

ABSTRACT
Small GTPases (e.g., Rac1) play key roles in glucose-stimulated insulin secretion (GSIS) in the β-cell. We investigated regulation by RhoGDIβ of glucose-induced activation of Rac1 and insulin secretion. RhoGDIβ is expressed in INS-1 832/13 cells, rodent and human islets. siRNA-mediated knockdown of RhoGDIβ in INS-1 832/13 cells significantly attenuated glucose-induced Rac1 activation without affecting its translocation and membrane association. Further, suppression of RhoGDIβ expression exerted minimal effects on GSIS at the height of inhibition of Rac1 activation, suggesting divergent effects of RhoGDIβ on Rac1 activation and insulin secretion in the glucose-stimulated β-cell. We provide the first evidence for the expression of RhoGDIβ in rodent and human β-cells, and its differential regulatory roles of this protein in G protein activation and GSIS.
Abbreviations: Arf6: ADP ribosylation factor; Cdc42: Cell Division Cycle; GAP: GTPase-activating protein; GDI: GDP dissociation inhibitor; GDIα: GDP dissociation inhibitorα; GDIβ: GDP dissociation inhibitorβ; GEF: Guanine nucleotide exchange factor; GSIS: Glucose-stimulated insulin secretion; Rac1: Ras-Related C3 Botulinum Toxin Substrate 1
KEYWORDS: RhoGDIβ, glucose-stimulated insulin secretion, Rac1, pancreatic beta-cell
Insulin secretion from pancreatic β-cells is principally regulated by ambient glucose concentrations. However, potential cellular mechanisms underlying the stimulus-secretion coupling of glucose-stimulated insulin secretion (GSIS) are only partially understood. GSIS occurs largely via the generation of soluble second messengers, such as cyclic nucleotides and biologically active lipids, as well as an increase in intracellular calcium concentrations [1,2]. It is well established that, following Glut‐2‐mediated entry into the β‐cell, glucose is metabolized via the glycolytic and tricarboxylic acid pathways with a resultant increase in intracellular ATP, which, in turn, mediates closure of ATP‐sensitive K+ channels localized on the plasma membrane resulting in membrane depolarization. These signalling steps promote influx of extracellular calcium through the voltage‐gated calcium channels. Increase in intracellular calcium has been shown to be essential for the transport of insulin‐containing secretory granules to the plasma membrane for fusion and release of insulin into circulation [1,2]. Importantly, in addition to adenine nucleotides, the guanine nucleotides (e.g., GTP) have been shown to play key regulatory roles in GSIS. For example, using selective inhibitors of the GTP biosynthetic pathway, Metz and associates have provided the first evidence for a permissive role for GTP in GSIS [3,4].
Although the precise mechanisms underlying the regulatory role(s) of GTP in GSIS remain elusive, emerging evidence indicates that they might involve activation of one (or more) G proteins [5–7]. At least two major groups of G proteins have been described in pancreatic β‐cells. The first group is trimeric in nature, which is comprised of α/β/γ subunits. These signalling proteins are involved in coupling of various G protein-coupled receptors to their intracellular effectors, such as adenylate cyclase, phosphodiesterase, or phospholipases. The second group is comprised of small molecular mass (monomeric) G proteins, which are involved in protein sorting as well as trafficking of secretory vesicles [5–7]. Both trimeric and small G-proteins (Arf6, Cdc42, Rac1, Rab) have been implicated in islet β-cell function including cytoskeletal remodelling, vesicular transport and GSIS [5–10]. The cycling of small G proteins (e.g., Rac1) between their inactive (GDP-bound) and active (GTP-bound) conformations is precisely mediated by specific regulatory proteins. Three major types of such regulatory proteins have been identified thus far. The first group is comprised of guanine nucleotide exchange factors (GEFs), which facilitate the conversion of the GDP-bound forms to their GTP-bound forms. Using pharmacological and molecular biological approaches, several recent studies have identified GEFs for small G-proteins in pancreatic islets (e.g., Tiam1, Vav2, ARNO, Epac; 5–7, 11). The second group represents the GTPase-activating proteins (GAPs), which promote the conversion of active G-proteins to their inactive form by activating their intrinsic GTPase function to complete the G-protein activation-deactivation cycle. Recent experimental evidence suggests regulation of islet β-cell function by a variety of GAPs including AS160, ARHGAP21, Stard13, TBC1D1 [11–16]. The third group of regulatory proteins are the GDP dissociation inhibitors (GDIs; GDIα, caveolin-1), which play key regulatory roles in G-protein function [5–7,17–19]. First, they inhibit dissociation of GDP from G-proteins, thus maintaining the GDP-bound G proteins in their inactive conformation and preventing their activation by GEFs. Second, they have been shown to interact with GTP-bound G proteins, thus preventing their inactivation by GAP proteins. Lastly, they regulate the cycling of G proteins from cytosol and membrane [5–7].
It is noteworthy that the (de)activation of Rho G proteins is regulated by a large number of GEFs (~60) and GAPs (~70), but by only three GDIs, namely RhoGDIα, RhoGDIβ, and RhoGDIγ [20–25]. The expression of RhoGDIα is ubiquitous and it has been shown to prevent GDP dissociation from RhoA, Cdc42, and Rac1. Its regulatory role in small G protein activation was well studied and we have also previously demonstrated a negative regulatory role for RhoGDIα in GSIS in pancreatic β-cells [17–19]. RhoGDIβ (also known as LyGDI or ARHGDIB) is believed to be exclusively expressed in hematopoietic lineage. RhoGDIγ is expressed in lung, brain and testis. Although RhoGDIβ shares 67% amino acid identity with RhoGDIα, it was found to be less efficient in its capacity to inhibit GDP/GTP exchange or to promote membrane dissociation of these proteins [20–25]. Studies have also demonstrated that RhoGDIβ is not able to form complexes in vivo with these proteins and suggested its association with some still uncharacterized Rho proteins [20–25].
The expression and the biochemical pathways regulated by RhoGDIβ, specifically in physiological insulin secretion, have not been examined in the pancreatic β-cell. Therefore, in the present study, to gain further insight into the biological role of RhoGDIβ in islet β-cell function, we investigated the protein expression of RhoGDIβ and its role in GSIS from the islet β-cell. INS-1 832/13 cells were used in the majority of the studies described herein. This cell line, originally developed in Newgard’s laboratory via stable transfection of rat INS-1 cells with a plasmid containing the human proinsulin gene, responds robustly to physiological glucose concentrations to elicit insulin secretion [26]. First, we determined the expression of RhoGDI family members in β-cells. Lysates from INS-1 832/13 cells were analyzed for expression of Rab GDIα/β, RhoGDIα, RhoGDIβ and Rho GDIγ by Western blot analysis. Data depicted in Figure 1a, demonstrate expression of all four GDIs in INS-1 832/13 cells. Since the primary objective of the current investigation is to assess the regulatory roles of RhoGDIβ, we further determined the RhoGDIβ expression in normal rat islets and human islets. Data in Figure 1b further affirm expression of RhoGDIβ in primary rodent and human islets.
Figure 1.
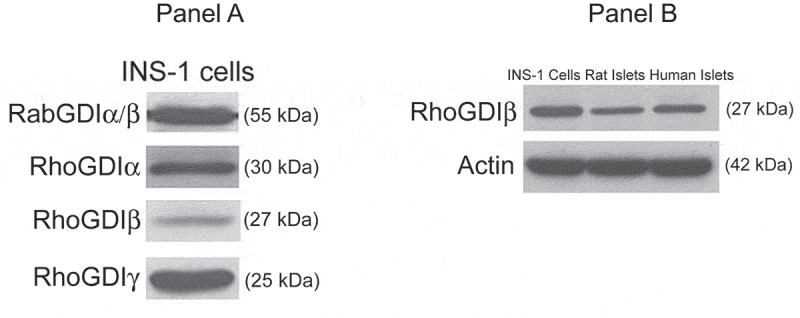
Expression of GDIs in pancreatic β-cells (a). INS-1 832/13 cell lysates were probed for the expression of Rab GDIα/β, Rho GDIα, Rho GDIβ and Rho GDIγ by Western blot analysis. Respective molecular weights of each of these proteins are indicated in parentheses (b) Lysates from INS-1 832/13 cells, rats and human islets were analyzed for RhoGDIβ protein expression by Western blot analysis
It is well established that small G-proteins, including, Rac1 and Cdc42, are cytosolic in their distribution and are translocated to the membrane following cellular activation [5–7]. Indeed, published evidence implicates that exposure of pancreatic β-cells to stimulatory glucose concentrations results in translocation of Rac1 to the membrane fraction [5–7]. Evidence is also available to suggest that such effects of glucose may be mediated via dissociation of RhoGDIα/Rac1 complex by lipid hydrolytic products of phospholipases [27]. Therefore, we next examined the subcellular distribution (i.e., cytosolic vs. membrane) of RhoGDIβ and Rac1 in INS-1 832/13 cells exposed to basal or stimulatory glucose. Data shown in Figure 2 (a and b) indicate that RhoGDIβ and Rac1 are predominantly localized in the cytosolic compartment under basal conditions. However, a significant amount of Rac1, but not RhoGDIβ, was seen associated with membrane fraction derived from INS-1 832/13 cells under glucose-stimulated conditions. Note that we have determined the purity of cytosol and membrane fractions by assessing the relative abundance of marker proteins for these fractions, namely GAPDH for cytosol and E-cadherin for the membrane. Data in Panel A indicate lack of E-cadherin in the cytosolic fraction suggesting that this fraction is devoid of membranous contamination. However, we did observe association of GAPDH with the membrane fraction. This is not surprising since GAPDH has been shown to bind specifically to certain integral membrane proteins in the membrane [28–30]. Together, these data suggest translocation and membrane association of Rac1, but not RhoGDIβ, in β-cells exposed to insulinotropic glucose concentration.
Figure 2.
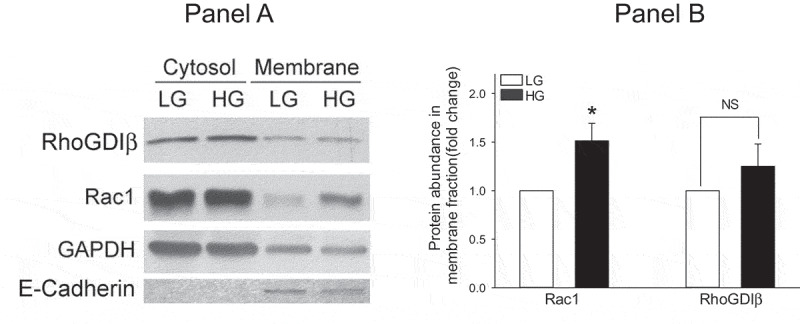
Glucose promotes translocation and membrane association of Rac1, but not RhoGDIβ, in INS-1 832/13 cells. INS-1 832/13 cells were exposed to low glucose (LG, 2.5 mM) or stimulatory glucose (HG, 20 mM) for 15 min. Cell lysates were subjected to subcellular fractionation and analyzed for RhoGDIβ and Rac1 protein expression levels in cytosolic and membrane fractions. GAPDH and E-Cadherin were used as loading controls for cytosol and membrane fractions, respectively. Representative blots from three independent studies are provided. (b) Densitometric quantitation of relative abundance of Rac1 and RhoGDIβ in the membrane fractions isolated from low and stimulatory glucose-treated INS-1 832/13 cells (a) is shown here. The data are expressed as fold change relative to LG-treated. (mean ± SEM; n = 3; * p < 0.05)
In order to affirm regulatory roles of RhoGDIβ in glucose-induced Rac1 activation and insulin secretion, in the next series of experiments, we investigated glucose-induced membrane association and activation of Rac1 as well as insulin secretion in INS-1 832/13 cells following siRNA-mediated depletion of endogenous RhoGDIβ . Data in Figure 3a indicate significant reduction (~50%) in the expression of RhoGDIβ in INS-1 832/13 cells transfected with siRNA- RhoGDIβ (RhoGDIβ-si), but not in cells transfected with scrambled siRNA (Scr-si). In addition, no significant effects on the expression of RhoGDIα were noted in these cells transfected with RhoGDIβ-si (Figure 3b), further demonstrating the specificity of RhoGDIβ knockdown under our current experimental conditions. Data presented in Figure 3c demonstrate no detectable effects of RhoGDIβ knockdown on glucose-induced translocation and membrane association of Rac1. Pooled data from multiple experiments are provided in Figure 3d. Interestingly, however, glucose-induced Rac1 activation is significantly attenuated in INS-1 832/13 cells following RhoGDIβ knockdown (Figure 4a and 4b). These findings are somewhat unexpected since knockdown of GDI is expected to relieve its inhibitory function on Rac1 activity. Together, our findings indicate that knockdown of RhoGDIβ results in significant reduction in Rac1 activation without affecting its translocation and membrane association in a glucose-stimulated β-cell.
Figure 3.
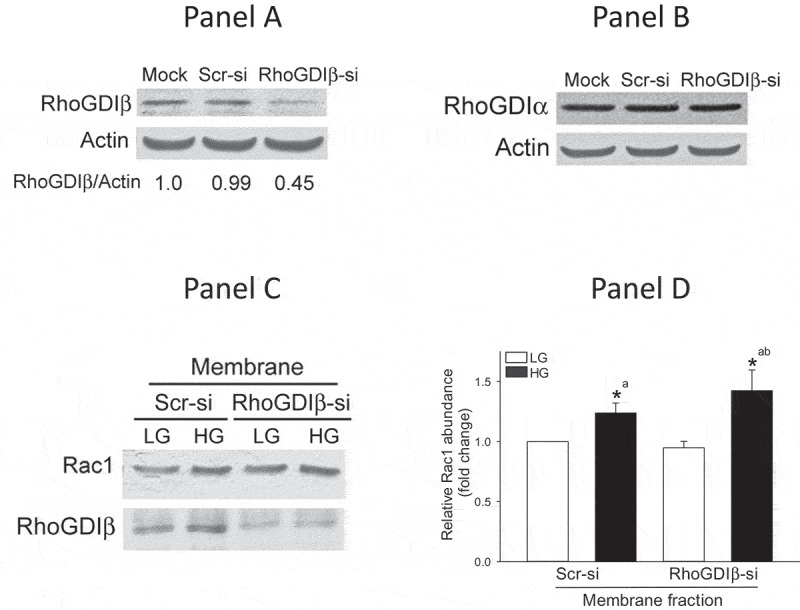
Glucose-induced translocation and membrane association of Rac1 is not altered in RhoGDIβ -depleted (RhoGDIβ siRNA) INS-1 832/13 cells. INS-1 832/13 cells were transfected with scrambled siRNA or siRNA targeted to RhoGDIβ. Cell lysates prepared after 48 h were analyzed by Western blotting for the expression of RhoGDIβ (a) or RhoGDIα (b; as a negative control to assess off-target effects). Actin was used as loading control. A representative blot from n = 3 independent studies is shown here. (c) Western blot data indicating that glucose-induced membrane association of Rac1 was not affected following RhoGDIβ depletion in INS-1 832/13 cells. (d) Densitometric quantitation of Rac1 expression in the membrane fraction depicted in Panel C is shown here. The results are presented as means ± SEM. The data are expressed as fold change relative to LG-treated Scr-si. (n = 4; * p < 0.05). Comparisons shown: a: significant compared with LG-treated Scr-si; b: significant compared with LG-treated RhoGDIβ-si
Figure 4.
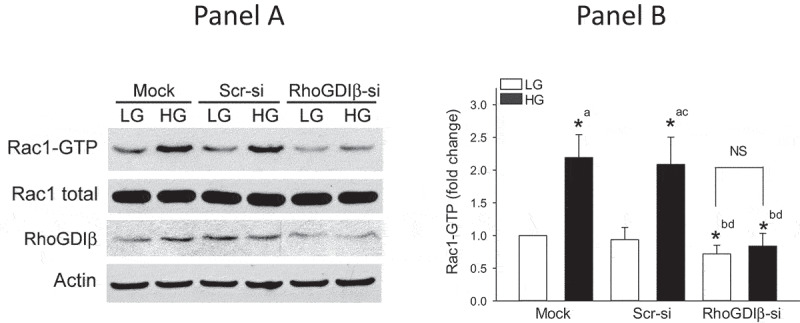
Glucose-induced Rac1 activation is attenuated in INS-1 832/13 cells following suppression of RhoGDIβ expression. a. INS-1 832/13 cells were transfected with scrambled siRNA or siRNA targeted to RhoGDIβ protein. After 48 h of transfection, cells were subjected to overnight starvation and then were treated with LG (2.5 mM) or HG (20 mM) for 15 min. Rac1 activation was quantified by Rac1 pull-down assay. Representative blots from four independent studies are provided. (b) Densitometric quantitation of activated Rac1 in Panel A is shown here. The results from four independent experiments are presented as means ± SEM. The data are expressed as fold change relative to LG-mock (n = 4; * p < 0.05). Comparisons shown: a: significant compared with LG-treated mock; b: significant compared with HG-treated mock; c: significant compared with LG-treated Scr-si. d: significant compared with HG-treated Scr-si; NS: Not significant
We next quantified GSIS in INS-1 832/13 cells in RhoGDIβ -depleted cells. Data in Figure 5 indicate a modest (but not statistically significant) increase in basal insulin secretion in RhoGDIβ depleted cells. Interestingly, the magnitude of GSIS remained unchanged in INS-1 832/13 cells following RhoGDIβ knockdown (Figure 5) even at the height of significant inhibition of glucose-induced Rac1 activation under these conditions (Figure 4A and 4B). Also, note that, RhoGDIβ knockdown modestly (and significantly) potentiated GSIS compared to Scr-si transfected cells (Figure 5). Together, these findings suggest paradoxical regulatory roles of RhoGDIβ in glucose-induced Rac1 activation and insulin secretion.
Figure 5.
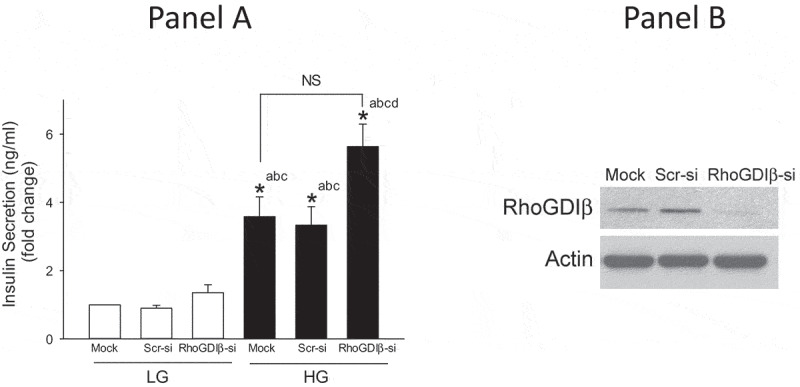
siRNA-mediated knockdown of RhoGDIβ modestly, but significantly, potentiates GSIS in INS-1 832/13 cells. A. INS-1 832/13 cells were transfected with scrambled siRNA or siRNA targeted to RhoGDIβ protein. After 48 h of transfection, cells were subjected to overnight starvation and then GSIS was quantified by ELISA as described under Methods. Data are mean ± SEM from four experiments. The data are expressed as fold change relative to LG-mock. (n = 4; * p < 0.05). Comparisons shown: a: significant compared with LG-treated mock; b:significant compared with LG-treated Scr-si; c: significant compared with LG-treated RhoGDIβ-si; d: significant compared with HG-treated Scr-si. NS: not significant. A representative blot demonstrating the efficiency of RhoGDIβ knockdown is shown in Panel B.
Previously published evidence suggests that GSIS involves sequential activation of Arf6 (~1 min), Cdc42 (2–3 min) and Rac1 (15–20 min), thus implicating Cdc42 activation as an upstream signalling event to Rac1 activation and insulin secretion [5–7,10]. Furthermore, evidence from the studies of Ngo and coworkers identified Cdc42 and Rac1 as target proteins for RhoGDIβ in the cascade of events leading to cytoskeletal remodelling in platelet function [31]. Therefore, we asked if inhibition of glucose-induced Rac1 activation in RhoGDIβ-depleted INS-1 832/13 cells (Figure 4 A and B) is due to inhibition of glucose-induced Cdc42 activation. Data in Figure 6 indicated no significant difference between the degrees of glucose-induced activation of Cdc42 in mock, Scr-si and RhoGDIβ-si transfected cells. Interestingly, however, we observed modest (but statistically insignificant) activation of Cdc42 under basal conditions in RhoGDIβ-si transfected cells. These data raise an interesting possibility that acute glucose-induced activation of Cdc42 in RhoGDIβ-si transfected cells may be adequate to promote insulin secretion even under conditions of inhibited Rac1 activation.
Figure 6.
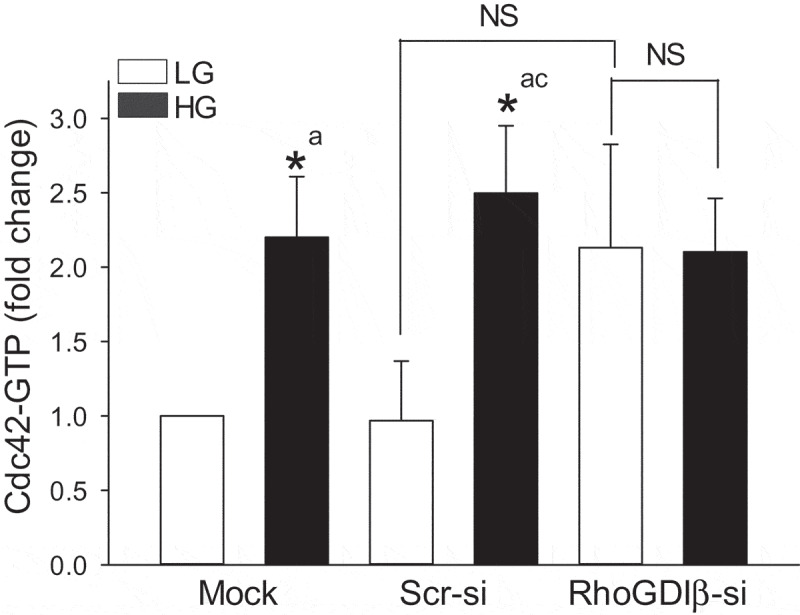
Suppression of RhoGDIβ expression modestly activate Cdc42 under basal condition but does not alter glucose-induced Cdc42 activation. INS-1 832/13 cells were transfected with scrambled siRNA or siRNA targeted to RhoGDIβ protein. After 48 h of transfection, cells were subjected to overnight starvation and then were treated with LG (2.5 mM) or HG (20 mM) for 3 mins. Cdc42 activation was quantified by Cdc42 pull-down assay. The results from four independent experiments are presented as means ± SEM. The data are expressed as fold change relative to LG-mock (n = 4; * p < 0.05). Comparisons shown: a: significant compared with LG-treated mock; b: significant compared with HG-treated mock; c: significant compared with LG-treated Scr-si; NS: Not significant
In summary, this brief report provides evidence for paradoxical roles for RhoGDIβ (which is expressed in INS-1 832/13 cells, normal rodent and human islets) in an acutely stimulated pancreatic β-cell. Selective knockdown of this protein appears to significantly attenuate glucose-induced Rac1 activation, without affecting its membrane association. Further, it seems to exert minimal effects on GSIS. Future studies will further assess roles of this regulatory protein in islet β-cell function, including the sustained activation of Rac1 under conditions of metabolic stress. Indeed, several recent investigations have demonstrated sustained activation of Rac1 under conditions of glucotoxicity, lipotoxicity and exposure to pro-inflammatory cytokines and biologically active sphingolipids, such as ceramide [5–7,32–36]. It is likely that such pathological conditions promote dissociation of RhoGDIβ/Rac1 complex thereby providing optimal conditions that are conducive for activation by various GEFs (e.g., Tiam1 and Vav2; ref. 11). Lastly, evidence from the studies of Groysman and associates [37,38] on interaction between RhoGDIβ and Vav proteins provides additional insights into potential cross-talk between GEFs and GDI in regulation of cell function. Methodical investigations are in progress to assess the roles of RhoGDIβ signalling in β-cell models of metabolic stress. These aspects on RhoGDIβ biology are subject of current investigations in our laboratory.
Materials and methods
Materials
Rab, RhoGDIα, RhoGDIβ, RhoGDIγ, Cdc42, and E-Cadherin antibodies were from Santa Cruz Biotechnology (CA, USA). GAPDH and HRP-conjugated secondary antibodies were from Cell Signaling (Danvers, MA, USA). Rac1 Antibody was purchased from EMD Millipore. Rat high range insulin ELISA was from ALPCO (Salem, NH, USA). The ON-TARGETplus SMARTpool and non- TARGETplus SMARTpool siRNAs as well as DharmaFect1 were from Dharmacon (Lafayette, CO, USA). Antibody for β-actin and all other reagents used in the current studies were purchased from Sigma Aldrich (St. Louis, MO, USA). Mem-PER Plus Membrane Extraction Kit was purchased from Thermo scientific. Pull-down assay kit used for the Rac1 and Cdc42 activation were from Cytoskeleton (Denver, CO, USA).
Cell culture and experimental conditions
INS-1 832/13 cells were cultured in RPMI-1640 medium containing 10% FBS supplemented with 100 IU/ml penicillin and 100 IU/ml streptomycin, 1 mM sodium pyruvate, 50 µM 2-mercaptoethanol and 10 mM HEPES (pH 7.4). The cultured cells were sub-cloned twice weekly following trypsinization. INS-1 832/13 cells were exposed to different concentrations of glucose (2.5–20 mM) for various time points, as indicated in the text.
Islet isolation
All protocols, including isolation of pancreatic islets from rats, were reviewed and approved by the Wayne State University and John D. Dingell VA Medical Center Institutional Animal Care and Use Committees. Islets from normal 10-week-old male Sprague–Dawley rats were isolated by the collagenase digestion method [9,10,33–35]. Human islets were from Prodo Labs (Aliso Viejo, CA). Studies involving human islets were conducted according to the guidelines established by the US Department of Health and Human Services/NIH and approved by the Biosafety Committee at the John D. Dingell VA Medical Center.
SiRNA-mediated knockdown of RhoGDIβ expression
Endogenous RhoGDIβ expression was knockdown by small interfering RNA (siRNA) transfection. Cells were transfected with siRNA at a final concentration of 100 nM using DhamaFect1 reagent. To assess specificity of RNA interference, control cells were transfected (as above) with nontargeting RNA (i.e., scrambled siRNA) duplexes specific for rat genome. Transfection was performed as per the Dharmacon transfection protocol. Transfected cells were maintained in complete growth medium for 48 h hrs. Efficiency of RhoGDIβ knockdown was determined by Western blots of lysates derived from scrambled siRNA and RhoGDIβ siRNA transfected cells.
Glucose-stimulated insulin secretion
GSIS was performed in Krebs Ringer’s bicarbonate (KRB) buffer. Following a 60 min pre-incubation at 37°C in glucose-free KRB, the cells were maintained at 2.5 mM or 20 mM glucose for 45 min at 37°C. An aliquot of the medium was collected after incubation. Insulin released was quantified by rat high range insulin ELISA kits according to manufacturer’s instruction. All the sample were tested in duplicates for quantification of insulin. Data were expressed as ng/ml insulin secreted in the medium as we described earlier [9,10].
Subcellular fractionation
The subcellular fractions were isolated using Mem-PER Plus Membrane Extraction Kit as per manufacturer instructions. Total membrane and cytosol fractions were separated and used for the determination of relative abundance of RhoGDIβ and Rac1 in these fractions by Western blotting. The purity of cytosolic and membrane fractions was assessed using specific protein markers, GAPDH and E-Cadherin, respectfully.
Rac1 and Cdc42 activation assay
The degree of glucose-induced activation of Rac1 and Cdc42 was determined using a pull-down assay kit as we described previously [10]. In brief, INS-1 cells were exposed to basal (2.5 mM) or stimulatory (20 mM) concentrations of glucose for 3 min (for Cdc42 activation) or 15 min (for Rac1 activation) in Krebs Ringer buffer medium. Lysates were clarified by centrifugation and p21-binding domain of p21-activated kinase beads were added to the supernatant. The mixture was then rotated for 1 h at 4°C and pelleted by centrifuging at 4,000g for 3 min. This pellet was washed once with wash buffer then reconstituted in Laemmli buffer and boiled for 5 min. Proteins were resolved by SDS-PAGE and transferred to a nitrocellulose membrane, and the relative abundance of Rac1 or Cdc42 was determined by Western blotting [10,33–35].
Statistical analysis
Data are presented as mean ± SEM of at least three independent experiments. Statistical analysis for differences between groups was analyzed by ANOVA and the significance of differences between groups was assessed by Student-Newman-Keuls post-hoc test. p < 0.05 was considered to be statistically significant.
Funding Statement
This work was supported by grants from the NIDDK/NEI, the US Department of Veterans Affairs (MERIT Review and Senior Research Career Scientist Awards); National Eye Institute [EY22230]; Department of VA [13S-RCS-006]; Department of VA [BX002801].
Data availability
Individual data points are shown in the Figures. Tabulated data are available upon request from the corresponding author.
Disclosure statement
No potential conflict of interest was reported by the authors.
Author contributions
VT performed the experiments, analyzed data and wrote the manuscript. AK supervised the project, designed the experiments, analyzed the data and wrote the manuscript.
References
- [1].Jitrapakdee S, Wutthisathapornchai A, Wallace JC, et al. Regulation of insulin secretion: role of mitochondrial signaling. Diabetologia. 2010;53:1019–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Prentki M, Matschinsky FM, Madiraju SR.. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185. [DOI] [PubMed] [Google Scholar]
- [3].Metz SA, Rabaglia ME, Pintar TJ. Selective inhibitors of GTP synthesis impede exocytotic insulin release from intact rat islets. J Biol Chem. 1992;267:12517–12527. [PubMed] [Google Scholar]
- [4].Metz SA, Meredith M, Rabaglia ME, et al. Small elevations of glucose concentration redirect and amplify the synthesis of guanosine 5ʹ-triphosphate in rat islets. J Clin Invest. 1993;92:872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kowluru A. Small G proteins in islet beta-cell function. Endocr Rev. 2010;31:52–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Wang Z, Thurmond DC. Mechanisms of biphasic insulin-granule exocytosis- roles of the cytoskeleton, small GTPases and SNARE proteins. J Cell Sci. 2009;122:896–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kowluru A. Role of G-proteins in islet function in health and diabetes. Diabetes Obes Metab. 2017;19(S1):63–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ljubicic S, Bezzi P, Brajkovic S, et al. The GTPase Rab37 participates in the control of insulin exocytosis. PLoS One. 2013;8:e68255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Arora DK, Syed I, Machhadieh B, et al. Rab-geranylgeranyl transferase regulates glucose-stimulated insulin secretion from pancreatic β-cells. Islets. 2012;4:354–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jayaram B, Syed I, Kyathanahalli CN, et al. Arf nucleotide binding site opener [ARNO] promotes sequential activation of Arf6, Cdc42 and Rac1 and insulin secretion in INS 832/13 beta-cells and rat islets. Biochem Pharmacol. 2011;81:1016–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kowluru A. Tiam1/Vav2-Rac1 axis: A tug-of-war between islet function and dusfunction. Biochem Pharmacol. 2017;132:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bouzakri K, Ribaux P, Tomas A, et al. Rab GTPase-activating protein AS160 is a major downstream effector of protein kinase B/Akt signaling in pancreatic beta-cells. Diabetes. 2008;57:1195–1204. [DOI] [PubMed] [Google Scholar]
- [13].Rosa LRO, Soares GM, Silveira LR, et al. ARHGAP21 as a master regulator of multiple cellular processes. J Cell Physiol. 2018;233:8477–8481. [DOI] [PubMed] [Google Scholar]
- [14].Naumann H, Rathjen T, Poy MN, et al. The RhoGAP Stard13 controls insulin secretion through F-actin remodeling. Mol Metab. 2018;8:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Paglialunga S, Simnett G, Robson H, et al. The Rab-GTPase activating protein, TBC1D1, is critical for maintaining normal glucose homeostasis and β-cell mass. Appl Physiol Nutr Metab. 2017;42:647–655. [DOI] [PubMed] [Google Scholar]
- [16].Rutti S, Arous C, Nica AC, et al. Expression, phosphorylation and function of the Rab-GTPase activating protein TBC1D1 in pancreatic beta-cells. FEBS Lett. 2014;588:15–20. [DOI] [PubMed] [Google Scholar]
- [17].Kowluru A, Veluthakal R. Rho guanosine diphosphate-dissociation inhibitor plays a negative modulatory role in glucose-stimulated insulin secretion. Diabetes. 2005;54:3523–3529. [DOI] [PubMed] [Google Scholar]
- [18].Wang Z, Thurmond DC. Differential phosphorylation of RhoGDI mediates the distinct cycling of Cdc42 and Rac1 to regulate second-phase insulin secretion. J Biol Chem. 2010;285:6186–6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Nevins AK, Thurmond DC. Caveolin-1 functions as a novel Cdc42 guanine nucleotide dissociation inhibitor in pancreatic beta-cells. J Biol Chem. 2016;281:18961–18972. [DOI] [PubMed] [Google Scholar]
- [20].Hodge RG, Ridley AJ. Regulating Rho GTPases and their regulators. Nat Rev Mol Cell Biol. 2016;17:496–510. [DOI] [PubMed] [Google Scholar]
- [21].DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15:356–363. [DOI] [PubMed] [Google Scholar]
- [22].Cherfils J, Zeghouf M. Regulation of small GTPases by GEFs, GAPs, and GDIs physiol. Rev. 2013;93:269–309. [DOI] [PubMed] [Google Scholar]
- [23].Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J. 2005;390:1–9.16083425 [Google Scholar]
- [24].Olofsson B. Rho guanine dissociation inhibitors: pivotal molecules in cellular signalling. Cell Signal. 1999;11:545–554. [DOI] [PubMed] [Google Scholar]
- [25].Garcia-Mata R, Boulter E, The BK. ‘invisible hand’: regulation of RHO GTPases by RHOGDIs. Nat Rev Mol Cell Biol. 2011;12:493–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hohmeier HE, Mulder H, Chen G, et al. Isolation of INS-1 derived cell lines with robust ATP-sensitive K+channel-dependent and –independent glucose-stimulated insulin secretion. Diabetes. 2000;49:424–430. [DOI] [PubMed] [Google Scholar]
- [27].McDonald P, Veluthakal R, Kaur H, et al. Biologically active lipids promote trafficking and membrane association of Rac1 in insulin-secreting INS 832/13 cells. Am J Physiol Cell Physiol. 2007;292:C1216–20. [DOI] [PubMed] [Google Scholar]
- [28].Taha MS, Nouri K, Milroy LG, et al. Subcellular fractionation and localization studies reveal a direct interaction of the fragile X mental retardation protein (FMRP) with nucleolin. PlosOne. 2014;9:e91465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Seidler NW. Compartmentalization of GAPDH. AdvExp Med Biol. 2013;985:61–101. [DOI] [PubMed] [Google Scholar]
- [30].Tisdale EJ, Kelly C, Artalejo CR. Glyceraldehyde-3-phosphate degydrogenase interacts with Rab2 and plays essential role in endoplasmic reticulum to Golgi transport excusive of its glycolytic activity. J Biol Chem. 2004;279:54046–54052. [DOI] [PubMed] [Google Scholar]
- [31].Ngo AT, Thierheimer ML, Ö B, et al. Assessment of roles for the Rho-specific guanine nucleotide dissociation inhibitor Ly-GDI in platelet function: a spatial systems approach. Am J Physiol Cell Physiol. 2017;312:C527–C536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kowluru A. Friendly, and not so friendly, roles of Rac1 in islet beta-cell function: lessons learnt from pharmacological and molecular biological approaches. Biochem Pharmacol. 2011;81:965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Syed I, Jayaram B, Subasinghe W, et al. Tiam1/Rac1 signaling pathway mediates palmitate-induced ceramide-sensitive generation of superoxides and lipid peroxides and loss of mitochondrial membrane potential in pancreatic beta-cells. Biochem Pharmacol. 2010;80:874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sidarala V, Veluthakal R, Syeda K, et al. Phagocyte-like NADPH oxidase (Nox2) promotes activation of p38MAPK in pancreatic β-cells under glucotoxic conditions: evidence for a requisite role for Ras-related C3 botulinum toxin substrate 1 (Rac1). Biochem Pharmacol. 2015;95:301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sidarala V, Kowluru A. Exposure to chronic hyperglycemic conditions results in Ras-related C3 botulinum toxin substrate 1 (Rac1)-mediated activation of p53 and ATM kinase in pancreatic β-cells. Apoptosis. 2017;22:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kowluru A, Kowluru RA. RACking up ceramide-induced islet β-cell dysfunction. Biochem Pharmacol. 2018;154:161–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Groysman M, Russek CSN, Katzav S. Vav, a GDP/GTP nucleotide exchange factor, interacts with GDIs, proteins that inhibit GDP/GTP dissociation. FEBS Lett. 2000;467:75–80. [DOI] [PubMed] [Google Scholar]
- [38].Groysman M, Hornstein I, Alcover A, et al. Vav1 and Ly-GDI two regulators of Rho GTPases, function cooperatively as signal transducers in T cell antigen receptor-induced pathways. J Biol Chem. 2002;277:50121–50130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Individual data points are shown in the Figures. Tabulated data are available upon request from the corresponding author.