

ABSTRACT
Rho proteins are signalling molecules that control cellular dynamics, movement and morphological changes. They are activated by Rho guanine-nucleotide exchange factors (Rho GEFs) that transduce upstream signals into Rho-mediated activation of downstream processes. Fgd5 is a Rho GEF involved in angiogenesis and its target Rho protein for this process has been linked to Cdc42 activation. Here, we examined the function of purified Fgd5, specifically, which Rho proteins it activates and pinpoint the structural domains required for enzymatic activity. Using a GEF enzyme assay, we found that purified Fgd5 showed preferential activation of Rac1 and direct binding of Rac1 in pull-down and co-immunoprecipitation assays. Structural comparisons showed that the Fgd5 DH domain is highly similar to the Rac1 GEF, TrioN, supporting a role for Fgd5 as a Rac1 GEF. Compounds that bind to purified Fgd5 DH-PH protein were identified by screening a small molecule library via surface plasmon resonance. The effects of eleven ligands were further examined for their ability to inhibit the Fgd5 GEF enzymatic activity and Rac1 interaction. From these studies, we found that the compound aurintricarboxylic acid, and to a lesser extent mitoxantrone dihydrochloride, inhibited both Fgd5 GEF activation of Rac1 and their interaction. Aurintricarboxylic acid had no effect on the activity or binding of the Rac1 GEF, TrioN, thus demonstrating the feasibility of selectively disrupting Rho GEF activators.
Abbreviations: a.a.: amino acid; ATA: aurintricarboxylic acid; DH: Dbl homology; DOCK: dictator of cytokinesis; Fgd: faciogenital dysplasia; GEF: guanine-nucleotide exchange factor; GST: glutathione S-transferase; LOPAC: library of pharmacologically active compounds; PH: pleckstrin homology; PDB: protein data bank; s.e.m.: standard error of the mean; SPR: surface plasmon resonance.
KEYWORDS: Rho GTPase, Rac1, guanine-nucleotide exchange factor, aurintricarboxylic acid, surface plasmon resonance
Introduction
Faciogenital dysplasia (Fgd) genes encode a family of six homologous proteins predicted to be Rho guanine-nucleotide exchange factors (Rho GEFs). They contain the typical Rho GEFs subdomain structures with a Dbl homology domain adjacent to a pleckstrin homology domain (DH-PH). In addition to the common structures, all Fgd proteins contain a C-terminal FYVE domain and a second PH domain, which may have a role in membrane localization. FYVE domains are known to bind the lipid, phosphatidylinositol 3-phosphate, whereas PH domains can bind di- or tri-phosphorylated phosphatidylinositol lipids. While all six Fgd proteins share the same C-terminal sub-domain structures, the N-terminal domain of each is unique.
There are several emerging roles for mutated FGD genes and Fgd proteins in human disorders. Fgd1, the founding member of the Fgd family, was originally identified as the gene defective in the Aarskog-Scott syndrome. This X-linked chromosome disorder presents with multiple developmental defects, including bone and body malformations [1]. Mutations in Fgd4, also known as Frabin, are associated with the peripheral nerve disorder Charcot-Marie-Tooth disease [2,3]. Several recent studies indicate that Fgd5 contributes to pro-angiogenesis processes in endothelial cells [4,5] with elevated expression leading to poor prognosis in breast cancer [6]. Fgd5 is abundantly expressed in endothelial cells [7]. While its activity does not seem to be altered by VEGF stimulation, it is known to be involved in the regulation of endothelial adhesion, survival, and angiogenesis by modulating phosphatidylinositol 3-kinase signalling [4,8]. These effects of the Fgd family of proteins is likely mediated through downstream signalling from Rho GTPase which are known regulators of cellular remodelling, cellular transport and gene expression [9]. In their active form, Rho GTPases bind and act on their downstream effector proteins to generate a cellular response [10]. Rho GEFs have emerged as important therapeutic targets due to the significance of the downstream actions of activated GTPases and their role in disease [11].
Rho GEFs catalyse the exchange of GDP for GTP which results in the activation of Rho proteins and subsequent downstream signalling. Rho GEFs transmit upstream signals into numerous Rho protein-control biological processing downstream which depends on their subcellular localization and specific Rho protein interaction. Many Rho GEFs show catalytic activity for multiple substrate GTPases, however a few Rho GEFs are specific for a single GTPase [11]. Structural determinants in the DH domain determine the specificity of coupling between Rho GEFs and Rho GTPases. The specificity of GEF activity for four Fgd family members, Fgd1-4, have been characterized and found to be nucleotide exchange factors for the GTPase Cdc42 [12–15]. It has been suggested that Fgd5 and Fgd6 are also Cdc42 GEFs based on similarities in subdomain architecture. The potential GEF activity of Fgd5 has been examined in cellular assays of endothelial cell barrier function and proliferation in response to growth factors [16–18]. These studies showed downstream functions of Fgd5 were likely mediated through interaction and activation of Cdc42. However, it has also been reported that Fgd5 effects in endothelial cells may be mediated by Rac1 [4,18]. Here we present a biochemical analysis of Fgd5 GEF activity and show that it selectively binds and activates the Rac1 GTPase. We also screened a small molecule library and identified compounds that bound to Fgd5 and interfered in Rac1 binding and activation.
Materials and methods
Protein production
His6-tagged DH-PH domains from the GEFs Itsn1 (a.a. 1229–1581) cloned into pPRoExHTb and Dbs (a.a. 623–967) cloned into pET28a were a gift from Dr. Kent Rossman (University of North Carolina, Chapel Hill). The Fgd5 DH-PH domains (a.a. 650–967) and DH-PH-FYVE domains (a.a. 650–1060) were cloned into pET11b from cDNA. An Fgd5-DH-PHF803K point mutant within the DH specificity patch was generated by site directed mutagenesis. The DH-PH domain of TrioN (a.a. 1292–1594) was cloned into pET15 from cDNA. GST-tagged RhoA, Rac1 and Cdc42 were purchased from cDNA.org. Plasmids were used to transform E. coli Rosetta DE3 cells and proteins were expressed by growing cultures at 22°C after inducing with 0.4 mM IPTG. Proteins were purified from E. coli lysates by affinity chromatography using Ni2+-bound chelating sepharose (GE Healthcare) in Ni-buffer (20 mM HEPES pH 7.5, 300 mM NaCl, 1 mM MgCl2, 0.1 mM ZnCl2, 30 mM imidazole) eluted by the addition of 300 mM imidazole to Ni-buffer, or using glutathione sepharose (GE Healthcare) in PBS, eluted by the addition of 10 mM reduced glutathione in 50 mM TrisCl pH 8. Typically, protein concentrations of 10–40 μM were obtained.
SDS-PAGE and immunoblot
SDS-PAGE and Coomassie Blue staining were performed to analyse proteins after purification processes. Immunoblotting was performed by transferring gels to nitrocellulose. Primary antibodies used were mouse monoclonal anti-His6 used at 1 μg/ml (Proteintech), rabbit polyclonal anti-GST used at 0.2 μg/ml (Thermo Fisher), rabbit polyclonal anti-Rac1 used at 0.4 μg/ml (C-14, Santa Cruz Biotechnology), mouse monoclonal Cdc42 at 0.2 μg/ml (B-8, Santa Cruz Biotechnology) and mouse monoclonal anti-RhoA at 0.4 μg/ml (26C4, Santa Cruz Biotechnology). Secondary antibodies used were DyLight 800 conjugated goat anti-mouse IgG used at 10 ng/ml (Thermo Fisher) and Alexafluor 680 goat anti-rabbit IgG used at 50 ng/ml (Invitrogen). Blots were scanned and quantified using a Licor Odyssey digital fluorescent scanner.
GEF assay
The nucleotide exchange activity of purified GEFs was determined by monitoring the relative increase in fluorescence of the fluorescent GTP analogue, MANT-GTP (Thermo Fisher), upon binding a GTPase [19,20]. GEF assays contained 1 μM of purified GST-Rho protein, 150 nM MANT-GTP in GEF buffer (20 mM Tris-Cl pH 7.5, 60 mM NaCl, 5 mM MgCl2, 1 mM DTT, 50 μg/ml BSA, 10% glycerol) in a total volume of 2 ml. Fluorescence measurements were taken using a fluorimeter (PTI), ex/em = 360/440 nm ± 5 nm, with a temperature-controlled cuvette holder set to 25°C. After 5 min of equilibration time, 10–200 nM GEF or buffer (control) was added and reactions were monitor for a further 20 min. GEF activity was calculated as the initial rate of fluorescence increase relative to buffer control. GEFs were pre-incubated with 450 nM drugs for 10 min to analyse the effect of Fgd5-binding compounds.
Binding assays
To examine Fgd5-Rho protein interactions, HEK293T cells were transfected with GFP-tagged full-length Fgd5 and a mutant lacking the DH domain (a.a. 650–842). Cells were lysed 24 h post transfection and GFP-Fgd5 was immunoprecipitated with goat anti-GFP antibodies bound to protein-G sepharose. Co-immunoprecipitation of Rho proteins was analysed by immunoblot. To analyse the direct binding of GEFs and Rho proteins, GST-tagged Rho proteins, and GST control, immobilized on glutathione resin (Sigma) were incubated with purified GEF. Each assay contain 5 μM GEF, 10 μl of protein saturated glutathione resin in binding buffer (20 mM HEPES pH 7.5, 100 mM NaCl, 40 μM GDP, 1 mM EDTA, 0.5% Trition X-100). Samples were incubated for 1 h at 4°C on a rotator, washed with the respective binding buffers and the resin bound sample prepared for SDS-PAGE and analysis of binding was done by immunoblot with anti-GST and anti-His6 antibodies.
Compound screening
The Library of Pharmacologically Active Compounds 1280 (LOPAC1280) (Sigma-Aldrich) was screened by surface plasmon resonance (SPR) for binding to the Fgd5 DH-PH-FYVE domain. The protein was cross-linked to the surface of a CM5 chip in channels 2 and 4, while channels 1 and 3 were used as control. The chips were loaded into a Biacore T200 which were primed with drug binding buffer (2% DMSO in PBS-P+ (PBS with 0.05% P20 surfactant)), followed by three control runs of the drug binding buffer then ten drugs diluted to 50 μM in PBS-P+ (the final concentration of DMSO was maintained at 2%). After every ten runs a solvent correction curve was generated contain 1.5% to 2.8% DMSO in PBS-P + . Binding was calculated from the channel 2–1 or 4–3 response units corrected for DMSO solvent effects. The average binding levels were calculated from each drug plate (16 plates total) and compounds showing signals 3 fold above average were deemed potential binding compounds.
Amino acid alignment and structural modelling
Sequences for human Fgd family members, TrioN, ITSN1 and Dbs were obtained from the National Center for Biotechnology Information. Boundaries of the DH domains were based on annotation by ProSite signature scanning. Structural coordinates were downloaded from the Protein Database (PDB) for Fgd5 (3MPX), and Rac1 co-crystals with TrioN (2NZ8) and Vav1 (2VRW). Ribbon structures were created using PyMol v2.3.2. The DH domains of GEFs were aligned via Clustal Omega (European Bioinformatics Institute) and the structures of Rac1 and Fgd5 were superimposed, leaving the relative positions of the PH domain and Rac1 with respect to the DH domain unchanged to allow comparison of the respective positions of the PH domains and Rac1.
Results
Comparison of Fgd5 to other Rho GEFs
The action of GEFs can be highly selective towards one GTPase, or they can have broad specificity for a class of GTPases [9]. For example, Fgd1 has been shown to be a specific GEF for Cdc42, while Vav1 can act as a GEF for Cdc42, Rac1, and RhoA [21]. We first determined the Rho GTPase specificity of Fgd5 by direct enzyme assays. The DH domains of GEFs are involved in the interaction and exchange activity with Rho proteins [20]. While the DH domains of Fgd1-4 are highly similar, the DH domains of Fgd5 and Fgd6 show divergence, particularly at critical amino acid residues in the specificity patch which define the interaction points with specific Rho proteins (Figure 1(a)) [19]. This suggests that they may have different GTPase selectivity. Alignment of the Fgd5 DH domain with the Rac1 GEF, TrioN, showed elevated sequence similarity here while alignment with the Cdc42 and RhoA GEFs, Itsn1 and Dbs, showed reduced similarity (Figure 1(b)), inferring that Fgd5 may be a Rac1 GEF. Similarity scores were calculated for the domains shown in Figure 1(a,b) based on identical or highly homologous (i.e. I/L/V, S/T, G/A, D/E, N/Q, K/R) amino acid pairs. Similarity scores between Fgd1-4 proteins are greater than 72%, but less than 38% for Fgd5. The similarity score between Fgd5 and the Rac1 GEF, TrioN, rises to 41%, but when comparing Fgd1 to TrioN, it is less than 36%. Similarity scores between Fgd5, Fgd1 and the Cdc42 GEF, Itsn1, are 23% and 37.5%, respectively, showing that Fgd1 more closely resembles a Cdc42 GEF. Similarity scores between Fgd5, Fgd1 and the RhoA/Cdc42 GEF, Dbs, are 31% and 27% respectively. This analysis suggests that Fgd5 may be more closely related to Rac1 GEFs because the similarity score is the highest.
Figure 1.
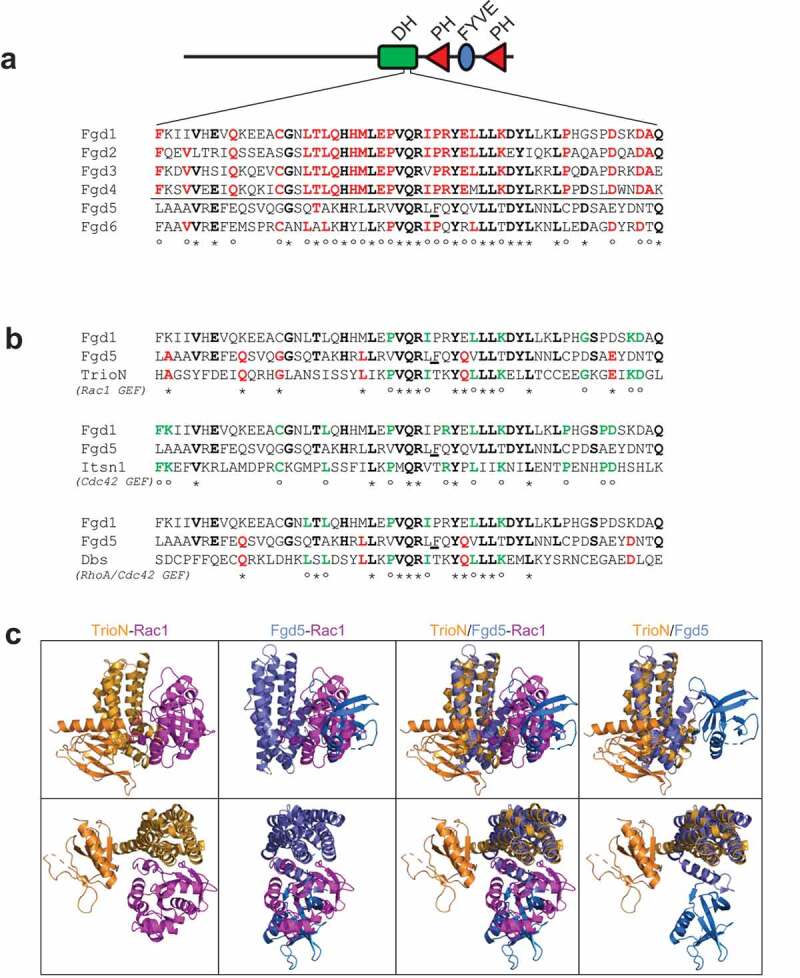
Protein sequence and structural comparison of Fgd5 DH domain. Amino acid sequence alignment of Fgd DH domain specificity patches within the Fgd family (a), or with GEFs of known Rho GTPase specificity as indicated (b). BLACK/BOLD amino acids marked with ‘*’indicate identical residues within at least four Fgd family members; RED/BOLD residues marked with ‘°’ indicate identical residues within the comparison group that are non-identical with Fgd5; GREEN/BOLD residues marked with ‘*’ indicate identical residues with the comparison sequence with Fgd5. (c) Superposition of the structures of the similar DH domains of Fgd5 (blue) and TrioN (orange), which is bound to Rac1 (magenta). The PH domains of Fgd5 (light blue) and TrioN (light orange) are oriented in opposite directions with Fgd5 exhibiting a distinctive bend in the final helix of its DH domain. Models were prepared in PyMol using structural coordinates from PDB. Structures between upper and lower panels are rotated 90° top to bottom and 90° right to left
Structural coordinates are available in the Protein Database (PDB) for an Fgd5 construct containing the DH-PH domains. The structure of the Fgd5 DH domain superimposes closely onto the structure of the TrioN DH domain. However, in Rac1 docking models, the PH domain of Fgd5 occupies a position that could encumber the docking of Rac1 (Figure 1(c), light blue ribbon). This is because the final helical bundle of the Fgd5 DH domain executes a turn that orients the PH domain towards the Rho binding interface, as depicted in Figure 1(c) (lower left panel) and Supplemental Data Figure S1 (left panels). When the structure of the Rho GEF, Vav1, was superimposed onto the Rac1 complex the PH domain extends away from the Rac1 docking site of the DH-domain (Supplemental Data Figure S1).
Biochemical analysis of Fgd5 GEF activity
We used an enzyme assay to characterize the GEF activity of purified Fgd5 and its Rho selectivity. A double domain construct containing DH-PH module was cloned, as was a triple domain construct which also contained the FYVE domain (DH-PH-FYVE). The Fdg5 double and triple domain constructs were cloned with a 6xHis tag, and purified from E. coli lysates. The purified proteins were tested for GEF activity by incubating them with multiple Rho proteins in the presence of MANT-GTP which increases fluorescence when bound to protein; thereby assessing the ability of a GEF to catalyse nucleotide exchange [20]. The DH-PH domains from TrioN, Itsn1 and Dbs were used as controls which showed activation of Rac1, Cdc42 and RhoA, respectively (Figure 2(a–c)). Fgd5 did not show activation of Cdc42 as expected (Figure 2(b)), but instead showed activation of Rac1 (Figure 2(a)). Specific activity was calculated from reactions that contained various concentration of Fgd5 and TrioN (Supplemental Data Figure S2), which showed that Fgd5 DH-PH-FYVE had 36% (± 6.2%) of the activity of TrioN (Figure 2(d)). Therefore, Fgd5 is significantly less active than TrioN, which has well-defined Rac1 activation properties [22,23]. The Fgd5 DH-PH double domain showed a 68% (± 9.8%) reduction in activity when compared to the Fgd5 DH-PH-FYVE triple domain (Figure 2(e)). An F803K point mutant was generated in the specificity patch of the Fgd5 DH domain (see underlined residue in Figure 1(a,b)). The F803K mutant failed to activate Rac1 (Figure 2(e)). These results suggest that the FYVE domain might facilitate Rac1 docking since molecular modelling showed the PH domain may block Rho interaction with the DH domain.
Figure 2.
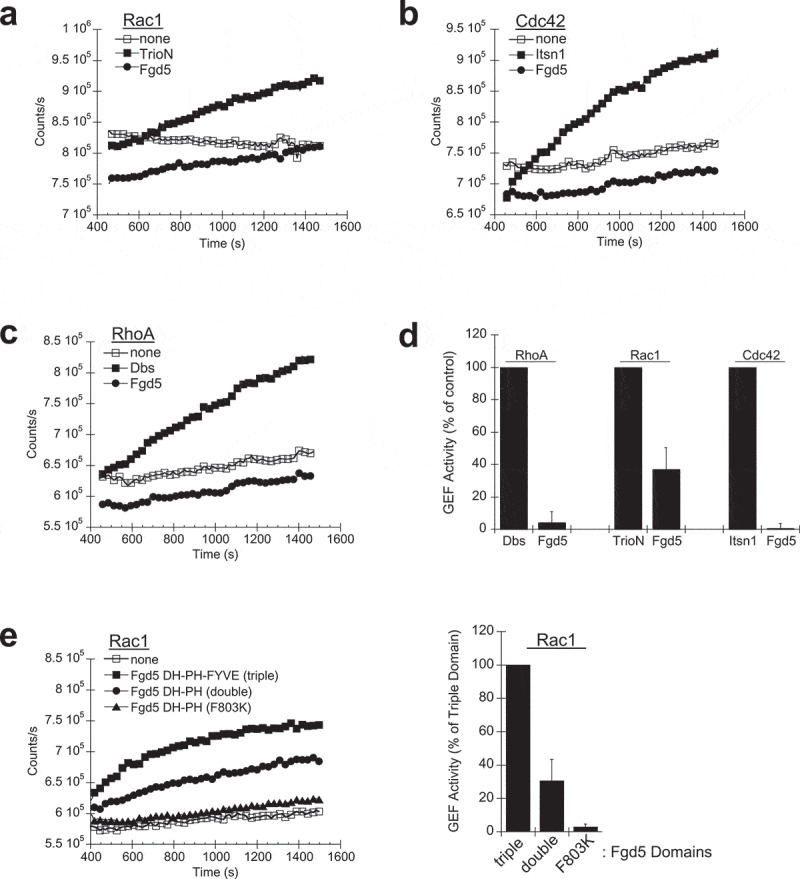
Nucleotide exchange assays. 200 nM purified Fgd5-DH-PH-FYVE was incubated in the presence of 1 mM purified GST-Rac1 (a), Cdc42 (b) and RhoA (c) and fluorescent MANT-GTP to examine nucleotide exchange activity. 50 nM of the DH-PH domains from TrioN (a), Itsn1 (b) and Dbs (c) were used as controls which showed activation of Rac1, Cdc42 and RhoA, respectively. (d) GEF activity was calculated from the initial slopes of activation assays normalized to controls. (e) Comparison of the Fgd5-DH-PH-FYVE triple domain, Fgd5-DH-PH double domain and a double domain F803K mutant. GEF activities at 200 nM in the presence of 1 mM purified GST-Rac1. Representative results from single GEF assays are shown in the line graphs (a, b, c, e-left panel), while average activities calculated from four experiments are shown in the bar graphs (d, E-right panel). Error bars = s.e.m
Analysis of Fgd5-Rho GTPase interactions
Next we examined which Rho proteins specifically interact with the Fgd5 DH domain. Direct binding of Rho GEFs was analysed via pull-down assays with GST-tagged Rho proteins immobilized on glutathione agarose. We found that the Fgd5 triple domain bound to immobilized Rac1, and not to Cdc42 or RhoA (Figure 3(a)). The Fgd5 double domain also bound selectively to Rac1, even though it showed minimal GEF activity (Figure 3(b)) while the F803K specificity patch mutant did not bind (Figure 3(c)). Control experiments were also carried out to show direct binding of Cdc42 with the Cdc42-specific GEF Itsn1, and Rac1 with the Rac1-specific GEF TrioN (Supplemental Data Figure S3A and S3B, respectively). Notably, Rac1 did not interact with Itsn1, and Cdc42 did not interact with TrioN which indicates binding specificity of this assay was maintained. The RhoA GEF, Dbs, showed binding to both RhoA and Cdc42, but not Rac1 (Supplemental Data Figure S3C). Interactions between Fgd5 and Rho proteins were also examined by co-immunoprecipitation experiments. HEK293T cells were transfected with GFP-tagged Fgd5-full-length or -DH domain deletion constructs. Both constructs were expressed at similar levels. Immunoprecipitation using GFP antibodies showed a small amount of endogenous Rac1 co-immunoprecipitated specifically with full length Fgd5 (Figure 3(d)). Cdc42 was not detected in the immunoprecipitate (Supplemental Data Figure S4). Therefore, Fgd5 showed selective GEF interaction and activation of Rac1.
Figure 3.
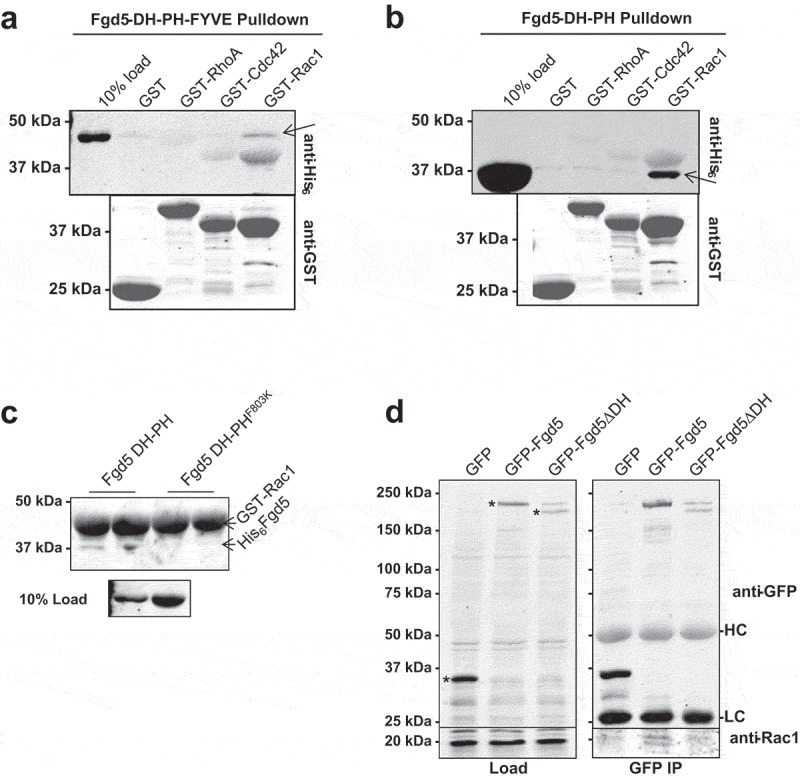
Analysis of Fgd5-Rho GTPase interaction. (a and b) Direct binding of Fgd5 to Rac1. Incubation of 5 μM Fgd5-DH-PH-FYVE triple domain (a), or the Fgd5-DH-PH double domain (b) with immobilized GST or GST-tagged RhoA, Cdc42 or Rac1. Arrows indicate the Fgd5 bands, while the GST probes can be seen in the background. (c) The Fgd5 DH-PHF803K double domain mutant does not bind Rac1. (d) Co-immunoprecipitation of Fgd5 and Rac1. Lysates (left panels) from HEK293T cells expressing GFP, GFP-Fgd5 (full length), GFP-Fgd5∆DH (deletion of the DH domain) were subjected to immunoprecipitation with GFP antibodies (right panels). Upper panels were immunoblotted with anti-GFP, * indicates GPF proteins in lysates. Lower panels were immunoblotted with anti-Rac1, showing equivalent levels of Rac1 in lysates (left) and its detection only in the GFP-Fgd5 immunoprecipitation (right). HC and LC indicates heavy and light chain IgG in the immunoprecipitate
Fgd5 compound screening by SPR
Compounds that interfere with specific Rho GEFs have recently been reported [24–26]. For example, the compound NSC 23,766 blocks the TrioN and Tiam1 activation of Rac1 while having no effect on Vav1 activation of Rac1 [24], although specific Fgd5 inhibitors have not yet been reported. In order to identify potential Fgd5-Rac1 ligands we screened the LOPAC1280 compound library for small drug-like molecules that bind the Fgd5 DH-PH-FYVE construct since its domains are most critical to GEF activity. This Fgd5 construct was crosslinked to a CM5 Biacore chip and interacting ligands were identified by surface plasmon resonance (SPR) (see Figure 4(a) for representative SPR curves). A total of 93 compounds showed binding levels greater than 3-fold above plate averages (Supplemental Data Table S1), and 20 compounds showed binding levels greater than 10-fold above plate averages (Table 1). From this subset, some compounds were removed due to repeated identification in unrelated screens (personal communications), leaving 11 compounds which were further tested for interference in Rac1 binding and exchange activity (Figure 4(b)).
Figure 4.
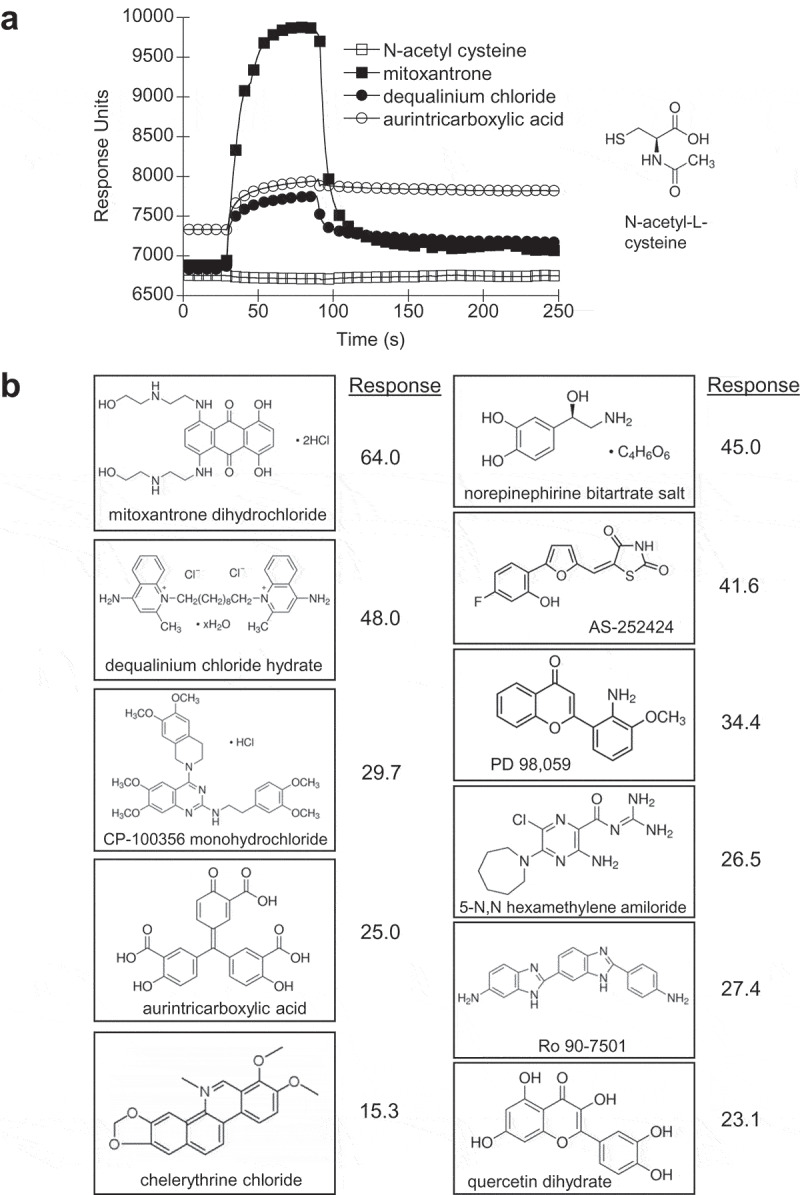
Compound screening for small molecule ligands of Fgd5. The Fgd5 triple domain was crosslinked to a CM5 Biacore chip and compounds from the LOPAC1280 library were screened for binding by surface plasmon resonance (SPR). (a) Representative SPR curves for compounds that showed no response (N-acetyl-L-cysteine) and high response (mitoxantrone, dequalinium chloride and aurintricarboxylic acid). Response calculations from compounds showing greater than 3-fold response over the average response calculated for each plate are provided in Table S1 (supplemental data). (b) Structures of the top 11 compounds selected for further analysis
Table 1.
Fgd5 Binding Compounds Identified by SPR
| Response (fold over plate avg) |
Sigma Cat No | Chemical Name |
|---|---|---|
| 64.03 | M6545 | Mitoxantrone dihydrochloride |
| 48.01 | D3768 | Dequalinium chloride hydrate |
| 45.04 | A0937 | Norepinephirine bitartrate salt |
| 41.57 | A8981 | AS-252,424 |
| 34.40 | p-215 | PD 98,059 |
| 29.68 | pz0171 | CP-100,356 monohydrochloride |
| 27.43 | R0529 | Ro 90-7501 |
| 26.54 | A9561 | 5-N,N hexamethylene amiloride |
| 24.99 | A1895 | Aurintricarboxylic acid |
| 23.05 | Q0125 | Quercetin dihydrate |
| 15.28 | C2932 | Chelerythrine chloride |
Biochemical analysis of compounds
Compounds that displayed significant binding to the Fgd5 DH-PH-FYVE construct were tested for inhibition of GEF activity. Only a subset were expected to be Fgd5 inhibitors since some could bind to sites unrelated to the active site. For cross-validation two assays were performed; one examined the inhibitory effect on GEF enzyme activity and the other whether compounds interfered in direct binding between Rac1 and Fgd5.
GEF enzyme assays were performed in the presence of each of the 11 Fgd5-binding compounds. Assays contained 200 nM Fgd5 and 450 nM of each compound that were pre-mixed before adding to GEF assays that contained 1 μM GST-Rac1 and 150 nM MANT-GTP. The percent inhibition was calculated as the difference in activity divided by the activity in the absence of inhibitor. Three of the 11 compounds tested showed a significant inhibition of Fgd5 GEF activity by Student’s t-test (Figure 5(a)). Of these, CP-100,356 had the most striking effect, resulting in negative activity values based on the negatively sloped GEF curves. However, addition of CP-100,356 to GEF assays containing TrioN also resulted in negative activity values, suggesting non-specifically effects. Aurintricarboxylic acid significantly inhibited Fgd5 GEF activity while 5-N,N hexamethylene amiloride also showed slight inhibition (Figure 5(a)), and neither compound showed any effect on TrioN-stimulated GEF activity (Figure 5(b)). Application of a Holm-Bonferroni statistical correction for multiple comparisons resulted in only aurintricarboxylic acid maintaining statistical significance. Hence we consider aurintricarboxylic acid a candidate specific inhibitor of Fgd5 GEF activity.
Figure 5.
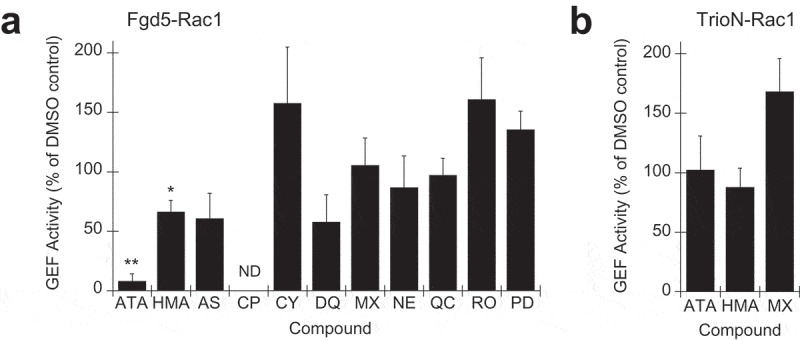
Effect of Fgd5-binding compounds on GEF enzyme assays. 200 nM of Fgd5 DH-PH-FYVE triple domain (a) or 50 nM of TrioN DH-PH domain (b) was preincubated with 450 nM of each of the 11 compounds selected for further analysis as indicated in Figure 4B prior to the addition of standard GEF assays (2 ml final volume). Each assay was normalized to a control experiment contain Fgd5 that was preincubated without compound but with an equivalent volume of DMSO (vehicle). Error bars = s.e.m.; n = 3; significance was calculated using an unpaired Students t-test (*, p < 0.1; **, p < 0.01). Compound abbreviations: Aurintricarboxylic acid (ATA), 5-N,N hexamethylene amiloride (HMA), AS-252,424 (AS), CP-100,356 (CP), chelerythrine (CY), dequalinium (DQ), mitoxantrone (MX), norepinephirine (NE), PD 98,059 (PD), quercetin (QC), Ro 90–7501 (RO)
Direct binding assays were also performed in the presence of potential Fgd5 inhibitors. This showed that the Fgd5 triple domain interaction with Rac1 could also be inhibited significantly by aurintricarboxylic acid and also slightly by mitroxantrone and chelerythrine chloride (Figure 6(a)). Control experiments showed that the Rac1-TrioN interaction was not affected by aurintricarboxylic acid, and only slightly by mitoxantrone (Figure 6(b)). We again applied a Holm-Bonferroni statistical correction for multiple comparisons which showed that aurintricarboxylic acid and chelerythrine maintained statistical significance. Taken together with GEF assays, these results suggest that aurintricarboxylic acid may be a relatively specific Fgd5 GEF inhibitor since it disrupts Fgd5 binding and activation of Rac1. Titration of aurintricarboxyic acid into Fgd5 GEF assays showed the approximate IC50 for GEF inhibition was 157 nM (Figure 7(a)). Similarly, titration of aurintricarboxylic acid into Fgd5 binding assays with GST-Rac1 showed a dose dependent inhibition of both Fgd5 DH-PH and DH-PH-FYVE domains (Figure 7(b), upper and middle panels, respectively), while having no effect on the binding of TrioN DH-PH domain (Figure 7(b), lower panel). Hence, the drug molecule aurintricarboxylic acid is an Fgd5 inhibitor that displays selectivity for this target over other GEFs.
Figure 6.
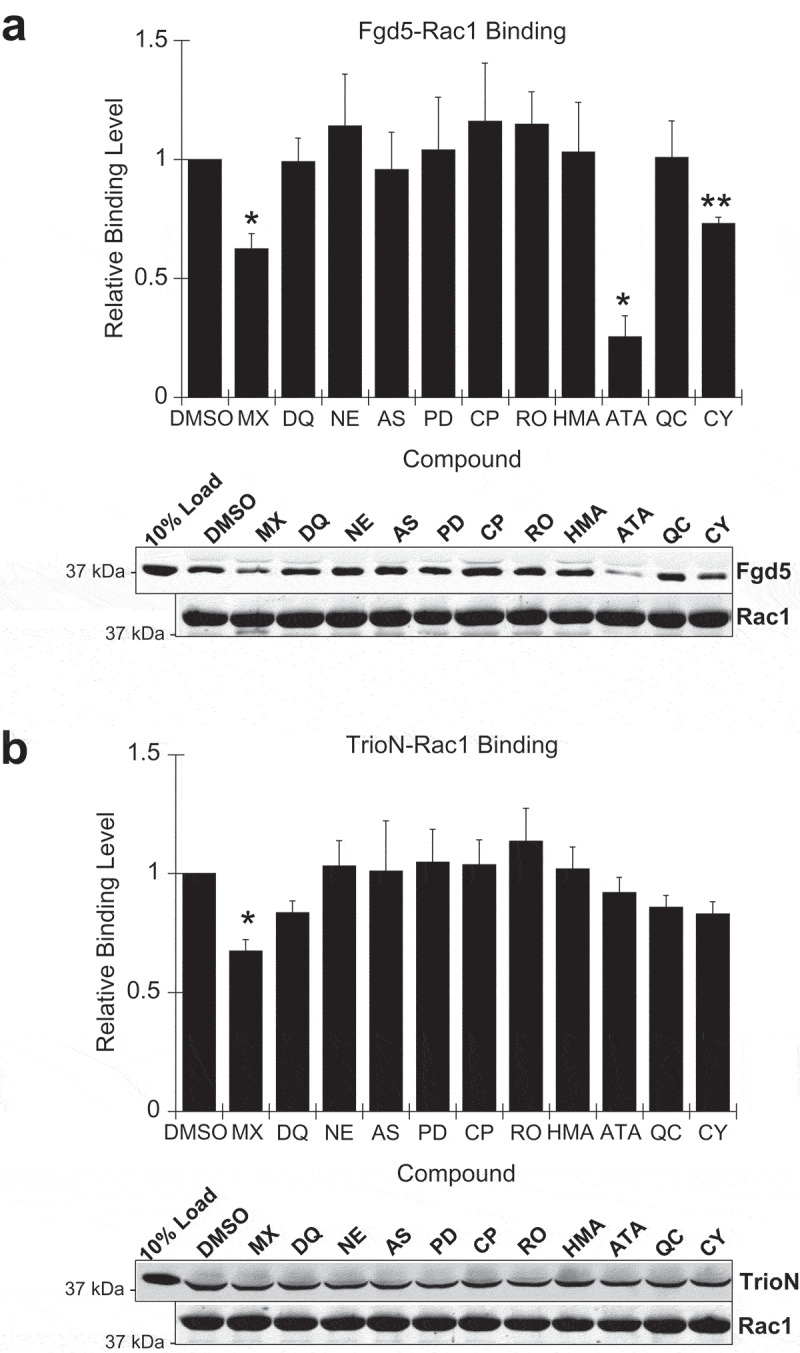
Effect of Fgd5-binding compounds on Fgd5-Rac1 interactions. (a) Inhibition of Rac1-Fgd5 binding was performed by pull-down assay in the presence of potential Fgd5 GEF inhibitors. Fgd5 triple domain (5 μM final concentration) was preincubated with the 11 compounds selected for further analysis (20 μM final concentration) then added to tubes containing immobilized GST-Rac1 (0.5 ml final volume). This showed that Fgd5 triple domain interaction with Rac1 could be significantly inhibited by aurintricarboxylic acid (ATA), mitoxantrone (MX) and chelerythrine (CY). (b) Specificity of compound inhibitory effect was examined by preincubation of TrioN with Fgd5-binding compounds then adding to tubes containing immobilized GST-Rac1. Mitoxantrone also inhibits TrioN-Rac1 binding and thus is not a selective inhibitor. Sample immunoblots from binding assays are shown below the histograms. (a and b) Error bars = s.e.m.; n = 3; significance was calculated using an unpaired Students t-test (*, p < 0.05; **, p < 0.01). Compound abbreviations: see legend to Figure 5
Figure 7.
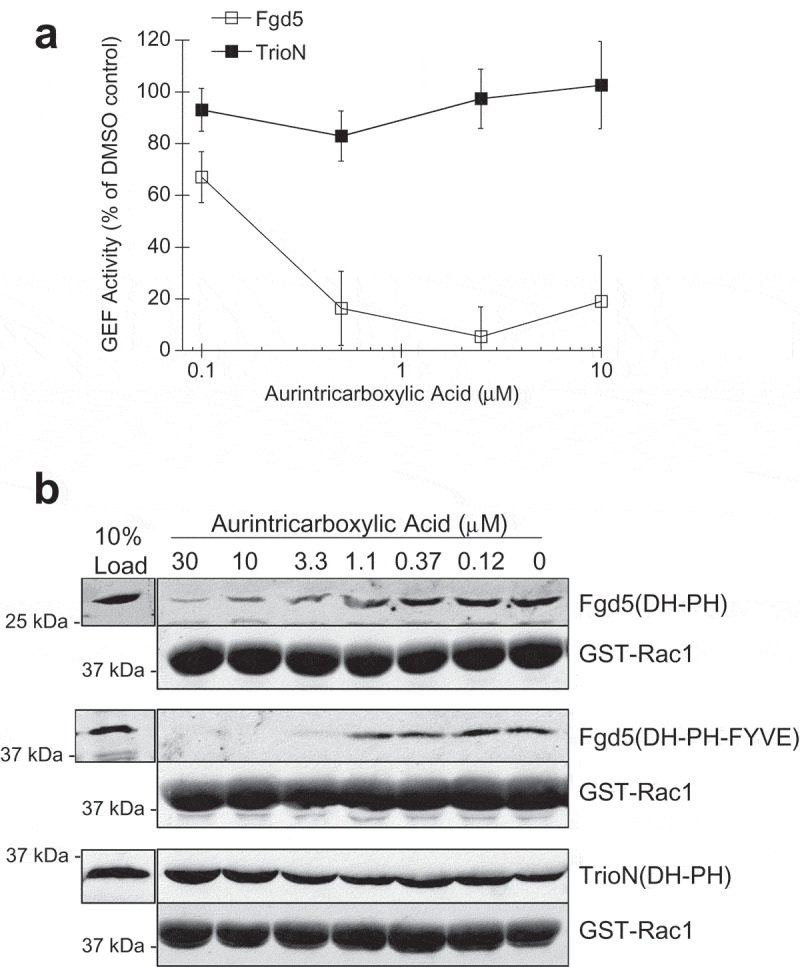
Determination of the Fgd5 inhibitory concentration of aurintricarboxylic acid. Fgd5 GEF assays (a) and GST-Rac1 binding assays (b) were performed in the presence of varying concentrations of aurintricarboxylic acid. (a) Aurintricarboxylic acid inhibited Fgd5 Rac1 GEF activity with a calculated IC50 of 157 nM. Error bars = s.e.m., n = 4. (b) The IC50 for inhibition of GST-Rac1 binding was 1.8 μM for the Fgd5 DH-PH domain (upper panel), and 1.1 μM for the Fgd5 DH-PH-FYVE domain (middle panel), while no inhibition was observed for binding the TrioN DH-PH domain (lower panel)
Discussion
Rho GEFs comprise a large family of 70 Dbl-like and 11 DOCK-like activators of Rho GTPase [11]. With minimal numbers of Rho proteins, this large family of GEFs is needed to produce highly selective activation events in different cell types to maintain their function. The FGD family of Rho GEFs comprise 6 members with highly similar domain architecture of adjacent DH-PH-FYVE domains followed by a second, C-terminal PH domain [27]. The human disorders, Aarskog-Scott syndrome and Charcot-Marie-Tooth disease have been linked to mutations in FGD1 and FGD4, respectively [1–3]. While these disorders are quite dissimilar, they do define the importance of Fgd proteins in human development. Fgd5 is a Rho GEF that is highly expressed in endothelial cells [7]. While genetic mutations of FGD5 have identified they have not been linked to human disease causation. Fgd5 is known to be is highly expressed in endothelial cells [7] and we and others have previously shown that it plays a role in endothelial cell sprouting [4,17].
Our results support a role for Fgd5 as Rho GEF for Rac1. This differentiates it from Fgd1-4, which have all been previously shown to bind and activate Cdc42 [13,15,28–31]. Our results are in disagreement with several prior reports that show Fgd5 plays a role in Cdc42 activation [16–18,32]. Fgd5 overexpression was shown to increased Cdc42 activation [16,17], however in endothelial cells, reduced Fgd5 expression still resulted in a modest increase in active Cdc42 when VEGF-stimulated [16]. There are additional conflicting results from other studies that show Fgd5 is required for both Rac1 and Cdc42 activation when endothelial cells are treated with AKB-9785, a phosphatase inhibitor that enhances tyrosine kinase signalling [18]. We have previously shown that Rac1 is required for VEGF-stimulated angiogenic sprouting, an event controlled by Fgd5 [4]. These discordant results in endothelial cells suggest that Fgd5 may have dual roles; the stimulation of endothelial cell sprouting via Rac1, and the maintenance of endothelial cells junctions via Cdc42. Indeed, active Cdc42 was shown to localize to endothelial cell junctions with other proteins required for barrier function [32].
Interaction between Fgd5 and Cdc42 was also shown by co-immunoprecipitation of overexpressed Fgd5 in endothelial cells with concomitant activation of Cdc42 and no activity towards Rac1 [17]. This is different from our results using purified proteins that show Fgd5 binds and activates Rac1 and lacks this activity for Cdc42 (Figures 2 and 3). We also examined for cellular interactions via expression of GFP-Fgd5 in HEK293T cells; this showed an interacted with Rac1 and not Cdc42 (Figure 3(d) and S4). Although the level of interaction in the co-immunoprecipitation was low, it is known that GEFs transiently bind their cognate Rho proteins to catalyse GTP loading; once this occurs, binding affinity decreases [19,20]. Differences in these results may arise from cellular models; our results used HEK293T cells for GFP-Fgd5 overexpression while other studies used endothelial cells. In endothelial cells it cannot be ruled out that Cdc42 activation may be connected to Rac1 activation. A recent report of Fgd5 function in endothelial cells also examined the effect of reduced Cdc42 expression on Rac1 activation [18]. While Fgd5 was required for both Cdc42 and Rac1 activation, it was found that Rac1 activation was not affected by reduced Cdc42 expression. Hence, Fgd5 may coordinately regulate the activation of Rac1 followed by Cdc42 in endothelial cells.
Structural studies show that Rho GEFs have a ‘specificity patch’, which is considered to be the decisive factor for Rho GTPases selectivity [19]. The specificity patch on Rho GEFs is created by a ‘U’ shaped structure of α helices which contributes to the interface that interacts with Rho GTPases [10]. We provide further analysis of the specificity of the Fgd family by homology analysis within the specificity patch. This showed that while Fgd1-4 have long stretches of identical amino acids, Fgd5 and Fgd6 are significantly divergent here (Figure 1(a)). Alignment of Fgd5 with the Rac1 GEF, TrioN, showed an elevated similarity (Figure 1(b)), which supports our results showing that Fgd5 is Rac1 GEF (Figure 2(b)).
Rho GEFs have a highly conserved Dbl homology (DH) domain which catalyzes the nucleotide exchange required to create the active Rho protein [10]. These domains are approximately 200 amino acids and contain GEF enzymatic activity. Within the DH domain, there are two α helices (CR1 and CR3) on the surface of the domain that are directly involved in the formation of the GTPase interaction pocket [18]. This area interacts with GDP-bound GTPases to catalyse exchange of GDP for a GTP [9]. The combined action of the tandem DH-PH domains is known to be the minimal unit needed to activate Rho GTPases [9]. PH domains are thought to facilitate Rho GTPase interaction by membrane localization through binding of phosphorylated phosphoinositides [33,34]. Binding of the PH domain to inositol phospholipids in the membrane has been suggested to expose the DH domain and allow interaction with GTPases [10]. Our structural model of the Fgd5 DH-PH domain suggests that the PH domain could orient in a way that occludes the interaction of the DH domain with Rac1 (Figure 1(c) and S1). While the final CR3 helical domain of other Rac1 GEFs, such as TrioN and Vav1, are straight and hence result in the projection of the PH domain away from the Rac1-binding interface, there is a significant bend in final CR3 helix of Fgd5 which orients the PH domain towards the Rac1-binding interface (Figure 1(c) and S1, lower left panel). In GEF assays, purified TrioN DH-PH domain showed significant GEF activity, while purified Fgd5 DH-PH domain showed little activity. This significant reduction may be due to the structural differences in the orientation of the PH domain. Inclusion of the FYVE domain increased activity (Figure 2(e)), which suggests that the FYVE domain may have a role in regulating the GEF activity of Fgd5, for example, by facilitating a conformation change upon ligand binding. This is supported by previous results that show overexpression of only the Fgd5 DH-PH domain in endothelial cells has no activity [16].
The uniqueness of the Fgd specificity patch provides opportunities for the development of highly specific pharmacological inhibitors [35]. Therefore we performed a limited compound library screen using SPR, which identified numerous small molecules that bind to the Fgd5 DH-PH-FYVE domain. Comparison of the compounds that bound significantly indicated that some chemical groups are shared between a few of the molecules. Different salts of norepinephrine were present on several different plates and binding to Fgd5 was detected each time (Supplemental Data Table S1). Additionally, R-(-)-apomorphine hydrochloride hemihydrate and R(-)-propyl norapomorphine hydrochloride both bound extremely well and have similar substructures. Common chemical structures that reappear in all four of these molecules are catechol structures (Figure 4). Pharmacologically, this suggests that catechol-like motifs exhibit affinity for the Fgd5 protein.
A subset of compounds were further examined in GEF activity and direct-binding assays. Some compounds, such as Ro-7501, increased Fgd5 activity which may be due to affecting the conformational changes that facilitate Fgd5 interaction with Rac1. Aurintricarboxylic acid was found to inhibit both the interaction with Rac1 and its GEF activity (Figures 5 and 6). This compound was originally reported to be a strong inhibitor of topoisomerases and ribonucleases [36]. It has since been shown to be an active inhibitor of viral pathogenesis [37,38] and expression of adhesion molecules [39]; however, the specific targets for these effects are unknown, although they may be mediated through inhibition of nucleases. Nuclease-independent targets of aurintricarboxylic acid include reports of binding and inhibition of phospholipase C [40] and more recently, inhibition of Staphylococcus aureus phosphatases involved in the regulation of their virulence [41]. Therefore, aurintricarboxylic acid may serve to inhibit multiple processes, with identification of its interactors being key to the design of more specific inhibitors.
The development of Fgd5 inhibitors is important for investigating its role in human disease. The Fgd5 gene was found to be highly expressed in human vascular endothelial cells [7]. Fgd5 may regulate VEGF actions through affecting Rac1 in endothelial cells through proliferation and pro-angiogenesis [4]. In support of this finding, we found that inhibition of Fgd5 expression was related to the reduction of endothelial cell sprout protrusion [8]. Furthermore, Fgd5 knockdown reduces adhesion to fibronectin and collagen IV and the remodelling of matrix proteins complexes. Fgd5 has also been implicated in cell survival, due to the increase in the expression of caspase-3 in endothelial cells lacking Fgd5. Adhesion and survival signal transduction pathways are both regulated by phosphatidylinositol 3-kinase/Akt pathway, which requires Fgd5 [4]. The development of chemical probes and specific inhibitors of Fgd5 will help to delineate its mechanism of action in these pathways and its utility as a target for cancer progression and the control of the growth and survival of blood vessels.
Supplementary Material
Acknowledgments
We would like to thank Kent Rossman (University of North Carolina, Chapel Hill) for providing Rho GEF plasmids that express the DH-PH domains of Dbs and Itsn1 and Luc Berthiaume (University of Alberta) for goat anti-GFP antibodies. This work was supported by The Natural Sciences and Engineering Research Council of Canada under grant number 327237 to GE; and the Canadian Cancer Society under grant number 703774 to GE and AGM. MO is funded by the Campus Alberta Innovation Program (RCP-12-002C), Alberta Prion Research Institute/Alberta Innovates Bio Solutions (201600018) and NSERC RGPIN-2018-04994, and the CFI and Genome Canada grants which support The Metabolomics Innovation Centre (TMIC). Sally Park is recipient of a summer studentship from Alberta Innovates Health Solutions. Yitian Guo is the recipient a studentship from the China Scholarship Council.
Funding Statement
This work was supported by the Canadian Cancer Society Research Institute [703774]; Natural Sciences and Engineering Research Council of Canada [327237].
Authorship Contributions
SP, GE, AKA and AGM participated in research design. SP, YG, JN and GE conducted experiments. AKA, CCS, MO, and GE contributed new reagents or analytic tools. SP, YG, JP and GE performed data analysis. SP, CCS, MO and GE contributed to the writing of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this can be accessed here.
References
- [1].Pasteris NG, Cadle A, Logle LJ, et al. Isolation and characterization of the faciogenital dysplasia (Aarskog-Scott Syndrome) gene: a putative Rho/Rac guanine nucleotide exchange factor. Cell. 1994;79:669–678. [DOI] [PubMed] [Google Scholar]
- [2].Delague V, Jacquier A, Hamadouche T, et al. Mutations in FGD4 encoding the Rho GDP/GTP exchange factor FRABIN cause autosomal recessive Charcot–marie–tooth type 4H. Am J Hum Genet. 2007;81:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Stendel C, Roos A, Deconinck T, et al. Peripheral nerve demyelination caused by a mutant Rho GTPase guanine nucleotide exchange factor, frabin/FGD4. Am J Hum Genet. 2007;81:158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Farhan MA, Azad AK, Touret N, et al. FGD5 regulates VEGF receptor-2 coupling to PI3 kinase and receptor recycling. Arterioscler Thromb Vasc Biol. 2017;37:2301–2310. [DOI] [PubMed] [Google Scholar]
- [5].Heldin J, O’Callaghan P, Hernández Vera R, et al. FGD5 sustains vascular endothelial growth factor A (VEGFA) signaling through inhibition of proteasome-mediated VEGF receptor 2 degradation. Cell Signal. 2017;40:125–132. [DOI] [PubMed] [Google Scholar]
- [6].Valla M, Engstrøm MJ, Ytterhus B, et al. FGD5 amplification in breast cancer patients is associated with tumour proliferation and a poorer prognosis. Breast Cancer Res Treat. 2017;162:243–253. [DOI] [PubMed] [Google Scholar]
- [7].Hernández-García R, Iruela-Arispe ML, Reyes-Cruz G, et al. Endothelial RhoGEFs: A systematic analysis of their expression profiles in VEGF-stimulated and tumor endothelial cells. Vascul Pharmacol. 2015;74:60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nakhaei-Nejad M, Haddad G, Zhang Q, et al. Facio-Genital dysplasia-5 regulates matrix adhesion and survival of human endothelial cells. Arterioscler Thromb Vasc Biol. 2012;32:2694–2701. [DOI] [PubMed] [Google Scholar]
- [9].Schmidt A, Hall A.. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Gen Dev. 2002;16:1587–1609. [DOI] [PubMed] [Google Scholar]
- [10].Rossman KL, Der CJ, Sondek J. GEF means Go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol. 2005;6:167–180. [DOI] [PubMed] [Google Scholar]
- [11].Cook DR, Rossman KL, Der CJ. Rho guanine nucleotide exchange factors: regulators of Rho GTPase activity in development and disease. Oncogene. 2014;33:4021–4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Zheng Y, Fischer DJ, Santos MF, et al. The faciogenital dysplasia gene product FGD1 functions as a Cdc42Hs-specific guanine-nucleotide exchange factor. J Biol Chem. 1996;271:33169–33172. [DOI] [PubMed] [Google Scholar]
- [13].Huber C, Mårtensson A, Bokoch GM, et al. FGD2, a CDC42-specific exchange factor expressed by antigen-presenting cells, localizes to early endosomes and active membrane ruffles. J Biol Chem. 2008;283:34002–34012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Pasteris NG, Nagata K, Hall A, et al. Isolation, characterization, and mapping of the mouse Fgd3 gene, a new Faciogenital Dysplasia (FGD1; Aarskog Syndrome) gene homologue. Gene. 2000;242:237–247. [DOI] [PubMed] [Google Scholar]
- [15].Ono Y, Nakanishi H, Nishimura M, et al. Two actions of frabin: direct activation of Cdc42 and indirect activation of Rac. Oncogene. 2000;19:3050–3058. [DOI] [PubMed] [Google Scholar]
- [16].Kurogane Y, Miyata M, Kubo Y, et al. FGD5 mediates proangiogenic action of vascular endothelial growth factor in human vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2012;32:988–996. [DOI] [PubMed] [Google Scholar]
- [17].Cheng C, Haasdijk R, Tempel D, et al. Endothelial cell-specific FGD5 involvement in vascular pruning defines neovessel fate in mice. Circulation. 2012;125:3142–3158. [DOI] [PubMed] [Google Scholar]
- [18].Braun LJ, Zinnhardt M, Vockel M, et al. VE-PTP inhibition stabilizes endothelial junctions by activating FGD5. EMBO Rep. 2019;20:e47046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Snyder JT, Worthylake DK, Rossman KL, et al. Structural basis for the selective activation of Rho GTPases by Dbl exchange factors. Nat Struct Biol. 2002;9:468–475. [DOI] [PubMed] [Google Scholar]
- [20].Leonard DA, Evans T, Hart M, et al. Investigation of the GTP-binding/GTPase cycle of Cdc42Hs using fluorescence spectroscopy. Biochem. 1994;33:12323–12328. [DOI] [PubMed] [Google Scholar]
- [21].Hart MJ, Eva A, Zangrilli D, et al. Cellular transformation and guanine nucleotide exchange activity are catalyzed by a common domain on the dbl oncogene product. J Biol Chem. 1994;269:62–65. [PubMed] [Google Scholar]
- [22].Debant A, Serra-Pagès C, Seipel K, et al. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc Natl Acad Sci USA. 1996;93:5466–5471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gao Y, Xing J, Streuli M, et al. Trp(56) of rac1 specifies interaction with a subset of guanine nucleotide exchange factors. J Biol Chem. 2001;276:47530–47541. [DOI] [PubMed] [Google Scholar]
- [24].Gao Y, Dickerson JB, Guo F, et al. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc Natl Acad Sci USA. 2004;101:7618–7623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Shutes A, Onesto C, Picard V, et al. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282:35666–35678. [DOI] [PubMed] [Google Scholar]
- [26].Montalvo-Ortiz BL, Castillo-Pichardo L, Hernández E, et al. Characterization of EHop-016, novel small molecule inhibitor of Rac GTPase. J Biol Chem. 2012;287:13228–13238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Eitzen G, Smithers CC, Murray AG, et al. Structure and function of the Fgd family of divergent FYVE domain proteins. Biochem Cell Biol. 2019;97:257–264. [DOI] [PubMed] [Google Scholar]
- [28].Estrada L, Caron E, Gorski JL. Fgd1, the Cdc42 guanine nucleotide exchange factor responsible for faciogenital dysplasia, is localized to the subcortical actin cytoskeleton and Golgi membrane. Hum Mol Genet. 2001;10:485–495. [DOI] [PubMed] [Google Scholar]
- [29].Liu HP, Chen CC, Wu CC, et al. Epstein-Barr virus-encoded LMP1 interacts with FGD4 to activate Cdc42 and thereby promote migration of nasopharyngeal carcinoma cells. PLoS Pathog. 2012;8:e1002690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hayakawa M, Matsushima M, Hagiwara H, et al. Novel insights into FGD3, a putative GEF for Cdc42, that undergoes SCF(FWD1/beta-TrCP)-mediated proteasomal degradation analogous to that of its homologue FGD1 but regulates cell morphology and motility differently from FGD1. Genes Cells. 2008;13:329–342. [DOI] [PubMed] [Google Scholar]
- [31].Nakanishi H, Takai Y. Frabin and other related Cdc42-specific guanine nucleotide exchange factors couple the actin cytoskeleton with the plasma membrane. J Cell Mol Med. 2008;12:1169–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ando K, Fukuhara S, Moriya T, et al. Rap1 potentiates endothelial cell junctions by spatially controlling myosin II activity and actin organization. J Cell Biol. 2013;202:901–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp. 2007;1:81–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Fuentes EJ, Karnoub AE, Booden MA, et al. Critical role of the pleckstrin homology domain in Dbs signaling and growth regulation. J Biol Chem. 2003;278:21188–21196. [DOI] [PubMed] [Google Scholar]
- [35].Nassar N, Cancelas J, Zheng J, et al. Structure–function based design of small molecule inhibitors targeting Rho family GTPases. Curr Top Med Chem. 2006;6:1109–1116. [DOI] [PubMed] [Google Scholar]
- [36].Hallick RB, Chelm BK, Gray PW, et al. Use of aurintricarboxylic acid as an inhibitor of nucleases during nucleic acid isolation. Nucleic Acids Res. 1977;4:3055–3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chen Y, Bopda-Waffo A, Basu A, et al. Characterization of aurintricarboxylic acid as a potent hepatitis C virus replicase inhibitor. Antivir Chem Chemother. 2009;20:19–36. [DOI] [PubMed] [Google Scholar]
- [38].Hashem AM, Flaman AS, Farnsworth A, et al. Aurintricarboxylic acid is a potent inhibitor of influenza A and B virus neuraminidases. PLoS One. 2009;4:e8350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kim JE, Lee S, Han KS, et al. Aurintricarboxylic acid inhibits the nuclear factor-κB-dependent expression of intercellular cell adhesion molecule-1 and endothelial cell selectin on activated human endothelial cells. Blood Coagul Fibrinolysis. 2011;22:132–139. [DOI] [PubMed] [Google Scholar]
- [40].Huang W, Barrett M, Hajicek N, et al. Small molecule inhibitors of phospholipase C from a novel high-throughput screen. J Biol Chem. 2013;288:5840–5848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Liu TT, Yang T, Gao MN, et al. The inhibitory mechanism of aurintricarboxylic acid targeting serine/threonine phosphatase Stp1 in Staphylococcus aureus: insights from molecular dynamics simulations. Acta Pharmacol Sin. 2019;40:850–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.