

Abstract
Aims
In light of recent data regarding inflammatory signalling pathways in cardiovascular disease and the recently demonstrated impact of pharmacologic inhibition of interleukin-1β (IL-1β) in heart failure, the primary aim was to assess the physiologic effects of cardiac resynchronization therapy (CRT) on the expression of systemic inflammatory, immune-modulatory, metabolic, and apoptotic genes in peripheral blood mononuclear cells (PBMCs) of patients with heart failure.
Methods and results
We used RNA sequencing (RNA-Seq) and reverse transcription quantitative real-time polymerase chain reaction (RT-qPCR) to identify gene expression changes in PBMCs in response to CRT. In total, 27 patients were analysed: 12 with heart failure undergoing CRT, 6 with heart failure undergoing standard implanted cardioverter defibrillators, and 9 with coronary artery disease but not heart failure. In CRT patients (median age 65.5 years, interquartile range 63.0–66.8 years, 33% female), RNA-Seq analysis identified 40 genes, including multiple genes associated with the IL-1β pathway, with significant correlations (false discovery rate < 0.05) with four key CRT response measures. CRT was associated with suppression of PBMC expression of IL-1β (1.80-fold decrease, P = 0.047), FOS proto-oncogene (FOS) (3.25-fold decrease, P = 0.01), dual specificity phosphatase 1 (DUSP1) (2.05-fold decrease, P = 0.001), and early growth response 1 (EGR1) (7.38-fold decrease, P = 0.03), and suppression was greater in responders vs. non-responders (P = 0.03 for IL-1β, P = 0.02 for FOS, P = 0.02 for DUSP1, and P = 0.11 for EGR1). Baseline FOS and DUSP-1 levels were greater in responders vs. non-responders (6.15-fold higher, FOS, P = 0.002; 2.60-fold higher, DUSP1, P = 0.0001). CRT responders but not non-responders showed higher baseline gene expression of FOS (P = 0.04) and DUSP1 (P = 0.06) compared with control patients without heart failure. Baseline serum high-sensitivity C-reactive protein levels were 3.47-fold higher in CRT responders vs. non-responders (P = 0.008).
Conclusion
Treatment of heart failure with CRT resulted in decreased PBMC expression of genes linked to inflammation. Moreover, CRT responders had higher expression of these inflammatory genes prior to CRT and greater suppression of these genes after CRT compared with non-responders.
Keywords: Cardiac resynchronization therapy, RNA-seq, PCR, Heart failure, Il-1β
Graphical Abstract
Graphical Abstract.

1. Introduction
While cardiac resynchronization therapy (CRT) has been shown to be an effective treatment for many patients with heart failure,1,2 response rates are in the range of 50–60%.3–8 Heart failure is a progressive disease, and straightforward methods such as blood tests to predict and monitor the durability of response after therapies such as CRT could greatly improve clinical care. More generally, there is an unmet need for biomarkers that can assess response, determine prognosis, and inform the need for other therapies. Inflammation and dysregulation of the immune response are key inter-related factors involved in heart failure progression, independent of the initial form of cardiac injury.9,10 Both innate and adaptive immune cells such as monocytes, T cells, and B cells regulate cellular processes in heart failure and display functional heterogeneity in a subset-dependent manner.11–13 Complex interactions between different immune cell subsets and inflammatory cytokines post-myocardial injury promote cardiac repair, eccentric cardiac remodelling, and ventricular dysfunction.9,11 In addition to the local immune-inflammatory activation, heart failure patients display increased levels of systemic inflammation, and peripheral immune cells with altered activation and inflammatory states are associated with heart failure severity and adverse clinical outcomes.10,13,14 Recently, a sub-analysis of the Canakinumab Anti-inflammatory Thrombosis Outcome Study (CANTOS) trial demonstrated that reducing inflammation by targeted inhibition of interleukin-1β (IL-1β) with a monoclonal antibody (Canakinumab) reduces hospitalization for heart failure and heart failure-associated mortality in a subset of patients with prior myocardial infarction and elevated levels of the inflammatory C-reactive protein (CRP).15 Furthermore, IL-1β blockade by both canakinumab and the IL-1 receptor antagonist, anakinra has been shown to improve peak oxygen consumption (VO2) and left ventricular ejection fraction in subjects with systolic heart failure.16,17
Studies have revealed that injured myocardium sends signals to other organs triggering leucocyte proliferation with increased numbers in the circulation.18,19 Circulating immune cells serve as messengers linking local and systemic inflammation18 and potentially mirror local pathophysiologic processes as evidenced by transcript signatures of peripheral blood mononuclear cells (PBMCs) in heart failure patients.20–23 Because CRT modulates the inflammatory response24,25 in heart failure, we hypothesized that CRT also modulates expression of key genes in circulating immune cells that regulate inflammation. Identification of these genes and gene pathways in circulating PBMCs has the potential to provide unique mechanistic insights into intracellular pathways modulated by CRT and identify novel biomarkers for safe and targeted immune therapies for heart failure. Additionally, such a gene expression profile could aid physicians in tailoring recommendations for patients with heart failure with or without CRT. The present investigation aims to assess variations in the transcriptomic profile of PBMCs before and 6 months after CRT using ribonucleic acid (RNA) sequencing (RNA-Seq) microarrays and validation of differentially expressed transcripts with quantitative real-time polymerase chain reaction (RT-qPCR). In addition, we also evaluated the associations of these peripheral RNA expression changes with standard CRT response measures such as improvement in left ventricular systolic function and serum levels of high-sensitivity CRP (hs-CRP).
2. Methods
2.1 Study cohort
Twelve heart failure patients with either ischaemic or non-ischaemic cardiomyopathy with a standard guideline-based indication for CRT and six heart failure patients with an indication for the implanted cardioverter defibrillator (ICD) were recruited from the University of Virginia Health System. Nine age- and sex-matched subjects from the Coronary Assessment in Virginia Cohort (CAVA)26 with ejection fraction >50 were used as control subjects with no heart failure. Informed consent was obtained from all participants, and the study protocol was approved by the Institutional Review Board for Human Subjects Research at the University of Virginia. This research study was performed in accordance with the principles of the Declaration of Helsinki.
2.2 Clinical data and determination of clinical response to CRT
The CRT and ICD procedures were performed using standard clinical methods. All patients had a comprehensive assessment with four clinical endpoints based on echocardiography, cardiopulmonary exercise tests, heart failure symptom scores (Minnesota Living with Heart Failure Questionnaire), and serum B-type natriuretic peptide (BNP) levels before and 6 months after CRT. Patients also had blood draws at these time points for RNA analysis and hs-CRP assessments. WBC counts were also determined for all patients before CRT and in all but two patients after CRT. In these two patients, the WBC counts post-CRT were imputed as the same values obtained pre-CRT. The percentage change in left ventricular end-systolic volume (LVESV) before and after CRT was calculated as: (post-CRT LVESV − pre-CRT LVESV)/pre-CRT LVESV. Changes in peak VO2 and heart failure scores post-CRT were evaluated as the net paired change per patient based on typical practice. BNP changes were reported as the fractional paired change per patient. Four-year survival free of cardiac transplantation and left ventricular assist device after CRT was assessed in all patients.
2.3 Total PBMC RNA preparation
Whole blood was drawn into citrated Cell Preparation Tubes (BD Biosciences # 362761, San Jose, CA, USA) and processed within 1 h of collection. PBMCs were separated from granulocytes and red blood cells by ficoll density-gradient centrifugation, and total PBMC RNA was purified using Qiagen RNeasy Plus kit, including a DNase digest following the manufacturer’s instructions (Qiagen, Valencia, CA, USA). RNA was quantified spectrophotometrically (Nanodrop, Thermo Scientific, Wilmington, DE, USA) and assessed for quality by capillary gel electrophoresis (Agilent 2100 Bioanalyzer; Agilent Technologies, Inc., Palo Alto, CA, USA). cDNA libraries were prepared using the Illumina TruSeq RNA Sample Preparation Kit following the manufacturer’s recommended procedures. Of the 12 heart failure patients, a cDNA library could not be generated successfully for one patient, resulting in RNA-Seq analysis in 11 patients, although RT-qPCR was subsequently performed from samples for all 12 patients. Libraries were sequenced using 100 paired-end reads on Illumina HiSeq 2500 instruments. Library construction and RNA-Seq were performed at the Hudson Alpha Institute for Biotechnology (Huntsville, AL, USA).
2.4 Validation of differentially expressed genes by RT-qPCR
Expression of important differentially expressed genes from RNA-Seq analysis was confirmed by quantitative RT-qPCR using Taqman probes from Applied Biosystems (Foster City, CA, USA). cDNA samples were reverse transcribed from RNA of all 12 heart failure patients using 15 ng of the cDNA per 10 μL of the Taqman RT-qPCR reaction mix (Applied Biosystems). Taqman components included 5 μL of the Taqman universal PCR master mix and 0.5 μL of the TaqMan gene expression assay probe. Results were normalized to 18S ribosomal RNA (rRNA), and relative fold changes in gene expression levels were calculated using the 2-ΔΔCt method.27
2.5 Data analysis and statistics
Baseline demographic and clinical characteristics of patients were expressed as medians and interquartile range (IQR) for continuous variables, while numbers and percentages were used to describe categorical variables. Bivariable logistic regression was used to evaluate differences in 4-year survival free of cardiac transplantation and left ventricular assist device in patients designated as responders or non-responders based on the change in the LVESV during the first 6 months after CRT. SAS 9.4 (Cary, NC, USA) was used for this statistical analysis.
With respect to analysis of RNA-Seq data, reads from Hudson Alpha were aligned using the Human Genome Build 38 project. A total of 22 375 transcripts were identified using Salmon and imported into R using tximport. A regularized log transformation was applied to the count data to perform quality assessment. The bioconductor package edgeR was used to identify differentially expressed genes before and after CRT using a paired design to fit a quasi-likelihood negative binomial generalized log-linear model to count data. We evaluated fold changes for differentially expressed genes before and after CRT with a Benjamini–Hochberg false discovery rate (FDR) of 0.05, which accounts for multiple comparisons. Each of the four clinical endpoints was used to perform a covariate analysis using edgeR. Changes in gene expression before and after CRT were correlated with changes in response measures before and after CRT.
Selection of genes for further analysis with RT-qPCR was based on a two-step process in which we first selected those genes with significant changes post-CRT and significant associations with CRT response, and second determined which of these genes appeared to have the greatest impact on key inflammatory pathways. In the first step, we performed an analysis of differences in gene expression for each patient before and after CRT and a correlation analysis to identify those genes most closely associated with the four clinical response measures (LVESV from echocardiography, heart failure scores, peak VO2 from cardiopulmonary exercise testing, and serum BNP levels). Those genes (N = 2853) with the strongest associations with these response measures were then subjected to a pathway enrichment analysis in order to identify genes associated with a broad array of systemic inflammatory, immune-modulatory, metabolic, and apoptotic pathways. A full summary of the analytic strategy and study design are provided in Figure 1.
Figure 1.
Summary of analytic strategy and major findings. The flow chart shows a summary of the analytic strategy for PBMC gene expression analysis using RNA-Seq and RT-qPCR for the 27 patients in the study. The locations in tables and figures of key findings are also specified. RNA-Seq, RNA sequencing; RT-qPCR, quantitative real-time polymerase chain reaction.
RT-qPCR gene expression levels, for which all measurements were obtained twice, were analysed using linear mixed effect models. The models included the timing of the measurement (pre-CRT vs. post-CRT), the patient’s responder status (as a binary variable), the interaction between the timing of the measurement and the responder status, and replicates of each measurement. The P-value for the interaction term in the model was used to determine whether there was a significant difference in gene expression post-CRT in responders vs. non-responders, while the P-value based on the timing variable (pre-CRT vs. post-CRT) was used to evaluate whether there was a significant difference in expression post-CRT. Comparisons of baseline gene expression in two groups were also performed using t-tests.
3. Results
3.1 Patient characteristics
The baseline characteristics of the 27 patients in the study (12 heart failure patients undergoing CRT therapy, 6 patients with ICDs, and 9 matched controls with coronary artery disease but no heart failure) are shown in Table 1. The median age of CRT patients was 65.5 years (IQR 63.0–66.8 years), while the median age of control coronary artery disease (CAD) patients was similarly 65.0 years (IQR 59.0–66.0 years), and 33.3% of both groups were female.
Table 1.
Baseline characteristics
| CRT all (N = 12) | CRT R (N = 8) | CRT NR (N = 4) | ICD (non-CRT) (N = 6) | CAD (N = 9) | |
|---|---|---|---|---|---|
| Demographics | |||||
| Age (years) | 65.5 (63.0–66.8) | 66.1 (55.6–67.5) | 65.0 (64.2–66.0) | 63.0 (56.0–65.0) | 65.0 (59.0–66.0) |
| Gender (female) | 4 (33.3) | 4 (50) | 0 (0) | 1 (16.7) | 3 (33.3) |
| Vital signs | |||||
| Body mass index (kg/m2) | 27.2 (24.3–31.0) | 29.7 (24.3–33.8) | 27.1 (23.8–27.2) | 26.5 (25.0–33.0) | 27.3 (27.1–28.7) |
| Systolic blood pressure (mmHg) | 115 (108.0–129.5) | 117.5 (106.0–129.5) | 115.0 (111.0–126.5) | 120.0 (118–121.5) | 124.0 (119.0–136.0) |
| Diastolic blood pressure (mmHg) | 64.0 (61.0–80.0) | 66.0 (60.0–86.5) | 64.0 (62.0–71.0) | 71.0 (70.0–75.0) | 63 (60–70) |
| Comorbid disease | |||||
| Ischaemic cardiomyopathy | 6 (58.3) | 4 (50.0) | 2 (50.0) | 3 (50.0) | 0 (0) |
| Diabetes mellitus | 6 (50.0) | 4 (50.0) | 2 (50.0) | 3 (50.0) | 2 (22.2) |
| Chronic kidney disease | 3 (25.0) | 1 (12.5) | 2 (50.0) | 2 (33.3) | 0 (0) |
| Hypertension | 6 (50.0) | 5 (62.5) | 1 (25.0) | 1 (16.7) | 7 (77.8) |
| CMR findings | |||||
| LVEF (%) | 17.0 (13.5–27.0) | 21.0 (10.5–27.0) | 16.0 (15.5–22.0) | – | – |
| LVEDVI (mL/m2) | 127.6 (122.1–154.5) | 127.6 (123.3–154.5) | 127.9 (120.5–168.6) | – | – |
| LVESVI (mL/m2) | 99.3 (92.2–135.0) | 99.3 (92.2–135.0) | 98.8 (60.3–137.0) | – | – |
| RVEF (%) | 35.5 (17.5–40.0) | 37.0 (17.5–42.0) | 32.0 (21.5–38.5) | – | – |
| RVEDVI (mL/m2) | 64.5 (50.2–79.3) | 64.5 (45.3–71.0) | 69.6 (54.3–104.6) | – | – |
| RVESVI (mL/m2) | 39.4 (28.5–57.7) | 45.5 (24.4–57.7) | 35.1 (32.1–72.4) | – | – |
| ECG and lab findings | |||||
| QRS (ms) | 158.0 (143.0–178.0) | 164.5 (145.0–180.0) | 154.0 (141.0–169.0) | 88.0 (78.5–97.0) | 88.0 (86.0–84.0) |
| Q-LV (ms) | 110.0 (80.0–130.0) | 130.0 (110.0–152.0) | 80.0a (50.0–80.0) | – | – |
| Sodium (mEq/L) | 139.5 (137.5–143.0) | 139.5 (136.5–143.0) | 139.0 (138.0–141.5) | 139.0 (137.0–139.0) | 139.0 (138.0–140.0) |
| Creatinine (mg/dL) | 1.1 (1.1–1.2) | 1.1 (1.1–1.2) | 1.1 (1.0–1.3) | 1.0 (0.9–1.4) | 0.8 (0.7–0.9) |
| GFR (mL/min/1.73 m2) | 70.7 (54.5.71.9) | 67.4 (54.5–71.4) | 71.4 (60.5–87.3) | 84.0 (65.0–92.0) | 89.0 (89.0–92.0) |
| Medications | |||||
| Beta-blocker | 12 (100) | 8 (100) | 4 (100) | 6 (100) | 5 (55.6) |
| ACE inhibitor or ARB | 12 (100) | 8 (100) | 4 (100) | 5 (83.3) | 3 (33.3) |
| Loop diuretic | 11 (91.7) | 7 (87.5) | 4 (100) | 5 (83.3) | 2 (22.2) |
| Digoxin | 3 (25) | 1 (12.5) | 2 (50) | 0 (0) | 0 (0) |
Continuous variables are presented as median (interquartile range). Categorical variables are presented as frequency (percentage).
ACE, angiotensin converting enzyme; ARB, angiotensin receptor blocker; CAD, coronary artery disease; CRT, cardiac resynchronization therapy; ICD, implantable cardioverter defibrillator; LVEDVI, left ventricular end-diastolic volume index; LVEF, left ventricular ejection fraction; LVESVI, left ventricular end-systolic volume index; NR, non-responder; Q-LV, QRS-to-left-ventricular-electrogram time; R, responder; RVEDVI, right ventricular end-diastolic volume index; RVEF, right ventricular ejection fraction; RVESVI, right ventricular end-systolic volume index.
P = 0.06 vs. CRT responders.
The response parameters of the CRT patients are shown in Table 2. The median net improvement in peak VO2 per patient (paired analysis) was 2.2 (IQR −0.4 to 3.6), and the median fractional improvement (decrease) in LVESV per patient (paired analysis) was −0.18 (IQR −0.30 to 0.04). Of the 12 patients, 8 had a reduction of the LVESV by at least 15% after 6 months of CRT. These eight patients were designated as CRT responders. This distinction between responders and non-responders was validated by differences in 4-year-survival free of heart transplantation and left ventricular assist device after CRT, which was 88% in responders and 25% in non-responders (P = 0.053).
Table 2.
Response characteristics (CRT patients, N = 12)
| B-type natriuretic peptide | |
| Baseline (pg/mL) | 673 (295–1145) |
| 6-Month (pg/mL) | 116 (81–931) |
| Paired fractional change | 0.63 (0.37–0.80) |
| Peak VO2 | |
| Baseline (mL/kg/min) | 14 (11–15) |
| 6-Month (mL/kg/min) | 15 (13–18) |
| Paired net change | 2.2 (−0.4 to 3.6) |
| Heart failure score | |
| Baseline | 44 (23–70) |
| 6-Month | 22 (9–38) |
| Paired net decrease | 23 (10–34) |
| Left ventricular end-systolic volume by echocardiography | |
| Baseline (mL) | 152 (124–195) |
| 6-Month (mL) | 139 (93–156) |
| Paired fractional change | −0.18 (−0.30 to 0.04) |
| Survival free of heart transplantation and left ventricular assist device | |
| 1-Year | 12 (100) |
| 4-Year | 8 (67) |
3.2 Significant down-regulation of multiple gene transcripts after CRT
RNA-Seq was performed genome wide, and 22 375 transcripts were analysed using the Human Genome Build 38. A total of 2853 genes were analysed because they showed significant correlations with clinical endpoints based on peak VO2, BNP levels, LVESV, and heart failure scores with an FDR of <0.05. As shown in Figure 2 and the Supplementary material online, Supplementary Table, analysis of differences in gene expression for each patient showed that 138 of these 2853 genes had significant differences in expression before and after CRT based on an FDR <0.05. Of these 138 genes, 107 had suppressed levels of expression after CRT, while 31 had increased expression after CRT. The distribution of log2 fold changes in the PBMC expression of these genes post-CRT may be visualized in Figure 2.
Figure 2.
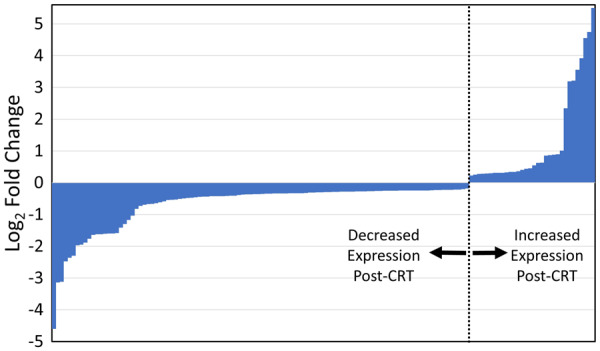
Log2 fold changes of genes with significant paired differences in PBMC expression post-CRT. This graphic displays the log2 fold changes for the 138 genes with significant (FDR < 0.05) paired differences in PBMC expression post-CRT relative to pre-CRT expression based on RNA-Seq in the 12 patients with CRT. The actual FDRs and a full list of the genes are shown in the Supplementary material online, Supplementary Table. CRT, cardiac resynchronization therapy; FDR, false discovery rate; RNA-Seq, RNA sequencing.
3.3 Selection of candidate genes of interest for RT-qPCR analysis
A pairwise correlation analysis identified 40 genes with the strongest association with all four CRT response measures (reduction in the LVESV, a more favourable heart failure symptom score post-CRT, reduction in BNP, and improvement in cardiopulmonary exercise capacity post-CRT), based on an FDR of 0.05 (Table 3). We then evaluated the significance of these 40 genes with respect to biological pathways by performing a gene set enrichment analysis using the Molecular Signatures Database. We identified 20 pathways significantly enriched among these 40 genes, and 14 genes were most significantly involved in these pathways, many of which were related to inflammation, cytokine signalling, immune cell functions, metabolism, apoptosis, and stress response associated with CRT treatment. There were nine genes associated with more than one of these pathways, and eight genes were associated with four or more pathways. These eight genes and the 20 associated pathways are shown in Figure 3.
Table 3.
RNA-Seq findings for 40 genes most strongly associated with CRT response
| Gene ID | Gene name | FDR for post-CRT expression | FDR for BNP correlation post-CRT | FDR for echo LVESV correlation post-CRT | FDR for HF score correlation post-CRT | FDR for peak VO2 correlation post-CRT |
|---|---|---|---|---|---|---|
| ENSG00000213934 | HBG1 | 1.64E−04 | 6.70E−08 | 1.57E−11 | 1.05E−04 | 2.29E−07 |
| ENSG00000214921 | NA | 9.11E−06 | 1.24E−13 | 2.01E−12 | 1.23E−03 | 5.74E−09 |
| ENSG00000254673 | AC110275.1 | 3.63E−02 | 2.97E−09 | 3.50E−06 | 6.11E−06 | 4.21E−03 |
| ENSG00000166947 | EPB42 | 4.75E−04 | 1.46E−08 | 2.78E−11 | 3.85E−07 | 1.45E−02 |
| ENSG00000004939 | SLC4A1 | 7.41E−05 | 1.88E−22 | 1.88E−55 | 2.85E−19 | 3.71E−08 |
| ENSG00000143416 | SELENBP1 | 3.23E−05 | 2.26E−15 | 7.05E−12 | 5.96E−06 | 1.42E−02 |
| ENSG00000133742 | CA1 | 4.70E−04 | 6.57E−19 | 7.82E−17 | 3.15E−04 | 2.01E−07 |
| ENSG00000122877 | EGR2 | 7.41E−05 | 1.03E−04 | 2.68E−14 | 3.04E−08 | 1.22E−05 |
| ENSG00000158578 | ALAS2 | 8.24E−04 | 1.34E−84 | 1.92E−90 | 2.29E−28 | 1.69E−17 |
| ENSG00000169877 | AHSP | 3.27E−03 | 3.42E−09 | 6.35E−06 | 1.54E−02 | 2.27E−02 |
| ENSG00000120738 | EGR1 | 2.87E−03 | 5.42E−39 | 3.71E−89 | 1.31E−62 | 3.76E−72 |
| ENSG00000223609 | HBD | 1.82E−02 | 1.48E−33 | 1.04E−16 | 4.60E−07 | 2.93E−13 |
| ENSG00000170345 | FOS | 2.06E−07 | 6.92E−120 | 8.27E−13 | 8.60E−17 | 1.51E−36 |
| ENSG00000147454 | SLC25A37 | 3.42E−02 | 2.48E−97 | 1.06E−65 | 1.14E−48 | 2.72E−56 |
| ENSG00000196730 | DAPK1 | 6.02E−03 | 2.94E−13 | 3.90E−20 | 3.41E−05 | 2.37E−08 |
| ENSG00000029534 | ANK1 | 2.57E−02 | 3.32E−07 | 3.76E−06 | 1.17E−08 | 3.82E−07 |
| ENSG00000120129 | DUSP1 | 4.85E−05 | 2.63E−70 | 2.54E−12 | 4.97E−18 | 1.24E−34 |
| ENSG00000162722 | TRIM58 | 3.00E−02 | 4.72E−33 | 4.76E−16 | 6.54E−15 | 2.00E−06 |
| ENSG00000183508 | FAM46C | 3.99E−02 | 3.07E−05 | 3.85E−16 | 2.57E−11 | 6.68E−15 |
| ENSG00000096060 | FKBP5 | 1.12E−04 | 1.42E−16 | 1.01E−36 | 9.47E−23 | 2.73E−16 |
| ENSG00000184557 | SOCS3 | 6.02E−03 | 2.75E−29 | 1.40E−04 | 3.73E−08 | 3.29E−07 |
| ENSG00000082146 | STRADB | 3.58E−02 | 2.96E−13 | 1.51E−05 | 2.44E−07 | 5.41E−14 |
| ENSG00000172331 | BPGM | 2.87E−02 | 5.31E−21 | 1.17E−03 | 1.09E−10 | 2.26E−09 |
| ENSG00000171552 | BCL2L1 | 6.58E−03 | 1.65E−32 | 4.24E−12 | 1.77E−18 | 1.39E−13 |
| ENSG00000173334 | TRIB1 | 6.02E−03 | 3.72E−02 | 6.27E−05 | 1.18E−02 | 3.02E−04 |
| ENSG00000153815 | CMIP | 3.11E−02 | 1.53E−14 | 1.39E−09 | 1.33E−02 | 4.31E−28 |
| ENSG00000128016 | ZFP36 | 1.50E−03 | 1.06E−10 | 8.88E−19 | 3.46E−02 | 9.55E−08 |
| ENSG00000165406 | MARCH8 | 3.10E−02 | 1.58E−06 | 5.53E−14 | 3.17E−06 | 4.51E−05 |
| ENSG00000154122 | ANKH | 4.83E−02 | 1.05E−06 | 4.33E−07 | 2.24E−04 | 4.15E−04 |
| ENSG00000159388 | BTG2 | 1.14E−02 | 3.01E−03 | 2.05E−24 | 1.28E−03 | 7.34E−11 |
| ENSG00000198876 | DCAF12 | 1.58E−02 | 2.13E−13 | 2.51E−04 | 2.56E−09 | 3.11E−10 |
| ENSG00000114738 | MAPKAPK3 | 7.03E−03 | 1.74E−02 | 1.44E−03 | 1.98E−02 | 1.90E−02 |
| ENSG00000171223 | JUNB | 3.58E−02 | 2.20E−31 | 1.84E−05 | 1.36E−05 | 1.89E−21 |
| ENSG00000124098 | FAM210B | 2.77E−02 | 6.63E−13 | 2.30E−03 | 3.32E−06 | 4.22E−05 |
| ENSG00000165671 | NSD1 | 4.72E−02 | 2.61E−05 | 3.41E−15 | 2.20E−02 | 9.27E−03 |
| ENSG00000103342 | GSPT1 | 1.80E−02 | 3.10E−13 | 3.62E−04 | 2.15E−09 | 1.25E−07 |
| ENSG00000090376 | IRAK3 | 4.07E−02 | 4.66E−02 | 1.23E−02 | 1.20E−02 | 2.92E−06 |
| ENSG00000147416 | ATP6V1B2 | 1.23E−02 | 2.00E−02 | 6.60E−03 | 1.01E−04 | 9.11E−03 |
| ENSG00000213145 | CRIP1 | 1.23E−02 | 2.19E−03 | 1.06E−07 | 7.36E−06 | 1.24E−06 |
| ENSG00000225748 | PRRC2A | 3.70E−02 | 4.66E−08 | 1.03E−28 | 7.59E−09 | 2.95E−43 |
The genes in bold represent the top genes enriched in the pathway analysis.
Figure 3.

Pathway analysis. Of the 14 genes enriched in the pathway analysis for the 12 CRT patients, the 8 genes with involvement of 4 or more pathways are shown with the associated pathways marked. Expression of these eight genes and IL-1β were evaluated with RT-qPCR. CRT, cardiac resynchronization therapy; FDR, false discovery rate; HF, heart failure; IL-1β, interleukin-1β; LVESV, left ventricular end-systolic volume; RT-qPCR, quantitative real-time polymerase chain reaction.
3.4 Confirmed down-regulation of inflammatory genes by RT-qPCR post-CRT
PBMC expression changes of the nine important genes including IL-1β (which has a significant role in the pathophysiology of heart failure15) and the genes identified by the pathway analysis [FOS proto-oncogene (FOS), JUNB proto-oncogene (JUNB), suppressor of cytokine signalling 3 (SOCS3), dual specificity phosphatase 1 (DUSP1), interleukin 1 receptor associated kinase 3 (IRAK3), early growth response 1 (EGR1), BCL2-like 1 (BCL2L1), and MAP-kinase activated protein kinase 3 (MAPKAPK3)] were evaluated using RT-qPCR, as these genes code for either transcription factor subunits or signalling regulatory proteins that control production of various inflammatory mediators or cellular apoptotic pathways. Moreover, based on the link of the above genes with the IL-1β signalling, we also evaluated whether CRT modulates expression of IL-1β mRNA. As shown in Figure 4, RT-qPCR confirmed post-CRT suppression of IL-1β (1.80-fold decrease, P = 0.047), FOS (3.25-fold decrease, P = 0.01), DUSP1 (2.05-fold decrease, P = 0.001 and EGR1 (7.38-fold decrease, P = 0.03) with a trend noted for SOCS3 (1.83-fold decrease, P = 0.14. The 1.92-fold decrease in JUNB was driven by a large decrease in one patient and was not statistically significant.
Figure 4.

Gene expression pre-CRT and post-CRT based on RT-qPCR. Measured gene expression levels normalized to 18S rRNA based on RT-qPCR in the 12 CRT patients are shown for the following genes: IL-1β, FOS, DUSP1, JUNB, SOCS3, IRAK3, EGR1, BCL2L1, and MAPKAPK3. Blue lines indicate CRT responders (R), while red lines indicate non-responders (NR). P-values were determined using linear mixed effect models, which account for the timing of the measurement (pre-CRT vs. post-CRT), the patient’s responder status (as a binary variable), the interaction between the timing of the measurement and the responder status, and replicates of each measurement. BCL2L1, BCL2-like 1; CRT, cardiac resynchronization therapy; DUSP1, dual specificity phosphatase 1; EGR1, early growth response 1; FOS, Fos proto-oncogene; IL-1β, interleukin-1β; IRAK3, interleukin 1 receptor associated kinase 3; JUNB, JunB proto-oncogene; MAPKAPK3, MAP-kinase activated protein kinase 3; RT-qPCR, quantitative real-time polymerase chain reaction; SOCS3, suppressor of cytokine signalling 3.
3.5 Association of baseline inflammatory gene expression and inflammatory gene suppression with improvement in left ventricular systolic function after CRT
Although the primary hypothesis was that expression changes of key genes would be associated with CRT response, additional analysis was performed based on the observation that expression of genes with significant reductions in responders tended to be expressed at higher levels in responders at baseline relative to baseline levels in non-responders. Baseline levels in CRT responders and non-responders, as well as in patients with standard ICDs and patients with coronary artery disease but no heart failure for reference, are shown in Figure 5. Baseline levels of FOS were 6.15-fold higher (P = 0.038), and baseline levels of DUSP1 were 2.60-fold higher (P = 0.013) in responders vs. non-responders (Figure 5). Baseline levels of IL-1β also trended to be higher in responders (2.61-fold higher P = 0.093) when compared with baseline levels in the non-responders (Figure 5). Interestingly, CRT responders but not CRT non-responders also showed higher baseline gene expression of FOS (P = 0.043) and DUSP1 (P = 0.01) compared with the control subjects with coronary artery disease but no heart failure, indicating an altered baseline inflammatory status in the responders. Moreover, baseline expression of FOS and DUSP1 were higher or trending higher in responders vs. patients with coronary artery disease and no heart failure (FOS P = 0.04; DUSP1 P = 0.06) (Figure 5). In addition, FOS (P = 0.15) and DUSP1 (P = 0.126) also trended to be higher in responders when compared with the heart failure subjects with an indication for a standard (non-CRT) ICD (Figure 5). As shown also in Figure 4, in which blue lines represent changes in responders and red lines represent changes in non-responders, the interactions between pre-CRT vs. post-CRT gene expression levels and responder status in linear mixed effect models were also statistically significant for IL-1β (P = 0.03), FOS (P = 0.02), and DUSP1 (P = 0.02), indicating greater suppression of PBMC expression of these genes post-CRT in responders vs. non-responders. A trend for suppression was noted for IRAK3 (P = 0.10) and EGR1 (P = 0.11). With respect to aetiology of cardiomyopathy, we found no statistically significant difference in gene expression based on ischaemic aetiology of cardiomyopathy.
Figure 5.

Gene expression changes at baseline based on RT-qPCR. Baseline levels of the 9 genes of interest based on RT-qPCR in the 27 study patients are shown in CRT responders (R), CRT non-responders (NR), patients with standard (non-CRT) ICDs, and patients with coronary artery disease but no heart failure. P-values reported for baseline expression differences between responders and another group were obtained using t-tests. BCL2L1, BCL2-like 1; CAD, coronary artery disease; CRT, cardiac resynchronization therapy; DUSP1, dual specificity phosphatase 1; EGR1, early growth response 1; FOS, Fos proto-oncogene; ICD, implantable cardioverter defibrillator; IL-1β, interleukin-1β; IRAK3, interleukin 1 receptor associated kinase 3; JUNB, JunB proto-oncogene; MAPKAPK3, MAP-kinase activated protein kinase 3; RT-qPCR, quantitative real-time polymerase chain reaction; SOCS3, suppressor of cytokine signalling 3.
3.6 Comparison with serum hs-CRP levels and WBC counts before and after CRT
For comparison, serum levels of hs-CRP were assessed before and after CRT. The pre-CRT hs-CRP levels were 3.47-fold higher in responders vs. non-responders (P = 0.008) (Figure 6). Pre-CRT hs-CRP was not significantly correlated with the pre-CRT PBMC expression of IL-1β, FOS, or DUSP1. Unlike IL-1β, FOS, or DUSP1, whose expression was significantly suppressed after CRT, serum hs-CRP levels post-CRT were not significantly different from baseline levels after CRT (P = 0.58). Also shown in Figure 6, WBC counts were not significantly different pre-CRT and post-CRT in the overall cohort, and no significant differences were observed in responders and non-responders.
Figure 6.
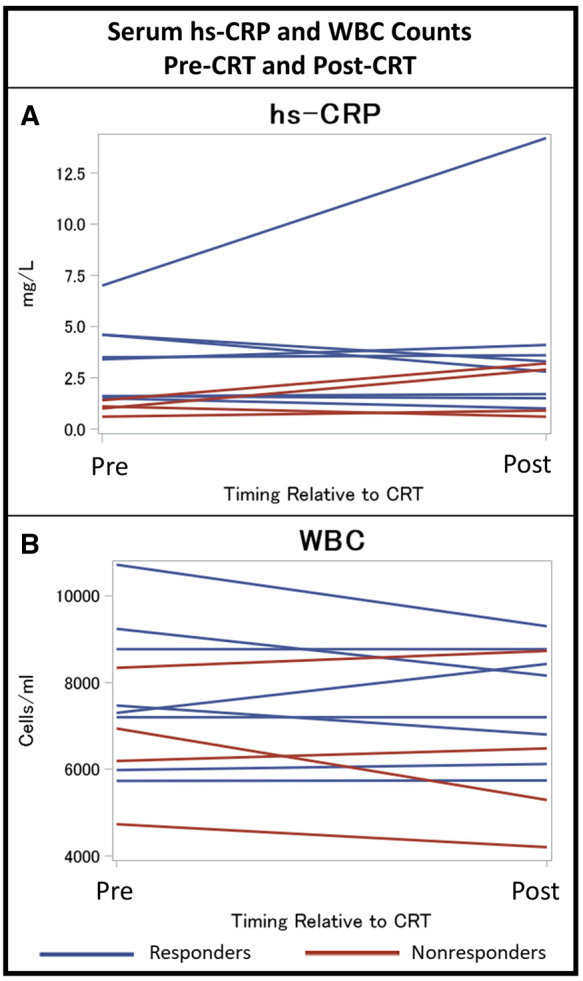
Serum hs-CRP and white blood cell counts pre-CRT and post-CRT. Levels of serum hs-CRP and white blood cell counts before and after CRT in the 12 CRT patients are shown for responders and non-responders. CRT, cardiac resynchronization therapy; hs-CRP, high-sensitivity C-reactive protein.
4. Discussion
4.1 Summary of findings
The main findings of this investigation are: (i) 138 genes, many related to inflammation, immune regulation, apoptosis, metabolism, and stress response showed significant differential expression in PBMCs post-CRT based on the RNA-Seq analysis; (ii) most of the differentially expressed genes were down-regulated after CRT; (iii) 40 of these genes were significantly correlated with all four CRT response measures, and eight in particular were noted to have prominent correlations with multiple inflammatory and other biologic pathways of interest; (iv) RT-qPCR confirmed CRT-associated suppression of many inflammatory transcripts, particularly those involved in IL-1β signalling (IL-1β, FOS, and DUSP1) and EGR1, a transcription factor that plays critical roles in various cardiovascular pathologies; (v) CRT responders had greater pre-CRT PBMC expression of these genes and greater suppression of PBMC expression of these genes post-CRT; and (vi) serum hs-CRP levels prior to CRT were higher in CRT responders compared with CRT non-responders; however, hs-CRP levels were not down-regulated post-CRT.
4.2 Associations with IL-1β and cardiovascular disease
IL-1β, an apical proinflammatory cytokine, is up-regulated in heart failure and associated with the degree of disease severity and unfavourable outcomes independent of the heart failure aetiology.28 Multiple preclinical studies and small clinical trials have indicated the potential benefit of IL-1β inhibition with anakinra in heart failure.29 Moreover, the recent CANTOS trial demonstrated that reducing inflammation by targeted inhibition of IL-1β reduces recurrent cardiovascular events and heart failure-associated mortality in patients with prior myocardial infarction and elevated levels of CRP.30,31 CRT has been shown to reduce inflammation associated with heart failure,24,25 and our data showing significant reduction of IL-1β mRNA in PBMCs with CRT further supports these findings. Of course, confirmation of changes at the protein level would strengthen this finding, and this is being evaluated through ongoing studies in our group. Of note, there are similarities and differences with respect to the effects of CRT on inflammation in heart failure and the effect of targeted pharmacologic inhibition of IL-1β in heart failure. First, while lower on-treatment hs-CRP levels were associated with better outcomes in the CANTOS trial, our findings with respect to CRT in heart failure indicate that greater suppression of IL-1β and associated inflammatory transcripts in PBMCs was strongly associated with response, although suppression of serum hs-CRP was not significantly different in CRT responders and non-responders. Even so, it is remarkable that baseline hs-CRP was higher in CRT responders vs. CRT non-responders. Second, whereas pharmacologic suppression of IL-1β in the CANTOS trial was associated with leucopenia in some patients, CRT-associated reductions in PBMC expression levels of IL-1β and associated transcripts were not associated with any significant changes in WBC counts.
4.3 PBMC transcripts associated with IL-1β signalling altered by CRT
FOS and JUNB are immediate-early proteins that form the proinflammatory transcription factor AP-1. Activation of AP-1 plays an essential role in cardiac remodelling, hypertrophy, and ventricular dysfunction.32–34 JUNB has been shown to modulate macrophage activation and expression of various proinflammatory mediators such as tumour necrosis factor-α (TNF-α), IL-1β, and IL-12,35 higher levels of which are present in heart failure patients and associated with unfavourable outcomes.14 Moreover, AP-1 and NF-κB activation underlie β-adrenoreceptor-mediated induction of IL-6,36 another prominent cytokine considered a biomarker as well as a potential therapeutic target for heart failure and other cardiovascular disorders.
DUSP1 plays a pivotal role in heart disease as a major negative modulator of the MAP kinase signalling pathways that co-ordinate diverse events including inflammation and cardiac remodelling.37,38 DUSP1 predominantly dephosphorylates and inhibits p38 and JNK, and to a lesser extent ERK.39 DUSP1 negatively regulates production of proinflammatory cytokines, and evidence suggests that it represses maladaptive cardiac hypertrophy.38,40 Proinflammatory mediators, such as LPS and IL-1β, growth factors, and stress could induce transient DUSP1 expression.39,41 Notably, DUSP1 expression was found to be up-regulated in the hearts of patients with end-stage heart failure and pressure-overloaded cardiomyopathy.42,43 Together, these indicate that induced expression of DUSP1 under chronic inflammatory state may serve as a feedback control mechanism to limit inflammation and pathological consequences.42
EGR1, an immediate early gene and a zinc finger transcription factor, plays critical roles in a myriad of cardiovascular pathological processes including atherogenesis, ischaemia–reperfusion damage, cardiac hypertrophy, angiogenesis, and intimal thickening after acute vascular injury.44 Immune cells and vascular cells such as endothelial cells, smooth muscle cells, cardiac fibroblasts, and cardiomyocytes express EGR1. Stress stimuli, proinflammatory cytokines, and growth factors are known to induce expression of EGR1, which, in turn, induces expression of a variety of proinflammatory mediators such as TNF-α and IL-6.45 Interestingly, IL-1β has been shown to induce EGR1 expression, promoting downstream production of IL-6, IL-8, and MCP-1.46,47 Higher expression of EGR1 transcripts as well as the EGR protein has been reported in PBMCs of chronic heart failure patients as compared with control subjects.20 Consistent with these observations, our data also showed trending higher baseline expression of EGR1 in CRT responders who seemed to have a more inflammatory state as compared with the non-responders and control subjects with no heart failure.
4.4 Comparison with studies of cardiac myocyte changes in response to CRT
Our approach represents a novel and complementary approach to previous studies in animals of cardiac cellular and gene expression changes in response to CRT.48–55 This prior work has been immensely informative in demonstrating how treating abnormalities in regional contraction patterns in heart failure can alter both regional and global cellular changes in cardiac myocytes. Application of cardiac tissue analysis in humans is less feasible because it requires cardiac tissue biopsy, which is invasive and associated with some procedural risks. In contrast, PBMC analysis offers a non-invasive assessment of global physiologic changes in response to CRT that can be easily repeated at serial time points.
4.5 Implications for CRT response
The assessment of PBMC gene expression changes offers a novel insight into a different measure of CRT response. While CRT response is often considered in terms of left ventricular functional measures, short-term symptom reduction, exercise capacity, and reduction in clinical events, PBMC gene expression changes may be considered to represent another dimension of CRT response. In this present analysis, we have demonstrated that CRT is associated with a reduction in key inflammatory pathways, particularly those associated with IL-1β signalling based on both RNA-Seq and RT-qPCR analysis of PBMCs. The strongest associations with CRT response and gene expression were noted for IL-1β, FOS, DUSP1, and EGR1. Of note, identification of responders and non-responders based on left ventricular reverse remodelling was validated by the greater long-term survival free of cardiac transplantation and left ventricular assist device (88% vs. 25%) 4 years after CRT in responders vs. non-responders.
In addition to being suppressed after CRT in the entire cohort, IL-1β, FOS, DUSP1, and EGR1 had both greater suppression in responders vs. non-responders and greater baseline levels pre-CRT in responders vs. non-responders. In addition, serum hs-CRP was also higher at baseline in responders vs. non-responders; however, hs-CRP was not significantly suppressed post-CRT. As a result, assessment of CRT patients based on PBMC expression of IL-1β, FOS, DUSP1, and EGR1 has an advantage over assessment based on serum hs-CRP. In summary, these findings support the existence of an inflammatory PBMC phenotype associated with CRT response based on PBMC gene expression of inflammation-promoting genes.
4.6 Limitations
As noted earlier, we studied a relatively small number of patients in this proof-of-concept study. Even so, with high-quality RNA-Seq, RT-qPCR data, and response data, it is remarkable that significant associations confirming our hypothesis were still demonstrated in our analysis (which included adjustments for multiple comparisons based on assessment of FDRs) in a cohort of this size. Furthermore, the conclusions are supported by biologic plausibility, with a recent role demonstrated for IL-1β in other larger cohorts of patients with cardiovascular disease. These findings provide the rationale for much needed larger cohort studies of CRT patients to confirm these findings and assess associations between PBMC expression of inflammatory genes and long-term survival after CRT in all populations including minorities. Based on these findings, we expect that PBMC gene expression findings may be helpful in risk stratification after CRT and early identification of patients who may benefit from advanced heart failure therapies.
5. Conclusions
Treatment of heart failure with CRT resulted in decreased PBMC expression of genes linked to inflammation. Additionally, baseline levels of inflammatory genes prior to CRT were higher in CRT responders. Moreover, CRT responders had greater suppression of these genes after CRT compared with non-responders.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
The authors acknowledge the contributions of clinical research co-ordinators Hollis Phillips, Susan Osmanzada, and Shawnette Gray to this research.
Conflicts of interest: K.B. receives grant support from Medtronic and Siemens Healthineers. All other authors have no declared conflicts of interest.
Funding
This work was supported by the American Heart Association [18TPA34170579 to K.B.].
Translational perspective
Assessment of peripheral blood mononuclear cell expression of inflammatory genes before and after cardiac resynchronization therapy (CRT) may be useful for characterizing CRT response at the gene expression level and identifying patients most likely to benefit from this therapy. As a more direct measure of the biologic response to CRT compared with typical response metrics such as left ventricular volumes and symptom scores, peripheral inflammatory gene expression offers an exciting window into molecular changes occurring in response to CRT. In the future, a peripheral inflammatory gene expression profile could be developed and checked at regular intervals to monitor inflammatory physiology in heart failure patients.
References
- 1. Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, DeMets D, White BG, DeVries DW, Feldman AM.. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. N Engl J Med 2004;350:2140–2150. [DOI] [PubMed] [Google Scholar]
- 2. Cleland JG, Daubert JC, Erdmann E, Freemantle N, Gras D, Kappenberger L, Tavazzi L.. The effect of cardiac resynchronization on morbidity and mortality in heart failure. N Engl J Med 2005;352:1539–1549. [DOI] [PubMed] [Google Scholar]
- 3. Chung ES, Leon AR, Tavazzi L, Sun JP, Nihoyannopoulos P, Merlino J, Abraham WT, Ghio S, Leclercq C, Bax JJ, Yu CM, Gorcsan J III, St John SM, De Sutter J, Murillo J.. Results of the predictors of response to CRT (PROSPECT) trial. Circulation 2008;117:2608–2616. [DOI] [PubMed] [Google Scholar]
- 4. Khan FZ, Virdee MS, Palmer CR, Pugh PJ, O'Halloran D, Elsik M, Read PA, Begley D, Fynn SP, Dutka DP.. Targeted left ventricular lead placement to guide cardiac resynchronization therapy: the TARGET study: a randomized, controlled trial. J Am Coll Cardiol 2012;59:1509–1518. [DOI] [PubMed] [Google Scholar]
- 5. Linde C, Ellenbogen K, McAlister FA.. Cardiac resynchronization therapy (CRT): clinical trials, guidelines, and target populations. Heart Rhythm 2012;9:S3–S13. [DOI] [PubMed] [Google Scholar]
- 6. Exner DV, Auricchio A, Singh JP.. Contemporary and future trends in cardiac resynchronization therapy to enhance response. Heart Rhythm 2012;9:S27–S35. [DOI] [PubMed] [Google Scholar]
- 7. Ramachandran R, Chen X, Kramer CM, Epstein FH, Bilchick KC.. Singular value decomposition applied to cardiac strain from MR imaging for selection of optimal cardiac resynchronization therapy candidates. Radiology 2015;275:413–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bilchick KC, Kuruvilla S, Hamirani YS, Ramachandran R, Clarke SA, Parker KM, Stukenborg GJ, Mason P, Ferguson JD, Moorman JR, Malhotra R, Mangrum JM, Darby AE, DiMarco J, Holmes JW, Salerno M, Kramer CM, Epstein FH.. Impact of mechanical activation, scar, and electrical timing on cardiac resynchronization therapy response and clinical outcomes. J Am Coll Cardiol 2014;63:1657–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang Y, Bauersachs J, Langer HF.. Immune mechanisms in heart failure. Eur J Heart Fail 2017;19:1379–1389. [DOI] [PubMed] [Google Scholar]
- 10. Van Linthout S, Tschöpe C.. Inflammation—cause or consequence of heart failure or both? Curr Heart Fail Rep 2017;14:251–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Honold L, Nahrendorf M.. Resident and monocyte-derived macrophages in cardiovascular disease. Circ Res 2018;122:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bansal SS, Ismahil MA, Goel M, Patel B, Hamid T, Rokosh G, Prabhu SD.. Activated T lymphocytes are essential drivers of pathological remodeling in ischemic heart failure. Circ Heart Fail 2017;10:e003688.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Epelman S, Liu PP, Mann DL.. Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat Rev Immunol 2015;15:117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gullestad L, Ueland T, Vinge LE, Finsen A, Yndestad A, Aukrust P.. Inflammatory cytokines in heart failure: mediators and markers. Cardiology 2012;122:23–35. [DOI] [PubMed] [Google Scholar]
- 15. Everett BM, Cornel J, Lainscak M, Anker SD, Abbate A, Thuren T, Libby P, Glynn RJ, Ridker PM.. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation 2019;139:1289–1299. [DOI] [PubMed] [Google Scholar]
- 16. Trankle CR, Canada JM, Cei L, Abouzaki N, Oddi-Erdle C, Kadariya D, Christopher S, Viscusi M, Del Buono M, Kontos MC, Arena R, Van Tassell B, Abbate A.. Usefulness of canakinumab to improve exercise capacity in patients with long-term systolic heart failure and elevated C-reactive protein. Am J Cardiol 2018;122:1366–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C, Abouzaki NA, Dixon D, Kadariya D, Christopher S, Schatz A, Regan J, Viscusi M, Del Buono M, Melchior R, Mankad P, Lu J, Sculthorpe R, Biondi-Zoccai G, Lesnefsky E, Arena R, Abbate A.. Interleukin-1 blockade in recently decompensated systolic heart failure: results from REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ Heart Fail 2017;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Libby P, Nahrendorf M, Swirski FK.. Leukocytes link local and systemic inflammation in ischemic cardiovascular disease: an expanded “cardiovascular continuum”. J Am Coll Cardiol 2016;67:1091–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ismahil MA, Hamid T, Bansal SS, Patel B, Kingery JR, Prabhu SD.. Remodeling of the mononuclear phagocyte network underlies chronic inflammation and disease progression in heart failure: critical importance of the cardiosplenic axis. Circ Res 2014;114:266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cappuzzello C, Napolitano M, Arcelli D, Melillo G, Melchionna R, Di Vito L, Carlini D, Silvestri L, Brugaletta S, Liuzzo G, Crea F, Capogrossi MC.. Gene expression profiles in peripheral blood mononuclear cells of chronic heart failure patients. Physiol Genomics 2009;38:233–240. [DOI] [PubMed] [Google Scholar]
- 21. Voellenkle C, van Rooij J, Cappuzzello C, Greco S, Arcelli D, Di Vito L, Melillo G, Rigolini R, Costa E, Crea F, Capogrossi MC, Napolitano M, Martelli F.. MicroRNA signatures in peripheral blood mononuclear cells of chronic heart failure patients. Physiol Genomics 2010;42:420–426. [DOI] [PubMed] [Google Scholar]
- 22. Szmit S, Jank M, Maciejewski H, Grabowski M, Glowczynska R, Majewska A, Filipiak KJ, Motyl T, Opolski G.. Gene expression profiling in peripheral blood nuclear cells in patients with refractory ischaemic end-stage heart failure. J Appl Genet 2010;51:353–368. [DOI] [PubMed] [Google Scholar]
- 23. Gupta MK, Halley C, Duan ZH, Lappe J, Viterna J, Jana S, Augoff K, Mohan ML, Vasudevan NT, Na J, Sossey-Alaoui K, Liu X, Liu CG, Tang WH, Naga Prasad SV.. miRNA-548c: a specific signature in circulating PBMCs from dilated cardiomyopathy patients. J Mol Cell Cardiol 2013;62:131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Stanciu AE, Vatasescu RG, Stanciu MM, Iorgulescu C, Vasile AI, Dorobantu M.. Cardiac resynchronization therapy in patients with chronic heart failure is associated with anti-inflammatory and anti-remodeling effects. Clin Biochem 2013;46:230–234. [DOI] [PubMed] [Google Scholar]
- 25. Lappegard KT, Bjornstad H, Mollnes TE, Hovland A.. Effect of cardiac resynchronization therapy on inflammation in congestive heart failure: a review. Scand J Immunol 2015;82:191–198. [DOI] [PubMed] [Google Scholar]
- 26. Manichaikul A, Rich SS, Perry H, Yeboah J, Law M, Davis M, Parker M, Ragosta M, Connelly JJ, McNamara CA, Taylor AM.. A functionally significant polymorphism in ID3 is associated with human coronary pathology. PLoS One 2014;9:e90222.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yuan JS, Reed A, Chen F, Stewart CN Jr.. Statistical analysis of real-time PCR data. BMC Bioinformatics 2006;7:85.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Szekely Y, Arbel Y.. A review of interleukin-1 in heart disease: where do we stand today? Cardiol Ther 2018;7:25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Van Tassell BW, Raleigh JM, Abbate A.. Targeting interleukin-1 in heart failure and inflammatory heart disease. Curr Heart Fail Rep 2015;12:33–41. [DOI] [PubMed] [Google Scholar]
- 30. Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, Kastelein J, Koenig W, Genest J, Lorenzatti A, Varigos J, Siostrzonek P, Sinnaeve P, Fonseca F, Nicolau J, Gotcheva N, Yong H, Urina-Triana M, Milicic D, Cifkova R, Vettus R, Anker SD, Manolis AJ, Wyss F, Forster T, Sigurdsson A, Pais P, Fucili A, Ogawa H, Shimokawa H, Veze I, Petrauskiene B, Salvador L, Cornel JH, Klemsdal TO, Medina F, Budaj A, Vida-Simiti L, Kobalava Z, Otasevic P, Pella D, Lainscak M, Seung K-B, Commerford P, Dellborg M, Donath M, Hwang J-J, Kultursay H, Flather M, Ballantyne C, Bilazarian S, Chang W, East C, Forgosh L, Harris B, Ligueros M.. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 2018;391:319–328. [DOI] [PubMed] [Google Scholar]
- 31. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida-Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ, Group CT.. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 32. Windak R, Muller J, Felley A, Akhmedov A, Wagner EF, Pedrazzini T, Sumara G, Ricci R.. The AP-1 transcription factor c-Jun prevents stress-imposed maladaptive remodeling of the heart. PLoS One 2013;8:e73294.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Takemoto Y, Yoshiyama M, Takeuchi K, Omura T, Komatsu R, Izumi Y, Kim S, Yoshikawa J.. Increased JNK, AP-1 and NF-kappa B DNA binding activities in isoproterenol-induced cardiac remodeling. J Mol Cell Cardiol 1999;31:2017–2030. [DOI] [PubMed] [Google Scholar]
- 34. Frantz S, Fraccarollo D, Wagner H, Behr TM, Jung P, Angermann CE, Ertl G, Bauersachs J.. Sustained activation of nuclear factor kappa B and activator protein 1 in chronic heart failure. Cardiovasc Res 2003;57:749–756. [DOI] [PubMed] [Google Scholar]
- 35. Fontana MF, Baccarella A, Pancholi N, Pufall MA, Herbert DR, Kim CC.. JUNB is a key transcriptional modulator of macrophage activation. J Immunol 2015;194:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Rohrbach S, Engelhardt S, Lohse MJ, Werdan K, Holtz J, Muller-Werdan U.. Activation of AP-1 contributes to the beta-adrenoceptor-mediated myocardial induction of interleukin-6. Mol Med 2007;13:605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li CY, Yang LC, Guo K, Wang YP, Li YG.. Mitogen-activated protein kinase phosphatase-1: a critical phosphatase manipulating mitogen-activated protein kinase signaling in cardiovascular disease (review). Int J Mol Med 2015;35:1095–1102. [DOI] [PubMed] [Google Scholar]
- 38. Liu R, Molkentin JD.. Regulation of cardiac hypertrophy and remodeling through the dual-specificity MAPK phosphatases (DUSPs). J Mol Cell Cardiol 2016;101:44–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Liu Y, Shepherd EG, Nelin LD.. MAPK phosphatases–regulating the immune response. Nat Rev Immunol 2007;7:202–212. [DOI] [PubMed] [Google Scholar]
- 40. Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T.. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol 2006;176:1899–1907. [DOI] [PubMed] [Google Scholar]
- 41. Shah S, King EM, Chandrasekhar A, Newton R.. Roles for the mitogen-activated protein kinase (MAPK) phosphatase, DUSP1, in feedback control of inflammatory gene expression and repression by dexamethasone. J Biol Chem 2014;289:13667–13679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Communal C, Colucci WS, Remondino A, Sawyer DB, Port JD, Wichman SE, Bristow MR, Singh K.. Reciprocal modulation of mitogen-activated protein kinases and mitogen-activated protein kinase phosphatase 1 and 2 in failing human myocardium. J Card Fail 2002;8:86–92. [DOI] [PubMed] [Google Scholar]
- 43. Ohki R, Yamamoto K, Ueno S, Mano H, Misawa Y, Fuse K, Ikeda U, Shimada K.. Transcriptional profile of genes induced in human atrial myocardium with pressure overload. Int J Cardiol 2004;96:381–387. [DOI] [PubMed] [Google Scholar]
- 44. Khachigian LM. Early growth response-1 in cardiovascular pathobiology. Circ Res 2006;98:186–191. [DOI] [PubMed] [Google Scholar]
- 45. Decker EL, Nehmann N, Kampen E, Eibel H, Zipfel PF, Skerka C.. Early growth response proteins (EGR) and nuclear factors of activated T cells (NFAT) form heterodimers and regulate proinflammatory cytokine gene expression. Nucleic Acids Res 2003;31:911–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hoffmann E, Ashouri J, Wolter S, Doerrie A, Dittrich-Breiholz O, Schneider H, Wagner EF, Troppmair J, Mackman N, Kracht M.. Transcriptional regulation of EGR-1 by the interleukin-1-JNK-MKK7-c-Jun pathway. J Biol Chem 2008;283:12120–12128. [DOI] [PubMed] [Google Scholar]
- 47. Sanchez-Guerrero E, Chen E, Kockx M, An SW, Chong BH, Khachigian LM.. IL-1beta signals through the EGF receptor and activates Egr-1 through MMP-ADAM. PLoS One 2012;7:e39811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bilchick KC, Saha SK, Mikolajczyk E, Cope L, Ferguson WJ, Yu W, Girouard S, Kass DA.. Differential regional gene expression from cardiac dyssynchrony induced by chronic right ventricular free wall pacing in the mouse. Physiol Genomics 2006;26:109–115. [DOI] [PubMed] [Google Scholar]
- 49. Spragg DD, Leclercq C, Loghmani M, Faris OP, Tunin RS, DiSilvestre D, McVeigh ER, Tomaselli GF, Kass DA.. Regional alterations in protein expression in the dyssynchronous failing heart. Circulation 2003;108:929–932. [DOI] [PubMed] [Google Scholar]
- 50. Aiba T, Barth AS, Hesketh GG, Hashambhoy YL, Chakir K, Tunin RS, Greenstein JL, Winslow RL, Kass DA, Tomaselli GF.. Cardiac resynchronization therapy improves altered Na channel gating in canine model of dyssynchronous heart failure. Circ Arrhythm Electrophysiol 2013;6:546–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sachse FB, Torres NS, Savio-Galimberti E, Aiba T, Kass DA, Tomaselli GF, Bridge JH.. Subcellular structures and function of myocytes impaired during heart failure are restored by cardiac resynchronization therapy. Circ Res 2012;110:588–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barth AS, Aiba T, Halperin V, DiSilvestre D, Chakir K, Colantuoni C, Tunin RS, Dimaano VL, Yu W, Abraham TP, Kass DA, Tomaselli GF.. Cardiac resynchronization therapy corrects dyssynchrony-induced regional gene expression changes on a genomic level. Circ Cardiovasc Genet 2009;2:371–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aiba T, Hesketh GG, Barth AS, Liu T, Daya S, Chakir K, Dimaano VL, Abraham TP, O'Rourke B, Akar FG, Kass DA, Tomaselli GF.. Electrophysiological consequences of dyssynchronous heart failure and its restoration by resynchronization therapy. Circulation 2009;119:1220–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Aiba T, Barth A, Tomaselli GF.. Deciphering gene expression profiling in cardiac resynchronization therapy. J Am Coll Cardiol 2008;52:1177; author reply 1177–8. [DOI] [PubMed] [Google Scholar]
- 55. Spragg DD, Akar FG, Helm RH, Tunin RS, Tomaselli GF, Kass DA.. Abnormal conduction and repolarization in late-activated myocardium of dyssynchronously contracting hearts. Cardiovasc Res 2005;67:77–86. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.