib did not inhibit mTORC1-dependent phosphorylation of these proteins (Figure 5A). These results demonstrate that knockout of TSC2 in MCF7 cells enabled sustained mTORC1 activity during palbociclib treatment.
Figure 5.
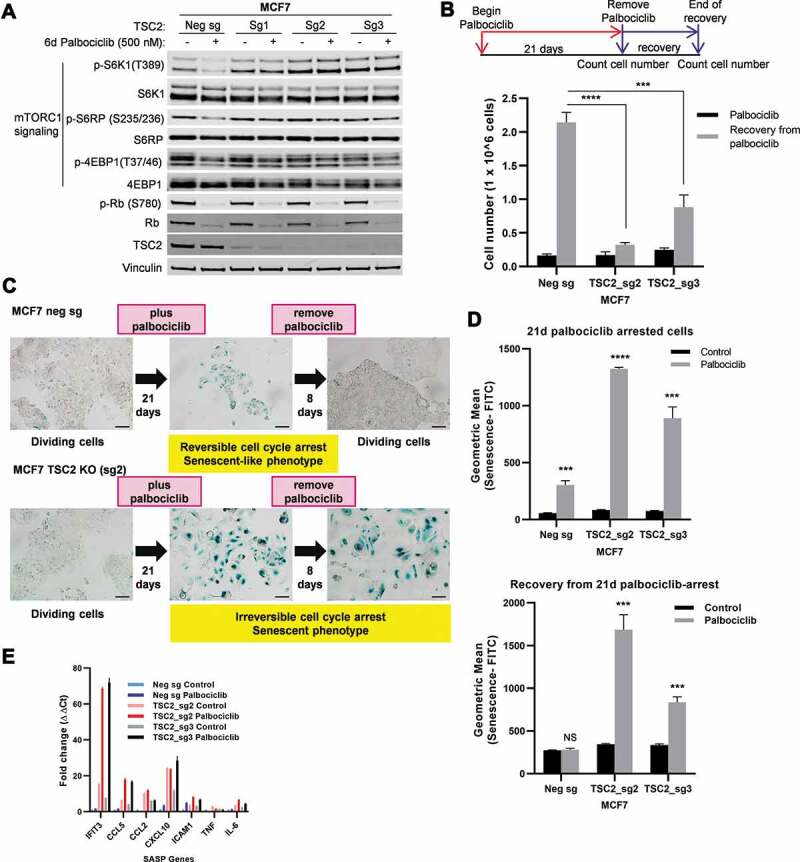
Knockout of TSC2 in MCF7 causes palbociclib-mediated complete senescence. (a) Western blot analyses of the indicated proteins in MCF7 negative control (Neg sg) and MCF7 TSC2 KO (Sg1, Sg2 and Sg3) cell lines treated with vehicle only (-) or palbociclib (500 nM) for 6 days. (b) Schematic diagram shows the treatment and recovery assay from palbociclib (500 nM)-induced arrest. Cells treated with palbociclib for 21 days (black bars) and allowed to recover for 8 days after palbociclib withdrawal (gray bars) were counted. (c) Qualitative SA-β-Gal assays and representative images of SA-β-Gal stained samples captured with 10× objective. Scale bars, 100 µm. (d) Quantitative detection of senescent cells by flow cytometry where values are mean ± s.d. of three biological replicates. (e) Mean ± s.d. of SASP gene expression by qRT-PCR analysis in the indicated cell lines treated with vehicle (control) or palbociclib (500 nM) for 21 days. Values are normalized to Actb and are shown as fold change to negative sg vehicle treatment. NS, not significant P> 0.05; ***P< 0.001; ****P< 0.0001 (two-tailed unpaired t-test) relative to the corresponding vehicle-treated cells (control) unless indicated
We tested whether this sustained mTORC1 activity in MCF7 during palbociclib treatment converts the MCF7 response from incomplete to complete senescence. We treated MCF7 TSC2 KO (sg2 and sg3) and MCF7 negative sg cells with palbociclib for either 14 or 21 days, then removed the drug, and allowed the cells to recover from palbociclib arrest (Figure 5B and Supplementary Figure 4, schematic diagram). Palbociclib treatment resulted in similar proliferative arrest in MCF7 TSC2 KO and MCF7 negative sg cells (Figure 5B black bars). Interestingly, palbociclib treatment of the TSC2 KO cells resulted in a flatter morphology and stronger SA-β-Gal activity than in the MCF7 negative sg cells (Figure 5C,Figure 5D upper panel). Importantly, while MCF7 negative sg cells recovered from either 14 or 21 days of palbociclib-mediated cell cycle arrest and resumed proliferation, MCF7 TSC2 KO cells remained irreversibly arrested (Figure 5B; Supplementary Figure 4). The inhibitory effect on the recovery in sg2 and sg3 populations correlated with the level of TSC2 protein depletion. Inhibition of recovery of the TSC2 KO cells from palbociclib arrest was strongest after 21 days of palbociclib treatment. Moreover, while the increased SA-β-Gal staining in MCF7 negative sg cells was reversed after palbociclib removal, MCF7 TSC2 KO cells remained SA-β-Gal-positive after drug removal following 21 days of palbocilcib treatment (Figure 5C). We also observed a partial induction of SASP genes in TSC2 KO cells in response to palbociclib (Figure 5E). Together, these results demonstrate that sustained mTORC1 activity during prolonged palbociclib arrest due to genetic inactivation of TSC2 drives MCF7 cells into complete senescence.
We also examined whether TSC2 depletion converts incomplete senescence into complete senescence in T47D cells treated with palbociclib. Similar to MCF7, while palbociclib inhibited mTORC1-dependent phosphorylation of S6K1, S6RP, and 4EBP1 in the negative sg transfected T47D, mTORC1 activity during palbociclib treatment was sustained in T47D TSC2 KO (sg1, sg2, and sg3) cells (Supplementary Figure 5A). However, in contrast to MCF7 TSC2 KO cells, 500 nM palbociclib treatment failed to block proliferation of the T47D TSC2 KO cells (Supplementary Figure 5B). Western blot analysis of phospho-Rb revealed less phospho-Rb inhibition by palbociclib treatment in the TSC2 KO cells versus negative sg cells (Supplementary Figure 5A). These results indicated that loss of TSC2 in T47D cells reduced their intrinsic sensitivity to palbociclib. We treated the T47D TSC2 KO cells with higher concentrations of palbociclib (1 and 3 µM), with similar results (Supplementary Figure 5C,D). Because T47D TSC2 KO cells failed to arrest in response to palbociclib treatment, we were unable to test in these cells whether sustained mTORC1 activity resulting from TSC2 depletion converts their response from reversible to irreversible cell cycle arrest. The results suggest that deregulated mTORC1 activity feeds positively into the pRB phosphorylation pathway in T47D but not MCF7 or CAMA1 cells.
To test this hypothesis, we focused on the cyclin E-CDK2 complex, which functions as an upstream protein kinase that contributes to multi-site Rb phosphorylation [40]. Overexpression of either cyclin E1 or cyclin E2 has been associated with palbociclib resistance presumably through CDK2 activation and multi-site phosphorylation of Rb [41,42]. To develop insights into the mechanism underlying T47D resistance to palbociclib, we investigated whether palbociclib treatment of the T47D TSC2 KO cells induced either Cyclin E1 or Cyclin E2 protein. Palbociclib treatment (1 and 3 µM) reduced the level of cyclin E2 in T47D negative sg as well as in T47D TSC2 KO cell lines (Supplementary Figure 5C). Importantly, while there was a modest increase in cyclin E1 in palbociclib-treated T47D negative sg cells, there was a substantial increase in level of cyclin E1 in T47D TSC2 KO cell lines. These results suggest that the upregulation of cyclin E1 in T47D TSC2 KO cells may contribute to their reduced sensitivity to palbociclib, which in turn, make it unable to test their recovery from palbociclib-induced cell cycle arrest.
Palbociclib treatment induces cellular senescence in TSC1 –/– MEFs
To further examine the impact of TSC1/2-dependent mTORC1 regulation, we asked whether palbociclib treatment induces cellular senescence in other cell models. TSC1 and TSC2 function as a protein complex, in which TSC1 stabilizes TSC2 by inhibiting the ubiquitin/proteasomal-degradation of TSC2 [43]. Various reports have shown that TSC1 –/– mouse embryonic fibroblasts (MEFs) display constitutive activation of mTORC1 [25,44]. We treated TSC1+/+ and TSC1 –/– MEFs with different concentrations of palbociclib for 7 days. Consistent with the results obtained with TSC2 KO MCF7 cells, palbociclib effectively inhibited phosphorylation of Rb in both TSC1 –/– and TSC1+/+ MEFs (Figure 6A). As expected, TSC1 –/– MEFs showed high basal levels of S6K1, S6RP, and 4EBP1 phosphorylation compared to TSC1+/+ cells, indicating higher basal activity of mTORC1 in TSC1 –/– MEFs (Figure 6A). At concentrations of palbociclib (3 and 4 µM) that resulted in potent suppression of phospho-Rb, we saw concomitant reductions of mTORC1-dependent phosphorylation of S6K1 and S6RP in TSC1+/+ MEFs, but not in TSC1 –/– MEFs (Figure 6A). Importantly, at these concentrations of palbociclib, TSC1 –/– cells exhibited features of senescent cells including flattening of cells, increased cell size and enhanced SA-β-Gal staining, whereas TSC1+/+ cells did not (Figure 6B). These results indicate that sustained mTORC1 activity in palbociclib-arrested MEFs due to loss of a negative regulator of mTORC1 also induces a stress response and pushes cells toward senescence when treated with CDK4/6 inhibitor, palbociclib.
Figure 6.

Palbociclib treatment induces senescence in TSC1 –/– MEFs. TSC1+/+ and TSC1 –/– MEFs were treated with vehicle only (-) or indicated palbociclib concentration for 7 days and subjected to western blotting analyses of the indicated proteins (A) or SA-β-Gal activity (B). Representative images of SA-β-Gal stained samples captured with 10× objective are shown. Scale bars, 100 µm
Discussion
In this study, we investigate the molecular mechanism underlying differential response to palbociclib-mediated CDK4/6 inhibition. Previous reports and our study show that palbociclib treatment generally induces a reversible, senescence-like phenotype in ER+ breast cancer cell lines such as MCF7 and T47D. However, we discovered that in a specific cellular context, represented by the CAMA1 cell model, a profound and irreversible senescence phenotype occurs with prolonged palbociclib treatment that ultimately leads to cell death.
Our examination into molecular mechanisms that underly these different responses to palbociclib uncovered a striking requirement for breast cancer cells to downregulate mTORC1 signaling during palbociclib treatment to escape complete senescence and maintain a reversible G1 cell cycle arrest. We showed that palbociclib treatment of the ER+ breast cancer cell lines, T47D and MCF7, results in a persistent reduction of mTORC1 activity throughout the duration of treatment, but this is not the case in CAMA1. We also found that palbociclib treatment stably reduced TSC2 phosphorylation at the ERK1/2 site, modification of which suppresses TSC2 function, in T47D and MCF7 cells, but not in CAMA1 cells. In addition, prolonged palbociclib exposure led to increased ERK1/2 activation as well as AKT activation only in CAMA1 cells. Although the exact contributions of ERK1/2 and AKT activation to the sustained mTORC1 activity in CAMA1 cells remain to be defined, these cells, unlike MCF7 and T47D cells, appear to inappropriately reinvigorate growth-stimulatory pathways leading to mTORC1 activation in the setting of prolonged CDK4/6 inhibition. We speculate that the clash between the cell proliferative arrest provoked by palbociclib and the persistent stimulation of anabolic metabolism induced by mTORC1 activation engenders this senescence-inducing stress stimulus. The suppression of autophagy by mTORC1 may also contribute to the onset of senescence, in that palbociclib appears to induce reactive oxygen species [8] resulting in proteotoxic and organellar stress that would normally be alleviated by autophagy. Therefore, it is possible that sustained mTORC1 activity during palbociclib-arrest antagonizes catabolic processes resulting in accumulation of ROS, stress and induction of complete senescence.
Consistent with differences in signaling pathways that regulate TSC2/mTORC1 activity as a key determinant of ER+ breast cancer response to palbociclib, genetic ablation of TSC2 in MCF7 cells caused sustained mTORC1 activity during the G1 arrest and resulted in an irreversible arrest with similar morphological changes as was seen in CAMA1 cells. Conversely, when mTORC1 activity was inhibited in CAMA1 cells during palbociclib arrest, either by pharmacologic (rapamycin) or genetic (Raptor knockdown) approaches, these cells no longer exhibited a complete senescence response. Taken together, our data support a feedback loop between CDK4/6 activity and upstream mitogenic signaling pathways that converge on the TSC2-mTORC1 axis. Alternatively, CDK4/6 may act directly on TSC2 as suggested by a recent report that showed CDK4/6 activates mTORC1 via phosphorylation and inhibition of TSC2 [14].
CDK4/6 inhibitors (palbociclib or abemaciclib) have been shown to reduce mTORC1 activity in multiple human cancer cell lines, including melanoma, glioblastoma, and breast cancer cell lines [14,22–24]. However, these reports did not demonstrate whether CDK4/6 inhibitor treatment of these cancer models resulted in irreversible proliferation arrest. Our studies support that while inhibition of Cyclin D – CDK4/6 may commonly suppress mTORC1 activity in several cancer subtypes, mTORC1 suppression is not a universal response to CDK4/6 inhibitors, and suggest that certain cancer cells might display a persistent mTORC1 activation response, and a consequent complete senescence response to these drugs. Our observation that palbociclib alone can induce complete senescence in breast cancer cells may relate to reports that elevated mTORC1 activity blocks recovery from cell cycle arrest in melanoma, retinal pigment epithelium, and fibrosarcoma cell lines [19,20,36]. Interestingly, other groups reported that combining CDK4/6 inhibition with MEK inhibition not only triggers complete senescence in KRAS-mutant lung cancer cells, but also promotes innate immune responses [45]. The failure to suppress mTORC1 activity during CDK4/6 inhibition may not only induce complete senescence but also promote innate immune responses.
PI-3-kinase and its downstream target, mTOR, are approved as anti-cancer agents; however, the rapid onset of clinical resistance, in addition to their tolerability profiles, have limited the clinical utility of these agents [46]. The identification of efficacious, well-tolerated combinations of PI-3-kinase/mTOR inhibitors with other drugs is an area of increasing interest. Recent studies demonstrated that mTOR inhibitors can be combined productively with the CDK4/6 inhibitor, palbociclib, in certain tumors [24,47,48]. One of these reports indicated that low concentrations of mTOR inhibitors combined with CDK4/6 inhibitors trigger a more profound G1 phase arrest in breast cancer cells than that provoked by either agent alone. The proposed mechanism involves more severe reductions in cyclin D levels and E2F activity by the drug combination [48]. Inhibition of PI-3-kinase also shows synergy with CDK4/6 inhibitors, particularly in PIK3CA mutant breast cancer [49]. However, the long-term cellular changes and reversibility of the associated phenotype resulting from this drug combination warrants further investigation. In one of the indicated studies, the combination of palbociclib with the mTOR inhibitor, vistusertib (AZD2014), led to enhanced inhibition of MCF7 cell proliferation, but antagonized the emergence of senescent phenotypes, including SA-β-Gal activity and large-flattened morphology [48]. Moreover, the effect of the palbociclib and vistusertib (AZD2014) combination against MCF7 cell proliferation was reversible since removal of the compounds restored cell division. Our study supports that deregulated mTORC1 activation may also enhance the cancer growth inhibition induced by single-agent CDK4/6 inhibitor by promoting complete senescence.
Supplementary Material
Acknowledgments
We thank the Q2 solutions (previously known as EA, North Carolina, USA) for performing RNA-Seq analysis, Brendan Veeneman for designing TSC2 Crispr guides and Max Follettie for insightful scientific discussions on this project. We also thank the Pfizer Postdoctoral Training Program for support.
Funding Statement
This work was supported by Pfizer Inc .
Disclosure of interest
All authors are currently employees or were employed by Pfizer during preparation of this manuscript. Reeja Maskey is a current employee of Ideaya Biosciences. Anthony Mazurek is a current employee of GlaxoSmithKline. Jeremy S. Myers and Guixian Jin are current employees of EvolveImmune Therapeutics. Robert T. Abraham is a current employee of Vividion Therapeutics. Natasha Emmanuel is a current employee of Tango Therapeutics. Elyssa Lehman is a current employee of Regeneron Pharmaceuticals. Wenyan Zhong is a current employee of AstraZeneca.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Nasrazadani A, Thomas RA, Oesterreich S, et al. Precision Medicine in Hormone Receptor-Positive Breast Cancer. Front Oncol. 2018;8:144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].American Cancer Society . Breast cancer facts & figures 2019-2020. Atlanta: American Cancer Society, Inc; 2019. [Google Scholar]
- [3].Pan H, Gray R, Braybrooke J, et al. 20-year risks of breast-cancer recurrence after stopping endocrine therapy at 5 years. N Engl J Med. 2017;377(19):1836–1846. DOI: 10.1056/NEJMoa1701830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Foster JS, Henley DC, Ahamed S, et al. Estrogens and cell-cycle regulation in breast cancer. Trends Endocrinol Metab. 2001;12(7):320–327. DOI: 10.1016/S1043-2760(01)00436-2. [DOI] [PubMed] [Google Scholar]
- [5].Toogood PL, Harvey PJ, Repine JT, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48(7):2388–2406. DOI: 10.1021/jm049354h. [DOI] [PubMed] [Google Scholar]
- [6].Turner NC, Ro J, André F, et al. Palbociclib in hormone-receptor–positive advanced breast cancer. N Engl J Med. 2015;373(3):209–219. DOI: 10.1056/NEJMoa1505270. [DOI] [PubMed] [Google Scholar]
- [7].Finn RS, Dering J, Conklin D, et al. PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res. 2009;11(5):R77. DOI: 10.1186/bcr2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vijayaraghavan S, Karakas C, Doostan I, et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun. 2017;8:15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Campisi J. Aging, cellular senescence, and cancer. Annu Rev Physiol. 2013;75:685–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Miller TW, Rexer BN, Garrett JT, et al. Mutations in the phosphatidylinositol 3-kinase pathway: role in tumor progression and therapeutic implications in breast cancer. Breast Cancer Res. 2011;13(6):224. DOI: 10.1186/bcr3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Knudsen ES, Witkiewicz AK. The strange case of CDK4/6 inhibitors: mechanisms, resistance, and combination strategies. Trends Cancer. 2017;3(1):39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020;21:183-203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Romero-Pozuelo J, Demetriades C, Schroeder P, et al. CycD/Cdk4 and discontinuities in dpp signaling activate TORC1 in the drosophila wing disc. Dev Cell. 2017;42(4):376–387 e5. doi: 10.1016/j.devcel.2017.07.019. [DOI] [PubMed] [Google Scholar]
- [14].Romero-Pozuelo J, Figlia G, Kaya O, et al. Cdk4 and Cdk6 couple the cell-cycle machinery to cell growth via mTORC1. Cell Rep. 2020;31(2):107504. DOI: 10.1016/j.celrep.2020.03.068. [DOI] [PubMed] [Google Scholar]
- [15].Martinez-Carreres L, Puyal J, Leal-Esteban LC, et al. CDK4 regulates lysosomal function and mTORC1 activation to promote cancer cell survival. Cancer Res. 2019;79(20):5245–5259. DOI: 10.1158/0008-5472.CAN-19-0708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kolesnichenko M, Hong L, Liao R, et al. Attenuation of TORC1 signaling delays replicative and oncogenic RAS-induced senescence. Cell Cycle. 2012;11(12):2391–2401. DOI: 10.4161/cc.20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Harrison DE, Strong R, Sharp ZD, et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460(7253):392–395. DOI: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Houssaini A, Breau M, Kanny Kebe SA, et al. mTOR pathway activation drives lung cell senescence and emphysema. JCI Insight. 2018;3(3):1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Leontieva OV, Blagosklonny MV. CDK4/6-inhibiting drug substitutes for p21 and p16 in senescence: duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle. 2013;12(18):3063–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Demidenko ZN, Zubova SG, Bukreeva EI, et al. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8(12):1888–1895. DOI: 10.4161/cc.8.12.8606. [DOI] [PubMed] [Google Scholar]
- [21].Herranz N, Gallage S, Mellone M, et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat Cell Biol. 2015;17(9):1205–1217. DOI: 10.1038/ncb3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Goel S, Wang Q, Watt AC, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell. 2016;29(3):255–269. DOI: 10.1016/j.ccell.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Yoshida A, Lee EK, Diehl JA. Induction of therapeutic senescence in vemurafenib-resistant melanoma by extended inhibition of CDK4/6. Cancer Res. 2016;76(10):2990–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Olmez I, Brenneman B, Xiao A, et al. Combined CDK4/6 and mTOR inhibition is synergistic against glioblastoma via multiple mechanisms. Clin Cancer Res. 2017;23(22):6958–6968. DOI: 10.1158/1078-0432.CCR-17-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Emmanuel N, Ragunathan S, Shan Q, et al. Purine nucleotide availability regulates mTORC1 activity through the Rheb GTPase. Cell Rep. 2017;19(13):2665–2680. DOI: 10.1016/j.celrep.2017.05.043. [DOI] [PubMed] [Google Scholar]
- [26].Ni Y, Schmidt KR, Werner BA, et al.. Death effector domain-containing protein induces vulnerability to cell cycle inhibition in triple-negative breast cancer. Nat Commun. 2019;10(1):2860. DOI: 10.1038/s41467-019-10743-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36(6):320–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Zhang S, Zeng X, Ren M, et al.. Novel metabolic and physiological functions of branched chain amino acids: a review. J Anim Sci Biotechnol. 2017;8(1):10. DOI: 10.1186/s40104-016-0139-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Puertollano R. mTOR and lysosome regulation. F1000Prime Rep. 2014;6:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Inoki K, Li Y, Xu T, et al. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. DOI: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Abraham RT. Regulation of the mTOR signaling pathway: from laboratory bench to bedside and back again. F1000 Biol Rep. 2009;1:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ma L, Chen Z, Erdjument-Bromage H, et al. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. 2005;121(2):179–193. DOI: 10.1016/j.cell.2005.02.031. [DOI] [PubMed] [Google Scholar]
- [33].Ma L, Teruya-Feldstein J, Bonner P, et al. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res. 2007;67(15):7106–7112. DOI: 10.1158/0008-5472.CAN-06-4798. [DOI] [PubMed] [Google Scholar]
- [34].Roskoski R Jr. Targeting ERK1/2 protein-serine/threonine kinases in human cancers. Pharmacol Res. 2019;142:151–168. [DOI] [PubMed] [Google Scholar]
- [35].Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J. 2008;412(2):179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle. 2008;7(21):3355–3361. [DOI] [PubMed] [Google Scholar]
- [37].Hara K, Maruki Y, Long X, et al. Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell. 2002;110(2):177–189. DOI: 10.1016/S0092-8674(02)00833-4. [DOI] [PubMed] [Google Scholar]
- [38].Kim DH, Sarbassov DD, Ali SM, et al. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell. 2002;110(2):163–175. DOI: 10.1016/S0092-8674(02)00808-5. [DOI] [PubMed] [Google Scholar]
- [39].Sarbassov DD, Ali SM, Kim D-H, et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14(14):1296–1302. DOI: 10.1016/j.cub.2004.06.054. [DOI] [PubMed] [Google Scholar]
- [40].Hwang HC, Clurman BE. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24(17):2776–2786. [DOI] [PubMed] [Google Scholar]
- [41].Turner NC, Liu Y, Zhu Z, et al. Cyclin E1 expression and palbociclib efficacy in previously treated hormone receptor–positive metastatic breast cancer. J Clin Oncol. 2019;37(14):1169–1178. DOI: 10.1200/JCO.18.00925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Milioli H, Alexandrou S, Lim E, et al.. Cyclins E1 and E2 in ER+ breast cancer: prospects as biomarkers and therapeutic targets. Endocr Relat Cancer. 2020. DOI: 10.1530/ERC-19-0501. [DOI] [PubMed] [Google Scholar]
- [43].Benvenuto G, Li S, Brown SJ, et al.. The tuberous sclerosis-1 (TSC1) gene product hamartin suppresses cell growth and augments the expression of the TSC2 product tuberin by inhibiting its ubiquitination. Oncogene. 2000;19(54):6306–6316. DOI: 10.1038/sj.onc.1204009. [DOI] [PubMed] [Google Scholar]
- [44].Kwiatkowski DJ, Zhang H, Bandura JL, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11(5):525–534. DOI: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- [45].Ruscetti M, Leibold J, Bott MJ, et al. NK cell-mediated cytotoxicity contributes to tumor control by a cytostatic drug combination. Science. 2018;362(6421):1416–1422. DOI: 10.1126/science.aas9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Hillmann P, Fabbro D. PI3K/mTOR pathway inhibition: opportunities in oncology and rare genetic diseases. Int J Mol Sci. 2019;20(22):1-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Song X, Liu X, Wang H, et al. Combined CDK4/6 and Pan-mTOR inhibition is synergistic against intrahepatic cholangiocarcinoma. Clin Cancer Res. 2019;25(1):403–413. DOI: 10.1158/1078-0432.CCR-18-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Michaloglou C, Crafter C, Siersbaek R, et al. Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long-term growth inhibition in estrogen receptor-positive breast cancer. Mol Cancer Ther. 2018;17(5):908–920. DOI: 10.1158/1535-7163.MCT-17-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Vora SR, Juric D, Kim N, et al. CDK 4/6 inhibitors sensitize PIK3CA mutant breast cancer to PI3K inhibitors. Cancer Cell. 2014;26(1):136–149. DOI: 10.1016/j.ccr.2014.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.