

ABSTRACT
The inositol 1,4,5-triphosphate receptor (InsP3R)-mediated calcium (Ca2+) transfer to mitochondria is important to maintain mitochondrial respiration and bioenergetics in normal and cancer cells, even though cancer cells have defective oxidative phosphorylation (OXPHOS). Here, we discuss how tumor mitochondria could become a feasible therapeutic target to treat tumors that depend on reductive carboxylation.
KEYWORDS: Calcium, OXPHOS, cell survival, autophagy, cancer
Approximately 100 years ago, Otto Warburg described that cancer cells exhibit a high glucose consumption with a high lactate secretion, even in the presence of oxygen, which led him to speculate that cancer originates from irreversible damage in mitochondrial oxidative phosphorylation (OXPHOS) followed by a compensatory increase of glycolysis for adenosine triphosphate (ATP) generation despite the aerobic conditions.1 This idea, known as the “Warburg effect”, caught rapidly and currently is widely accepted as a hallmark of cancer.1,2 Although Warburg’s observation was accurate, his misconception about the role of mitochondria in cancer rooted deeply in the cancer field despite being immediately challenged. Today, increasing evidence indicates that most cancer cells rely on mitochondrial metabolism to stay viable and use a significant fraction of glucose-derived pyruvate for the generation of metabolic intermediates, reducing equivalents and ATP from the tricarboxylic acid (TCA) cycle to meet their anabolic demands.3
Since Hajnóczky’s seminal work in the middle 1990s, it is understood that calcium (Ca2+) release from the endoplasmic reticulum (ER) through the inositol 1,4,5-triphosphate receptors (ITPRs, best known as InsP3Rs) sustains the activation of mitochondrial metabolism by stimulating the activity of Ca2+-sensitive mitochondrial dehydrogenases such as the pyruvate dehydrogenase (PDH), isocitrate dehydrogenase (IDH) and α-ketoglutarate dehydrogenase (αKGDH).4
In 2010, we revealed that in unstimulated conditions, spontaneous InsP3R-mediated Ca2+ release events are essential to maintain mitochondrial respiration and normal cellular bioenergetics. Ca2+ stimulates the PDH, a key enzyme that links glycolysis to the TCA cycle.5 Upon interruption of the Ca2+ flux between the ER and mitochondria by using molecular and pharmacological interventions, cells enter a bioenergetic crisis characterized by the activation of the adenosine monophosphate (AMP)-activated protein kinase (AMPK) and a mechanistic target of rapamycin kinase (MTOR) complex 1-independent pro-survival macroautophagy (hereafter termed autophagy) even in nutrient-rich conditions.5 In this scenario, given the mitochondrial dependence that many cancer cells show, we wondered about the role of ER-to-mitochondria Ca2+ transfer in cell bioenergetics and survival of cancer cells. In 2016, we found that the interruption of the constitutive InsP3R-mediated Ca2+ transfer to mitochondria generates a bioenergetic crisis in transformed primary human fibroblasts, breast and prostate cancer cells as in non-tumorigenic cells.6 This bioenergetic crisis leads to selective cell death in the cancer cells, that cannot be rescued by the activation of autophagy and AMPK as happens in non-tumorigenic cells.6 Metabolic intermediates, such as pyruvate and α-ketoglutarate (αKG), rescue the lethal effect caused by inhibiting the InsP3R mediated Ca2+ transfer to the mitochondria, suggesting that cell death is generated by a compromise in bioenergetics.6 Notably, nucleotides also prevent the effects caused by the lack of Ca2+ transfer to mitochondria, suggesting that the role of mitochondrial Ca2+ goes beyond bioenergetics.6
This last observation, together with work from DeBerardinis’ lab elegantly showing that αKGDH sustains reductive carboxylation in OXPHOS defective cells,7 encouraged us to explore whether the inhibition of InsP3R would affect cancer cells that harbor defective OXPHOS and rely on reductive carboxylation to meet their metabolic demand; e.g., renal cell carcinoma, paraganglioma, pheochromocytoma, cancer cells under hypoxia or pseudohypoxia caused by activation of hypoxia inducible factor 1 subunit alpha (HIF1A, best known as HIF1α).8 We used cybrids derived from 143B osteosarcoma cells in which the mitochondrial DNA (mtDNA) was depleted and then reconstituted with either wild-type (wt) mtDNA (143Bwt) or with mtDNA containing a mutation in the cytochrome b (CYTB) gene (143BΔCYTB), which disables OXPHOS,9 and forces cells to use reductive carboxylation for growth.7 Interestingly, when constitutive Ca2+ transfer from the ER to the mitochondria was inhibited, OXPHOS-defective 143BΔCYTB cells died significantly more than the OXPHOS-competent 143Bwt (see Figure 1).9 We confirmed that the lack of ER to-mitochondria Ca2+ transfer decreased the activity of αKGDH causing an overall reduction of the TCA cycle activity in 143Bwt and limited the availability of reducing equivalents necessary for reductive carboxylation in 143BΔCYTB cells.7,9 In both cell types, the change in the nicotinamide adenine dinucleotide oxidized (NAD+)/nicotinamide adenine dinucleotide reduced (NADH) ratio activated the NAD+-dependent deacetylase sirtuin 1 (SIRT1), which in turn activated a pro-survival autophagy. Despite that both cell types activated the same survival response, this was clearly less effective in 143BΔCYTB, where cell death occurred more quickly and to a larger extent than in 143Bwt. This observation made us wonder about the mechanism responsible for this difference. Previous work showed that mitochondrial respiration is necessary for proper lysosome function.10 Since degradation of autophagosome content needs the lysosome, we looked into the autophagic flux and found that no acidification was occurring in 143BΔCYTB, further reducing the amount of intermediate metabolites needed to maintain cellular homeostasis (Figure 1).9 The question of whether this phenomenon is the result of the lack of fusion between the autophagosome and the lysosome or a lack of acidification of the lysosome still remains. We believe that further experiments are necessary to determine whether there is any direct role of InsP3R-mediated Ca2+ inhibition on the function of the lysosome.
Figure 1.
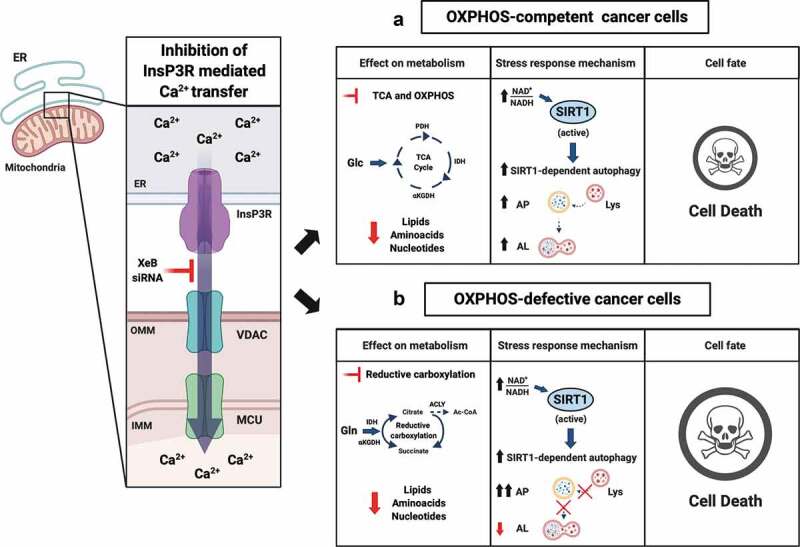
Effects of the inhibition of InsP3R-mediated Ca2+ transfer to mitochondria on OXPHOS-competent and OXPHOS-defective cancer cell viability. (a) The interruption of the inositol 1,4,5-triphosphate receptors (InsP3R)-mediated calcium (Ca2+) transfer to mitochondria in oxidative phosphorylation (OXPHOS)-competent cells decreases the tricarboxylic acid (TCA) cycle activity and OXPHOS, creating a bioenergetic crisis characterized by an increase in the nicotinamide adenine dinucleotide oxidized (NAD+)/nicotinamide adenine dinucleotide reduced (NADH) ratio. Consequently, the high NAD+/NADH ratio allows the activation of sirtuin 1 (SIRT1) which in turn activates autophagy, providing some protection against cell death. (b) In OXPHOS defective cells, the interruption of the InsP3R-mediated Ca2+ transfer to mitochondria reduces the activity of the α-ketoglutarate dehydrogenase (αKGDH) and in turn reduces the levels of NADH necessary to sustain reductive carboxylation. As in OXPHOS-competent cells, the increase in the NAD+/NADH ratio activates SIRT1 and autophagy. However, the impaired lysosome function in these cells enhances their death. Abbreviations: ER: endoplasmic reticulum; VDAC: voltage dependent anion channel; MCU: mitochondrial calcium uniporter; XeB: xestospongin B; siRNA: small interfering RNA; OMM: outer mitochondrial membrane; IMM: inner mitochondrial membrane; PDH: pyruvate dehydrogenase; IDH: isocitrate dehydrogenase; ACLY: ATP citrate lyase; Ac-CoA: acetyl-CoA; Glc: glucose; Gln: glutamine; AP: autophagosome; AL: autolysosome; Lys: Lysosome. Created with BioRender.com
In summary, we show that in OXPHOS-defective cancer cells the constitutive InsP3R-mediated Ca2+ transfer to mitochondria is necessary to maintain the activity of αKGDH which in turn provides the reducing equivalents necessary to support reductive carboxylation. Thus, the inhibition of this signal in addition to impairing lysosomal function causes a massive cell death. We believe that the interruption of InsP3R-mediated Ca2+ transfer to the mitochondria may offer a real therapeutic opportunity in the future for cancers that depend on reductive carboxylation.
Funding Statement
This work was supported by NIH P30NS047243 (AL), ANID/FONDECYT no. 1160332 and 1200255 (CC) and ANID/FONDAP no. 15150012 (CC); fondecyt [1160332]; fondap [15150012]; fondecyt [1200255].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Warburg O. Injuring of respiration the origin of cancer cells. Science. 1956;123(3191):1–3. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA.. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Vyas S, Zaganjor E, Haigis MC.. Mitochondria and Cancer. Cell. 2016;166:555–566. doi: 10.1016/j.cell.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hajnóczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of Cytosolic Calcium Oscillations in the Mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- 5.Cardenas C, Miller RA, Smith I, Bui T, Molgo J, Muller M, Vais H, Cheung KH, Yang J, Parker I, et al. Essential regulation of cell bioenergetics by constitutive InsP(3) receptor Ca(2+) transfer to mitochondria. Cell. 2010;142:270–283. doi: 10.1016/j.cell.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cárdenas C, Müller M, McNeal A, Lovy A, Jaňa F, Bustos G, Urra F, Smith N, Molgó J, Diehl J, et al. Selective vulnerability of cancer cells by inhibition of Ca2+ transfer from endoplasmic reticulum to mitochondria. Cell Rep. 2016;14:2313–2324. doi: 10.1016/j.celrep.2016.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mullen AR, Hu Z, Shi X, Jiang L, Boroughs L, Kovacs Z, Boriack R, Rakheja D, Sullivan L, Linehan W, et al. Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep. 2014;7(5):1679–1690. doi: 10.1016/j.celrep.2014.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hollinshead and Tennant. Mitochondrial metabolic remodeling in response to genetic and environmental perturbations. WIREs. 2016;8(4):272–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cardenas C, Lovy A, Silva-Pavez E, Urra F, Mizzoni C, Ahumada-Castro U, Bustos F, Jana F, Cruz P, Farias P, et al. Cancer cells with defective oxidative phosphorylation require endoplasmic reticulum – to – mitochondria Ca 2 + transfer for survival. Sci Sign. 2020;13(640):eaay1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baixauli F, Acín-Pérez R, Villarroya-Beltrí C, Mazzeo C, Nuñez-Andrade N, Gabandé-Rodriguez E, Ledesma M, Blázquez A, Martin M, Falcón-Pérez J, et al. Mitochondrial respiration controls lysosomal function during inflammatory t cell responses. Cell Metab. 2015;22(3):P485–498. doi: 10.1016/j.cmet.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]