

ABSTRACT
In human nutrition randomized controlled trials (RCTs), planning, and careful execution of clinical data collection and management are vital for producing valid and reliable results. In this article, we provide an overview of best practices for biospecimen collection and analyses, and for the fundamentals of clinical data management, including preparation and study startup; data collection, entry, cleaning, and authentication; and database lock. The reader is also referred to additional resources for information to assist in the planning and conduct of human RCTs. The tools and strategies described are expected to improve the quality of data produced in human nutrition research that can, therefore, be used to support food and nutrition policies.
Keywords: dietary interventions, human nutrition, clinical trials, best practices, biospecimens, laboratory methods, data management
Introduction
The foundation of good clinical nutrition research practice is a comprehensive and detailed plan that defines the study design (1), documentation, and regulatory procedures (2), as well as data collection and management (discussed herein). Maintaining data integrity is essential for generating valid and reproducible results, which are linchpins for use of study results to inform evidence-based nutrition guidance. Compromised data are the “death knell” for meaningful scientific interpretation. With careful planning, training, and appropriate oversight, much can be done to minimize the risk of errors in data collection, cleaning, and analyses. In this article, we review best practices for ensuring biospecimen integrity and provide an overview of appropriate clinical data management (preparation and study startup; data collection, entry, cleaning, and authentication; database lock) for human nutrition randomized controlled trials (RCTs).
Biospecimen Integrity
The validity of a human nutrition RCT is dependent, to a great extent, on the quality control (QC) measures implemented for biospecimens. Before beginning a human nutrition RCT, it is essential to consider all steps in the process and have a detailed plan for biospecimen collection, processing, transport, storage, and assay. Key to this plan is anticipating all steps and contingencies, while balancing the practical needs and limitations of the study design, participants, infrastructure, and costs. It is also important to standardize procedures for “chain-of-custody” to minimize variability of results for both single-center and multicenter studies. Because most investigations involving human participants are expensive to conduct, it is also important to anticipate potential future uses and include plans and additional funding that might be necessary to support long-term storage that maintains biospecimen integrity. It cannot be emphasized enough that plans for all steps involving biospecimens require significant time and preparation, and thus should be completed well before the first participant is recruited. A breakdown in any step in the process can reduce validity or lead to loss of critical data. Guidelines for human biospecimen storage, tracking, sharing, and disposal have been provided by the NIH (3). All study personnel involved in biospecimen collection or analysis must complete appropriate institutional safety training in universal precautions of bloodborne pathogens.
Assays
A protocol for collecting, storing, shipping, and assaying biospecimens should start with the final laboratory outcome in mind, that is, the results of the assays. Specific assay requirements will dictate acceptable procedures for sample collection, processing, transport, and storage. Importantly, the requirements for each assay must be considered separately. The information below focuses on whole blood, serum, and plasma, but other biospecimens are frequently collected with specific assay requirements, including blood spots, buffy coats, spot and 24-h urine, stool, cerebrospinal fluid, saliva, biopsies, and buccal cells, among others. For many analytes, good sources of assay requirements are available online from national clinical laboratories (4–6), as well as academic medical centers (7). For assays not conducted in service clinical laboratories, literature searches can identify references that describe appropriate assays to use and specific requirements for sample collection and storage. Inserts in commercial assay kits also often provide guidance on appropriate sample collection and storage.
Sample volume or amount
Although each assay is optimized for a specific minimum volume or amount of a biospecimen, a greater volume must be provided to the laboratory running the assay. This is due to losses during sample transfer prior to and during an assay, dead volumes within automated systems, the possibility that an assay must be run again if there is a problem with the measurement, or to run in duplicate or triplicate. In some cases, although they are optimized for specific volumes or amounts, assays must be scaled down to accommodate reduced availability of sample. However, the scaled down assay must be tested before running actual study samples to be sure that the change in procedures produces valid (i.e., precise and accurate) results. Multiple aliquots can be necessary when multiple assays will be conducted in order to prevent the compromise of multiple freeze/thaw cycles. It is also beneficial to have additional aliquots available in case the assay needs to be rerun, which usually happens when the data are reviewed and there is a question about the validity of an outlying value.
Fresh compared with refrigerated compared with frozen
Some analytes are sensitive to freezing and thawing, and therefore must be performed on fresh, never-frozen samples. A good example is the complete blood count, which includes determinations of the numbers of intact RBC and white blood cells, which will burst after freezing and thawing. If an analyte is to be measured without cryopreservation, but not immediately (i.e., within hours to days), refrigeration or temporary storage on ice can be necessary, but there are limits on how long the sample is viable. Most analytes are amenable to cryopreservation, but actual temperature is sometimes an important consideration. Storage at −70°C to −80°C is the default standard. Some analytes can be stored at −20°C but might not be viable after extended storage. Another important consideration is that some samples, even if stored at −80°C, can deteriorate over time, whereas others will be stable. For some analytes, preservative(s) can be added to lengthen viability, such as metaphosphoric acid used to stabilize serum that will be analyzed for vitamin C, or a solution containing sulfamic acid plus a surfactant as a preservative for urine samples to prevent loss of mercury (8). Also, some analytes deteriorate with multiple freeze/thaw cycles, such as plasma fatty acids. Examples of common analytes in diet and nutrition studies and their viability under different temperature conditions are provided in Table 1.
TABLE 1.
Examples of analytes commonly used in nutrition research and viability under different temperature conditions1
| Analyte | RT | 4°C | −20°C |
|---|---|---|---|
| Alanine aminotransferase (plasma/serum) | 3 d | 1 wk | 2 wk |
| Aspartate aminotransferase (plasma/serum) | 24 h | 1 wk | 1 mo |
| Basic metabolic panel (plasma/serum)2 | 4 h | 1 wk | 2 wk |
| Calcium (plasma/serum) | 4 h | 3 wk | 8 mo |
| Complete blood count (whole blood)3 | 24 h | 48 h | Not viable |
| Creatinine (plasma/serum) | 1 wk | 1 wk | 3 mo |
| C-reactive protein (plasma/serum) | 1–2 wk | 2 mo | 3 y |
| Folate (RBC) | 2 h | 4 h | 2 mo |
| Folate/vitamin B-12 (plasma/serum) | 2 h | 8 h | 3 mo |
| Glucose (plasma/serum) | 24 h | 1 wk | 1 y |
| Glycated hemoglobin (HbA1c; whole blood) | 72 h | 1 wk | 3 mo |
| Insulin (plasma/serum) | 8 h | 1 wk | 1 mo |
| Iron (plasma/serum) | 1 wk | 3 wk | 3 mo |
| Lipid panel (plasma/serum)4 | 24 h | 5 d | 3 mo |
| Magnesium (plasma/serum) | 1 wk | 1 wk | 1 y |
| Retinol/retinyl palmitate (plasma/serum) | Not viable | 1 mo | 1 y |
| 25-Hydroxyvitamin D (serum) | 72 h | 1 wk | 6 mo |
| α-/γ-Tocopherol (plasma/serum) | Not viable | 1 mo | 1 y |
| TNF-α (plasma) | 30 min | Not viable | 1 y |
| Vitamin K-1 (plasma/serum) | Not viable | 1 mo | 6 mo |
| Zinc (plasma/serum) | Indefinitely | Indefinitely | Indefinitely |
These are the guidelines of a commercial clinical laboratory (ARUP Laboratories). Guidelines can vary among commercial laboratories. Some samples can be viable for longer times, particularly when stored at −80°C. However, viability information when stored at −80°C is not readily available for most analytes. RT, room (ambient) temperature.
Basic metabolic panel consists of carbon dioxide, chloride, creatinine, glucose, potassium, sodium, and urea nitrogen.
Complete blood count consists of hematocrit, hemoglobin, RBC count, white blood cell count, red cell distribution width, mean platelet volume, mean corpuscular volume, mean corpuscular hemoglobin, mean corpuscular hemoglobin concentration, nucleated RBC percentage and number, and platelets.
A lipid panel typically consists of total cholesterol, triglycerides, HDL cholesterol, LDL cholesterol (calculated), and VLDL cholesterol (calculated).
Plasma compared with serum
Plasma and serum are obtained from blood collected with or without an anticoagulant, respectively. Examples of color-coded tubes that are used to designate the type of sample to be collected, additives, and potential clinical uses, are shown in Table 2. Blood collection tubes for serum collection can contain a procoagulant, which hastens the clotting process to allow the tube to be centrifuged more quickly. To obtain serum, whole blood collected without anticoagulant must sit at room temperature for 30–60 min to clot before centrifugation to separate the serum. This can lead to artifactual changes in analyte concentrations due to release from, or metabolism by, RBCs and/or white blood cells. To limit this phenomenon, whole blood collected into an anticoagulant can be placed on ice or refrigerated before removing the plasma. An example of this effect is total homocysteine, the concentration of which tends to be ∼20% higher in serum than in plasma.
TABLE 2.
Common blood collection tube coding1
| Blood collection type | Color coding | Additive | Clinical use |
|---|---|---|---|
| Serum | Red | Plain (no additive) OR clot activator | Serum biochemistry, drug monitoring, and serum immunology test |
| Serum | Yellow | Clot activator with gel | Serum biochemistry, drug monitoring, and serum immunology test |
| Whole blood | Lavender | K3 EDTA or K2 EDTA | Hematology test |
| Whole blood | Black | Sodium citrate | Sedimentation rate test |
| Plasma | Gray | Sodium fluoride + potassium oxalate | Glucose test (analysis of blood sugar) |
| Plasma | Light blue | Sodium citrate | Coagulation test |
| Plasma | Green | Lithium heparin | Emergency biochemistry and plasma biochemistry test |
Refer to the “Plasma compared with serum” section for a discussion about instances when certain additives would be contraindicated.
Many analyte assays require specifically plasma or serum, whereas some are viable using either. Moreover, for plasma, there are several types of anticoagulants that are used, including K2 EDTA, sodium or lithium heparin, sodium citrate, and oxalate. Choice of anticoagulant is an important consideration because some can interfere with the assay to be performed. For example, EDTA is also a chelator of cationic metals, and thus is not appropriate for measurement of minerals such as iron, calcium, and magnesium. Other additives can also be included, such as sodium fluoride, which prevents glycolysis, thereby preserving the glucose concentration in a sample while it awaits analysis. Grossly hemolyzed samples are often not acceptable because RBC contents can have significantly higher concentrations of some analytes than plasma or serum or can otherwise interfere with some assays. Hemolyzed samples can have artifactually elevated concentrations for analytes measured using colorimetric assays such as iron, folate, and glucose.
Fasting compared with nonfasting
The prandial state of the participant can also be important for the validity or interpretation of an assay. The classic example is blood glucose, which fluctuates in serum or plasma depending on the timing and content of the prior meal. Typically, if fasting samples are required, individuals are instructed to refrain from food or drink (other than water) after 22:00 or midnight prior to the morning blood draw. Special considerations might be needed to take into account the different circadian rhythms of people with nontraditional job hours (e.g., health care or law enforcement). In many studies, fasting samples are collected as a default irrespective of the assay requirements, which is often a good practice to minimize variability. However, participant burden and other practical considerations can affect the decision to collect fasting samples (see below).
QC samples
Another important, but underappreciated, consideration with respect to assays is QC. The statistical power of a study to detect significant differences between or among groups and significant correlations within groups depends on the precision and accuracy of each assay. Researchers should also be aware when purchasing assay kits of potential variation among different lots or batches of assay kits, as well as the potential short half-life of certain kits. Therefore, it is important to include a plan to assess precision and accuracy.
Precision (sometimes referred to as imprecision) is typically determined by measuring an analyte in a single sample multiple times within the same assay (intra-assay variability) and between assays (interassay variability). A CV is then calculated using the following formula:
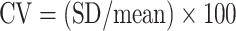 |
(1) |
where SD = SD of the mean, and mean = arithmetic average of the assay values for the QC sample.
The CV values are then reported as a percentage. Many scientific journals require reporting of CV values for each assay. Commercial and medical center clinical laboratories should provide CV values upon request. If running assays in a research laboratory, a plan for producing CV values must be in place and included when considering how many samples will be run in an assay. An acceptable CV varies according to the specific characteristics of each assay and needs to be evaluated for each analyte based on medical significance. A good rule of thumb for CV values is that they should be <10%, with values <5% considered to be excellent. CV values >10% can still be acceptable, but the power to detect small effect sizes will be diminished (9, 10).
Accuracy requires inclusion of a sample of known analyte concentration in all runs of an assay (referred to as a “QC” sample). Commercial and medical center clinical laboratories have programs for regularly determining the accuracy of their assays using QC samples measured by gold standard methodologies. A source of many QC samples is the National Institute of Standards and Technology (11). Importantly, a priori rules should be in place to determine if an assay is valid. For example, if the measured QC concentration within any given assay is outside a prespecified range around the actual value of the QC sample, then the entire run is invalid, and the samples must be run again. If multiple laboratories are to be used for the same assay, a plan for QC assessment must be in place whereby all the laboratories measure a shared QC to assure that each laboratory is producing values within the a priori established range around the actual value. The QC process should include an investigation of reasons that a set of results are outside of the acceptable range, such as the assay was run by a new person unfamiliar with the protocol, an expired assay kit was used, a different batch of the assay kit was used, and so forth.
Biospecimen collection
Although the default often is to require an overnight fast before blood collection, this can be a burden for the participants, particularly if blood will be drawn at a time of day other than first thing in the morning. This can be particularly true for young children, older adults, and patients who take medications for conditions such as diabetes or hypertension. If assays are not affected by prandial status, then not requiring an overnight fast can be considered. Any study design issue that reduces the participant burden has the potential to increase the number of willing participants and also maximize participant adherence to all aspects of the study, but there is a trade-off with potential increased variability due to circadian variations in certain assays. In any case, participants should be instructed to be hydrated to facilitate phlebotomy. Hydration status can also affect some blood analytes, such as hematocrit, hemoglobin, and blood urea nitrogen due to hemoconcentration or hemodilution.
The amount and frequency of blood drawing also affects participant burden. Total volume of blood to be drawn should be calculated taking into consideration the factors described in “Sample volume or amount” above. The frequency of blood draws should be minimized. If multiple blood draws are required within a short period (e.g., at various times during a single day), an indwelling catheter can be considered, although this increases risk for hemolysis and potential for variation from the scheduled collection times. Institutional review boards (IRBs) typically require justification for the total amount and frequency of draws to minimize the risk of excessive blood collection. The US Department of Health and Human Services Office for Human Research Protections has published the following guidance regarding limitations on blood volume collection by finger stick, heel stick, ear stick, or venipuncture (12):
(a) from healthy, nonpregnant adults who weigh at least 110 pounds. For these participants, the amounts drawn may not exceed 550 mL in an 8 week period and collection may not occur more frequently than 2 times per week; or (b) from other adults and children, considering the age, weight, and health of the participants, the collection procedure, the amount of blood to be collected, and the frequency with which it will be collected. For these participants, the amount drawn may not exceed the lesser of 50 mL or 3 mL per kg in an 8 week period and collection may not occur more frequently than 2 times per week.
Guidelines have also been published by various clinical research centers (13–15). The US NIH also provide guidelines for blood draws in children. These include ≤5% of total blood volume per day (i.e., 5 mL/kg body weight), and ≤11% of total blood volume over any 8-wk period (i.e., 9.5 mL/kg body weight) (16). There can be institution-specific IRB restrictions for total blood draw amounts within a day, within a month, and for the duration of the study. The investigator should also investigate whether potential research participants are enrolled in another clinical trial where blood samples are collected and whether they donate blood and/or plasma to determine eligibility for additional blood draws.
Ideally, blood draws occur in centralized clinical locations that allow for either efficient processing of biospecimens on site or are within easy transport distance of the location where the biospecimens will be processed, analyzed, and/or stored, for example, a clinical or research laboratory. Phlebotomy sites staffed by experienced and certified phlebotomists also provide a sense of confidence in the process among the participants, which can reduce participant dropout and loss to follow-up. If phlebotomy is conducted in the field, standardized storage, transport, and processing procedures become more critical, particularly with respect to time and temperature (see below). Point-of-care test devices, such as those used to measure cholesterol or lipids with a finger stick, can also be used in certain situations, such as remote locations or if data are collected at home.
A unique issue for metabolic studies involving feeding of participants is the proximity of food preparation and consumption to where biospecimens are collected, transported, processed, and stored. Ideally, such studies should be conducted in clinical research facilities where food preparation, dining facilities, phlebotomy stations, and laboratories are physically separated. When space is limited, some activities can be colocalized with appropriate precautions to maintain safety of participants and investigators and the integrity of the study by using appropriate physical barriers and clean-up procedures, and appropriate precautions for handling biological specimens (e.g., gloves, goggles). Activities that should not share physical space are food preparation with phlebotomy and blood processing, transport, and storage. Also, food should never be stored in refrigerators and freezers in which biological samples or laboratory reagents are stored. Breaches of these safety procedures are in violation of federal law and could result in institutional fines.
An important consideration when taking blood samples for different analytical purposes is the order in which they are collected during phlebotomy. Additives in the first blood tubes drawn (e.g., anticoagulants, glycolysis inhibitors) can cross-contaminate blood collected into subsequent tubes and potentially cause invalid analyte measurements. Typically, serum tubes (without anticoagulant) are collected first, followed by those containing anticoagulants, and then those containing glycolysis inhibitors. For a more detailed description, see reference 17.
Biospecimen processing
Because it is often not practical to analyze samples immediately after collection, protocols should describe how samples will be stored to preserve their integrity. (For the purposes herein, “processing” refers to the handling and manipulation of biospecimens from collection to the time of assay or storage for future use, and includes attention to time and temperature, exposure to light, fractionation, and preparing aliquots for archiving.)
Time and temperature
The importance of time and temperature relative to sample processing is discussed, in part, in the “Assays” section above. The clinical laboratory websites cited above (4–6) provide information on how long a sample remains stable for a particular assay when it is stored at room (ambient) temperature, refrigerated, exposed to light, or frozen at −20°C and −80°C. Processing protocols must provide standardized collection procedures that limit variance in time and temperature between participants within the practical constraints of the study parameters, facilities, and staffing. For example, blood drawn at an academic medical center, clinical laboratory, or dedicated clinical research facility can allow for control of temperature and processing of samples within a short time window, such as <75 min. Studies in which samples are collected more remotely might require longer windows between collection and processing. A typical time frame, particularly if samples can be refrigerated or kept on ice, is within 4 h of collection; however, the specific time frame depends on the analyte and assay being used. With longer time windows, specific assays, such as glucose, might have to be excluded from a study because they will not provide accurate results.
Exposure to light
Many analytes are light sensitive. Examples include B vitamins, such as folate and vitamin B-6, and vitamin A and other carotenoids. Instructions regarding special precautions needed for light-sensitive analytes include:
Collect blood directly into tubes wrapped in aluminum foil, or wrap tubes in aluminum foil immediately after collection.
Transport samples in sealed, opaque secondary containment.
Process samples in subdued light (minimum required to work safely and efficiently). For some analytes, exchanging full-spectrum light for yellow or red light in the work space can provide adequate protection of the samples, while providing sufficient lighting for laboratory activities.
Avoid exposure to direct light. Use curtains or blinds to limit sunlight in the workspace.
If available, designate a dark room that is protected from external light and has a controllable light source within. If a room is not available, a box of sufficient size for sample processing can be lined with black plastic to limit external light while allowing for ample light within the surrounding laboratory space.
When performing assays in the lab, keep samples and reagents in opaque brown or amber bottles or tubes, and cover clear glassware or plasticware with foil.
For some analytes, protection from light exposure should occur when samples are still in the form of whole blood due to the chromophores of the blood cells. After serum or plasma is separated from the blood cells, the effects of light exposure become more pronounced.
Centrifugation
Serum and plasma are separated from blood cells via centrifugation. Use of a refrigerated centrifuge is recommended so that samples can be kept cold during the process. A fairly wide range of relative centrifugal force (RCF), between 500 × g and 2000 × g, is tolerated by blood samples. An RCF <500 × g might not provide complete separation of cells from the plasma or serum; an RCF >2000 × g could damage the blood cells and alter the concentrations of analytes in the plasma or serum. Typically, samples are centrifuged for 10–15 min.
Transfer of plasma and serum
After centrifugation, plasma and serum are removed from the cell layers by pipette. Care must be taken to prevent contamination of the plasma and serum when pipetting close to the cell layer to avoid disturbing the buffy coat. Use of serum and plasma separator tubes is recommended if this is potentially an issue; these have a gel that physically separates the serum and plasma from the cells, thus maximizing the volume that can be collected.
Transport and storage
For cryostorage, freezers should ideally be connected to emergency backup power and on a remote monitoring system. Alarms on the freezers are also helpful to alert when the temperature drops, and an emergency contact number should be posted on the freezer identifying who should be called if this occurs. Backup freezers (although an added expense) should be available to temporarily house samples if there is a freezer failure. In addition, procedures should be in place to monitor temperature fluctuations. On rare occasions, proof that the samples were maintained at constant temperature might be requested. It also is important not to use frost-free freezers. Although very convenient because they do not need to be defrosted periodically, they can cause sublimation of frozen samples and thus affect assay results. An account of an unfortunate experience with shipping frozen samples from a large clinical trial, and tips to prevent such a disaster, are shown in Box 1.
Box 1. Shipping samples: a cautionary tale.
Several years ago, frozen samples from a large clinical intervention trial were shipped to one of the authors on dry ice from the East Coast to the West Coast where the samples were to be assayed for various analytes. The package was shipped on a Thursday to arrive the following day. Major storms hit the East Coast that evening, extensively disrupting air travel across the country. The samples made it to a central distribution hub of the shipping company, where they remained at ambient temperature for most of the weekend before finally being shipped to the West Coast destination. The samples arrived on the following Monday, completely thawed with no dry ice left. Needless to say, no valid data were collected from those samples.
Lessons learned from this experience include:
Always ship samples early in the work week (avoiding holidays);
Include enough dry ice to last ≥3 d; and
Check the weather before shipping.
Note that these issues become even more acute when shipping samples to other countries, where packages might be held up in customs. This, and other unforeseen problems, such as freezer failures and power outages, are potentially catastrophic for any study, and pose distinct threats to data quality, integrity, and sample size. Therefore, contingency plans (including having backup samples) must be in place. Also, some courier services will chaperone samples and add dry ice to prevent thawing during shipment. Although such services are relatively expensive, the cost is often justified by the reduction in risk of data loss, particularly when shipping between countries.
It is good practice for biospecimens to be stored in a “double-containment” container that will prevent contamination of other aliquots in case of tube leakage, and in a way that is organized so that samples can be easily identified. Storage volumes must balance factors discussed above that can affect assays with practical considerations, such as availability of freezer space. If possible, samples should not be stored in bulk volumes, but rather in separate tubes in volumes that are sufficient for specific assays to minimize freeze-thaw cycles. Cryovials with screw caps and rubber gaskets designed to withstand ultralow freezer temperatures should always be used. Eppendorf tubes should be avoided for cryostorage because the caps are prone to opening under cold storage and subsequent contamination with ice crystals. Glass tubes should also be avoided because they are prone to breakage at very cold temperatures and during thawing.
Labeling of cryovials is of paramount importance, and should be done with self-stick printed labels specifically designed for cryostorage, if possible. Hand-printed labels can become smeared or be hard to read if an individual has poor handwriting, particularly when a sample is thawed and condensation on the outside of the tube occurs. If ink labeling is used, it should be done with an indelible ink that will maintain clarity under frozen conditions. Using tape is not recommended because it often falls off at cold temperatures or when thawing. Lastly, labels should include all relevant information for identifying the source and kind of sample, including participant identification, date, type of sample (e.g., serum, EDTA plasma, etc.), and volume in the tube. Also, if a sample is to be frozen and thawed multiple times, a record of the number (and dates) of freeze/thaw cycles is necessary.
It is useful to archive biological samples from an intervention study in a biobank for potential subsequent analyses when additional funding is obtained and/or when new hypotheses are generated that can be addressed with stored samples. Study participants must be informed about planned uses of their biological samples. Therefore, it is helpful to include a statement about general categories of analyses that might be considered for later evaluation in the informed consent document for the trial. Additionally, investigators considering performing analyses from archived samples under most circumstances will be required to obtain additional IRB approval before proceeding. Analyses of DNA and other genetic materials typically require a specific consent separate from the regular consent form.
International Air Transport Association hazardous material (i.e., “hazmat”) shipment training is required for all persons who are involved with shipping laboratory-related hazardous materials, including biospecimens.
In summary, the integrity of data produced from biospecimen assays is highly dependent on the conditions of collection, processing, transport, storage, and assay. Careful attention to these details is essential to ensure the quality and validity of the study findings.
Clinical Data Management
The goals of the data management process for a human nutrition RCT are to facilitate complete and efficient collection of all data specified in the study protocol (and any amendments) and to prepare an accurate trial database for statistical analyses. The process involves 3 broad categories of activities: 1) preparation and study startup; 2) data collection, entry, cleaning, and authentication; and 3) database lock.
The Society for Clinical Data Management (18) has published Good Clinical Data Management Practice guidelines to provide a reference for clinical data managers, which are mainly oriented toward clinical data management for studies intended to support regulatory filings for pharmaceutical and medical device applications. Similarly, the Clinical Data Interchange Standards Consortium (19), which is a multidisciplinary nonprofit organization, has published standards to support development, sharing, submission, and archiving of clinical research data with a focus on harmonization of data management practices for regulatory submissions.
Although many human nutrition RCTs are conducted for scientific purposes and are not intended to support regulatory applications, many of the principles employed in data management for clinical trials intended to support regulatory submissions are relevant to best practices for use in human nutrition RCTs.
Preparation and study startup
The key initial step in the data management process is the development of case report forms (CRFs), which are the paper or electronic forms into which data are entered directly or transcribed from source documents (Supplemental Figure 1). Source documents are all original records of clinical findings, observations, and other activities for a clinical trial. In some instances, a paper or electronic CRF will also act as the source document. However, more often, information is extracted from source documents for transcription into a CRF (the term CRF will be used henceforth for both electronic and paper versions, unless otherwise specified). Frequently, more information will be collected in source documents than is transcribed into the CRF. Thus, it is important to remember during CRF design that any information not included in the CRF or transferred from another source, such as a laboratory dataset, will not be in the trial database and will therefore be inaccessible for statistical analyses and archiving. Many journals now request a statement of whether raw data are available if requested, and can require that the data be posted publicly before the paper is accepted for publication.
Data management plan
Larger studies will usually have a data management plan that covers CRF design and completion guidelines, database design, procedures for data flow, data cleaning and validation, and database lock. For smaller studies, a data management plan might not be developed, but, regardless, procedures need to be in place in the form of standard operating procedures and/or a general procedural handbook or manual to cover each of these areas.
CRF design
It is recommended that each header page of the CRF contains key information such as the protocol identifier, site code, and a participant number. Electronic CRFs can be set up so that when the header has been completed, the relevant fields will populate for all pages for a given participant's CRFs.
The CRF should be organized in a manner that follows, to the degree practical, the order in which the data appear in the source documents. Annotation should be included so that the computer interface or paper CRF shows appropriate categories (e.g., current cigarette smoker, 0 = no, 1 = yes) next to the transcription or entry fields, and the allowable number of characters is shown for text fields. Use of coding should be consistent; if 0 = no and 1 = yes is used in one location, there should be no cases where a different coding system is employed for the same type of information, such as 1 = no and 2 = yes.
Measurement units should be specified (e.g., height in centimeters and weight in kilograms). Check boxes are used to indicate that a procedure was completed (e.g., Was blood sample obtained? check Yes___ or No___) and explanations should be provided for information that could not be obtained (e.g., If No, why not? ___________________________________). Free text fields should only be included to the degree necessary to explain information that cannot be easily reduced to a brief, but exhaustive, list of categories.
Redundancy should be avoided so that a given data field is entered only 1 time. Information that will not be relevant to evaluation of the study objectives should be omitted from the CRF. For example, the source document might collect the name and contact information for a participant's personal physician, but because these are not needed for statistical analyses, the CRF would not contain these identifiers and they would not be included in the CRF. When practical, derived values should not be included in the CRF to avoid uncertainties related to rounding or miscalculations. For example, BMI can be calculated within the software program in many cases so that only the components (height and weight) need be entered into the CRF.
For fields where signatures or initials are required for paper or electronic CRFs, the CRF should clearly state who is allowed to sign. For example, some pages might require the signature of the Principal Investigator, whereas other pages can be signed by the Principal Investigator or a designee such as a Sub- or Co-Investigator or Clinical Research Coordinator. If the signature or initials must be provided by an individual with specific qualifications, such as a physician, nurse, or certified rater, this should be specified.
CRF completion guide
A CRF completion guide is typically developed to facilitate consistency in CRF completion. Examples of information that might be included are:
Date formatting (e.g., MM-DD-YYYY or MM-DD-YY).
Acceptable abbreviations.
Definitions for terms such as hypertension and obesity.
How to handle missing information (e.g., mark as UNK for unknown data, NA for data that are not applicable; enter 999 for numeric data fields that are missing).
The preferred method for recording pharmaceutical names (e.g., generic, brand name, either is acceptable).
Instructions for handling data corrections (e.g., single line through with initials and date if a paper CRF).
A statement of the general principle that all CRF fields should be completed unless there is clear documentation in the CRF for the missing data, such as termination of participation, data unavailable, or data not applicable.
Personnel training
All parties involved in data collection, review, and management should have proper training and qualifications, and training for specific functions performed for the trial should be documented—see Weaver et al. (2) for more information. Personnel in data management might include head of data management, lead and assistant data managers, validation programmer, QC/quality assurance (QA) coordinator, and data entry operator. Key functions of these individuals include database creation, updating, validation, and locking; data entry; data QC and QA; data clarification form generation; coordination with operations team to resolve queries; clinical data management software training and validation.
Tracking of data corrections and clarifications
Good clinical practice (20) requires that data management systems are designed, either manually or via appropriate software, to permit data changes in such a way that these are documented, and that there is no deletion of entered data without an audit trail to allow a third party to determine who changed what, when, and for what reason. This will be discussed further in the section on procedures for “Data validation and cleaning.”
Data management software
Various options are available for data management software. Small studies can be managed using Microsoft Excel, Microsoft Access, or a statistical analysis package such as IBM SPSS Statistics, SAS, or R. Larger studies will require a data management software package such as ORACLE CLINICAL, CLINTRIAL, RedCap, eClinical Suite, OpenClinica, OpenCDMS, or TrialDB. Some of these are open source programs and available free of cost. The programs listed are shown as examples, and listing them does not imply endorsement or a preference for one program over another.
Most human nutrition RCTs do not require full 21 Code of Federal Regulations (CFR), Part 11 compliance, which is necessary for clinical trials intended to support pharmaceutical or medical device regulatory submissions, but good processes should always be followed. In 21 CFR Part 11, the manner in which electronic records are to be created, modified, maintained, archived, retrieved, and transmitted is described. Fully compliant studies will have validation of all elements of the data management process and any software used to ensure integrity, accuracy, and confidentiality of data. The data management system validation process is beyond the scope of this article. The US FDA has published guidance for industry regarding validation of computerized systems for use in clinical trials (21).
Database design
The overarching goal of the clinical database is to provide a complete and accurate set of clinical trial data for statistical analyses. The database will be set up in such a way that allows for efficient data entry and meets the needs of the biostatistician(s) (Supplemental Figure 2).
Each data field needs to be defined regarding the type of data to be entered, most commonly numeric, date, or text. For numeric fields, the type of number (integer or numbers with decimals), allowable range, and number of digits before and after the decimal place should be defined. Special considerations should also be addressed, such as what to do with a value that is mainly numeric, but for which nonnumeric values are possible (e.g., trace or below the lower limit of detection). Systems must also be in place to allow for missing data, such as using 999 for numeric values that are missing, and UNK or UK for unknown portions of a date (e.g., 05-UK-1985 for a MM-DD-YYYY format where the day is unknown). Frequently, the CRF will contain a place for a comment to explain why a value is missing, and this comment is its own variable.
Variable abbreviations and coding should be specified, such as in the example below:
Variable: HGHT
Description: Participant height
Units: cm
Response options: Numeric
Response values: Range of expected responses specified (e.g., 137 to 224 cm)
Edit checks are programmed or can be done manually by data management personnel to inspect data and identify discrepancies. Most databases allow each data field to be set up with a range of acceptable values, with each data point outside that range requiring entry by a supervisor with administrative privileges, or with entry being allowed if accompanied by flagging for evaluation by the data manager. Values can also be flagged if the difference from a prior value is outside an expected range, such as body weight differing by >4.0 kg between adjacent study visits. Some ranges will be determined by the protocol entry criteria. For example, the protocol might specify that participants aged 18 to 64 y can participate. An age outside that range could be flagged to alert the data manager that an approved protocol deviation should be on file for that participant to explain why he or she was allowed to enroll in the study. Calculations can be programmed to prevent entry of, or flag, logical inconsistencies such as diastolic blood pressure greater than systolic blood pressure, a date of birth error resulting in an unrealistic value for participant age, a male participant with a positive value recorded for pregnancy test result instead of NA, and so forth.
The underlying structure of a database can be set up so that data tables have fewer columns and more rows (taller and skinnier) or vice versa (shorter and wider), as illustrated in the examples below.
| Weight (kg) | |
| Visit 1 | 54.2 |
| Visit 2 | 54.5 |
| Visit 3 | 54.4 |
| Visit 4 | 53.7 |
| Weight (kg) | |||
| Visit 1 | Visit 2 | Visit 3 | Visit 4 |
| 54.2 | 54.5 | 54.4 | 53.7 |
The preference for the structure of data tables might depend, in part, on the statistical analysis program to be used. Some menu-driven programs, such as SPSS, are easiest to employ with a particular data structure. Therefore, the biostatistician for the trial should be consulted in advance to avoid additional programming to prepare datasets for analysis.
QC for database design
The database design should be reviewed by both a second data manager and a biostatistician. Dummy data should be entered to allow evaluation of the expected performance of the database during the data entry process.
Data entry, monitoring, and cleaning
With paper CRFs, data must be transcribed from the source document into the CRF document. With electronic CRFs, data are entered directly into the data management system from source documents. At present, CRFs for most clinical trials, except very small trials, use an electronic data capture system.
Data entry and monitoring
The standard employed for data entry in most large clinical trials with paper CRFs is blinded, independent, double data entry (2 separate individuals) with third party review and arbitration of discrepancies. As more trials have moved to use of electronic CRFs, it has become more common to employ single data entry with 100% verification against the source documents by a second person, or verification of key variables by a second person. Some hybrid systems exist, such as those in which paper CRFs are sent via facsimile to a data management system that digitizes the entries and converts them into numeric or text values in the trial database. Regardless of what type of system is used, procedures should be defined in advance regarding how data will make their way into the database, and how the accuracy of the process will be verified. Data authentication, or the process of confirming the origin and integrity of data, is done, in part, by the clinical monitor and also during the data management cleaning process.
For larger studies, clinical monitoring is done to verify that the information transcribed to, or entered into, the CRF accurately reflects the content of the source documents. When formal clinical monitoring is not a part of the study, QC procedures should be in place to ensure accuracy, such as review of key variables by a supervisor. Processes should also be defined so that any corrections or changes to data that have been entered into a CRF are documented with creation of a proper audit trail (i.e., who made the change, when and why the change was made).
Data flow
Whether paper or electronic CRFs are used, data management must track the flow of data. For paper CRFs, no-carbon-required duplicate or triplicate forms can be used, with ≥1 copy kept at the research site and the original sent to data management. Tracking forms are employed to document which pages have been sent to data management and which remain at the research site. This process is not required with electronic CRFs, because the data manager can access the trial database at any time.
Database backup
Data management systems for larger studies typically have systems in place for daily (or more frequent) backup of the study database, typically in multiple locations, such as 2 servers located in different buildings, or even different cities. In some instances, one server could be a physical server and the second server could be a “cloud” storage server. This provides protection against loss of data due to events such as system crashes or natural disasters. If such procedures are not in place, it is prudent to perform periodic manual backups, with the medium used for the backup stored in a location physically separate from the main system.
Datasets that comprise the trial database
A clinical trial database will consist of several datasets. Some of these will come from data entered in CRFs and others might be from sources such as analytical and bioimaging laboratories, as well as services or programs that score questionnaires or test batteries, such as FFQs and various psychometric scales or test batteries. The database might also include derived datasets with variables calculated, such as BMI or the domains on a psychometric scale. Processes should be in place to ensure that the data from different datasets can be “married” so that a complete set of variables is generated for all data collected from each participant. For example, laboratory datasets will generally have multiple identifiers such as participant number (e.g., randomization or screening number) and date of collection. This parallels the identifiers used for other types of data, which allows data management to ensure that laboratory results are mapped to the correct participant. The data need to be deidentified to ensure that the participants’ identities cannot be linked to their data. The types of identifiers that should be removed or recoded in order to prevent the risk of association between clinical trial participants and their data are listed below (22):
Names and initials.
All elements of dates (except year) that could be directly associated with a specific individual (e.g., birthdate, date of death, adverse event date, admission date, discharge date).
Kit numbers (diagnostic kits) and device numbers (devices used in the trials).
Geographic information (e.g., place of work, trial site location, addresses, zip codes).
Telephone numbers, email addresses, or fax numbers.
Account numbers, social security numbers, health plan beneficiary numbers, or medical record numbers.
Vehicle identifier or license plate numbers.
Certificate/license numbers (e.g., marriage license).
Biometric identifiers (e.g., MRI).
Photographic or similar images that show the full face.
Web Universal Resource Locators or Internet Protocol addresses.
Data validation and cleaning
Clinical data management plays an important role in ensuring the accuracy and consistency of the trial database. This includes generation and tracking of data queries. Queries or data clarification forms are generated for various reasons, including verification that each participant met the trial entry criteria, ensuring proper documentation for any protocol deviations/violations, provision of adequate documentation for data that are missing, ensuring completeness of study procedures (e.g., review of all adverse events with adequate documentation of any treatment or follow-up required), and identification of outlying values that appear implausible or otherwise suspect. Data transferred from sources other than CRFs must also be evaluated for completeness, accuracy, and the presence of implausible or duplicate values. Queries are generated and sent to the research site to request additional documentation when needed. These need to be tracked to create an audit trail with adequate documentation for any changes made to the database and to ensure that all requests for data clarification are addressed and resolved.
A combination of programmed checks and manual review are employed by data managers to identify implausible or outlying values. Descriptive statistics can be used to identify suspect values that are in the tails of the observed distribution. For example, if the mean change in body weight from the time of randomization to a given trial visit is 0.2 kg, a value that shows a change of 20 kg might be flagged for verification. Such a value might have resulted from an error of recording weight in the wrong units, transposition of digits, or an error in the transcription of the results from the source document.
Calculations, such as those for compliance with the study intervention (e.g., percentage of expected servings of study product consumed, or number of supplement pills returned), are verified during the data validation and cleaning process. Data managers also check items to assess internal consistency; for example, if there is a treatment listed for an adverse event, this treatment should also be documented in the list of concomitant medications used. Also, if an adverse event is listed, but is of a type that had been recorded in the participant's medical history, the data manager can issue a query to verify whether or not the event was truly an adverse event. If the reported adverse event was a pre-existing condition, and not reflective of worsening, it might need to be reclassified. Part of the data management process includes verifying that adverse events that require reporting in a certain time frame (e.g., serious adverse events and adverse events of special interest) are reported to the appropriate parties (e.g., IRB or data and safety monitoring board) in the appropriate time frame.
A key function of data management is to ensure that any clinical decisions made during a trial are adequately documented. Therefore, queries can be generated to collect additional information to ensure that documentation is present regarding who made the clinical decision, when the decision was made, the rationale for the decision, the recommended course of action, and all follow-up with the study participant and their health care provider(s), as appropriate, until resolution of the situation that led to the clinical decision.
Data coding
Coding dictionaries are available for standardization of terminology for adverse events, medical history, and medications. For pharmaceutical and medical device studies intended to support regulatory approval, the Medical Dictionary for Regulatory Activities (MedDRA) is required for coding of adverse events and medical history so that the myriad of terms used to describe such events can be mapped to a limited set of standardized terms. The World Health Organization-Drug Dictionary Enhanced (WHO-DDE) is generally used in such studies to code medications (23). There is also a dictionary available from the World Health Organization for coding of Adverse Reaction Terminology (WHO-ART) that can be used for adverse event reporting (24). Coding of adverse events, medical history, and concomitant medications might not be necessary for small human nutrition RCTs, but is appropriate for larger trials.
Preparation for, and execution of, database lock
After the data have been cleaned, the last step before finalizing and “locking” the database is to define the analysis populations. It is especially important to complete this step prior to breaking the treatment code for a blinded trial to avoid bias, or the appearance of bias, in any decisions about exclusion of data from statistical analyses. Examples of analysis populations can include intention-to-treat, safety, and per protocol.
Documentation of decisions prior to analysis
For large studies with hundreds or thousands of participants, it is not practical to review a detailed set of summaries for every participant to identify those for whom decisions must be made about inclusion/exclusion from analysis populations, and for dealing with implausible or outlying values, some of which can be set to missing if physiologically impossible or unlikely. In such cases, decisions are documented and handled through programming. For smaller studies, there can be a data review meeting that includes ≥1 investigator(s), data manager(s), biostatistician(s), and sometimes others, to carefully review data summaries (listings) for all participants so that contemplated decisions can be discussed, and consensus reached and documented. Decisions about values that are below the lower limits of detection can also be documented at this stage, or earlier. Some investigators set such values to the midpoint between zero and the lower limit, and others set them to zero or to the lower limit of detection.
Database lock
When all decisions about analysis populations and suspect values have been made and documented, the database is ready to be locked, which means that final datasets are prepared for delivery to the biostatistician. No more changes are allowed without a formal unlocking and subsequent relocking of the trial database, with the rationale for these actions and other audit trail requirements (who, when, why) documented. Often the first set of statistical analyses for a blinded trial will be completed on data for which the treatment code has not been fully broken (e.g., A, B, and C rather than actual treatment group names). Therefore, the unblinded treatment conditions are sometimes not included in the final, locked database. For large studies, interim analysis datasets can be generated and analyzed for review by a data and safety monitoring board to make decisions about whether the trial should be stopped for 1 of the following reasons: 1) clear evidence of harm, 2) futility, or 3) overwhelming evidence of benefit of the treatment.
All of the issues below must be addressed for database lock to proceed:
All CRFs have been received and verified.
All external data have been received and reconciled (e.g., analytical laboratory, bioimaging, dietary analysis, and psychometric testing scales).
Medical history, adverse events, and concomitant medication coding (if applicable) have been completed, reviewed, and approved.
All adverse events, particularly serious adverse events, have been resolved or classified as ongoing with appropriate documentation.
Database QC and audit procedures have been completed to ensure accuracy (usually a predefined verification of values for a percentage of data fields or participants, with an expansion required if a predefined level of errors is exceeded) with all discrepancies resolved.
Approval has been obtained to lock from all appropriate parties (e.g., investigator, sponsor, if applicable, biostatistician).
Treatment codes for unblinding have been loaded unless the initial statistical analysis will be completed with the code unbroken, in which case this step occurs later.
After each of the bullet points above has been addressed, database lock can proceed, at which time all records are marked as locked and permissions are set to read only for each dataset. For some smaller studies, such as pilot studies, a formal database lock might not occur. Nevertheless, procedures should be in place to address and document the issues described above.
Transfer to the biostatistician
When the database has been locked, it is ready for transfer to the biostatistician for analysis. One important element of this process is verification that no errors or uncertainties have been introduced during the transfer process. Statistical analysis programs such as SAS, SPSS, or R have the ability to import data from any type of file that has been created by the program used for data management. Values must be formatted in a manner that allows the program to import them correctly. For example, values that have been coded as 999 for missing can create problems in the analysis if they are transferred as numeric values. Similarly, nonnumeric values in a numeric field, such as those listed as BLQ (below limits of quantitation), can show up as missing or even zeros in the statistical analysis program. Therefore, data managers and biostatisticians must collaborate to avoid introduction of errors or uncertainties during the transfer process. Also, as described previously, the datasets should not contain any information that could allow identification of the participant.
Data archiving and storage
After the trial has been completed and the data analyzed, the trial datasets and output from statistical analyses will need to be stored. It is important to maintain security and confidentiality so that there is no unauthorized access to the clinical trial data. Consideration should be given to the media and programs used for storage. If it becomes necessary to access the data years after study completion, it is helpful to have a set of generic digital files stored on a medium that can be read by multiple programs, because the program or program version used for data management or analysis might no longer be supported.
Summary and Conclusions
This article has summarized key steps for biospecimen collection and analysis, as well as clinical data management for human nutrition RCTs. To ensure data integrity, it is vital that a detailed plan is in place prior to the initiation of the trial that maps out all the conditions that must be met for the collection, processing, transport, storage, and assay of all biospecimens. Processes for confirming that laboratory and other data are accurately collected, recorded, and assessed for consistency are also described to allow creation of a clean and accurate study database for statistical analyses. Best practices outlined in this article will help to ensure data integrity and quality for human nutrition RCTs so that results can be used with confidence to inform the development of clinical public health guidelines.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank all the organizations that provided financial support, resource support, and leadership on this project including the Tufts Clinical and Translational Science Institute (CTSI), Indiana CTSI, and Penn State CTSI. The authors are also grateful to Tufts CTSI for providing overall leadership on this project, organizing all writing group meetings, providing project management support, and hosting the writing workshop that initiated this project.
The authors’ responsibilities were as follows—KCM, JWM, GPM, GR, and PKE: were responsible for the design, writing, and final content of the manuscript; and all authors: read and approved the final manuscript.
Notes
This project was funded by National Institutes of Health Clinical and Translational Science Awards to Tufts Clinical and Translational Science Institute (UL1TR002544), Indiana Clinical and Translational Sciences Institute (UL1TR002529), and Penn State Clinical and Translational Science Institute (UL1TR002014). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Author disclosures: The authors report no conflicts of interest.
Perspective articles allow authors to take a position on a topic of current major importance or controversy in the field of nutrition. As such, these articles could include statements based on author opinions or point of view. Opinions expressed in Perspective articles are those of the author and are not attributable to the funder(s) or the sponsor(s) or the publisher, Editor, or Editorial Board of Advances in Nutrition. Individuals with different positions on the topic of a Perspective are invited to submit their comments in the form of a Perspectives article or in a Letter to the Editor.
Supplemental Figures 1 and 2 are available from the “Supplementary data” link in the online posting of the article and from the same link in the online table of contents at https://academic.oup.com/advances.
Abbreviations used: CFR, Code of Federal Regulations; CRF, case report form; CTSI, Clinical and Translational Science Institute; IRB, institutional review board; NA, not applicable; QA, quality assurance; QC, quality control; RCF, relative centrifugal force; RCT, randomized controlled trial; UNK or UK, unknown.
Contributor Information
Kevin C Maki, Department of Applied Health Science, Indiana University School of Public Health, Bloomington, IN, USA.
Joshua W Miller, Department of Nutritional Sciences, Rutgers University, New Brunswick, NJ, USA.
George P McCabe, Department of Statistics, Purdue University, West Lafayette, IN, USA.
Gowri Raman, Institute for Clinical Research and Health Policy Studies, Center for Clinical Evidence Synthesis (CCES), Tufts Medical Center, Boston, MA, USA.
Penny M Kris-Etherton, Department of Nutritional Sciences, The Pennsylvania State University, University Park, PA, USA.
References
- 1. Lichtenstein AH, Petersen K, Barger K, Hansen KE, Anderson CAM, Baer DJ, Lampe JW, Rasmussen H, Matthan NR. Perspective: Design and conduct of human nutrition randomized controlled trials. Adv Nutr. 2020. doi: 10.1093/advances/nmaa109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weaver CM, Fukagawa NK, Liska D, Mattes RD, Matuszek G, Nieves JW, Shapses SA, Snetselaar LG. Perspective: US Documentation and regulation of human nutrition randomized controlled trials. Adv Nutr. 2020. doi: 10.1093/advances/nmaa118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. National Institutes of Health . Guidelines for human biospecimen storage, tracking, sharing, and disposal within the NIH Intramural Research Program. NIH; 2019. [Google Scholar]
- 4. Quest Diagnostics. Test directory. [Internet]. [cited 2020 Feb 23]. Available from: https://testdirectory.questdiagnostics.com/test/home. [Google Scholar]
- 5. Mayo Clinic Laboratories . Testing home page. [Internet]. [cited 2020 Feb 23]. Available from: https://www.mayocliniclabs.com/.
- 6. ARUP Laboratories . ARUP test directory. [Internet]. 2020; [cited 2020 Feb 23]. Available from: https://www.aruplab.com/testing.
- 7. UC Davis Health, Department of Pathology and Laboratory Medicine . Laboratory test directory. [Internet]. [cited 2020 Feb 23]. Available from: https://www.testmenu.com/ucdavis.
- 8. US Department of Health and Human Services, Centers for Disease Control and Prevention . Improving the collection and management of human samples used for measuring environmental chemicals and nutrition indicators. [Internet]. Version 1.3; 2018. [cited 2020 Feb 23]. Available from: https://www.cdc.gov/biomonitoring/pdf/Human_Sample_Collection-508.pdf.
- 9. Reed GF, Lynn F, Meade BD. Use of coefficient of variation in assessing variability of quantitative assays. Clin Diagn Lab Immunol. 2002;9(6):1235–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. ClinLab Navigator . Precision. [Internet]. [cited 2020 Feb 23]. Available from: http://www.clinlabnavigator.com/precision.html.
- 11. National Institute of Standards and Technology, US Department of Commerce . Homepage. [Internet]. [cited 2020 Feb 23]. Available from: https://www.nist.gov/.
- 12. US Department of Health and Human Services, Office for Human Research Protections . OHRP expedited review categories (1998) [Internet]. [cited 2020 Feb 23]. Available from: https://www.hhs.gov/ohrp/regulations-and-policy/guidance/categories-of-research-expedited-review-procedure-1998/index.html.
- 13. Dana-Farber/Harvard Cancer Center . DF/HCC document library [Internet]. [cited 2020 Feb 23] Available from: https://www.dfhcc.harvard.edu/research/clinical-research-support/document-library/.
- 14. Duke University Health System . Blood drawing for human subject research [Internet]. December 2012 [cited 2020 Feb 23]. Available from: https://irb.duhs.duke.edu/sites/irb.duhs.duke.edu/files/Blood_Collect_Policy_Statement_12-13-2012.pdf.
- 15. Children's Hospital of Philadelphia Research Institute . Guidelines for limits of blood drawn for research purposes in the Clinical Center. [Internet]. [cited 2020 Feb 23]. Available from: https://irb.research.chop.edu/sites/default/files/documents/g_nih_blooddraws.pdf. [Google Scholar]
- 16. Peplow C, Assfalg R, Beyerlein A, Hasford J, Bonifacio E, Ziegler AG. Blood draws up to 3% of blood volume in clinical trials are safe in children. Acta Paediatr. 2019;108(5):940–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Becton Dickinson and Company . Order of draw for multiple tube collections. 2013. [Internet]. [cited 2020 Mar 28]. Available from: https://www.bd.com/a/36000. [Google Scholar]
- 18. Society for Clinical Data Management . Homepage. [Internet]. [cited 2019 Aug 26]. Available from: https://scdm.org/.
- 19. Clinical Data Interchange Standards Consortium . CDISC Standards in the Clinical Research Process. [Internet]. [cited 2019 Dec 13]. Available from: https://www.cdisc.org/standards.
- 20. US Food and Drug Administration . Regulations: good clinical practice and clinical trials. [Internet]. [cited 2019 Aug 25]. Available from: https://www.fda.gov/science-research/clinical-trials-and-human-subject-protection/regulations-good-clinical-practice-and-clinical-trials.
- 21. US Food and Drug Administration . Guidance for industry: computerized systems used in clinical trials. [Internet]. [cited 2019 Aug 25]. Available from: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/fda-bioresearch-monitoring-information/guidance-industry-computerized-systems-used-clinical-trials.
- 22. Sanofi . Clinical trial data sharing: data de-identification guidelines. [Internet]. [cited 2020 Feb 23]. Available from: https://www.clinicalstudydatarequest.com/Documents/Sanofi-DeIdentification-Guide.pdf.
- 23. Uppsala Monitoring Centre . WHODrug global. [Internet]. [cited 2020 Feb 23]. Available from: https://www.who-umc.org/whodrug/whodrug-portfolio/whodrug-global/.
- 24. Uppsala Monitoring Centre . WHODrug portfolio. [Internet]. [cited 2020 Feb 23]. Available from: https://www.who-umc.org/whodrug/whodrug-portfolio.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.