

ABSTRACT
Sepsis is a life-threatening organ dysfunction triggered by a dysregulated host immune response attempting to eliminate the infection. After hospital discharge, half of the sepsis survivors recover, one-third of the patients die the following year, and one-sixth have a long-term cognitive impairment, including memory dysfunction, anxiety, depression, and post-traumatic stress disorder. The infection triggers the host immune response, and both can cause vascular endothelial damage, interrupting tight junctions proteins; consequently, the blood-brain barrier (BBB) breaks down, allowing and facilitating the entry of peripheral immune cells into the brain, which triggers or exacerbates the activation of glial cells and neuroinflammation. The focus of this review is to identify biochemical abnormalities induced by sepsis, which is associated with BBB dysfunction; provide evidence of biomarkers involved in the tight junction disruption and BBB damage, and draw attention to the role of the BBB as a bridge between systemic infection and brain inflammation.
KEYWORDS: BBB, tight junction protein, neuroinflammation, sepsis
Background
Sepsis is a syndrome that triggers an organic dysfunction with life-threatening, triggered by a host’s unregulated immune response in a tentative to eliminate the infection.1 The Center for Disease Control (CDC) estimates that sepsis affects approximately 1.7 million adults, causing the death of 270,000 individuals and being responsible for one in three hospital deaths per year in the United States of America.2 After hospital discharge, half of the sepsis survivors recover, one-third of the patients die the following year, and one-sixth of them have a long-term cognitive impairment, including memory dysfunction, anxiety, depression, and post-traumatic stress disorder.3–6
In most cases, sepsis is triggered by bacterial infections, but other conditions, including viruses, parasites, and fungi, cause sepsis. The pathogen can replicate and release its compounds during the systemic infectious progression, known as pathogen-associated molecular patterns (PAMPs).7,8 These PAMPs are recognized by pattern recognition receptors (PRRs) and non-PRRs, which are essential constituents of the immune system.9,10 The detection of PAMPs by immune receptors triggers a cascade of signaling pathways that activate several transcription factors and promotes the production of proinflammatory molecules to eliminate invading pathogens.11 The host’s immune response and pathogen virulence factors may also trigger cellular damage; these cells begin to release endogenous molecules, causing an exacerbation of the inflammatory response. These endogenous molecules are known as damage-associated molecular patterns (DAMPs) and also are recognized by PRRs.12,13 In the sequence, the immune response can trigger vascular endothelial cells damage, interrupting tight junctions proteins; consequently, the blood-brain barrier (BBB) breaks down, allowing and facilitating the entry of peripheral immune cells into the brain, which triggers or exacerbates the activation of glial cells and neuroinflammation.14–17 The focus of this review is to 1. Identify biochemical abnormalities induced by sepsis, which is associated with BBB dysfunction; 2. Provide evidence of biomarkers involved in the tight junction disruption and BBB damage; and 3. Draw attention to the function of the BBB as a bridge between systemic infection and brain inflammation.
Sepsis pathophysiology on the blood-brain barrier
The BBB is formed by brain endothelial cells that line the cerebral microvasculature and is a vital mechanism to protect the brain from changes in the composition of plasma and circulating compounds capable of disrupting the neuronal function.18,19 Anatomically the BBB is a component of the neurovascular unit. The neurovascular unit (NVU) is formed by a complex cellular system, including neurons, interneurons, astrocytes foot, microglia, oligodendrocytes, basal lamina covered by vascular smooth muscle cells and pericytes, endothelial cells, and extracellular matrix in addition to components of circulating blood. However, the NVU composition varies among different brain regions.20 Sepsis is associated with changes in the BBB, NVU, and astrocytes. In preclinical models, lipopolysaccharide (LPS) triggered pericyte loss and microvascular dysfunction.21 The knockout mice of miR-145a submitted to CLP surgery presented increased inflammatory molecules and deaths rate compared to the control group.22 Both astrocytes and pericytes cell types are essential to maintain the BBB integrity; with the dysfunction of these cells, occurs the BBB disruption,23 however, the pericyte cell damage was identified in experimental research.
Sepsis triggers immune cells to express PRR that provides initial discrimination between self and non-self. After PPRs activation, tumor necrosis factor-alpha (TNF-α), IL (interleukin)-1β, and IL-6 are secreted, amplifying the production of inflammatory mediators, including C-reactive protein (CRP), IL-6, serum amyloid protein (SAP), fibrinogen, mannose-biding lectin (MBL), surfactants proteins (SP)-A, and SP-D, soluble receptors, among others. These inflammatory mediators present deleterious effects on the vascular endothelium cells, including disassembling the intracellular junctions by modifying the cytoskeletal structure or causing damage to the cell monolayer resulting in microvascular leaking and tissue edema usually found during sepsis.24 Also, endothelial cells increase the expression of the intracellular adhesion molecules (ICAMs), including ICAM-1, ICAM-2, vascular cell adhesion protein 1 (VCAM-1), and platelet endothelial cell adhesion molecule (PECAM) on the vascular endothelium. Then, the leukocytes ligands stick to the endothelial cells and migrate in direction to the chemoattractant mediators, for example, C-X-C motif chemokine ligand (CXCL)-8 (IL-8) and C-C motif chemokine ligand (CCL)-2.25,26
The immune cells present antimicrobial mechanisms of phagocytosis enhanced by the fMet-Leu-Phe (fMLF) and C5a receptors. Also, the most critical antimicrobial products released are reactive nitrogen species (RNS) such as nitric oxide (NO), and reactive oxygen species (ROS), including superoxide anion (O2−), hydrogen peroxide (H2O2), singlet oxygen (1O2•), hydroxyl radical (•OH), and hypohalite OCL−. The ROS and RNS contribute to vascular endothelial cell dysfunction during sepsis by direct damage or modulating innate immune by activating the transcription factor nuclear factor-kappa B (NF-κB) enhancing TNF-α, IL-1β, IL-6, IL-8, IL-17, and IL-18 production.27,28 The other mechanism that can cause vascular endothelial cell damage is the production of the matrix metalloproteinases (MMPs). The expression of MMPs can be mediated by many factors, such as vascular growth factors, including angiopoietin (Ang)-2, which levels are increased in the septic state and are associated with poor prognosis.29 Also, interferon-gamma (IFN-γ), IL-1β, IL-6, adhesion of immune cells on the extracellular matrix, PAMPs, and DAMPs can increase the expression of MMPs. The expression of MMPs is usually higher in tissues that are damaged, inflamed, or undergoing repair or remodeling, compared to the lesser expression in healthy tissue.30 Plasma levels of MMP-3, MMP-7, MMP-8, and MMP-9 were increased more than 3-fold in severe sepsis in the intensive care unit (ICU).31
In response to stimulation by inflammatory mediators, tight junction proteins are disrupted, which leads to increased vascular permeability and edema. In a clinical study, the occludin (OCLN) and zonula occludens 1 (ZO)-1 levels were elevated according to sepsis severity, demonstrating a positive correlation with the acute physiology and chronic health evaluation II (APACHE II) score, Sequential Organ Failure Assessment (SOFA) score, and lactate levels of the patients.17 In fatal sepsis, the BBB impairment was associated with a loss of cerebral endothelial expression of OCLN.32 The activation of glial cells gives rise to a less robust interaction between astrocytes and the neuronal unit, leading to low maintenance of neuronal synapses. The activation of astrocytes can cause these cells to release neurotoxins, causing the demyelination of oligodendrocytes, axonal damage, and loss of neurons. Thus, the complex pathophysiology of sepsis, triggered by infectious and inflammatory processes, which include a network of cytokines, chemokines, proteolytic enzymes, oxidants, immune cells, microglia, and astrocytes, can be a double-edged sword. It is necessary to eliminate the invading pathogen; still, on the other hand, the exacerbated immune response causes damage to the BBB, which can be the gateway to the cognitive sequelae in which the surviving patients experience in the long-term. For more details, see Figure 1.
Figure 1.
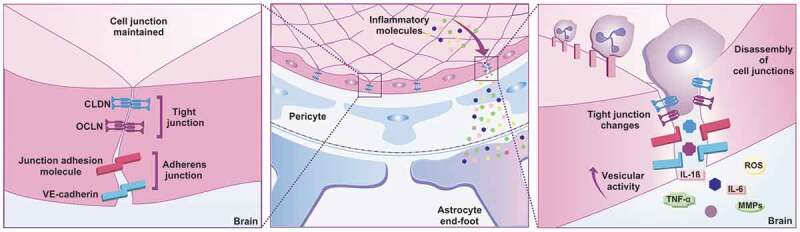
Sepsis pathophysiology on the BBB. The BBB junctions have transmembrane proteins, such as claudin (CLND), occluding (OCLN), and junctional adhesion molecules (JAMs) that interact with each other and seal the spaces that exist between neighboring cells. Adherens junctions have vascular endothelial cadherin (VE-cadherin), which facilitates the adhesion of endothelial cells to each other. The BBB assists in interacting with other glial cells including astrocytes end-foot protecting the CNS against the invasion of pathogens, toxins, and inflammation. During sepsis, infection and the host immune response promote the release of inflammatory molecules that can damage the vascular endothelium cells, interrupt tight junction proteins, and lead to BBB dysfunction, allowing peripheral immune cells to enter the CNS by altering their homeostasis with consequent neuronal damage. CLDN: claudin; IL: interleukin; JAM: junction adhesion molecule; MMPs: matrix metalloproteinases; OCLN: occludin; ROS: reactive oxygen species; TNF-α: tumor necrosis factor-alpha; VE-cadherin: vascular endothelial-cadherin
Preclinical sepsis model and blood-brain barrier dysfunction
Sepsis preclinical models have been used to study the complex syndrome that affects the NVU and, consequently, the BBB integrity. The gold standard model to study sepsis is laparotomy surgery with cecal ligation and puncture (CLP).33,34 However, there are other models, including colon ascendens stent peritonitis (CASP).35 and intraperitoneal fecal material administration.36 The inoculation of Gram (-) bacteria LPS is an endotoxemia model that triggers inflammation instead of an infection; also mice are less sensitive to this endotoxin than humans.37
The polymicrobial sepsis induced by CLP demonstrated that the leukocyte-endothelium rolling started at 6 hours on the BBB vessels. The leukocyte interaction in the cerebral microvasculature started at 6 hours and remained until 24 hours after CLP surgery. Both mechanisms of leukocytes rolling and interaction of this study were evaluated by intravital microscopy. The sepsis group increased the rolling and adhesion of leukocytes in the brain microvasculature, followed by an increase in brain myeloperoxidase (MPO) activity and the BBB breakdown 24 hours after CLP surgery.38 The proinflammatory cytokines, IL-1β, TNF-α, and IL-6, had higher levels 24 hours after CLP surgery concomitant with the BBB breakdown. The IL-1β receptor antagonist (IL1ra) administered after the CLP surgery prevents BBB integrity and decreases the levels of IL-1β.39
Also, the MMP-2 and MMP-9 were detected in the cortex and hippocampus microvessels at 12 hours after CLP surgery, and the MMP-2/MMP-9 inhibitors treatment prevented the BBB disruption. In this study, sepsis-induced by CLP released MMP-2 and MMP-9 before the BBB breakdown that occurred at 24 hours after the surgery.40 The BBB integrity and tight junction proteins were evaluated in mice after seven days of the LPS administration. This study demonstrated that LPS triggered BBB disruption and decreased the expression of claudin-5 (CLDN)-5 and OCLN. Also, administration of apocynin (5-aminoimidazole-4-carboxamide ribonucleotide) that has been used to upregulate the AMP-activated protein kinase (AMPK) activity eliminated the ROS production, prevented the BBB disruption and CLDN-5 and OCLN reduction triggered by LPS administration.41 LPS stimulates leukocytes to produce proinflammatory cytokines that trigger endothelial ROS production. Using LPS as a mice endotoxic model, the researchers found a decrease in sphingosine-1-phosphate (S1P) levels in the mice blood, microvessels, and brain tissue. Also, they found an increased expression of the S1P receptor type 1 and sphingosine kinase-1.
The researchers added LPS to primary mouse brain microvascular endothelial cells (PMBME) culture in the second experiment. The LPS did not change the transendothelial electrical resistance of the cells. However, when they added the serum of LPS-treated mice to the PMBME culture, it triggered a breakdown of the barrier compared to serum from vehicle-treated mice.42 In this present work, rats were subjected to the CLP surgery to evaluate the cerebral blood flow using arterial spin labeling and gray and white matter structure with T2- and diffusion magnetic resonance imaging. The CLP surgery triggered deficits in the righting reflex. It resulted in higher T2-weighted contrast intensities in the cortex, striatum, and the brain’s base decreased blood perfusion distribution to the cortex. Increased water diffusion parallels the corpus callosum fibers compared to sham surgery.43 In this study, the researchers demonstrated the effect of anti-C5a complement protein on BBB integrity. Using the CLP model, rats were treated with anti-C5a antibody or placebo. The placebo CLP group had a severe BBB dysfunction accompanied by a significant upregulation of pituitary C5a receptor and proinflammatory cytokine expression.44 Magnetic resonance imaging technique was used to assess brain morphology and metabolism in the CLP model. CLP mice group had an accumulation of vasogenic edematic fluid at the brain’s base observed in the T2-weighted image at 6 and 24 hours after surgery. The water apparent diffusion coefficients in both hippocampus and cortex were decreased, suggesting cytotoxic edema in the brains of nonsurvival septic animals.45 This study evaluated the effect of statin treatment to prevent microvascular cell dysfunction and reduce neuroinflammation during sepsis. Experimental sepsis was triggered by intraperitoneal injection of fecal material in mice. The statin prevented long-term cognitive impairment, reduced the brain lipid peroxidation and myeloperoxidase levels. The septic group had a decreased functional capillary density, an augment of rolling and adhesion of leukocytes on the vessels, and blood flow impairment, which were reversed by treatment with statins.46 In CLP survivors mice, the mesenchymal stromal cell therapy prevented the BBB disruption, decreased astrocyte activation and neuroinflammation, and improved cognition and behavior.47
Brain tight junction protein biomarkers in sepsis
Sepsis triggers a peripheral inflammation and activation of the cerebral endothelium, promoting an increase in the permeability of the BBB, and these abnormalities result in the release of bloodstream endothelial markers. The present study reported findings from magnetic resonance imaging of the brain of nine septic patients. The brain imaging was standard in two patients. Two patients had multiple ischemic strokes. Five patients had white matter lesions around of Virchow-Robin space, ranging from numerous small to diffuse lesions, and characterized by hyperintensity on fluid-attenuated inversion recovery images. The white matter lesions worsened with increasing duration of shock and were correlated with Glasgow Outcome Score. This study demonstrated that sepsis triggers brain lesions predominated in the white matter, suggesting increased BBB permeability and poor outcome.15
The proteins or peptides are biomarkers of endothelial cells damage and BBB disruption and maybe a helpful tool to predict mortality, multiple organ dysfunction syndromes (MODS), SOFA, and identify the sepsis-associated encephalopathy (SAE) patients.17,32,48 In an observational study, the researchers investigated if plasma levels of OCLN, CLDN-5, and ZO-1 could predict sepsis’s severity and clinical outcome. The OCLN, CLDN-5, and ZO-1 levels were evaluated in the blood of 51 septic patients. The OCLN and ZO-1 had higher levels in patients with severe sepsis and septic shock than sepsis; OCLN and ZO-1 had higher levels in the septic shock than severe sepsis. The OCLN plasma levels correlated positively with the severity of sepsis. The OCLN had an area under the curve (AUC) of 0.773 to predict mortality. The ZO-1 plasma levels correlated positively with the SOFA and APACHE II scores. The ZO-1 had an AUC of 0.748 to diagnose MODS and an AUC of 0.856 to predict mortality. The study demonstrated that OCLN and ZO-1 could be used as a biomarker in early prognosis in patients suffering from sepsis.17 A prospective cohort study investigated the endothelial vascular reactivity to predict prolonged acute brain dysfunction, BBB injury, and delirium duration during critical illness. The plasma levels of plasminogen activator inhibitor-1 (PAI-1), E-selectin, Ang-2, and calcium-binding protein B (S100B) were evaluated. Among 134 included patients, delirium occurred in 94 patients. The high levels of PAI-1, E-selectin, and S100B were associated with fewer delirium/coma-free days. Also, high levels of PAI-1 and S100B concentrations were associated with longer delirium duration in survivors.49 A prospective, observational study was organized to investigate the association of soluble fms-like tyrosine kinase 1 (sFlt-1), soluble E-selectin (sE-Selectin), soluble intercellular adhesion molecule 1 (sICAM-1), soluble vascular cell adhesion molecule-1 (sVCAM-1), and PAI-1 plasma levels of septic patients with organ dysfunction and in-hospital mortality. One hundred sixty-six patients were enrolled in a study, including 63 with sepsis, 61 with severe sepsis, and 42 with septic shock. The endothelial biomarkers included in this study had a positive correlation with sepsis severity. The sFlt-1 had the strongest association with SOFA score; also, the sFlt-1 and PAI-1 had the highest area under the operating receiver characteristic curve for mortality of 0.87. This multicenter study concluded that endothelial biomarker, sFlt-1, PAI-1, sICAM-1, and sVCAM-1 were associated with dysfunction of an organ, severity of sepsis, and mortality in patients with sepsis.50 Plasma levels of endothelial markers, including Ang-1, Ang-2, sVCAM-1, and inflammatory markers were evaluated in 943 critically ill patients that had systemic inflammatory response syndrome (SIRS) to investigate associations between each marker and 28-day mortality, shock, and SOFA score. Higher levels of all biomarkers were associated with increased 28-day mortality, but Ang-1 was associated with lower mortality. Ang-1 was higher in survivors, 5719 pg/ml versus non-survivors, 2504 pg/ml, while Ang-2 was lower in survivors, 11934 pg/ml, versus non-survivors, 42063 pg/ml. sVCAM-1 was also lower in survivors, 536 pg/ml versus non-survivors, 819 pg/ml.51 Syndecan-1 is a structural component of the endothelium. Antithrombin, PAI-1, syndecan-1, VCAM-1, E-selectin, IL-1β, IL-6, IL-8, high mobility group box 1 (HMGB-1), histone-H3 were evaluated in 39 septic patients, and 15 healthy controls. Syndecan-1 levels were increased in the septic patients compared with the healthy controls. Deceased patients had higher syndecan-1 levels compared with survivors’ patients. On day one in ICU were found significant correlations between syndecan-1 levels, APACHE II score, and SOFA score. The disseminated intravascular coagulation (DIC), hemostatic markers, IL-1β, IL-8, and PAI-1, were positively correlated with syndecan-1 levels. Also, syndecan-1 levels predicted mortality.52
The biomarker, S100B in serum, is a maker that can reflect the BBB disruption, glial cells injury, and activation. S100B also is a vital maker to evaluate brain injury severity and predict the outcomes of stroke, traumatic brain injury, encephalopathy, and delirium.53 A prospective cohort study included 104 adult patients with sepsis, 59 SAE patients, 45 non-SAE patients, and evaluated serum S100B on days 1 and 3 after ICU admission. The SAE group presented day 1 of 0.226 μg/l and day 3 of 0.144 μg/l of S100B levels. The AUC for S100B on day 3 for predicting 180-day mortality was more extensive than for S100B on day 1 (0.731 vs. 0.611).54 A total of 22 patients with septic shock were included in an observational study. Delirium was present in 10 of 22 patients, and the serum S100β levels had a cutoff of 0.15 μg/l in 59.1% of the patients. The cases with an S100β > 0.15 μg/l had an odds ratio of 18 for the risk of developing delirium. Patients who had delirium presented higher plasma levels of IL-6, 138.3 pg/ml, compared to those without delirium, 53.6 pg/ml. Also, S100 β and IL-6 levels presented a positive correlation.55
A case series study evaluated 47-brain tissue samples from patients deceased by sepsis. Thirty-eight percent of patients did not present the OCLN expression in the endothelium of cerebral microvessels; moreover, 34% of the patients before death had multiple organ failure, and 74.5% of the patients had septic shock. The patients deceased by sepsis that had a BBB damage also had a higher SOFA score and high levels of procalcitonin in the bloodstream during their stay in the ICU. CLDN-5 and ZO-1 staining were absent from the endothelial cells, and the endothelial OCLN expression in the telencephalon was present in 62% of cases and in the cerebellum in only 32% of patients samples. In this study, the BBB damage was associated with elevated systemic inflammation and organ dysfunction.32 For more details, see Table 1.
Table 1.
Endothelial cells and BBB biomarkers to predict mortality, MODS, and organ dysfunction in septic patients
| Biomarkers | Sample | Demographic | Specificity (%) | Sensitivity (%) | Cutoff | R-squared (R2) | AUC | Clinical relevance | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Prognostic values of tight-junction proteins levels for in-hospital mortality in septic patients | |||||||||
| Ang-1 | Plasma | SIRS, n = 943 | - | - | 5719 pg/ml | - | - | ↑ Ang-1 was associated with low 28-day mortality OR = 0.71. | 1 |
| Ang-2 | Plasma Serum |
SIRS, n = 943 Severe sepsis, n = 48 Sepsis shock, n = 54 |
-- | -- | 42063 pg/ml- | -- | -- | ↑ Ang-2 was associated with ↑ high 28-day mortality OR = 2.06. ↑ Ang-2/Ang-1 ratio increased from day 1 to day 7 in non-survivors (B = 1.965 and p = .000). |
1,2 |
| CLDN-5 | Plasma | Sepsis, n = 51 | - | - | - | - | 0.534 | CLDN-5 levels did not present a correlation with APACHE II score. No, predict mortality. | 3 |
| OCLN | Plasma Brain tissue |
Sepsis, n = 51 Sepsis, n = 47 |
80.49%- | 70.00%- | 509.06 pg/ml- | -- | 0.773- | ↑ OCLN correlated positively with APACHE II. Predict mortality. ↑ Sepsis severity. The deceased patients with BBB damage, less OCLN, had higher SOFA scores. |
3,4 |
| PAI-1 | Plasma | Sepsis, n = 166 | - | - | - | - | 0.85 | ↑ Sepsis severity. Predict mortality. | 5 |
| S100B | Serum | SAE, n = 59 non-SAE, n = 45 |
80.0% 84,44% |
66.1% 69,49% |
0.226 μg/l 0.144 μg/l |
-- | 0.611 0.731 |
S100B, on day-1 to predict 180-day mortality. S100B, on day-3 to predict 180-day mortality. |
6 |
| sE-selectin | Plasma | Sepsis, n = 166 | - | - | - | - | 0.77 | Predict mortality. | 5 |
| sFlt-1 | Plasma | Sepsis, n = 166 | - | - | - | - | 0.85 | ↑ Sepsis severity. Predict mortality. | 5 |
| sICAM | Plasma Serum |
Sepsis, n = 166 Severe sepsis, n = 48 Sepsis shock, n = 54 |
-- | -- | -Day-1 1599 ng/ml Day-7 2058 ng/ml | -- | 0.71- | Predict mortality. ↑ ICAMs levels on day-1 and on day-7 were associated with 90-day mortality. |
5,2 |
| sVCAM-1 | Plasma Plasma Plasma |
Sepsis, n = 166 SIRS, n = 943 Severe sepsis, n = 48 Sepsis shock, n = 54 |
--- | --- | -819 pg/ml Day-1 416 ng/ml Day-7 495 ng/ml |
--- | 0.78-- | Predict mortality. ↑ sVCAM-1 was associated with increased 28-day mortality, OR = 2.71. ↑ sVCAM-1 levels on day-1 and on day-7 were associated with 90-day mortality. |
5,1,2 |
| ZO-1 | Plasma | Sepsis, n = 51 | 87.80% | 90% | 354.3 pg/ml | - | 0.856 | ↑ Sepsis severity. ↑ ZO-1 correlated positively with APACHE II. Predict mortality. |
3 |
| Prognostic values of tight-junction proteins levels for MODS diagnostic in septic patients | |||||||||
| CLDN-5 | Plasma | Sepsis, n = 51 | - | - | - | - | 0.625 | CLDN-5 was not associated with MODS. | 3 |
| OCLN | Plasma | Sepsis, n = 51 | - | - | - | - | 0.633 | OCLN was not associated with MODS. | 3 |
| ZO-1 | Plasma | Sepsis, n = 51 | 79.31% | 63.64% | 272.36 pg/ml | - | 0.748 | ↑ ZO-1 levels were higher in patients for MODS diagnostic. | 3 |
| Prognostic values of tight-junction proteins levels for organ dysfunction in septic patients | |||||||||
| Ang-1 | Plasma | Shock, n = 206 | - | - | - | −0.67 | - | ↑ Ang-1 was associated with low organ dysfunction. | 1 |
| Ang-2 | Plasma Serum |
Shock, n = 206 Severe sepsis, n = 48 Sepsis shock, n = 54 |
-- | -- | -- | 0.97- | -- | ↑ Ang-2 was associated with high organ dysfunction. ↑ Ang-2/Ang-1 ratio ↑ SOFA score. |
1,2 |
| OCLN | Plasma | Sepsis, n = 51 | 80.49% | 70% | 509.06 pg/ml | - | 0.773 | ↑ OCLN correlated positively with SOFA scores. | 3 |
| Brain tissue | Sepsis, n = 47 | - | - | - | - | - | OCLN absence was associated with the maximum SOFA score. OCLN absence was associated with ↑ blood PCT (> 10 μg/l). |
4 | |
| PAI-1 | Plasma | Sepsis, n = 166 | - | - | - | 0.15 | - | ↑ PAI-1 correlated with the SOFA score. | 5 |
| S100B | Serum Serum |
Severe sepsis, n = 21 Shock, n = 22 Delirium, 45% MOF, 50% |
-- | -- | -0.15 μg/l | -- | 0.124- | S100B levels did not correlate with the SOFA score. Patients with delirium had elevated S‐100B levels and more severe organ dysfunction. |
7,8 |
| sE-selectin | Plasma | Sepsis, n = 166 | - | - | - | 0.18 | - | ↑ sE-selectin correlated with SOFA score. | 5 |
| sICAM | Plasma | Sepsis, n = 166 | - | - | - | 0.10 | - | ↑ ICAM correlated with the SOFA score. | 5 |
| sVCAM-1 | Plasma Plasma Serum |
Sepsis, n = 166 Shock, n = 206 Severe sepsis, n = 48 Sepsis shock, n = 54 |
--- | --- | --- | 0.24 1.51- |
--- | ↑ sVCAM-1 correlated with the SOFA score. ↑ sVCAM-1 associated with SOFA score. The sVCAM-1 levels, B = 0.008 p = .001, was a predictor of Δ SOFA score in septic patients. |
5,1,2 |
| sFlt-1 | Plasma | Sepsis, n = 166 | - | - | - | 0.36 | - | ↑ sFlt-1 had a higher association with organ dysfunction. | 5 |
| ZO-1 | Plasma | Sepsis, n = 51 | 87.80% | 90% | 354.3 pg/ml | - | 0.856 | ↑ ZO-1 correlated positively with SOFA scores. | 3 |
Abbreviation: Ang-1 (angiopoietin-1), Ang-2 (angiopoietin-2), APACHE II (acute physiology and chronic health evaluation II), BBB (blood-brain barrier), CLDN-5 (claudin-5); MODS (multiple organ dysfunction syndrome), MOF (multiple organ failure), OCLN (occluding), OR (odds ratio), PAI-1 (plasminogen activator inhibitor 1), qSOFA (quick sequential organ failure assessment), S100B (calcium-binding protein B), SAE (sepsis-associated encephalopathy), sE-Selectin (soluble E-selectin), sFlt-1 (soluble fms-like tyrosine kinase 1), sICAM-1 (soluble intercellular adhesion molecule 1), SIRS (systemic inflammatory response syndrome), SOFA (sequential organ failure assessment), sVCAM-1 (soluble vascular cell adhesion molecule 1), ZO-1 (zonula occluden 1).
Fang Y, et al. 2018; Piazza O, et al. 2007.
Conclusion
Sepsis is a life-threatening organ dysfunction triggered by an imbalance of the host immune response to eliminate the infection. Recent studies have suggested the importance of evaluating tight-junction markers and identify patients with a possible BBB injury. Also, several soluble tight junction proteins biomarkers presented prognostic values for in-hospital mortality. They were associated with SOFA score, identified MODS, and SAE, demonstrating the high importance of these biomarkers. The endothelial cells and BBB biomarkers should include in the routine of sepsis patients admitted to the ICU.
Acknowledgments
This work was supported by the Faillace Department of Psychiatry and Behavioral Sciences, McGovern Medical School, The University of Texas Health Science Center at Houston (UTHealth), USA (TB); the Graduate Program in Health Sciences, University of Southern Santa Catarina (UNESC) (TB, JSG, AC, and FDP), Brazil; The Graduate Program in Health Sciences, University of South Santa Catarina (UNISUL) (FP), Brazil; and the Alzheimer’s Association Grant number AARGDNTF-19-619645 (TB).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Disclosure of interest
The authors report no conflict of interest.
References
- 1.Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, Bellomo R, Bernard GR, Chiche J-D, Coopersmith CM, et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). Jama. 2016;315(8):1–10. Epub 2016/02/24. doi: 10.1001/jama.2016.0287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.CDC. Data & Reports . 2020. [updated August20, 2020]; Available from: cdc.gov/sepsis/datareports/index.html.
- 3.Prescott HC, Angus DC.. Enhancing recovery from sepsis: a review. Jama. 2018;319(1):62–75. doi: 10.1001/jama.2017.17687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prescott HC, Carmichael AG, Langa KM, Gonzalez R, Iwashyna TJ. Paths into sepsis: trajectories of presepsis healthcare use. Ann Am Thorac Soc. 2019;16(1):116–123. Epub 2018/09/14. doi: 10.1513/AnnalsATS.201806-391OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prescott HC, Osterholzer JJ, Langa KM, Angus DC, Iwashyna TJ. Late mortality after sepsis: propensity matched cohort study. BMJ (Clinical Research Ed). 2016;353:i2375. Epub 2016/05/18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. Jama. 2010;304(16):1787–1794. doi: 10.1001/jama.2010.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kumar S, Ingle H, Prasad DV, Kumar H. Recognition of bacterial infection by innate immune sensors. Crit Rev Microbiol. 2013;39(3):229–246. doi: 10.3109/1040841X.2012.706249. [DOI] [PubMed] [Google Scholar]
- 8.Heckenberg SG, Brouwer MC, van de Beek D. Bacterial meningitis. Handb Clin Neurol. 2014;121:1361–1375. Epub 2013/12/25. [DOI] [PubMed] [Google Scholar]
- 9.Sellner J, Täuber MG, Leib SL. Chapter 1 - Pathogenesis and pathophysiology of bacterial CNS infections. Karen LR, Allan RT editors. Handbook of Clinical Neurology. Elsevier, Netherlands; 2010, 1–16. [DOI] [PubMed] [Google Scholar]
- 10.Mook-Kanamori BB, Geldhoff M, van der Poll T, van de Beek D. Pathogenesis and pathophysiology of pneumococcal meningitis. Clin Microbiol Rev. 2011;24(3):557–591. Epub 2011/07/08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science (New York, NY). 2010;327(5963):291–295. Epub 2010/01/16. doi: 10.1126/science.1183021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiersinga WJ, Leopold SJ, Cranendonk DR, van der Poll T. Host innate immune responses to sepsis. Virulence. 2014;5(1):36–44. Epub 2013/06/19. doi: 10.4161/viru.25436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu B, Wang C, Wang M, Li W, Chen F, Tracey KJ, Wang H. Molecular mechanism and therapeutic modulation of high mobility group box 1 release and action: an updated review. Expert Rev Clin Immunol. 2014;10(6):713–727. Epub 2014/04/22. doi: 10.1586/1744666X.2014.909730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Annane D, Sharshar T. Cognitive decline after sepsis. Lancet Respir Med. 2015;3(1):61–69. doi: 10.1016/S2213-2600(14)70246-2. [DOI] [PubMed] [Google Scholar]
- 15.Sharshar T, Carlier R, Bernard F, Guidoux C, Brouland J-P, Nardi O, de la Grandmaison GL, Aboab J, Gray F, Menon D, et al. Brain lesions in septic shock: a magnetic resonance imaging study. Intensive Care Med. 2007;33(5):798–806. Epub 2007/03/23. doi: 10.1007/s00134-007-0598-y. [DOI] [PubMed] [Google Scholar]
- 16.Westhoff D, Engelen-Lee JY, Hoogland ICM, Aronica EMA, van Westerloo DJ, van de Beek D, van Gool WA. Systemic infection and microglia activation: a prospective postmortem study in sepsis patients. Immunity Ageing. 2019. Epub 2019/08/07;16(1):18. doi: 10.1186/s12979-019-0158-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao G-JG-J, Li D, Zhao Q, Lian J, Hu -T-T, Hong G-L, Yao Y-M, Lu Z-Q. Prognostic value of plasma tight-junction proteins for sepsis in emergency department: an observational study. Shock. 2016;45(3):326–332. doi: 10.1097/SHK.0000000000000524. [DOI] [PubMed] [Google Scholar]
- 18.Banks WA. From blood-brain barrier to blood-brain interface: new opportunities for CNS drug delivery. Nat Rev Drug Discov. 2016;15(4):275–292. Epub 2016/01/23 [DOI] [PubMed] [Google Scholar]
- 19.Erickson MA, Banks WA. Neuroimmune axes of the blood-brain barriers and blood-brain interfaces: bases for physiological regulation, disease states, and pharmacological interventions. Pharmacol Rev. 2018;70(2):278–314. Epub 2018/03/03. doi: 10.1124/pr.117.014647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Barichello TCA, Hasbun R, Morales R. An overview of the blood-brain barrier. In: Barichello T, editor. Blood-brain barrier. New York (NY): Humana Press; 2019. p. 1–8. [Google Scholar]
- 21.Zeng H, He X, Tuo QH, Liao DF, Zhang GQ, Chen JX. LPS causes pericyte loss and microvascular dysfunction via disruption of Sirt3/angiopoietins/Tie-2 and HIF-2α/Notch3 pathways. Sci Rep. 2016;6:20931. Epub 2016/02/13.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wu Y, Li P, Goodwin AJ, Cook JA, Halushka PV, Zingarelli B, Fan H. miR-145a regulation of pericyte dysfunction in a murine model of sepsis. J Infect Dis. 2020;222(6):1037–1045. Epub 2020/04/15. doi: 10.1093/infdis/jiaa184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danielski LG, Giustina AD, Badawy M, Barichello T, Quevedo J, Dal-Pizzol F, Petronilho F. Brain barrier breakdown as a cause and consequence of neuroinflammation in sepsis. Mol Neurobiol. 2018;55(2):1045–1053. Epub 2017/01/17. doi: 10.1007/s12035-016-0356-7. [DOI] [PubMed] [Google Scholar]
- 24.Lee WL, Slutsky AS. Sepsis and endothelial permeability. N Engl J Med. 2010;363(7):689–691. Epub 2010/09/08. doi: 10.1056/NEJMcibr1007320. [DOI] [PubMed] [Google Scholar]
- 25.Barichello T, Generoso JS, Silvestre C, Costa CS, Carrodore MM, Cipriano AL, Michelon CM, Petronilho F, Dal-Pizzol F, Vilela MC, et al. Circulating concentrations, cerebral output of the CINC-1 and blood–brain barrier disruption in Wistar rats after pneumococcal meningitis induction. Eur J Clin Microbiol Infect Dis. 2012;31(8):2005–2009. Epub 2012/02/04. doi: 10.1007/s10096-011-1533-2. [DOI] [PubMed] [Google Scholar]
- 26.Giridharan VV, Collodel A, Generoso JS, Scaini G, Wassather R, Selvaraj S, Hasbun R, Dal-Pizzol F, Petronilho F, Barichello T, et al. Neuroinflammation trajectories precede cognitive impairment after experimental meningitis-evidence from an in vivo PET study. J Neuroinflammation. 2020;17(1):5. Epub 2020/01/07. doi: 10.1186/s12974-019-1692-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biswal S, Remick DG. Sepsis: redox mechanisms and therapeutic opportunities. Antioxid Redox Signal. 2007;9(11):1959–1961. Epub 2007/08/23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barichello T, Generoso JS, Simões LR, Elias SG, Quevedo J. Role of oxidative stress in the pathophysiology of pneumococcal meningitis. Oxid Med Cell Longev. 2013;2013:371465. Epub 2013/06/15. doi: 10.1155/2013/371465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang RY, Zhang H, Huang J, Qu H-P, Tang Y-Q. Angiogenic factors in sepsis: are we ready for the new therapeutic era? Critical Care (London, England). 2014;18(1):403. Epub 2014/01/31. doi: 10.1186/cc13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masciantonio MG, Lee CKS, Arpino V, Mehta S, Gill SE. The balance between metalloproteinases and TIMPs: critical regulator of microvascular endothelial cell function in health and disease. Prog Mol Biol Transl Sci. 2017;147:101–131. Epub 2017/04/18. [DOI] [PubMed] [Google Scholar]
- 31.Yazdan-Ashoori P, Liaw P, Toltl L, Webb B, Kilmer G, Carter DE, Fraser DD. Elevated plasma matrix metalloproteinases and their tissue inhibitors in patients with severe sepsis. J Crit Care. 2011;26(6):556–565. Epub 2011/03/29. doi: 10.1016/j.jcrc.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 32.Erikson K, Tuominen H, Vakkala M, Liisanantti JH, Karttunen T, Syrjälä H, Ala-Kokko TI. Brain tight junction protein expression in sepsis in an autopsy series. Critical Care (London, England). 2020;24(1):385. Epub 2020/07/01. doi: 10.1186/s13054-020-03101-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wichterman KA, Baue AE, Chaudry IH. Sepsis and septic shock–a review of laboratory models and a proposal. J Surg Res. 1980;29(2):189–201. doi: 10.1016/0022-4804(80)90037-2. [DOI] [PubMed] [Google Scholar]
- 34.Hubbard WJ, Choudhry M, Schwacha MG, Kerby JD, Rue LW, Bland KI, Chaudry IH. Cecal ligation and puncture. Shock (Augusta, Ga). 2005;24(Suppl 1):52–57. Epub 2005/12/24. doi: 10.1097/01.shk.0000191414.94461.7e. [DOI] [PubMed] [Google Scholar]
- 35.Zantl N, Uebe A, Neumann B, Wagner H, Siewert J-R, Holzmann B, Heidecke C-D, Pfeffer K. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect Immun. 1998;66(5):2300–2309. Epub 1998/05/09. doi: 10.1128/IAI.66.5.2300-2309.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weinstein WM, Onderdonk AB, Bartlett JG, Gorbach SL. Experimental intra-abdominal abscesses in rats: development of an experimental model. Infect Immun. 1974;10(6):1250–1255. Epub 1974/12/01. doi: 10.1128/IAI.10.6.1250-1255.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deitch EA. Animal models of sepsis and shock: a review and lessons learned. Shock (Augusta, Ga). 1998;9(1):1–11. Epub 1998/02/18. doi: 10.1097/00024382-199801000-00001. [DOI] [PubMed] [Google Scholar]
- 38.Comim CM, Vilela MC, Constantino LS, Petronilho F, Vuolo F, Lacerda-Queiroz N, Rodrigues DH, da Rocha JL, Teixeira AL, Quevedo J, et al. Traffic of leukocytes and cytokine up-regulation in the central nervous system in sepsis. Intensive Care Med. 2011;37(4):711–718. doi: 10.1007/s00134-011-2151-2. [DOI] [PubMed] [Google Scholar]
- 39.Mina F, Comim CM, Dominguini D, Cassol-Jr OJ, Dall`Igna DM, Ferreira GK, Silva MC, Galant LS, Streck EL, Quevedo J, et al. Il1-β involvement in cognitive impairment after sepsis. Mol Neurobiol. 2014;49(2):1069–1076. Epub 2013/11/16. doi: 10.1007/s12035-013-8581-9. [DOI] [PubMed] [Google Scholar]
- 40.Dal-Pizzol F, Rojas HA, Dos Santos EM, Vuolo F, Constantino L, Feier G, Pasquali M, Comim CM, Petronilho F, Gelain DP, et al. Matrix metalloproteinase-2 and metalloproteinase-9 activities are associated with blood–brain barrier dysfunction in an animal model of severe sepsis. Mol Neurobiol. 2013;48(1):62–70. Epub 2013/03/13. doi: 10.1007/s12035-013-8433-7. [DOI] [PubMed] [Google Scholar]
- 41.Yu H-YH-Y, Cai Y-B, Liu Z. Activation of AMPK improves lipopolysaccharide-induced dysfunction of the blood–brain barrier in mice. Brain Injury. 2015;29(6):777–784. Epub 2015/03/21. doi: 10.3109/02699052.2015.1004746. [DOI] [PubMed] [Google Scholar]
- 42.Vutukuri R, Brunkhorst R, Kestner R-I, Hansen L, Bouzas NF, Pfeilschifter J, Devraj K, Pfeilschifter W. Alteration of sphingolipid metabolism as a putative mechanism underlying LPS-induced BBB disruption. J Neurochem. 2018;144(2):172–185. doi: 10.1111/jnc.14236. [DOI] [PubMed] [Google Scholar]
- 43.Griton M, Dhaya I, Nicolas R, Raffard G, Periot O, Hiba B, Konsman JP. Experimental sepsis-associated encephalopathy is accompanied by altered cerebral blood perfusion and water diffusion and related to changes in cyclooxygenase-2 expression and glial cell morphology but not to blood-brain barrier breakdown. Brain Behav Immun. 2020;83:200–213. doi: 10.1016/j.bbi.2019.10.012. [DOI] [PubMed] [Google Scholar]
- 44.Flierl MA, Stahel PF, Rittirsch D, Huber-Lang M, Niederbichler AD, Hoesel LM, Touban BM, Morgan SJ, Smith WR, Ward PA, et al. Inhibition of complement C5a prevents breakdown of the blood-brain barrier and pituitary dysfunction in experimental sepsis. Crit Care. 2009;13(1):6. doi: 10.1186/cc7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bozza FA, Garteiser P, Oliveira MF, Doblas S, Cranford R, Saunders D, Jones I, Towner RA, Castro-Faria-Neto HC. Sepsis-associated encephalopathy: a magnetic resonance imaging and spectroscopy study. J Cereb Blood Flow Metab. 2010;30(2):440–448. doi: 10.1038/jcbfm.2009.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reis PA, Alexandre PCB, D’Avila JC, Siqueira LD, Antunes B, Estato V, Tibiriça EV, Verdonk F, Sharshar T, Chrétien F, et al. Statins prevent cognitive impairment after sepsis by reverting neuroinflammation, and microcirculatory/endothelial dysfunction. Brain Behav Immun. 2017;60:293–303. doi: 10.1016/j.bbi.2016.11.006. [DOI] [PubMed] [Google Scholar]
- 47.Silva AYO, Amorim ÉA, Barbosa-Silva MC, Lima MN, Oliveira HA, Granja MG, Oliveira KS, Fagundes PM, Neris RLS, Campos RMP, et al. Mesenchymal stromal cells protect the blood-brain barrier, reduce astrogliosis, and prevent cognitive and behavioral alterations in surviving septic mice. Crit Care Med. 2020;48(4):e290–e8. doi: 10.1097/CCM.0000000000004219. [DOI] [PubMed] [Google Scholar]
- 48.Barichello T, Sayana P, Giridharan VV, Arumanayagam AS, Narendran B, Della Giustina A, Petronilho F, Quevedo J, Dal-Pizzol F. Long-term cognitive outcomes after sepsis: a translational systematic review. Mol Neurobiol. 2019;56(1):186–251. Epub 2018/04/25. [DOI] [PubMed] [Google Scholar]
- 49.Hughes CG, Pandharipande PP, Thompson JL, Chandrasekhar R, Ware LB, Ely EW, Girard TD. Endothelial activation and blood-brain barrier injury as risk factors for delirium in critically Ill patients. Crit Care Med. 2016;44(9):e809–17. Epub 2016/04/19. doi: 10.1097/CCM.0000000000001739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Skibsted S, Jones AE, Puskarich MA, Arnold R, Sherwin R, Trzeciak S, Schuetz P, Aird WC, Shapiro NI. Biomarkers of endothelial cell activation in early sepsis. Shock (Augusta, Ga). 2013;39(5):427–432. Epub 2013/03/26. doi: 10.1097/SHK.0b013e3182903f0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mikacenic C, Hahn WO, Price BL, Harju-Baker S, Katz R, Kain KC, Himmelfarb J, Liles WC, Wurfel MM. Biomarkers of endothelial activation are associated with poor outcome in critical illness. PloS One. 2015;10(10):e0141251. Epub 2015/10/23. doi: 10.1371/journal.pone.0141251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ikeda M, Matsumoto H, Ogura H, Hirose T, Shimizu K, Yamamoto K, Maruyama I, Shimazu T. Circulating syndecan-1 predicts the development of disseminated intravascular coagulation in patients with sepsis. J Crit Care. 2018;43:48–53. Epub 2017/08/28. doi: 10.1016/j.jcrc.2017.07.049. [DOI] [PubMed] [Google Scholar]
- 53.Bloomfield SM, McKinney J, Smith L, Brisman J. Reliability of S100B in predicting severity of central nervous system injury. Neurocrit Care. 2007;6(2):121–138. doi: 10.1007/s12028-007-0008-x. [DOI] [PubMed] [Google Scholar]
- 54.Wu L, Feng Q, Ai ML, Deng SY, Liu ZY, Huang L, Ai YH, Zhang L. The dynamic change of serum S100B levels from day 1 to day 3 is more associated with sepsis-associated encephalopathy. Sci Rep. 2020;10(1):7718. Epub 2020/05/10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Erikson K, Ala-Kokko TI, Koskenkari J, Liisanantti JH, Kamakura R, Herzig KH, Syrjälä H. Elevated serum S-100β in patients with septic shock is associated with delirium. Acta Anaesthesiol Scand. 2019;63(1):69–73. Epub 2018/08/07. doi: 10.1111/aas.13228. [DOI] [PubMed] [Google Scholar]