

ABSTRACT
Mineralocorticoid and androgen receptor antagonist, spironolactone, was recently identified as an inhibitor of nucleotide excision repair (NER), acting via induction of proteolysis of TFIIH component Xeroderma Pigmentosum B protein (XPB). This activity provides a strong rationale for repurposing spironolactone for cancer therapy. Here, we report that the spironolactone-induced XPB proteolysis is mediated through ubiquitin-selective segregase, valosin-containing protein (VCP)/p97. We show that spironolactone induces a dose- and time-dependent degradation of XPB but not XPD, and that the XPB degradation is blocked by VCP/p97 inhibitors DBeQ, NMS-873, and neddylation inhibitor MLN4924. Moreover, the cellular treatment by VCP/p97 inhibitors leads to the accumulation of ubiquitin conjugates of XPB but not XPD. VCP/p97 knockdown by inducible shRNA does not affect XPB level but compromises the spironolactone-induced XPB degradation. Also, VCP/p97 interacts with XPB upon treatment of spironolactone and proteasome inhibitor MG132, while the VCP/p97 adaptor UBXD7 binds XPB and its ubiquitin conjugates. Additionally, ATP analog-mediated inhibition of Cdk7 significantly decelerates spironolactone-induced XPB degradation. Likewise, engaging TFIIH to NER by UV irradiation slows down spironolactone-induced XPB degradation. These results indicate that the spironolactone-induced XPB proteolysis requires VCP/p97 function and that XPB within holo-TFIIH rather than core-TFIIH is more vulnerable to spironolactone-induced proteolysis. Abbreviations NER: nucleotide excision repair; TFIIH: transcription factor II H; CAK: Cdk-activating kinase (CAK) complex; XPB: Xeroderma Pigmentosum type B; VCP/p97: valosin-containing protein/p97; Cdk7: cyclin-dependent kinase 7; NAE: NEDD8-activating enzyme; IP: immunoprecipitation
KEYWORDS: Nucleotide excision repair, transcription factor II H, xeroderma pigmentosum type B, spironolactone, proteasome, VCP/p97, Cdk-activating kinase complex, cyclin-dependent kinase 7, neddylation
Introduction
Nucleotide excision repair (NER), one of the major DNA repair pathways, removes a broad variety of double-helix-distorting DNA lesions, including UV-induced cyclobutane pyrimidine dimers (CPD), 6–4 photoproducts (6–4PP), and polyaromatic compound-induced bulky adducts, as well as cisplatin-generated DNA lesions [1,2]. NER consists of two subpathways: global genomic NER (GG-NER) that removes DNA damage from the entire genome, and transcription-coupled NER (TC-NER) that eliminates lesions located on actively transcribed genes. These subpathways differ in how DNA lesions are recognized in the early steps of NER. In GG-NER, the damage-induced DNA distortion is sequentially recognized by DDB and Xeroderma Pigmentosum (XP) C (XPC) protein complexes [3,4]. Whereas in TC-NER, lesions are detected by RNA polymerase II (RNAPII) in coordination with the recognition of stalled RNAPII by Cockayne syndrome protein A (CSA) and B (CSB) [5–7]. In both subpathways, transcription factor II H (TFIIH) protein complex is recruited via its interaction with XPC or stalled RNAPII to damage sites [7,8]. Other NER factors, such as XPA and RPA are believed to join the TFIIH-containing repair complex to verify the nature of DNA alteration[9]. The two endonucleases XPG and XPF-ERCC1 are recruited to TFIIH and are responsible for the dual incision to remove lesion-containing oligo[10]. Subsequently, the gap-filling DNA synthesis is performed by the concerted action of pol δ or ε, which are enlisted by the cofactors PCNA, RF-C, and RPA.
Mammalian TFIIH (also referred to as holo-TFIIH) is comprised of a core-TFIIH complex, containing seven subunits XPB, XPD, p62, p52, p44, p34, and p8/TTD-A, and a Cdk-activating kinase (CAK) complex, containing three subunits Cdk7, cyclin H, and MAT1 [11–14]. TFIIH participates in transcription, NER, and cell cycle control. In NER, XPB and XPD helicases of TFIIH are involved in unwinding the DNA duplex around the lesion[13]. During transcription, TFIIH, together with other basal transcription factors, e.g. TFIIB, TFIID, TFIIE, and TFIIF, functions in transcription initiation and promoter escape[15]. In the latter processes, Cdk7 of CAK mediates, at least partially, the phosphorylation of the carboxyl-terminal domain (CTD) of Rpb1, the largest subunit of RNAPII[16]. However, CAK is dispensable for NER reaction [17–19]. In cellular NER, CAK appears to be released from the holo-TFIIH during NER and the release of the CAK promotes dual incision[20].
Despite its importance in NER and transcription, very little is known about how TFIIH is regulated within the cells. Alekseev et al. has reported an ubiquitin-mediated XPB degradation by cellular treatment of spironolactone[21], which is a mineralocorticoid and androgen receptor antagonist clinically used to treat hypertension, congestive heart failure[22], and various dermatological conditions [23,24]. The spironolactone-induced XPB degradation impairs NER and consequently, potentiates the cytotoxicity of platinum derivatives toward cancer cells. Recently, it was found that NER inhibition by spironolactone increases the sensitivity and overcomes resistance to alkylating agents in multiple myeloma[25]. It is also reported that spironolactone depletes XPB and inhibits DNA damage response in human skin[26]. These studies, with the observations that spironolactone inhibits DNA damage response in cancer stem cells and homology-directed DNA repair[27], provide a strong rationale for repurposing spironolactone as an adjuvant in cancer therapy. The mechanisms underlying spironolactone’s action in DNA damage response and DNA repair pathways, however, are currently unknown.
The ubiquitin-mediated protein degradation often requires the assistance of ubiquitin-specific segregase, valosin-containing protein (VCP)/p97, when the degradation-destined proteins are tightly bound to their partners within multiprotein complexes, membranes, and chromatin [28,29]. VCP/p97 is an ATP-driven molecular chaperone that belongs to ATPase-associated with various cellular activities (AAA) family [28,30]. VCP/p97 has two characteristic properties, the ATPase activity, and the ability to bind to ubiquitin[31]. These properties enable VCP/p97 to segregate ubiquitinated proteins from their tightly bound partners and present ubiquitinated proteins to the proteasome for proteolysis. For different cellular functions, VCP/p97 cooperates with different sets of mutually exclusive cofactors and additional adaptors, e.g. VCP/p97 cooperates with many adaptors from the UBX (ubiquitin regulatory X) family. Among them, UBXD7 has been shown to participate in ubiquitin-mediated degradation of hypoxia-inducible factor 1α[32], NER factors DDB2[33], XPC [33,34] and CSB[35]. The UBXD7 possesses ubiquitin-associated (UBA) domain, ubiquitin-associating (UAS) domain, ubiquitin-interacting motif (UIM), and ubiquitin regulatory X (UBX) domain, and these domains enable UBXD7 to link VCP/p97 to ubiquitinated clients and specific E3 ligases [29,32].
We have recently demonstrated that VCP/p97 is involved in CSB and human RNAPII degradation in damaged chromatin [35,36]. In the present work, we have investigated the molecular mechanism of selective destabilization of XPB by spironolactone. We show that inhibitors of the proteasome, neddylation and VCP/p97 all compromise spironolactone-induced degradation, and that the VCP/p97 complex is involved in such a process by selectively binding to ubiquitin-conjugated XPB and presenting it to the proteasome for proteolysis. Moreover, we showed that the spironolactone-induced degradation requires Cdk7 ATPase activity from the CAK complex of holo-TFIIH. The core TFIIH, pre-engaged with NER, is resistant to spironolactone-induced degradation. Our results suggest a model in which the spironolactone induces degradation of the XPB within holo-TFIIH, impacting TFIIH’s function in NER, transcription, and other DNA damage responses.
Results
Inhibition of proteasome, VCP/p97 or neddylation prevents spironolactone-induced XPB degradation
As shown in Figure 1(a), a dose-dependent XPB loss was seen 2 h after treatment with 10 µM spironolactone. The degree of XPB loss proceeded with a time of exposure and the XPB signal mostly disappeared after 4 to 6 h post-treatment. The XPB loss was intensified upon increasing the concentration of spironolactone from 10 to 30 µM. Simultaneous treatment with proteasome inhibitor MG132 prevented spironolactone-induced XPB proteolysis (Figure 1(b)). To some extent, DBeQ, a selective and reversible inhibitor of VCP/p97 ATPase reduced the spironolactone-induced XPB loss at both 4 and 6 h at 10 µM concentration (Figure 1(b)). We also examined the effect of neddylation inhibitor MLN4924. This chemical inhibits neddylation of cullins thereby disables ubiquitination mediated through cullin-based E3 ubiquitin ligases. As shown in Figure 1(b), MLN4924 effectively overcomes the spironolactone-induced XPB loss. By contrast, the cellular level of XPD, another integral component of TFIIH, was unaffected by spironolactone as well as MG132, DbeQ or MLN4924.
Figure 1.
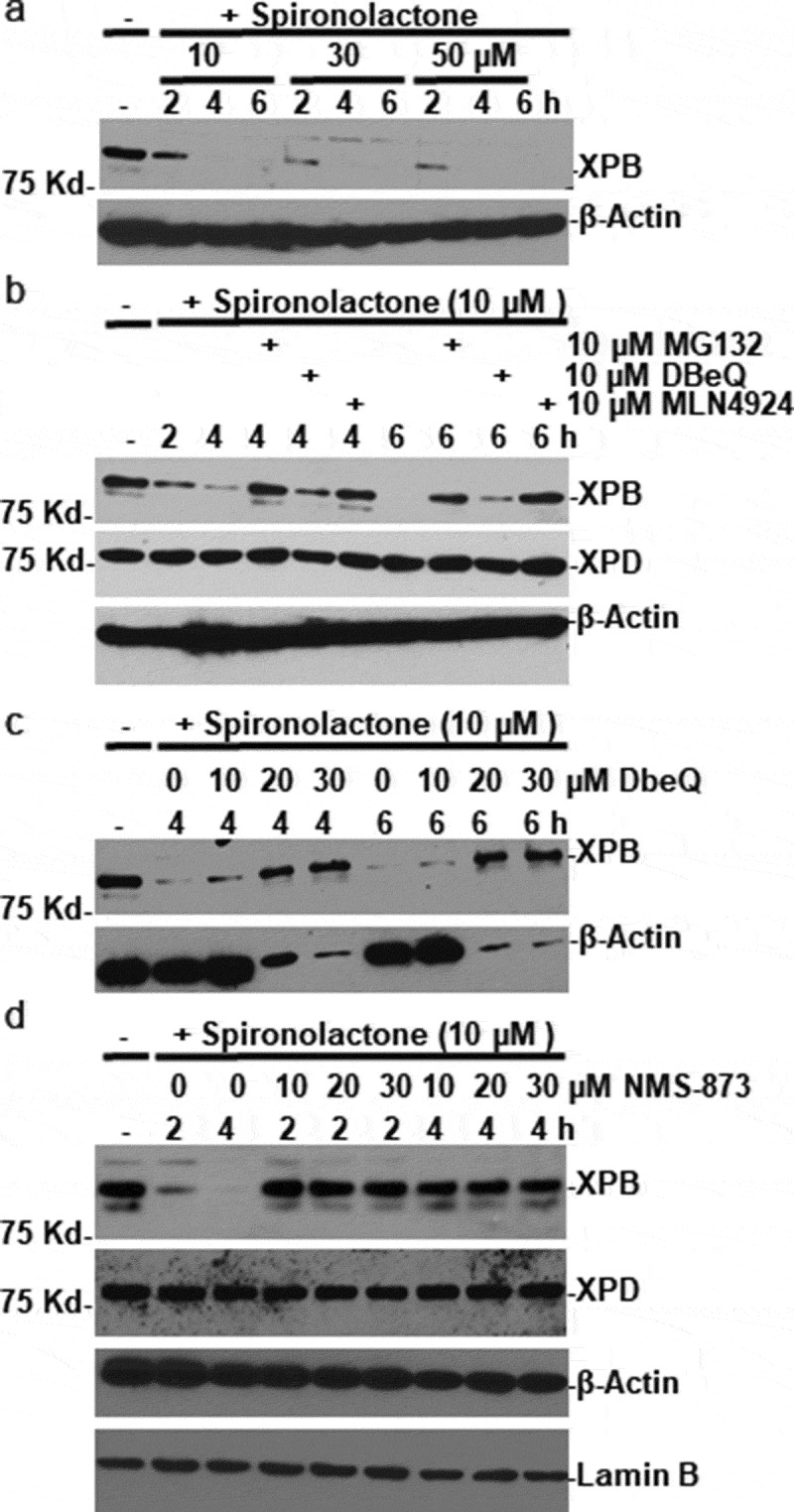
VCP/p97 inhibitors prevent spironolactone-induced XPB degradation. (a) HeLa cells were treated with spironolactone at 10, 30 or 50 M and harvested at indicated time points. The XPB levels in cell extracts were detected by Western blotting. The β-Actin blots serves as loading control. (b) HeLa cells were treated with spironolactone at 10 M alone or with proteasome inhibitor MG132 (10 M), or VCP/p97 inhibitor DBeQ (10 M) or NAE/neddylation inhibitor MLN4924 (10 M) for indicated time period. The XPB and XPD levels in cell extracts were detected. (c) HeLa cells were treated with spironolactone at 10 M alone or with increasing dose of DBeQ at 10, 20 and 30 M. The XPB and β-Actin levels in cell extracts were detected as Figure 1(a). (d) HeLa cells were treated with spironolactone at 10 M alone or with increasing dose of VCP/p97 inhibitor NMS-873 at 10, 20 and 30 M. The XPB, XPD, β-Actin and Lamin B proteins in cell extracts were detected by Western blotting. The Lamin B blots serve as loading control
To ascertain whether VCP/p97 is involved in spironolactone-induced XPB degradation, we examined the dose-dependent response of DBeQ inhibitor. As shown in Figure 1(c), spironolactone-induced XPB loss was effectively inhibited by increased DBeQ doses of 20 and 30 µM at both 4 and 6 h. Oddly, a significant loss of unrelated β-actin was also seen at these DBeQ doses. Thus, we tested an additional inhibitor NMS-873, which is a more potent and selective allosteric inhibitor of VCP/p97. As shown in Figure 1(d), simultaneous treatment with NMS-873 for 2 and 4 h effectively prevented spironolactone-induced XPB proteolysis at 10 µM, with no obvious effect on β-actin. Importantly, the cellular level of XPD was not affected by either spironolactone or NMS-873 treatments. Thus, we hypothesized that VCP/p97 is involved in spironolactone-induced XPB degradation.
Inhibition of VCP/p97 leads to accumulation of spironolactone-induced ubiquitin conjugates of XPB but not XPD
VCP/p97 complex usually acts as an ubiquitin-specific segregase to extract its ubiquitinated clients from their binding partners and presents the clients to the proteasome for proteolysis. To examine if VCP/p97 complex acts accordingly in the case of spironolactone-induced XPB degradation, we analyzed ubiquitin conjugates of XPB under different conditions. The HeLa-Ub5 cells that stably express 6xHis-tagged ubiquitin were treated with spironolactone alongside with VCP/p97 inhibitor NMS-873 or proteasome inhibitor MG132 for 6 h, and the whole-cell extracts were subjected to Ni-NTA agarose-based pulldown, followed by Western blotting for XPB-ubiquitin conjugates. As shown in Figure 2(a), spironolactone induced the typical XPB degradation in HeLa-Ub5 cells. The degradation was prevented by simultaneous treatment with VCP/p97 or proteasome inhibitors. As expected, the XPB-ubiquitin conjugates, as detected by XPB antibody, appeared in samples with simultaneous spironolactone and NMS-873 or MG132 treatment (Figure 2(a), upper panel), indicating that both NMS-873 and MG132 treatments allowed the XPB-ubiquitin conjugates to accumulate. The XPB-ubiquitin conjugates in NMS-873 treated cells appeared to be a wide range of molecular mass, while the ubiquitin conjugates in MG132 treated cells appeared to be of densely modified high molecular mass.
Figure 2.
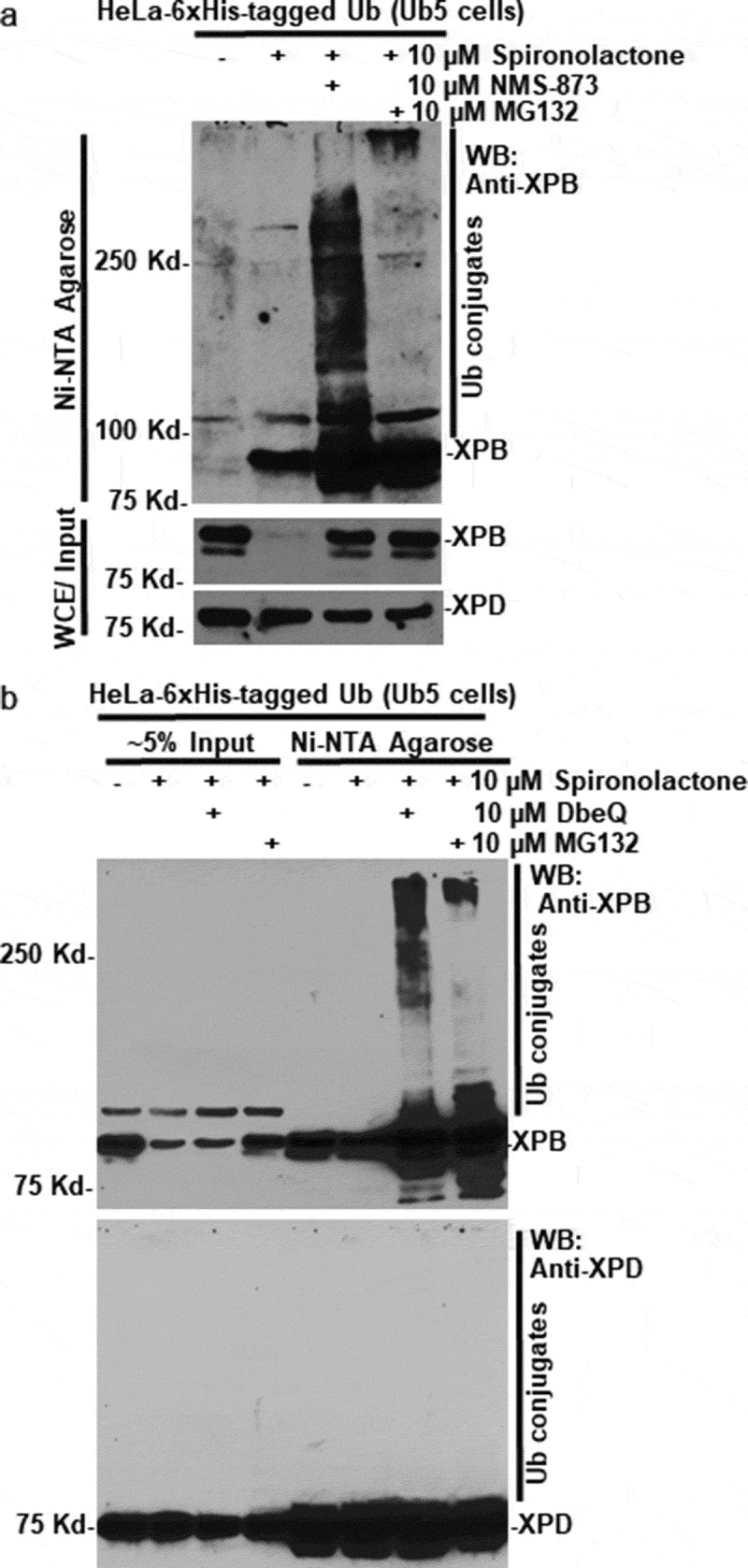
VCP/p97 inhibition leads to accumulation of spironolactone-induced ubiquitin conjugates of XPB. (a) HeLa-Ub5 cells, harboring a 6xHis-tagged ubiquitin transgene, were mock-treated or treated with 10 µM spironolactone alone, or together with VCP/p97 inhibitor NMS-873 at 10 µM or proteasome inhibitor MG132 at 10 µM. Whole-cell extracts (WCE) in RIPA buffer were subjected to a pulldown assay by Ni-NTA agarose. XPD, XPB and XPB-ubiquitin conjugates in Ni-NTA precipitates and in WCE were detected by western blotting. (b) as in Figure 2(a), HeLa-Ub5 cells were mock-treated or treated with spironolactone alone, or together with VCP/p97 inhibitor DbeQ at 10 µM or proteasome inhibitor MG132. XPD, XPB and XPB-ubiquitin conjugates in Input and in Ni-NTA precipitates were detected by Western blotting
We performed similar experiments with VCP/p97 inhibitor DbeQ (Figure 2(b)). The XPB-ubiquitin conjugates distinctly appeared in the samples treated with spironolactone along with DbeQ or MG132 treatment (Figure 2(b), upper panel). Interestingly, the XPB-ubiquitin conjugates in DBeQ-treated cells also appeared to be a wide range of molecular mass different than that in MG132 treated cells. In contrast, the XPD-ubiquitin conjugates were not detected under the same condition (Figure 2(b), lower panel). The appearance of XPB and XPD in mock-treated pulldown samples is likely due to overexposure, which was necessary for clearly detecting XPB or XPD-ubiquitin conjugate. We concluded that XPB but not XPD specifically undergoes spironolactone-induced ubiquitination, and VCP/p97 inhibition allows the accumulation of XPB-ubiquitin conjugates.
VCP/p97 knockdown prevents spironolactone-induced XPB degradation
We next examined the effect of VCP/p97 ablation on spironolactone-induced XPB degradation by shRNA-mediated VCP/p97 knockdown. In HEK293-V1-shVCP cells that have a stably integrated doxycycline-inducible VCP shRNA, doxycycline treatment provoked a progressive decline of VCP/p97 within 6 days of induction. On day 6, VCP/p97 protein level was significantly reduced (Figure 3(a)). Meanwhile, the levels of XPB and XPD were not noticeably affected by VCP shRNA induction, suggesting the native cellular XPB and XPD levels were not tightly controlled by VCP/p97. As expected, the level of β-actin was unaffected by VCP/p97 knockdown.
Figure 3.
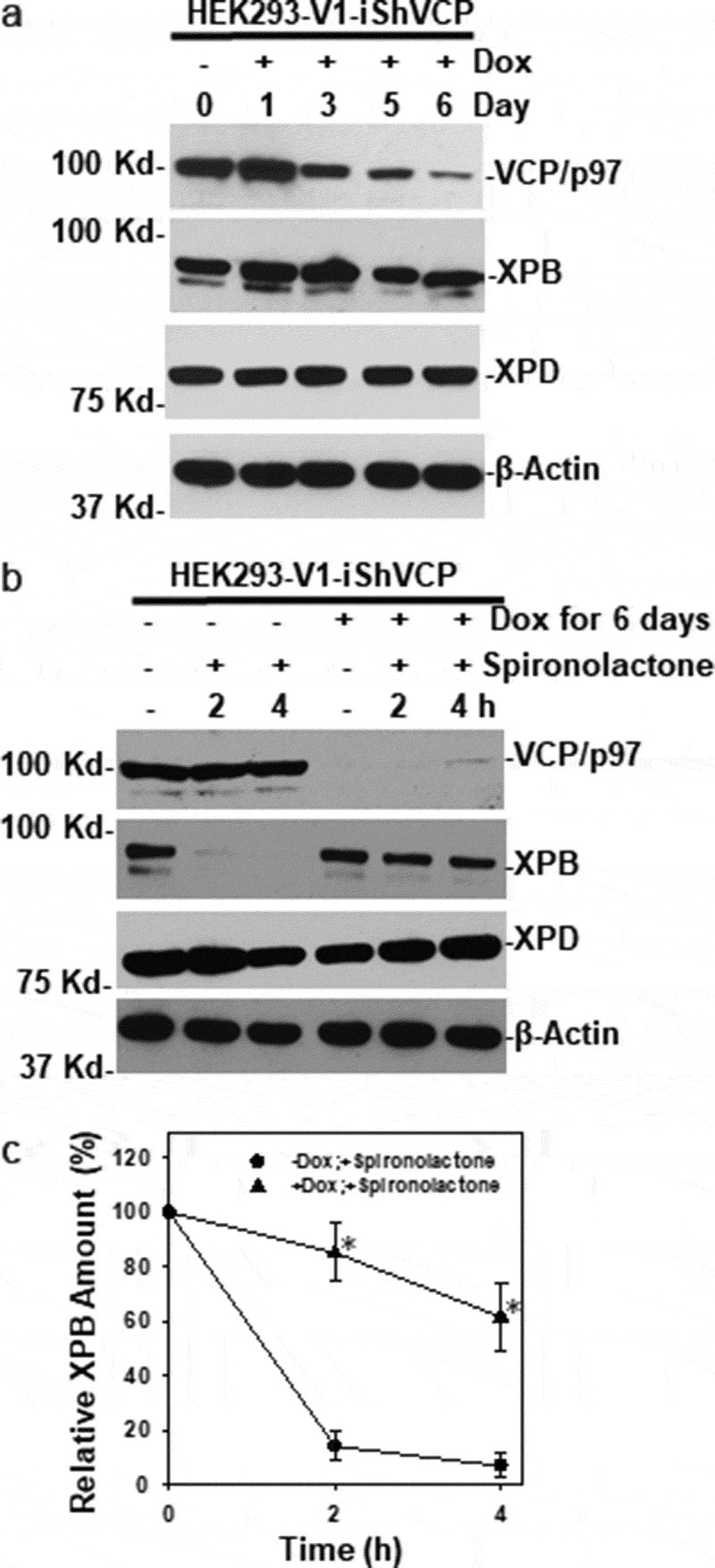
VCP/p97 knockdown compromises spironolactone-induced XPB degradation. (a) HEK293-V1-iShVCP cells that have a stably integrated, doxycycline-inducible VCP shRNA transgene were induced with doxycycline (1 µg/ml) for 1 to 6 days to express shRNA targeting VCP/p97. The cell extracts were made and the levels of VCP/p97, XPB, XPD and β-Actin were detected by Western blotting. (b) HEK293-V1-iShVCP cells were induced with doxycycline at 1 µg/ml for 6 days for knocking down VCP/p97. The cells were then treated with spironolactone at M for 2 or 4 h. The cell extracts from doxycycline and spironolactone treated or mock-treated cells were detected for VCP/p97, XPB, XPD and β-Actin. (c) Western blotting images from 3 independent experiments were quantitatively examined by ImageJ. The relative amount of XPB was calculated in comparison to the control without doxycycline induction and spironolactone treatment. Symbols (*) indicate p ≤ 0.05 in paired student’s-t-tests
Next, we investigated the spironolactone-induced XPB degradation upon cellular VCP/p97 ablation. On day 6 of doxycycline induction, HEK293-V1-shVCP cells were further treated with spironolactone at 10 µM or mock-treated for 2 or 4 h, and cellular XPB levels were examined in protein extracts. As shown in Figure 3(b), spironolactone caused significant loss of cellular XPB after 2 and 4 h spironolactone treatment in HEK293-V1-shVCP cells without doxycycline induction. Whereas VCP/p97 ablation by doxycycline induction largely prevented the loss of cellular XPB caused by spironolactone (Figure 3(b,c)). For example, at 4 h after spironolactone treatment, ~60% cellular XPB was still detected upon doxycycline induction, while only ~10% cellular XPB could be seen in the absence of doxycycline induction. Notably, a fraction of spironolactone-induced XPB degradation still occurred at 4 h under VCP/p97 ablation, and this partial degradation is likely independent of VCP/p97. Under the same conditions, the XPD level was not significantly changed. Taken together, our results indicated that the VCP/p97 function is primarily required for the spironolactone-induced XPB degradation.
Spironolactone induces interaction between XPB and VCP/p97 complex
We next examined the physical interaction between XPB and VCP/p97 complex. As shown in Figure 4(a), VCP/p97 was present in anti-XPB immunoprecipitates from cells treated with spironolactone alone and in combination with MG132 but was not present in anti-XPB immunoprecipitates from mock-treated cells. As expected, XPD was present in anti-XPB immunoprecipitates from mock-treated cells, and from the cells with dual spironolactone and MG132 treatment, but was less detectable in anti-XPB immunoprecipitates from the cells treated solely with spironolactone, where XPB loss was evident (Figure 4(a), Input panel).
Figure 4.
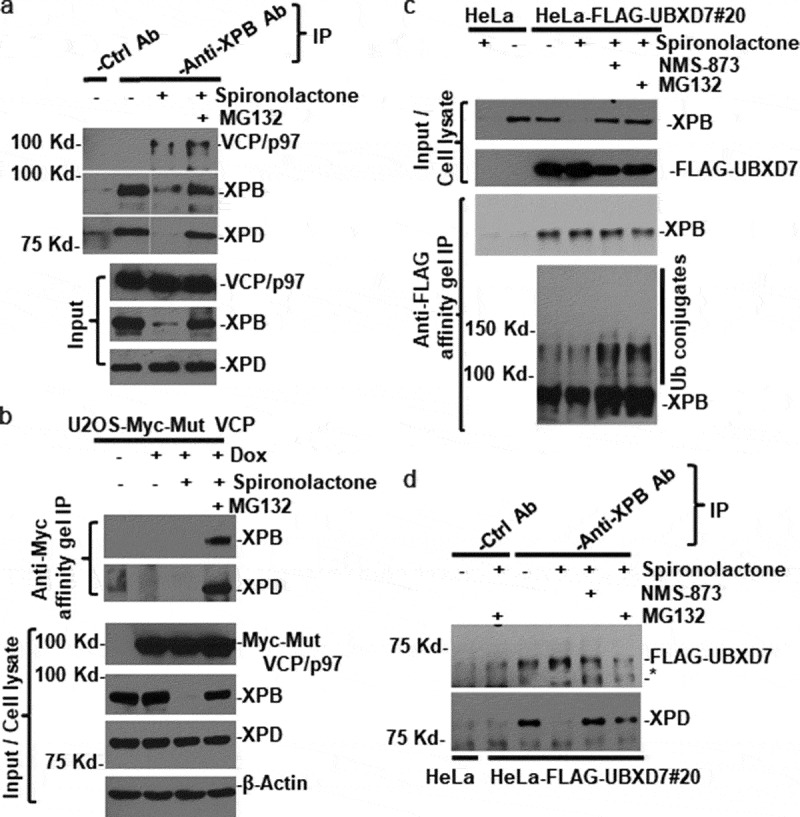
VCP/p97 physically interacts with XPB in spironolactone-treated cells. (a) HeLa cells were mock-treated or treated with spironolactone at 10 M or together with proteasome inhibitor MG132 at 10 M for 6 h. The cell extracts were made in RIPA buffer and subjected to immunoprecipitation with anti-XPB specific or control antibodies. The immunoprecipitates and input cell extracts were detected for presence of VCP/p97, XPB and XPD. All Western blotting analyses were done with single polyacrylamide gel. Nonessential lanes were digitally removed from XPB and XPD original diagrams. (b) U2OS-Myc-Mut VCP cells that have a stably integrated, doxycycline-inducible Myc epitope-tagged mutant VCP/p97 were induced with doxycycline (1 µg/ml) for 20 h to express epitope-tagged VCP/p97. The cells were then treated with mock-treated or treated with spironolactone as that in Figure 4(a). The cell extracts were subjected to immunoprecipitation with anti-Myc affinity gels. The immunoprecipitates and input cell lysates were detected for Myc-tagged VCP/p97, XPB and XPD as well as β-Actin. (c) HeLa cells and HeLa-derived HeLa-FLAG-UBXD7#20 cells that stably express FLAG epitope-tagged UBXD7 were mock-treated or treated for 6 h. with 10 M spironolactone, NMS-873 and MG132 alone or in combination. The cell extracts were made in Buffer A and subjected to immunoprecipitation with anti-FLAG agarose gels. The immunoprecipitates and cell extracts were detected for XPB and FLAG-tagged UBXD7. In lower image panel of anti-FLAG gel immunoprecipitation, anti-XPB blotting image was over-exposed to show ubiquitin (Ub) conjugates of XPB. (d) the same cell extracts as in Figure 4(c) were subjected to immunoprecipitation with anti-XPB and control antibody and the immunoprecipitates were detected for FLAG-UBXD7 and XPD by Western blotting. Asterisk (*) marks nonspecific band in anti-FLAG Western blotting
We further investigated the spironolactone-induced binding of VCP/p97 to XPB, using a genetically engineered U2OS cell line which harbors a doxycycline-inducible Myc-tagged mutant (E578Q/EQ) VCP/p97 transgene. The EQ mutant is deficient in ATP-hydrolysis activity of VCP/p97, but still able to bind ubiquitinated substrates[37]. Following induction, the engineered U2OS cells were mock-treated or treated with spironolactone alone or in combination with MG132. As expected, doxycycline induced a robust expression of mutant VCP/p97 in these cells, and such an induction did not affect spironolactone-induced XPB degradation and XPD expression (Figure 4(b), lower panel). In anti-Myc affinity gel immunoprecipitates, XPB and XPD were only detected in the immunoprecipitates from cells with dual spironolactone and MG132 treatments, suggesting that the mutant VCP/p97 is associated with XPB in the form of TFIIH in such-treated cells.
We next explored the physical interaction between XPB and UBXD7 adapter of the VCP/p97 segregase complex. The UBXD7 was FLAG-tagged and stably expressed in a HeLa-FLAG-UBXD7#20 cell line, and the cells were treated with spironolactone alone or in combination with NMS-873 or MG132, and immunoprecipitation was conducted using anti-FLAG affinity gels. As shown in Figure 4(c), XPB protein was detected in immunoprecipitates from HeLa-FLAG-UBXD7#20 cells but not in control HeLa cells. The XPB-UBXD7 association was seen to be independent of spironolactone, NMS-873, or MG132 treatments. Nevertheless, the ubiquitinated XPB forms, appearing as higher molecular masses than native XPB, were seen in immunoprecipitates from HeLa-FLAG-UBXD7#20 cells subjected to dual treatment with spironolactone and NMS-873 or MG132 (Figure 4(c), bottom panel). In a reciprocal immunoprecipitation using the same cell extracts from HeLa-FLAG-UBXD7#20 cells, FLAG-UBXD7 was detected in anti-XPB immunoprecipitates in cells regardless of spironolactone and NMS-873 or MG132 treatment (Figure 4(d)). Thus, UBXD7 binds to both XPB and its ubiquitinated forms.
Spironolactone-induced XPB degradation requires Cdk7 ATPase activity from CAK complex of TFIIH
As mentioned above, the holo-TFIIH contains XPB, XPD in core TFIIH and Cdk7 kinase in CAK complexes. We asked whether Cdk7 plays a role in the process of spironolactone-induced XPB degradation. We utilized a HCT116-Cdk7as/as cell line, in which the two wild-type alleles of the Cdk7 gene were replaced with analog-sensitive (as) mutant alleles engineered to accommodate bulky, unnatural ATP analogs in their enzymatic active sites[38]. We pretreated HCT116-Cdk7as/as cells with 10 µM 1-NMPP1 for 14 h to inhibit Cdk7’s ATPase activity. In our previous study, the same 1-NMPP1 treatment effectively inhibited phosphorylation of Cdk2, a biological downstream target of Cdk7[39]. In this study, 1-NMPP1 significantly slowed down spironolactone-induced XPB degradation in HCT116-Cdk7as/as cells, as depicted by the comparisons of the time course of XPB levels in Figure 5(a,b).
Figure 5.
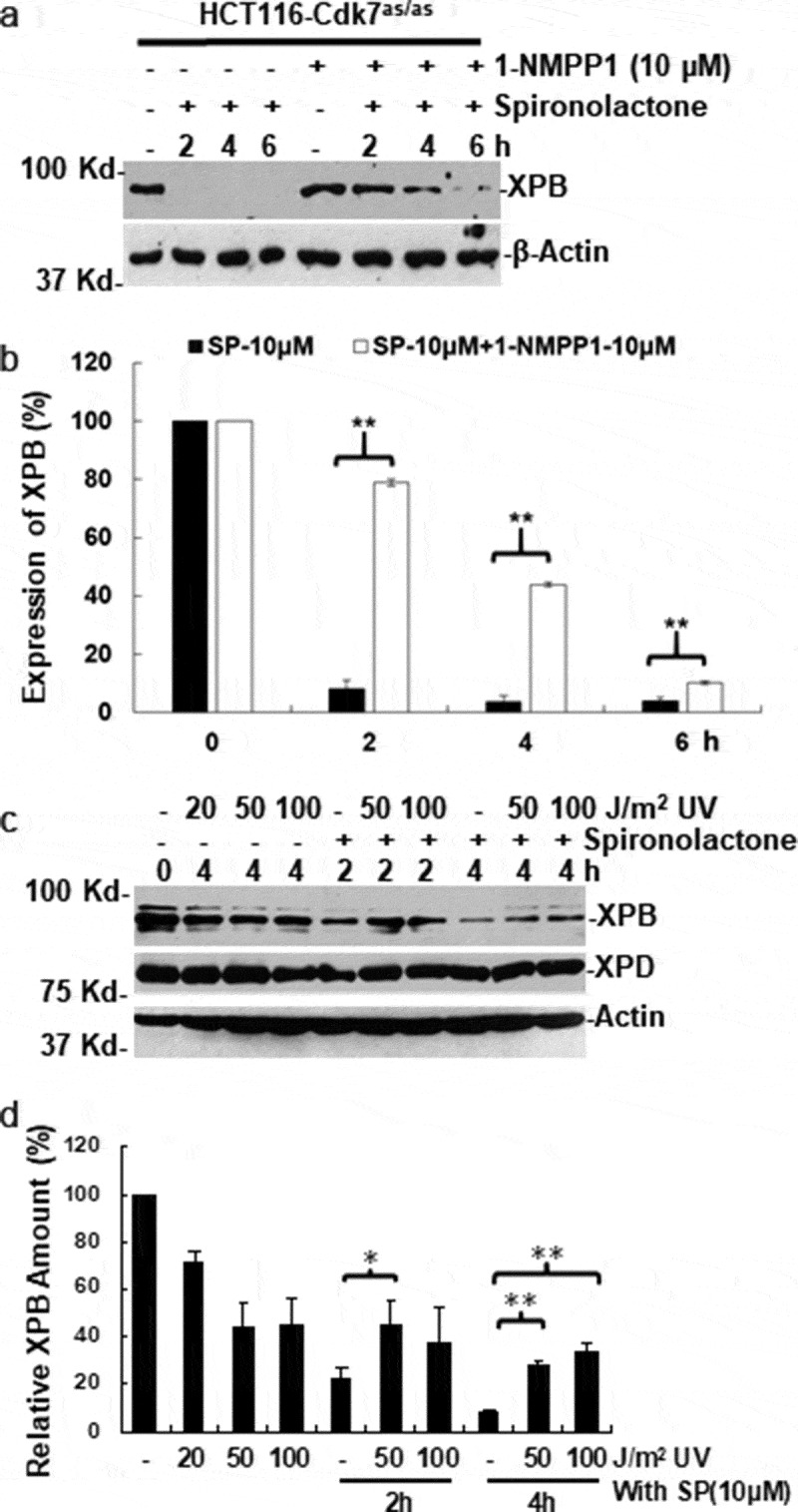
Spironolactone‐induced XPB degradation requires Cdk7 ATPase activity form CAK complex. (a) HCT116-Cdk7as/as cells, harboring homologous ATP analog-sensitive (as) Cdk7 gene, were pretreated with 10 µM 1-NMPP1 or mock-treated for 14 h. The cells were then further treated with 10 µM spironolactone for 2, 4 or 6 h or mock-treated. The XPB levels in cell lysates were detected by Western blotting. The β-Actin blots serves as loading control. (b) the Western blotting images from 3 independent experiments in Figure 5(a) were quantitatively examined by ImageJ. The relative amount of XPB was calculated in comparison to the control without 1-NMPP1 and spironolactone treatment. (c) HeLa cells were pretreated with UV radiation at 50 or 100 J/m2, and allowed to repair DNA photolesions for 1 h. The cells were further treated with spironolactone or mock-treated and harvested at indicated time points. The XPB, XPD and β-Actin levels were detected by Western blotting. (d) the Western blotting images from 4 independent experiments in Figure 5(c) were quantitatively examined by ImageJ. Asterisk symbols * and ** indicate p ≤ 0.05 and in p ≤ 0.01 paired student’s-t-tests, respectively
It has been reported that TFIIH changes its subunit composition in response to DNA damage[20]. The CAK complex is released from holo-TFIIH during NER and reassembled with core TFIIH into holo-TFIIH following the completion of NER. We, therefore, questioned if the core TFIIH engaged in NER is resistant to spironolactone-induced XPB degradation. We pretreated the HeLa cells with UV and allowed the cells to repair for 1 h, and then treated the cells with spironolactone to examine XPB degradation. As shown in Figure 5(c,d), increasing doses of UV alone caused a fair decrease in XPB levels. However, the pre-UV treatment at 50 J/m2 blocked the spironolactone-induced XPB degradation at 2 h of spironolactone treatment. This effect was more pronounced upon pre-UV treatment with 50 and 100 J/m2 at 4 h. These results are consistent with the notion that Cdk7 kinase is required for spironolactone-induced XPB degradation. We surmise that due to the release of CAK during engagement of TFIIH in NER, XPB within core TFIIH becomes resistant to spironolactone-induced degradation.
Discussion
Spironolactone-induced XPB degradation was first reported by Alekseev et al.[21], where spironolactone was identified as an inhibitor of NER of UV-induced 6–4PP, independent of spironolactone’s aldosterone antagonist function. A more recent study reported that SCFFBXL E3 ligase was identified as an E3 ligase in such spironolactone-induced XPB degradation[40]. In this study, we further revealed that ubiquitin-specific segregase VCP/p97 plays a crucial role in this XPB proteolytic process. Our observations are based on studies of spironolactone-induced XPB degradation, regarding (a) specific VCP/p97 inhibition, (b) inducible shRNA-mediated VCP/p97 knockdown, (c) accumulation of XPB-ubiquitin conjugates, and (d) interaction between XPB and VCP/p97 complex. In our experiments, both VCP/p97 inhibition and VCP/p97 knockdown compromise spironolactone-induced XPB degradation (Figures 1 and 3). The VCP/p97 inhibition also leads to the accumulation of XPB-ubiquitin conjugates (Figure 2). These results are consistent with the general role of VCP/p97 in extracting ubiquitinated clients and presenting them to the proteasome for degradation. On the other hand, XPD, another component of TFIIH is neither degraded by spironolactone induction nor do XPD-ubiquitin conjugates accumulate upon VCP/p97 or proteasome inhibitions. These results strongly suggest that the selective XPB degradation by spironolactone induction occurs as a result of XPB ubiquitination, subsequent extraction by VCP/p97, and proteolysis by proteasome. We, however, do not know if the architecture of TFIIH remains intact after XPB extraction.
UBXD7 is a member of the UBA-UBX protein family, which acts as an adapter of the VCP/p97 complex [29]. The UBA, UAS, UIM, and UBX structural domains in UBXD7 enable its interaction with VCP/p97, ubiquitinated clients, and E3 ubiquitin ligases, including those contain cullin 1, 2, 3, or 4[32]. More specifically, UBXD7 associates with activated cullin-RING ubiquitin ligases, via binding to neddylated/activated cullins [41,42]. In our experiments, neddylation inhibition by MLN4924 effectively blocked spironolactone-induced XPB degradation. In addition, our results revealed the interaction of UBXD7 and XPB as well as its ubiquitinated forms (Figure 4). Taken together, these results suggest that UBXD7 may link cullin-based E3(s) to VCP/p97 in spironolactone-induced XPB degradation. In accord with this notion, a recent study identified a SCFFBXL18 complex as an E3 ligase responsible for spironolactone-induced XPB poly-ubiquitination[40]. SCFFBXL18 contains F-box protein FBXL18, Skp1, cullin 1, and Rbx1. It is interesting to know if neddylation of cullin 1 links SCFFBXL18 to the VCP/p97 pathway via UBXD7 in spironolactone-induced XPB degradation.
Our observations of the impact of Cdk7as/as inhibition and UV on spironolactone-induced XPB degradation suggested that the integrity of holo-TFIIH as well as Cdk7 activity is required for such a rapid XPB degradation. In our experiment, Cdk7as/as inhibition by 1-NMPP1 effectively slowed down spironolactone-induced XPB degradation (Figure 5(a,b)). In this Cdk7as/as chemical-genetic system, inhibition of Cdk7as/as ATPase by 1-NMPP1 is very specific and effective[38]. In our previous study, Cdk7as/as inhibition abolished the preexisting Ser5-phosphorylation of RNAPII large subunit Rpb1 in HCT116-Cdk7as/as cells without UV irradiation but delayed disappearance of Rpb1 after UV irradiation[39]. Given that the CAK within holo-TFIIH is known to phosphorylate Ser5 and Ser7 in the CTD repeat of Rpb1, we speculate that CAK within holo-TFIIH is involved in spironolactone-induced XPB degradation via Cdk7-mediated phosphorylation. It was previously reported that the CAK complex is released during productive NER[20]. In this study, we found that pre-engagement of TFIIH with NER of photolesions renders XPB resistant to spironolactone-induced XPB degradation (Figure 5(c,d)). These observations suggest a plausible mechanistic model where the XPB within holo-TFIIH, rather than the XPB within core TFIIH is particularly vulnerable to spironolactone-induced XPB degradation (Figure 6), where VCP/p97 is involved in extracting ubiquitinated XPB from holo-TFIIH for proteolysis.
Figure 6.
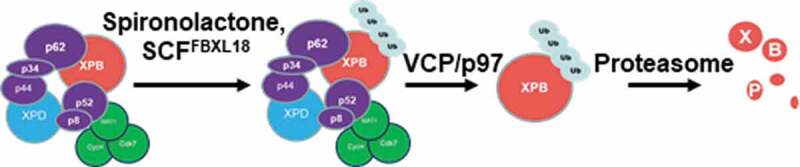
Hypothesized mechanistic model for the spironolactone‐induced XPB degradation. Spironolactone induces ubiquitination of XPB within holo-TFIIH by a putative cullin-based E3 ubiquitin ligase SCFFBXL18, in manner dependent on integrity of holo-TFIIH and ATPase activity of Cdk7 in CAK complex. The ubiquitinated XPB is selectively extracted from holo-TFIIH by VCP/p97 complex and presented to proteasome for degradation
Ueda et al. recently found that Ser90 in XPB is potentially phosphorylated by Cdk7[40]. Mutant XPB containing serine to alanine mutation at amino acid 90 was resistant to spironolactone-induced XPB degradation. XPB phosphorylation is proposed to facilitate spironolactone-induced XPB degradation[43]. Yet, how Cdk7 within CAK contributes to spironolactone-induced XPB degradation remains largely unclear. It would be desirable to characterize XPB as CAK or Cdk7 phosphorylation target. Moreover, it would be also interesting to learn if spironolactone induces the XPB phosphorylation or induces ubiquitination of the phosphorylated XPB by a specific E3 ubiquitin ligase, e.g. SCFFBXL18. Further characterization of the degron within XPB should reveal the detailed mechanisms of spironolactone-induced XPB degradation.
Several inhibitors of XPB and Cdk7 have been developed to curb transcription addiction in cancer cells and pinpoint these activities as potential targets in cancer chemotherapy[44]. Among them, spironolactone acts distinctively by inducing specific degradation of XPB via the ubiquitin-proteasome system. Our demonstration of the involvement of the VCP/p97 pathway in spironolactone-induced XPB degradation adds another regulatory layer to the phenomena. It provides an additional explanation of the selectivity of spironolactone-induced proteolysis toward XPB. Our finding that the XPB within holo-TFIIH is particularly vulnerable to spironolactone-induced XPB degradation implies that spironolactone may be used to target both basic transcription and NER functions of TFIIH to enhance the effectiveness of platinum-based therapy.
Material and methods
Cell lines
HCT116-Cdk7as/as cells were provided by Dr. Robert Fisher (Memorial Sloan–Kettering Cancer Center, NY). The cells were established by replacement of both wild-type Cdk7 alleles with mutant versions, in which the mutation of Phe91 to Gly in Cdk7 leads to an expansion of the ATP binding pocket, rendering the kinase analog selective and sensitive (as) [38]. HEK293-V1-iShVCP cell line, a derivative from HEK293 cells, with a stably integrated, doxycycline-inducible VCP shRNA (DTC204), was a gift from Raymond Deshaies laboratory [45]. U2OS cell lines, stably transfected with the doxycycline-inducible DNA constructs expressing Myc-tagged EQ (E578Q) mutant VCP/p97 were provided by Conrad C. Weihl laboratory [37]. The EQ mutant is deficient in ATP-hydrolysis activity of VCP/p97 but is still able to bind ubiquitinated substrates. HeLa-FLAG-UBXD7#20 cells, stably expressing FLAG-epitope-tagged UBXD7, were generated from HeLa cells by transfection of p3XFLAG-CMV™ vector-based expression construct (Sigma–Aldrich, St. Louis, MO 63,103), selected with G418, and further subcloned by single-cell dilution.
Chemical reagents and antibodies
VCP/p97 inhibitor, N2, N⁴-Dibenzylquinazoline-2,4-diamine (DBeQ), allosteric VCP/p97 inhibitor NMS-873, NEDD8-activating enzyme (NAE) inhibitor MLN4924, CAS 951,950–33-7, and spironolactone, CAS 52–01-7 were all obtained from Sigma–Aldrich (St. Louis, MO 63,103). 1-NMPP1, CAS 221,244–14-0, was purchased from Cayman Chemical (Ann Arbor, Michigan 48,108). Proteasome inhibitor MG132 was purchased from EMB Millipore (Billerica, MA 01821).
Anti-FLAG M2 and anti-Myc antibodies and affinity gels were purchased from Sigma–Aldrich. Anti-VCP/p97 was purchased from Abcam. Antibodies against XPB, XPD, β-actin, lamin B, were from Cell Signaling (Danvers, MA, 01923). Ni-NTA agarose was obtained from Thermo Fisher Scientific (Waltham, MA 02451).
Cell culture, chemical treatment and UV irradiation
HCT116 cells and their derivative cell lines were grown in McCoy’s 5A medium. HeLa, U2OS, and HEK293 cells as well as their derivative cell lines were grown in DMEM medium. All cell growth medium was supplemented with 10% FBS, antibiotics, and proper selective chemicals, and the cultures were kept in a humidified atmosphere of 5% CO2.
The cells in monolayer with 70–80% confluency were supplied with fresh medium and the cells were then mock-treated with vehicle DMSO or treated with spironolactone alone or in combination with VCP/p97 inhibitor DBeQ, or NMS-873, or NAE inhibitor MLN4924 or proteasome inhibitor MG132 at desired concentration for the chosen time period. For pre-UV treatment, the cells in a monolayer were washed twice with PBS and the UV-C was delivered at a dose rate of 0.5 J/m2/sec as measured by Model UVX Digital Radiometer. The UV-irradiated cells were supplied with fresh medium and allowed DNA repair for 1 h, and the cells were then further treated with spironolactone. At the end of cell treatment, the cells were rinsed twice with cold phosphate-buffered saline (PBS) and harvested in 1x SDS lysis buffer, or RIPA buffer, or in Buffer A[42].
Doxycycline (dox) induced knockdown of VCP/p97 and expression of Myc-tagged EQ (E578Q) mutant VCP/p97
HEK293-V1-iShVCP cells were seeded at ~20-30% confluency. Sixteen hours after seeding, the cells were supplied with fresh medium containing 1 µg/ml doxycycline for 3 days. On the third day, the cells were re-seeded at ~20-30% confluency, and induction continued for another 3 days. On day 6, HEK293-V1-iShVCP cells were collected for examination, or the cells were further treated with 10 µM spironolactone in presence of doxycycline for the desired time period. After spironolactone treatment, the cell extracts were made in 1x SDS lysis buffer for further analysis. For examination of the time course of VCP knockdown, samples for day 1 to 3 were collected in the first 3 days of doxycycline induction, while the samples for day 4 to 6 were collected during the second around doxycycline induction after cell re-seeding.
For induction of expression of mutant VCP/p97, the U2OS derived cell lines, expressing doxycycline-inducible Myc-tagged EQ (E578Q) mutant VCP/p97, were seeded at ~50% confluency. Twenty-four hours after seeding, the cells were supplied with fresh medium containing 1 µg/ml doxycycline for 20 h. The cells were further treated with spironolactone at 10 µM for the desired time period, the cell extracts were made in RIPA buffer for immunoprecipitation or Western blotting analysis.
Immunoprecipitation (IP) and Western blotting analysis
The cell extracts were made in RIPA buffer (50 mM Tris-HCl [pH 7.8], 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 10 mM 2-mercaptoethanol, and a complete protease inhibitor cocktail) (Roche Diagnostics, Indianapolis, IN) were used for immunoprecipitation with anti-XPB antibody and with anti-Myc affinity gels or Ni-NTA agarose. In anti-XPB IP, the cell extracts containing ~1 mg protein was precleared with protein A/G agarose beads (Calbiochem, San Diego, CA) and then incubated with 1 µg of specific anti-XPB antibodies in 1 ml RIPA buffer at 4°C overnight, followed by addition of 25 µl (bed volume) protein A/G agarose beads and incubation for another 2 h. In IP with anti-Myc affinity gels and Ni-NTA agarose, the cell extracts containing ~1 mg protein were incubated with 25 µl of anti-Myc affinity gel or 50 µl Ni-NTA agarose in 1 ml RIPA buffer at 4°C overnight. The immunoprecipitates were collected and washed 4 times with RIPA buffer, resuspended in Laemmli sample buffer, and boiled for 10 min, then subjected to Western blotting.
The IP experiments with anti-FLAG affinity gels for detecting UBXD7 associated proteins were conducted using HeLa-FLAG-UBXD7#20 cell extracts made in Buffer A (50 mM N-2-hydroxyethyl piperazine N’-2-ethanesulfonic acid (HEPES)/KOH, pH 7.2; 5 mM Mg(OAc)2, 70 mM KOAc, 0.2% Triton X-100,10% glycerol, 0.2 mM ethylenediaminetetraacetic acid (EDTA), and protease inhibitor cocktail). The cell extracts containing ~1 mg protein was incubated with 25 µl of anti-FLAG affinity gel in 1 ml Buffer A at 4°C overnight. The immunoprecipitates were collected and washed 4 times with Buffer A, resuspended in 1x Laemmli sample buffer, and boiled for 10 min before Western blotting analysis.
The Western blotting was applied to samples made from cell extracts and immunoprecipitates. The proteins in cell extracts were quantitated by DC Bio-Rad Protein Assay, and loading samples were made in 1x SDS loading buffer and boiled for 10 min. The samples were separated by SDS polyacrylamide gel electrophoresis and transferred to PVDF or nitrocellulose membrane. The immunoblotting was performed with incubations of appropriate primary and secondary antibodies. The Western blotting images were developed using enhanced chemiluminescent detection. The quantitative analysis was done on digitalized images using ImageJ software and the relative protein amounts were calculated based on gray density. The Student’s t-tests for paired comparison were performed on the data from experiments repeated 3 times, using SigmaPlot software.
Acknowledgments
We would like to thank Dr. Conrad C. Weihl (Department of Neurology and cell Biology and Physiology, Washington University of Medicine (St. Louis, Missouri 63110)) for providing Myc-tagged VCP/p97 U2OS cell lines. The authors are grateful to Dr. Robert Fisher (Memorial Sloan–Kettering Cancer Center, NY) for providing HCT116-Cdk7as/as cells. The authors also want to thank Dr. Raymond Deshaies (Division of California Institute of Technology, Pasadena, CA 91125) for providing the HEK293-V1-iShVCP cell line.
Funding Statement
This work was supported by the National Institute of Health grants (ES012991 to AAW and QZZ).
Disclosure statement
All authors have no actual or perceived conflict of interest with the contents of this article.
References
- [1].Wood RD. DNA repair in eukaryotes. Annu Rev Biochem. 1996;65:135–167. [DOI] [PubMed] [Google Scholar]
- [2].Wood RD. Nucleotide excision repair in mammalian cells. J Biol Chem. 1997;272:23465–23468. [DOI] [PubMed] [Google Scholar]
- [3].Volker M, Mone MJ, Karmakar P, et al. Sequential assembly of the nucleotide excision repair factors in vivo. Mol Cell. 2001;8:213–224. [DOI] [PubMed] [Google Scholar]
- [4].Fitch ME, Nakajima S, Yasui A, et al. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J Biol Chem. 2003;278:46906–46910. [DOI] [PubMed] [Google Scholar]
- [5].Evans E, Moggs JG, Hwang JR, et al. Mechanism of open complex and dual incision formation by human nucleotide excision repair factors. Embo J. 1997;16:6559–6573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Brueckner F, Hennecke U, Carell T, et al. CPD damage recognition by transcribing RNA polymerase II. Science. 2007;315:859–862. [DOI] [PubMed] [Google Scholar]
- [7].Sarker AH, Tsutakawa SE, Kostek S, et al. Recognition of RNA polymerase II and transcription bubbles by XPG, CSB, and TFIIH: insights for transcription-coupled repair and Cockayne Syndrome. Mol Cell. 2005;20:187–198. [DOI] [PubMed] [Google Scholar]
- [8].Yokoi M, Masutani C, Maekawa T, et al. The Xeroderma pigmentosum group C protein complex XPC-HR23B plays an important role in the recruitment of transcription factor IIH to damaged DNA. J Biol Chem. 2000;275:9870–9875. [DOI] [PubMed] [Google Scholar]
- [9].Sugasawa K, Okamoto T, Shimizu Y, et al. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001;15:507–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Evans E, Fellows J, Coffer A, et al. Open complex formation around a lesion during nucleotide excision repair provides a structure for cleavage by human XPG protein. Embo J. 1997;16:625–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Coin F, Egly JM. Ten years of TFIIH. Cold Spring Harb Symp Quant Biol. 1998;63:105–110. [DOI] [PubMed] [Google Scholar]
- [12].Coin F, Bergmann E, Tremeau-Bravard A, et al. Mutations in XPB and XPD helicases found in xeroderma pigmentosum patients impair the transcription function of TFIIH. Embo J. 1999;18:1357–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst). 2011;10:714–721. [DOI] [PubMed] [Google Scholar]
- [14].Giglia-Mari G, Coin F, Ranish JA, et al. A new, tenth subunit of TFIIH is responsible for the DNA repair syndrome trichothiodystrophy group A. Nat Genet. 2004;36:714–719. [DOI] [PubMed] [Google Scholar]
- [15].Bradsher J, Coin F, Egly JM. Distinct roles for the helicases of TFIIH in transcript initiation and promoter escape. J Biol Chem. 2000;275:2532–2538. [DOI] [PubMed] [Google Scholar]
- [16].Feaver WJ, Svejstrup JQ, Henry NL, et al. Relationship of CDK-activating kinase and RNA polymerase II CTD kinase TFIIH/TFIIK. Cell. 1994;79:1103–1109. [DOI] [PubMed] [Google Scholar]
- [17].Araujo SJ, Tirode F, Coin F, et al. Nucleotide excision repair of DNA with recombinant human proteins: definition of the minimal set of factors, active forms of TFIIH, and modulation by CAK. Genes Dev. 2000;14:349–359. [PMC free article] [PubMed] [Google Scholar]
- [18].Svejstrup JQ, Wang Z, Feaver WJ, et al. Different forms of TFIIH for transcription and DNA repair: holo-TFIIH and a nucleotide excision repairosome. Cell. 1995;80:21–28. [DOI] [PubMed] [Google Scholar]
- [19].Mu D, Park C-H, Matsunaga T, et al. Reconstitution of human DNA repair excision nuclease in a highly defined system. J Biol Chem. 1995;270:2415–2418. [DOI] [PubMed] [Google Scholar]
- [20].Coin F, Oksenych V, Mocquet V, et al. Nucleotide excision repair driven by the dissociation of CAK from TFIIH. Mol Cell. 2008;31:9–20. [DOI] [PubMed] [Google Scholar]
- [21].Alekseev S, Ayadi M, Brino L, et al. A small molecule screen identifies an inhibitor of DNA repair inducing the degradation of TFIIH and the chemosensitization of tumor cells to platinum. Chem Biol. 2014;21:398–407. [DOI] [PubMed] [Google Scholar]
- [22].Funder JW. Mineralocorticoid receptor antagonists: emerging roles in cardiovascular medicine. Integr Blood Press Control. 2013;6:129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Canavan TN, Chen E, Elewski BE. Optimizing non-antibiotic treatments for patients with acne: a review. Dermatol Ther (Heidelbs). 2016;6:555–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Endly DC, Miller RA. Oily skin: a review of treatment options. J Clin Aesthet Dermatol. 2017;10:49–55. [PMC free article] [PubMed] [Google Scholar]
- [25].Szalat R, Samur MK, Fulciniti M, et al. Nucleotide excision repair is a potential therapeutic target in multiple myeloma. Leukemia. 2018;32:111–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kemp MG, Krishnamurthy S, Kent MN, et al. Spironolactone depletes the XPB protein and Inhibits DNA damage responses in UVB-irradiated human skin. J Invest Dermatol. 2019;139:448–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shahar OD, Kalousi A, Eini L, et al. A high-throughput chemical screen with FDA approved drugs reveals that the antihypertensive drug Spironolactone impairs cancer cell survival by inhibiting homology directed repair. Nucleic Acids Res. 2014;42:5689–5701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Meyer H, Bug M, Bremer S. Emerging functions of the VCP/p97 AAA-ATPase in the ubiquitin system. Nat Cell Biol. 2012;14:117–123. [DOI] [PubMed] [Google Scholar]
- [29].Meyer H. p97 complexes as signal integration hubs. BMC Biol. 2012;10:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Stolz A, Hilt W, Buchberger A, et al. Cdc48: a power machine in protein degradation. Trends Biochem Sci. 2011;36:515–523. [DOI] [PubMed] [Google Scholar]
- [31].Ye Y. Diverse functions with a common regulator: ubiquitin takes command of an AAA ATPase. J Struct Biol. 2006;156:29–40. [DOI] [PubMed] [Google Scholar]
- [32].Alexandru G, Graumann J, Smith GT, et al. UBXD7 binds multiple ubiquitin ligases and implicates p97 in HIF1alpha turnover. Cell. 2008;134:804–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Puumalainen MR, Lessel D, Ruthemann P, et al. Chromatin retention of DNA damage sensors DDB2 and XPC through loss of p97 segregase causes genotoxicity. Nat Commun. 2014;5:3695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].He J, Zhu Q, Wani G, et al. Ubiquitin-specific protease 7 regulates nucleotide excision repair through deubiquitinating XPC protein and preventing XPC protein from undergoing ultraviolet light-induced and VCP/p97 protein-regulated proteolysis. J Biol Chem. 2014;289:27278–27289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].He J, Zhu Q, Wani G, et al. Valosin-containing protein (VCP)/p97 segregase mediates proteolytic processing of cockayne syndrome group B (CSB) in damaged chromatin. J Biol Chem. 2016;291:7396–7408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].He J, Zhu Q, Wani G, et al. UV-induced proteolysis of RNA polymerase II is mediated by VCP/p97 segregase and timely orchestration by Cockayne syndrome B protein. Oncotarget. 2016;8:11004–11019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ju JS, Miller SE, Hanson PI, et al. Impaired protein aggregate handling and clearance underlie the pathogenesis of p97/VCP-associated disease. J Biol Chem. 2008;283:30289–30299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Larochelle S, Merrick KA, Terret ME, et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol Cell. 2007;25:839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Arab HH, Wani G, Ray A, et al. Dissociation of CAK from core TFIIH reveals a functional link between XP-G/CS and the TFIIH disassembly state. PLoS One. 2010;5:e11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ueda M, Matsuura K, Kawai H, et al. Spironolactone-induced XPB degradation depends on CDK7 kinase and SCF(FBXL18) E3 ligase. Genes Cells. 2019;24:284–296. [DOI] [PubMed] [Google Scholar]
- [41].den BW VR, Kleiger G, Oania RS, et al. NEDD8 links cullin-RING ubiquitin ligase function to the p97 pathway. Nat Struct Mol Biol. 2012;19(511–6):S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bandau S, Knebel A, Gage ZO, et al. UBXN7 docks on neddylated cullin complexes using its UIM motif and causes HIF1alpha accumulation. BMC Biol. 2012;10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Gabbard RD Spironolactone and XPB: An Old Drug with a New Molecular Target; 2020. [DOI] [PMC free article] [PubMed]
- [44].Berico P, Is CF. TFIIH the new Achilles heel of cancer cells? Transcription. 2018;9:47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Xue L, Blythe EE, Freiberger EC, et al. Valosin-containing protein (VCP)-adaptor interactions are exceptionally dynamic and subject to differential modulation by a VCP inhibitor. Mol Cell Proteomics. 2016;15:2970–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]