

We developed a NASH‐on‐a‐chip where human primary hepatocytes (HCs), Kupffer cells (KCs), and hepatic stellate cells (HSCs) were cultured in the 3D hydrogel scaffold, while a blood vessel mimetic endothelialized inlet channel was engineered using human liver sinusoidal endothelial cells (LSECs), via which the system was exposed to metabolic and inflammatory stressors. Exposing liver chips to a lipotoxic environment led to the gradual development of NASH phenotypic characteristics, including intracellular lipids accumulation, hepatocellular ballooning, HSC activation, and elevation of inflammatory and profibrotic molecular markers. We demonstrate that the NASH‐on‐a‐chip can be used to test the efficacy of pharmacological agents to inhibit the development of NASH.

Abstract
Nonalcoholic steatohepatitis (NASH), an advanced stage of nonalcoholic fatty liver disease (NAFLD), is a rapidly growing and global health problem compounded by the current absence of specific treatments. A major limiting factor in the development of new NASH therapies is the absence of models that capture the unique cellular structure of the liver microenvironment and recapitulate the complexities of NAFLD progression to NASH. Organ‐on‐a‐chip platforms have emerged as a powerful approach to dynamically model diseases and test drugs. Herein, we describe a NASH‐on‐a‐chip platform. Four main types of human primary liver cells (hepatocytes [HCs], Kupffer cells, liver sinusoidal endothelial cells, and hepatic stellate cells [HSCs]) were cocultured under microfluidic dynamics. Our chip‐based model successfully recapitulated a functional liver cellular microenvironment with stable albumin and urea secretion for at least 2 weeks. Exposing liver chips to a lipotoxic environment led to gradual development of NASH phenotypic characteristics, including intracellular lipid accumulation, hepatocellular ballooning, HSC activation, and elevation of inflammatory and profibrotic markers. Further, exposure of the chip to elafibranor, a drug under study for the therapy of NASH, inhibited the development of NASH‐specific hallmarks, causing an ~8‐fold decrease in intracellular lipids, a 3‐fold reduction in number of ballooned HCs, a significant reduction in HSC activation, and a significant decrease in the levels of inflammatory and profibrotic markers compared with controls. Conclusion: We have successfully developed a microfluidic NASH‐on‐a‐chip platform that recapitulates the main NASH histologic endpoints in a single chip and that can emerge as a powerful noninvasive, human‐relevant, in vitro platform to study disease pathogenesis and develop novel anti‐NASH drugs.
Abbreviations
- 2D/3D
two dimensional/three dimensional
- ANOVA
analysis of variance
- COL1A1
collagen 1A1
- ECM
extracellular matrix
- FFA
free fatty acid
- HC
hepatocyte
- Hh
hedgehog
- HSC
hepatic stellate cell
- KC
Kupffer cell
- LDH
lactate dehydrogenase
- LPS
lipopolysaccharide
- LSEC
liver sinusoidal endothelial cell
- MCP1
monocyte chemoattractant protein 1
- MIP1α
macrophage inflammatory protein 1α
- NAFL
nonalcoholic fatty liver
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NF‐κB
nuclear factor kappa B
- OA
oleic acid
- OPN
osteopontin
- PA
palmitic acid
- SHh
sonic hedgehog
- TGF‐β
transforming growth factor β
- TIMP‐1
tissue inhibitor metalloproteinase‐1
- TNF‐α
tumor necrosis factor α
- α‐SMA
α‐smooth muscle actin
Nonalcoholic fatty liver disease (NAFLD), recently redefined as metabolic (dysfunction)‐associated fatty liver disease is a widespread rising epidemic affecting more than 25% of the world’s population.( 1 ) NAFLD is now recognized as the most frequent cause of chronic liver diseases around the world and is expected to be the main cause of liver transplantation in the United States by 2030.( 2 ) NAFLD constitutes a wide spectrum of liver pathologies starting from benign fat accumulation (hepatic steatosis) to a more complex and severe liver pathology known as nonalcoholic steatohepatitis (NASH), which can ultimately progress to cirrhosis and liver cancer.( 2 ) Around 10%‐20% of patients with NAFLD develop NASH, characterized by marked fatty change (hepatocyte [HC] ballooning), inflammation, fibrosis, and liver damage( 3 ) (Fig. 1A). The increased prevalence of NASH is paralleling the dramatic rise in obesity, metabolic syndrome, and sedentary lifestyle because lipid accumulation is the main engine for disease onset and progression resulting in both morphologic and functional disruption of liver tissue.( 4 ) However, some drugs and chemotherapeutics can also cause NASH through different mechanisms.( 5 )
FIG. 1.
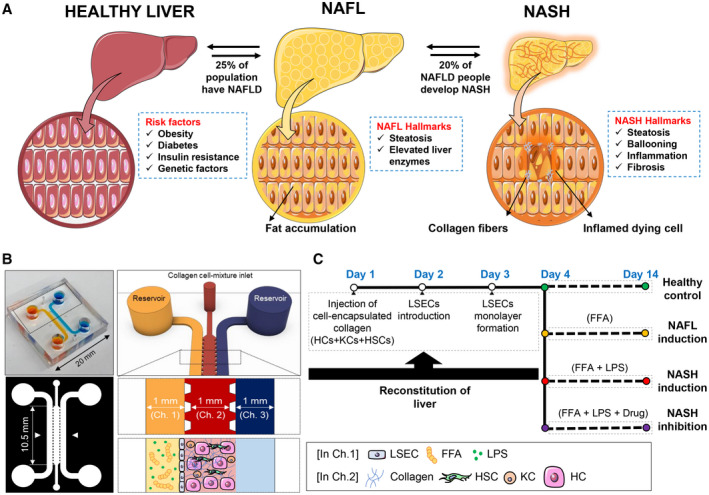
NASH‐on‐a‐chip. (A) Schematic representation illustrating the risk factors and characteristic hallmarks of NASH development. (B) Design of the microfluidic NASH‐on‐a‐chip model, which is composed of a hydrogel channel containing HCs, KCs, and HSCs surrounded by LSEC‐coated inlet and outlet channels. (C) Time frame of reconstruction of liver‐on‐chips and disease induction. Abbreviations: Ch., channel.
Despite significant progress in understanding its pathophysiology, there is no U.S. Food and Drug Administration‐approved therapy for the treatment of NASH.( 6 ) The financial burden of NAFLD is currently estimated at $100 billion in the United States alone, and the NASH drug market is expected to rise from $1 billion in 2015 to $25 billion by 2025.( 7 ) While there is an urgent and unmet medical need to develop effective NASH therapies, there are no robust in vitro models of the disease currently available. This is a major obstacle for drug discovery, and the need for a human‐relevant in vitro model of the disease is compelling and would unquestionably open new avenues for drug development.
Rodent models of NASH have been widely used, but significant discrepancies were found between rodent models and human in the transcriptomic profile of the liver tissue, the mechanism of fat accumulation, and degree of liver fibrosis.( 8 ) On the other hand, two‐dimensional (2D) in vitro culture models fail to recapitulate the disease as they do not capture the cellular complexity of the liver tissue and the contribution of multiple cell types to disease progression. Initial attempts to address the limitations of 2D cultures have led to the development of 3D models based on immortalized cell lines,( 9 ) precision‐cut liver slices,( 10 ) or primary human HCs( 11 ), but these models are limited by viability issues that prevent long‐term studies.( 12 ) More recently, 3D spheroidal cultures of human HCs with hepatic stellate cells (HSCs), endothelial cells, and Kupffer cells (KCs) have been developed and allow long‐term studies.( 13 ) However, the lack of dynamic perfusion is a major drawback of these static culture systems because the activity of HCs under dynamic flow is considerably higher than in static cultures owing to the continuous delivery of oxygen and nutrients and clearance of metabolites and toxic intermediates.( 14 )
Organ‐on‐a‐chip platforms have emerged as a powerful approach to dynamically model disease and test drugs. Miniaturized 3D organs have been engineered replicating the key physiological features of specific human microenvironments with precise structural, fluidic, and mechanical control.( 15 ) Despite the great promise of microfluidic‐based 3D organ‐on‐a‐chip models as robust platforms for preclinical studies,( 16 ) few studies have addressed NASH. For example, exposing HepG2 cells to free fatty acid (FFA) overload for 48 hours under microfluidic dynamics led to a significant accumulation of intracellular lipids.( 17 ) However, to the best of our knowledge, there is no dynamic 3D on‐chip model integrating the complex cellular interactions between parenchymal (HCs) and nonparenchymal human liver cells (liver sinusoidal endothelial cells [LSECs], KCs, HSCs) to model the development and progression of NASH.
Here, we describe a NASH‐on‐a‐chip in which we coculture the four major types of human primary liver cells (HCs, HSCs, LSECs, and KCs) under microfluidic dynamics, and NASH can be induced by perfusing the disease triggers through an endothelialized inlet microfluidic channel mimicking blood vessels. We demonstrate that our microstructured liver tissue can be maintained under disease‐inducing conditions for at least 10 days, simulating the key hallmarks of human NASH, including accumulation of intracellular lipids, hepatocellular ballooning, and production of inflammatory and profibrotic markers. Finally, as a proof of concept, we tested the effectiveness of elafibranor, a potential NASH therapeutic in inhibiting NASH in our liver‐on‐a‐chip model culture.
Materials and Methods
Cell Culture
Four types of primary human hepatic cells (HCs, HSCs, KCs, LSECs) and their corresponding media were purchased from Zen‐Bio Inc. Media composition is detailed in the Supporting Information.
Fabrication of the Microfluidic Device
Triplet microchannel devices, consisting of a middle hydrogel channel surrounded by two side channels, were fabricated with standard photolithography. The fabrication steps of the devices are detailed in the Supporting Information.
Construction of Liver‐on‐a‐Chip
First, rat tail collagen type I (2 mg/mL; Corning) was prepared according to the manufacturer’s protocol. HSCs (5 × 104 cells/mL), KCs (15 × 104 cells/mL), and HCs (5 × 105 cells/mL) were mixed with the collagen and then injected into the middle channel of the microfluidic chip. The chips were incubated in the incubator (37°C, 5% CO2) for 1 hour until the collagen formed a gel. The chips were then conditioned by a mixture of HC‐plating media:HSC‐growth media:KC‐plating media at their corresponding cell ratio (10:1:3) through the inlet channel. The chips were then incubated in the incubator for 4 hours. After that, the HC‐ and KC‐plating media were changed to their maintenance media and kept in the incubator for 24 hours. On day 2, LSECs were suspended in LSEC‐growth media at a density of 2 × 106 cells/mL. Existing media in the chip was removed from the reservoir of the outlet channel, and then 50 μL of LSEC suspension was flowed through the inlet channel. The microfluidic chip was tilted by 90 degrees and kept in the incubator for 1 hour to enable LSECs to adhere to the collagen surface. Then, the reservoirs were filled with a mixture of HC‐maintenance media:HSC‐growth media:LSEC‐growth media:KC‐maintenance media at their corresponding cell ratio and incubated for 1 day before disease induction. The time frame of liver constitution and disease induction is shown in Fig. 1C.
Induction of Steatosis, NASH, and Drug Application on a Chip
The chips were treated with various disease triggers and treatments once the LSECs were well attached and completely spread onto the collagen surface. Liver steatosis was induced by exposing the liver cells to an overload of long‐chain free fatty acids (FFAs) by flowing oleic acid (OA; 0.66 mM) (Sigma) and palmitic acid (PA; 0.33 mM) (Sigma) in the endothelialized channel. We induced NASH by similarly exposing the cells on a chip to OA (0.66 mM), PA (0.33 mM), and lipopolysaccharides (LPS; 10 µg/mL) (Sigma). Here, LPS was added to induce inflammation. To test whether the NASH‐on‐a‐chip platform can be used to detect the efficacy of potential pharmacologic interventions, we exposed the cells to NASH triggers in addition to elafibranor (30 µM) (MedChemExpress LLC). For the preparation of FFAs to apply on a chip, a stock solution of FFAs in dimethyl sulfoxide (DMSO) was diluted in HC‐maintenance media enriched with bovine serum albumin (BSA; 1%). In the control group, liver on chips were exposed to HC‐maintenance media with the same BSA and DMSO concentration (1% and 0.1%, respectively). All chips were put on the rocker and incubated at 5% CO2 and 37°C. Media of all chips was refreshed every 2 days to maintain constant head pressure during the culture period (14 days). The chips were monitored daily, and any changes in cell morphology were imaged using brightfield microscopy (Nikon Instruments Inc.). Live/dead assay (calcein AM‐ethidium homodimer [EthD‐1]) (Invitrogen) was performed for assessing cell viability. Additionally, viability of cells in culture was validated using immunolabeling for cleaved‐caspase 3. Protocols for biomarker quantification, other experimental procedures, and statistical analysis are detailed in the Supporting Information.
Results
Recapitulating Liver Function on a Chip
We used a triplet microchannel chip to coculture the four types of primary human liver cells under microfluidic dynamics, mimicking the multicellular liver microenvironment (Fig. 1B). First, the three types of human primary liver cells, HCs, HSCs, and KCs, were mixed with collagen type I hydrogel, which resembles the extracellular matrix (ECM) in human liver tissue, and then injected into the middle channel of the chip to create a 3D coculture. The LSEC monolayer was subsequently established on the lateral surface of the collagen hydrogel, mimicking the perforated endothelial lining in an in vivo liver sinusoid (Fig. 1C). The four types of liver cells could communicate with each other by direct cell to cell contact and factors secreted in the milieu. In our study, the HCs and nonparenchymal cells, including HSCs, LSECs and KCs, were cocultured at a ratio of 10:1:20:3 where the ratio between HCs, HSCs, and KCs (10:1:3) was similar to the cellular composition of in vivo liver.( 18 ) LSECs were cultured in our system at a relatively higher ratio to ensure formation of a continuous monolayer on the whole lateral surface of the ECM.( 19 )
The key features of the four hepatic cells in the 3D liver chip were characterized by immunofluorescence staining. After 1 day, LSECs formed a monolayer expressing their characteristic platelet endothelial cell adhesion molecule/cluster of differentiation (CD)31 (Fig. 2Ai, red). In the collagen matrix, the KCs attained a rounded structure expressing their characteristic CD68 protein (blue; insert in Fig. 2Aii), while HSCs showed their characteristic stellate‐like shape with branched structures like astrocytes (insert in Fig. 2Aii; the green color corresponded to the vimentin staining). HCs showed their distinct polygonal shape with red‐stained cytokeratin (insert in Fig. 2Aii). A brightfield microscope image of the orientation of HCs (red), HSCs (green), and KCs (blue) in the collagen matrix (gray) is shown in Fig. 2B, while a representative confocal microscope image of the 3D arrangement of HCs (red), HSCs (green), and KCs (blue) in a collagen matrix is shown in Fig. 2C. These cells in the collagen matrix showed high viability (>85%) even after 14 days of initial culture (Fig. 2D), indicating that our chip‐based liver tissue modeling system enabled adequate exchange of nutrients and gases to the cells for the long‐term culture.
FIG. 2.
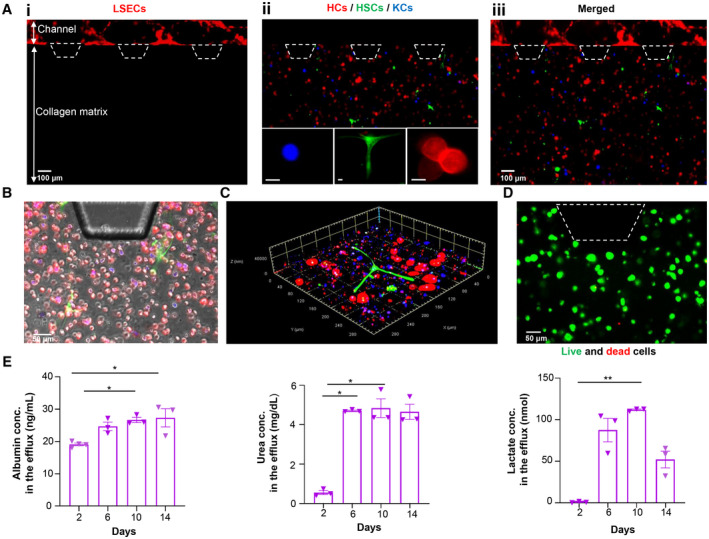
Construction of liver‐on‐a‐chip. (A) Recapitulation of the liver cellular microenvironment on a chip. (Ai) LSECs forming a continuous monolayer in the lateral channel. (Aii) Collagen compartment containing KCs (blue), HSCs (green), and HCs (red); scale bars are 25 µm. (Aiii) Merged image showing both endothelialized channel and collagen compartments. (B) Representative brightfield microscope image showing the orientation of HCs (red), HSCs (green), and KCs (blue) in the collagen matrix (gray). (C) Representative confocal microscope image of the 3D arrangement of HCs (red), HSCs (green), and KCs (blue) in a collagen matrix. (D) Live/dead assay of liver cells after 14 days of initial culture. (E) Functional activity of liver‐on‐chips, including albumin, urea, and lactate production. Data are presented as mean ± SEM; n = 3. Significance was calculated using ANOVA followed by the Kruskal‐Wallis test, *P < 0.05 and **P < 0.01. Trapezoids with dashed lines indicate the posts in the device for the collagen boundary; thus, cells are not present over the posts. Abbreviation: conc., concentration.
To further assess the synthetic, metabolic, and detoxification function of cells in our liver‐on‐a‐chip, cellular production of albumin, urea, and lactate were measured in the effluent culture media collected from chip reservoirs over 14 days. Albumin and urea secreted by the HCs increased gradually until reaching a plateau at day 6 (Fig. 2E). The average ± SEM values of albumin and urea detected at day 14 in the culture media were 27.33 ± 4.89 ng/mL and 4.65 ± 0.67 mg/dL, respectively. Assessment of energy metabolism by measurement of lactate secretion by HCs was also performed. An initial lactate concentration of 0.76 ± 0.87 nmol was detected in the culture media; this increased gradually until reaching a constant value. Afterwards, significant reduction in lactate production was detected, and a total lactate concentration of 52.09 ± 17.51 nmol was detected at the end of the experiment. These results showed that our chip‐based model successfully recapitulated a functional liver microenvironment with stable urea and albumin secretion by coculture of the four types of primary human liver cells in a 3D collagen matrix under dynamic flow for 2 weeks.
Induction and Morphological Characterization of NASH‐on‐a‐Chip
After establishing the viability and metabolic functionality of the cells in the dynamic 3D chip, we next exposed the liver cells to a lipotoxic stimuli to induce NASH‐on‐a‐chip (Fig. 1C). As described earlier, we perfused the liver cells with a high concentration of FFAs with and without LPS through the LSEC‐monolayered microfluidic channel for 10 days. We then analyzed the progression of NASH hallmarks, including intracellular lipid accumulation, hepatocellular ballooning, inflammation, and fibrosis, qualitatively and quantitatively (Fig. 3A). The exposure of liver cells to a high concentration of FFAs for 10 days led to a significant accumulation of neutral lipids inside the HCs that manifested as a ~3‐fold increase in red fluorescence intensity (HCS LipidTOX RED staining) compared to cells cultured under healthy conditions (Fig. 3B), indicating steatosis development. Similarly, we could visualize significant changes in the morphologies of HCs due to lipotoxic lethal and sublethal injury and apoptosis at different time points on a chip (Fig. 4A). Liver cell apoptosis and cytotoxicity were further examined biochemically by cleaved caspase‐3 staining and lactate dehydrogenase (LDH) activity assessment, respectively.( 20 ) Liver cells cultured under NASH triggers (FFAs + LPS) showed a significantly higher number of cleaved caspase‐3‐positive cells (~3‐fold) and significantly elevated levels of LDH activity compared with controls as early as day 4 when the chip was challenged with lipotoxic stress (Fig. 4); this indicated the onset of liver injury associated with NASH development on our chip model. Indeed, LDH levels are classically considered a biomarker for the viability of cells. We observed a significant increase in the levels of LDH activity in the control arm on day 10 (but not at other time points) compared with the day 2 control arm (analysis of variance [ANOVA] followed by the Kruskal‐Wallis test; adjusted P = 0.01). This increase in LDH levels is consistent with that normally associated with primary cell‐based long‐term cultures( 21 , 22 ); interestingly, the total LDH release was maintained at minimal values (below 2 U/L) compared to the levels seen in these previous studies. However, we validated the viability of our system using two additional approaches (cleaved‐caspase expression analysis and calcein AM/EthD‐1 labeling, which is a live/dead assay). A minimal cleaved caspase 3 signal was evident in the control arm on day 10 (Fig. 4B). Live cells enzymatically convert nonfluorescent cell‐permeant calcein AM to the intensely fluorescent calcein. EthD‐1 enters cells with damaged membranes (but is excluded by intact plasma membrane of live cells) and undergoes a 40‐fold enhancement of fluorescence following binding to nucleic acids, thereby producing a bright red fluorescence in dead cells. The ratio of live to dead cells in the control arm was found to be 90.4% (viable) to 9.6% (dead) on day 5, and ~85% cell viability was observed after 10 days of initial culture (Fig. 2D; Supporting Fig. S1A). Together, with good functionality evidenced by stable urea and albumin secretions at late time points (Fig. 2E), this supports that the chip was functionally viable during the study period. However, this baseline increase in LDH over time means the models still need to evolve to truly mimic the human liver.
FIG. 3.
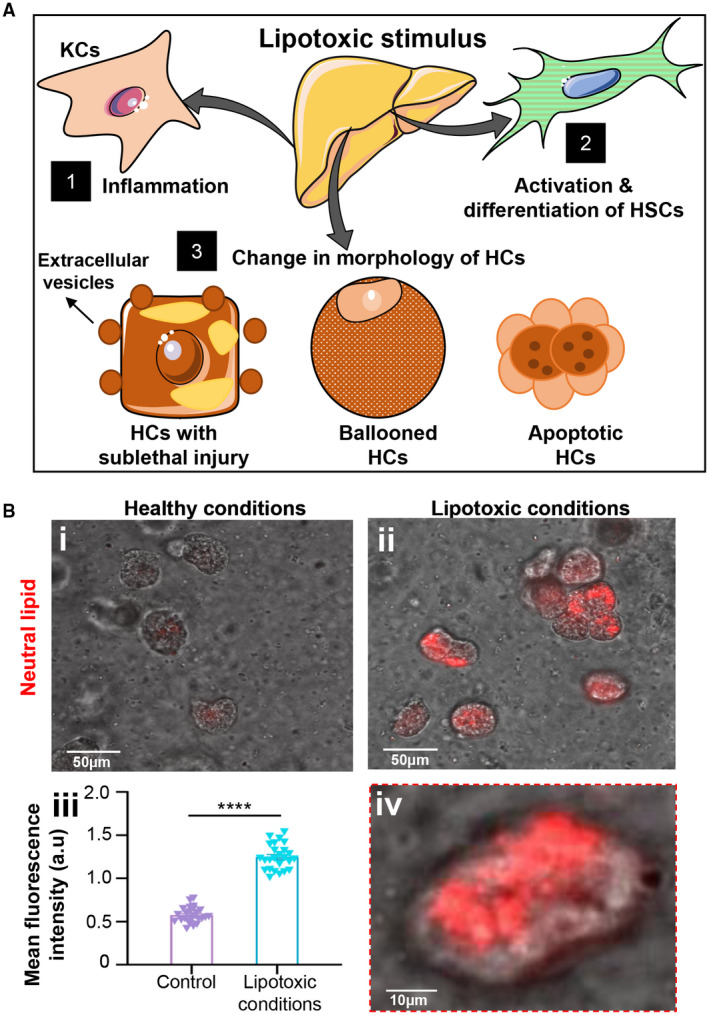
Histologic hallmarks of NASH. (A) Schematic representation of histologic hallmarks of NASH, including inflammation, HSC activation, and lethal and sublethal injury in HCs cultured under lipotoxic stress. (B) Steatosis on‐a‐chip. Representative images of HCs cultured under (Bi) healthy or (Bii) lipotoxic conditions for 10 days. (Biii) Mean fluorescence intensity of neutral lipid staining of HCs was quantified and plotted against healthy controls after 10 days of disease induction. (Biv) Magnified image of HCs cultured under lipotoxic stress. Data are presented as mean ± SEM; n ≥ 20. Significance was calculated by the nonparametric Mann‐Whitney test, ****P < 0.0001. Abbreviation: a.u., arbitrary unit.
FIG. 4.
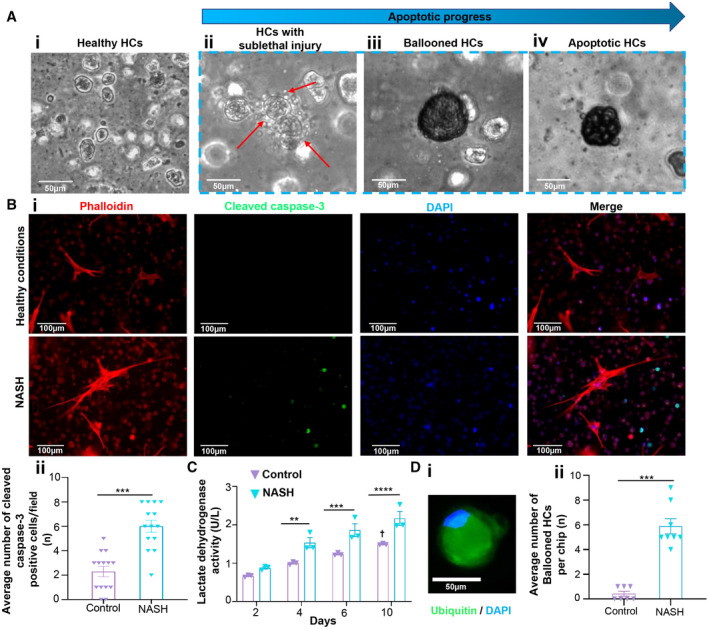
Morphologic changes of liver cells cultured under lipotoxic stimuli. (A) Apoptotic progress in HCs as a result of lipid‐induced lethal and sublethal signaling. Compared with (Ai) healthy controls, (Aii) sublethal proapoptotic signaling leads to release of extracellular vesicles from HCs and/or emergence of (Aiii) ballooned HCs while (Aiv) lethal signaling leads to lipoapoptosis (apoptotic cell death). (B) (Bi) Cleaved caspase‐3 staining images and (Bii) number of cleaved caspase‐3‐positive cells under healthy and NASH conditions from the imaging analysis. Data are presented as mean ± SEM; n ≥ 8. Significance was calculated by the nonparametric Mann‐Whitney test, ***P < 0.005. (C) LDH activity under healthy and NASH conditions. Data are presented as mean ± SEM; n = 3. Significance was calculated using two‐way ANOVA followed by the Bonferroni test, **P < 0.01, ***P < 0.005, and ****P < 0.0001; †control (day 10) versus control (day 2) (adjusted P = 0.01; ANOVA followed by the Kruskal‐Wallis test). (D) Development of ballooned HCs under lipotoxic stimulation. (Di) Representative fluorescence microscope image of ballooned HC with accumulated ubiquitinated proteins and displaced nucleus. (Dii) Average number of ballooned HCs under both healthy and NASH conditions after 10 days of disease induction. Data are presented as mean ± SEM; n ≥ 8. Significance was calculated by the nonparametric Mann‐Whitney test, ***P < 0.005. Abbreviation: DAPI, 4′,6‐diamidino‐2‐phenylindole.
Hepatocellular ballooning is a hallmark of NASH, and ballooned HCs are greatly involved in the disease pathogenesis by releasing profibrogenic factors, such as hedgehog (Hh) ligands, that induce hepatic fibrogenesis by paracrine signaling.( 23 ) A representative image of immunostained ballooned HCs with its characteristic features is shown in Fig. 4Di.( 24 ) The ballooned cells showed their distinct swollen and rounded morphology, displaced nucleus (blue), and accumulation of ubiquitinated proteins (green). Based on these unique features of ballooned HCs, emergence and the incidence rate of hepatocellular ballooning can be used for measuring the disease progression stage of NASH.( 25 ) Chips that were cultured under the NASH triggers for 10 days showed a significantly higher number of ballooned HCs (~14.5‐fold) compared to the control group (Fig. 4Dii), which further confirmed disease induction and progression in our NASH‐on‐a‐chip platform.
HSCs are the main contributors to liver fibrosis. During NASH progression, quiescent HSCs undergo rapid activation and transdifferentiation into myofibroblast‐like cells with fibrogenic, proliferative, and contractile characteristics. Myofibroblasts exhibit a characteristic morphology of large bundles of microfilaments extending along the cell axis. Increased expression of α‐smooth muscle actin (α‐SMA) is one of the indicative markers of HSC activation and the transdifferentiated phenotype.( 26 ) In this respect, we observed the morphology of the HSCs (Fig. 5Ai‐iv) and quantified the expression level of α‐SMA after immunofluorescence staining after 10 days of disease induction. Under NASH conditions, HSCs exhibited a significantly higher level (~7.7‐fold) of α‐SMA with a striated morphology compared to the control HSCs (Fig. 5Av). Additionally, the number of activated HSCs was found to be ~8.3‐fold and 2.4‐fold higher than that of the control group and chips exposed to FFAs alone, respectively (Fig. 5Avi). The secretion of excess ECM proteins by HSCs is another characteristic sign of NASH progression and can potentially provoke liver fibrosis.( 27 ) We quantified the expression of fibrosis‐related proteins (collagen 1A1 [COL1A1] and tissue inhibitor metalloproteinase‐1 [TIMP‐1]) in the cells by immunofluorescence staining and compared the results between groups. We used specific human antibody to distinguish COL1A1 secreted by HSCs from rat tail collagen type I that constituted the liver ECM on our chip design. The expression levels of both profibrotic proteins COL1A1 and TIMP‐1 were significantly up‐regulated in chips exposed to FFAs and LPS compared with control and chips exposed to FFAs alone (Fig. 5B,C).
FIG. 5.
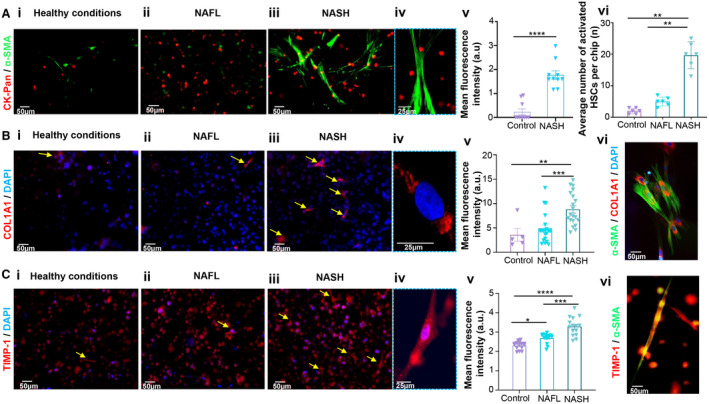
Fibrosis on‐a‐chip. (A) Representative fluorescence images of HSCs after ɑ‐SMA staining under (Ai) healthy, (Aii) NAFL, and (Aiii) NASH conditions. (Aiv) Magnified image of activated HSCs under the NASH condition. (Av) Mean fluorescence intensity of ɑ‐SMA‐stained HSCs and (Avi) average number of activated HSCs under healthy, NAFL, and NASH conditions after 10 days of disease induction. (B) Representative fluorescence images of COL1A1 staining under (Bi) healthy, (Bii) NAFL, and (Biii) NASH conditions. (Biv) Magnified image showing COL1A1 expression in HSCs under the NASH condition. (Bv) Mean fluorescence intensity of COL1A1 under healthy, NAFL, and NASH conditions after 10 days of disease induction. (Bvi) Representative fluorescence image showing COL1A1 and ɑ‐SMA expression in activated HSCs. (C) Representative fluorescence images of TIMP‐1 staining under (Ci) healthy, (Cii) NAFL, and (Ciii) NASH conditions. (Civ) Magnified image showing TIMP‐1 expression in HSCs under the NASH condition. (Cv) Mean fluorescence intensity of TIMP‐1 under healthy, NAFL, and NASH conditions after 10 days of disease induction. (Cvi) Representative fluorescence image showing TIMP‐1 and ɑ‐SMA expression in activated HSCs. Data are presented as mean ± SEM; n ≥ 10. Significance was calculated using ANOVA followed by the Kruskal‐Wallis test, *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.0001. Abbreviations: a.u., arbitrary units; CK‐Pan, pan‐cytokeratin; DAPI, 4′,6‐diamidino‐2‐phenylindole.
As mentioned previously, injured HCs play an essential role in activation of HSCs and induction of fibrosis by secretion of Hh ligands that promote liver fibrosis.( 28 ) We assessed the Hh signaling pathway as a possible mechanism of HSC activation in the perspective of multicellular communication with injured HCs on our NASH model (Fig. 6A). Injured HCs, including apoptotic and ballooned HCs, showed a significant increase (~1.4‐fold; Fig. 6B) in the expression of sonic Hh (SHh), which leads to activation of the Hh signaling pathway in HSCs as evidenced by nuclear accumulation of GLI family zinc finger 2 (GLI2; a marker of Hh signaling) in activated HSCs.
FIG. 6.
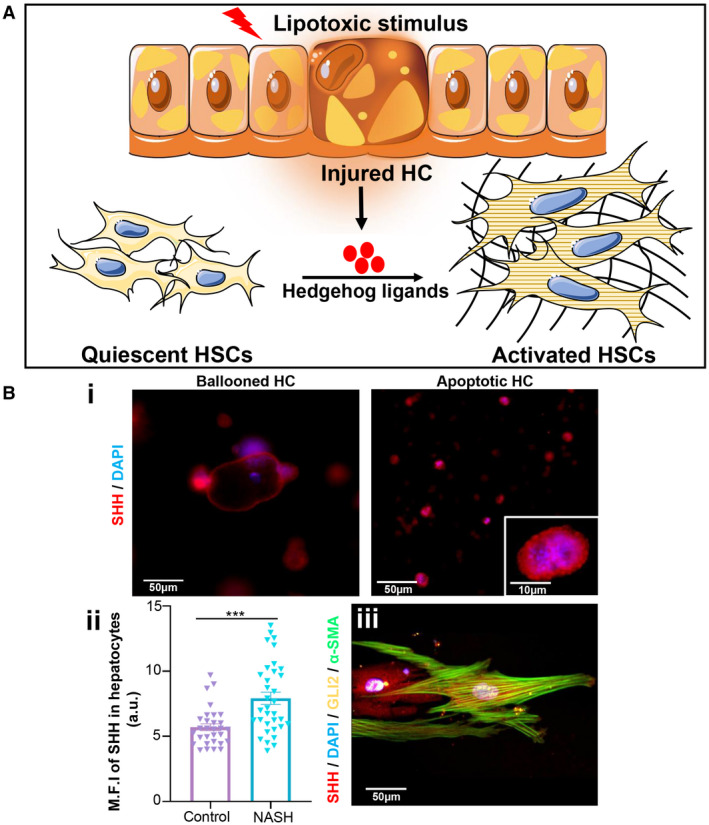
Mechanism of HSC activation. (A) Schematic representation of SHh‐mediated activation of HSCs under the NASH condition. (B) (Bi) Expression of SHh (red) in ballooned and apoptotic HCs. (Bii) Mean fluorescence intensity of SHh expression in HCs. (Biii) Expression of GLI2 (yellow), α‐SMA (green), and SHh (red) in activated HSCs. Data are presented as mean ± SEM; n ≥ 20. Significance was calculated by the nonparametric Mann‐Whitney test, ***P < 0.005. Abbreviations: a.u., arbitrary units; DAPI, 4′,6‐diamidino‐2‐phenylindole; M.F.I, mean fluorescence intensity.
Inflammatory and Profibrotic Biomarkers in NASH‐on‐a‐Chip
To assess NASH‐associated inflammation in our microfluidic chip, media effluents from liver tissue cultured on chips were collected at day 5 and day 10 after exposure to lipotoxic conditions to analyze inflammatory markers that are reported to be elevated during NASH development, including monocyte chemoattractant protein 1 (MCP1), macrophage inflammatory protein 1α (MIP1α), and tumor necrosis factor α (TNF‐α) (Fig. 7). Mean ± SEM MCP1 concentrations in media effluent collected from FFA‐treated (nonalcoholic fatty liver [NAFL]) and FFA + LPS‐treated (NASH) groups were 3.23 ± 0.69 ng/mL and 3.22 ± 0.35 ng/mL, respectively, which were significantly higher than the MCP1 concentration (mean ± SEM, 1.25 ± 0.22 ng/mL) detected in media from controls (Fig. 7A). MIP1α concentration in the NASH group (mean ± SEM, 282.21 ± 26.45 pg/mL) was significantly higher compared with both the NAFL group (mean ± SEM, 174.52 ± 65.83 pg/mL) and control groups (mean ± SEM, 153.85 ± 14.55 pg/mL) (P < 0.05). A similar trend was also observed for TNF‐α, where the NASH group showed the highest concentration of TNF‐α (mean ± SEM, 1.69 ± 0.50 ng/mL) while the NAFL and control groups were 0.95 ± 0.22 ng/mL and 0.67 ± 0.06 ng/mL, respectively (Fig. 7C). Such a significant difference among groups was not observed at day 5, which again confirmed the gradual NASH progression in our chip in a physiologically relevant manner.
FIG. 7.
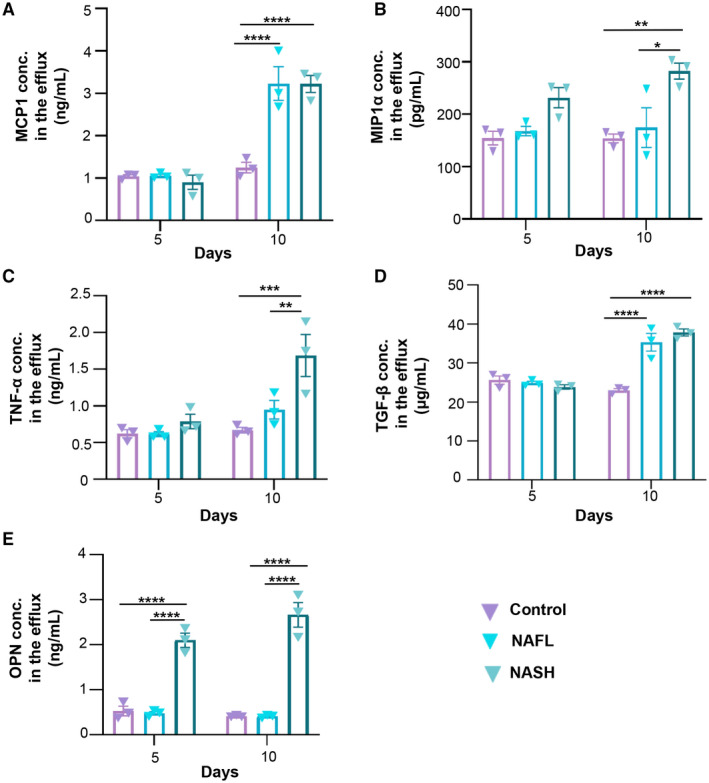
Assessment of inflammatory and profibrotic makers of liver cells cultured under healthy and lipotoxic conditions. Concentration levels of inflammatory markers (A) MCP1, (B) MIP1α, and (C) TNF‐α and profibrotic markers (D) TGF‐β and (E) OPN in the effluent media of control, NAFL, and NASH groups after 5 and 10 days. Data are presented as mean ± SEM; n = 3. Significance was calculated using two‐way ANOVA followed by the Bonferroni test, *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.0001. Abbreviation: conc., concentration.
Furthermore, we measured the expression levels of transforming growth factor β (TGF‐β) and osteopontin (OPN) (Fig. 7D,E). TGF‐β is a well‐known regulator of fibrosis as it promotes excessive secretion of ECM proteins by HSCs, especially collagens, and inhibits the production of matrix‐degrading proteases, thus preventing ECM degradation. OPN is another important biomarker that has been proven to have a positive correlation with liver fibrosis because it interacts with HSC surface integrins (αvβ3) resulting in activation of the phosphoinositide 3‐kinase (PI3K)/phosphorylated AKT serine/threonine kinase 1 (pAkt)/nuclear factor kappa B (NF‐κB) signaling pathway and subsequent up‐regulation of collagen type I and modulation of the HSC profibrogenic phenotype.( 29 ) It was found that concentration levels of both TGF‐β and OPN detected in the media collected from chips cultured under lipotoxic conditions were significantly higher than those cultured under healthy conditions. The mean ± SEM TGF‐β concentration in media effluent collected from NAFL and NASH groups was 35.33 ± 3.92 µg/mL and 37.83 ± 1.55 µg/mL, respectively, while significantly lower TGF‐β concentration (mean ± SEM, 23.17 ± 0.80 µg/mL) was detected in the media of the control group. The concentration of OPN in chips that were exposed to NASH triggers showed the highest concentration (mean ± SEM, 2.66 ± 0.47 ng/mL) compared with the control group (mean ± SEM, 0.41 ± 0.02 ng/mL) or NAFL group (mean ± SEM, 0.41 ± 0.03 ng/mL).
Testing Drug Efficacy Using the NASH‐on‐a‐Chip Platform
We next tested the feasibility of our NASH‐on‐a‐chip as a drug‐testing platform. Elafibranor is a dual agonist for peroxisome proliferator‐activated receptors (PPAR‐α/δ), which are highly involved in regulation of fatty acid metabolism in HCs. It attenuates the inflammation and fibrosis mediated by NF‐κB and activator protein 1 signaling pathways (Fig. 8A).( 30 ) We applied the drug through the endothelialized microfluidic inlet channel to mimic drug circulation in blood vessels in vivo. Treatment was started simultaneously with lipotoxic stress (NASH group), as described earlier. We then compared the expression level of NASH hallmarks, including lipid accumulation, hepatocellular ballooning, inflammation, and fibrosis, with and without application of drugs. Exposure of FFA + LPS‐treated liver cells to elafibranor significantly prevented fat accumulation within the HCs, which was manifested as a ~7.7‐fold decrease in the fluorescence intensity of stained neutral lipids compared to the control NASH group (LPS + FFA only, no elafibranor) (Fig. 8B). Our results are consistent with the effect of elafibranor on reducing lipid accumulation, as reported by Boeckmans et al.( 12 ) in hepatic cells differentiated from human skin‐derived precursors and match the antisteatotic effects of elafibranor that have been reported in clinical trials.( 30 ) In addition, the application of elafibranor protected liver cells against injury associated with NASH development, evidenced by the significantly lower number of ballooned (~3‐fold; Fig. 8D) and apoptotic and dead HCs (2.7‐fold) as well as reduced levels of LDH activity (Supporting Fig. S1). Furthermore, we observed that elafibranor significantly prevented the transdifferentiation of HSCs to myofibroblasts, evidenced by a ~13‐fold lower expression of ɑ‐SMA and a ~6‐fold lower number of activated HSCs in the elafibranor group compared with the NASH group (Fig. 8C). The expression of fibrosis‐related proteins (TIMP‐1, COL1A1) and the number of TIMP‐1+ and COL1A1+ HSC cells were significantly decreased when elafibranor was added to the NASH chips (Supporting Fig. S2), consistent with the antifibrotic action of elafibranor observed in clinics.( 31 )
FIG. 8.
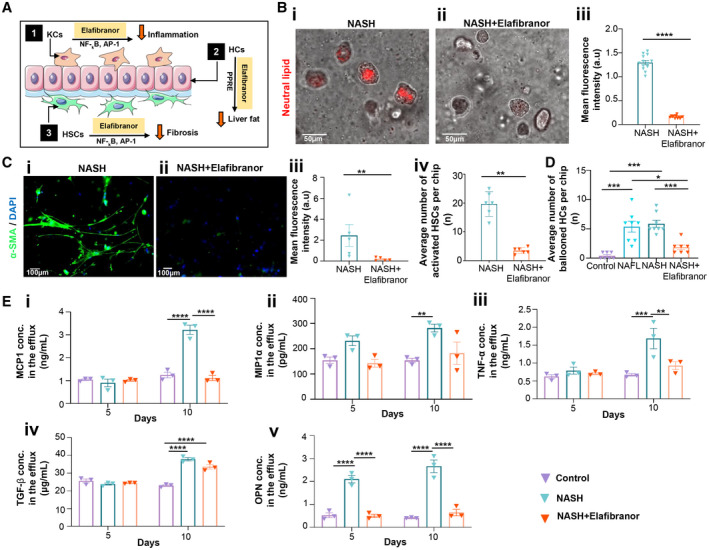
Effect of elafibranor on NASH characteristic hallmarks. (A) Schematic representation of the anti‐NASH mechanism of elafibranor. (B) Representative images of neural lipid staining of NASH chips in (Bi) absence and (Bii) presence of elafibranor. (Biii) Difference in mean fluorescence intensity of neural lipid staining between NASH chips and NASH chips exposed to elafibranor. Data are presented as mean ± SEM; n ≥ 10. Significance was calculated by the nonparametric Mann‐Whitney test, ****P < 0.0001. (C) Effect of elafibranor on HSC activation. Representative images of HSCs under a NASH condition (Ci) without elafibranor exposure and (Cii) in the presence of elafibranor. (Ciii) Mean fluorescence intensity of α‐SMA‐stained HSCs in NASH chips in the absence and presence of elafibranor. Data are presented as mean ± SEM; n = 5. Significance was calculated by the nonparametric Mann‐Whitney test, **P < 0.01. (Civ) Average number of activated HSCs under a NASH condition without elafibranor exposure and in the presence of elafibranor after 10 days of disease induction. Data are presented as mean ± SEM; n = 6. Significance was calculated by the nonparametric Mann‐Whitney test, **P < 0.01. (D) Average number of ballooned HCs in control, NAFL, NASH, and NASH chips exposed to elafibranor. Data are presented as mean ± SEM; n ≥ 7. Significance was calculated using ANOVA followed by the Kruskal‐Wallis test, *P < 0.05 and ***P < 0.005. (E) Effect of elafibranor on expression levels of inflammatory and profibrotic markers of liver cells under a NASH condition. Concentration levels of inflammatory markers (Ei) MCP1, (Eii) MIP1α, and (Eiii) TNF‐α and profibrotic markers (Eiv) TGF‐β and (Ev) OPN in the effluent media of control, NASH, and NASH with elafibranor groups after 5 and 10 days of culturing. Data are presented as mean ± SEM; n = 3. Significance was calculated using two‐way ANOVA followed by the Bonferroni test, *P < 0.05, **P < 0.01, ***P < 0.005, and ****P < 0.0001. Abbreviations: AP‐1, activator protein 1; a.u., arbitrary units; conc., concentration; DAPI, 4′,6‐diamidino‐2‐phenylindole; PPRE, peroxisome proliferator response element.
The effect of elafibranor on NASH‐associated inflammation was assessed by quantification of representative inflammatory chemokines in the media collected from NASH‐on‐chips. The application of elafibranor reduced the NASH‐associated increased expression of inflammatory chemokines, like MCP1 (Fig. 8Ei), MIP1ɑ (Fig. 8Eii), and TNF‐ɑ (Fig. 8Eiii). For example, the concentration of MCP1 in the elafibranor group (mean ± SEM, 1.11 ± 0.19 ng/mL) was significantly lower than the concentration detected in the NASH group (mean ± SEM, 3.22 ± 0.35 ng/mL). Additionally, the expression of other fibrosis mediators, including TGF‐β (Fig. 8Eiv) and OPN (Fig. 8Ev), in the NASH groups was also attenuated when elafibranor was added. For example, the mean ± SEM expression level of OPN was dramatically decreased in the elafibranor group to 0.65 ± 0.03 ng/mL, with no significant difference to the baseline controls (mean ± SEM, 0.41 ± 0.02 ng/mL).
Discussion
Modeling complex diseases using organ‐on‐a‐chip platforms is an emerging and general paradigm.( 32 ) By leveraging human primary cells, ECM, and microfluidics, we have successfully extended this approach to NASH and developed a “NASH‐on‐a‐chip” culture system capable of (i) maintaining primary human liver cells viable and functional over a period of at least 2 weeks (Fig. 2); (ii) recapitulating the morphologic and biochemical hallmarks of the human disease (Figs. 3, 4, 5, 6, 7); and (iii) validating the activity of elafibranor, a lead therapeutic in advanced stages of development for NASH therapy (Fig. 8). The NASH‐on‐a‐chip described here is a clear advance over previous static, 3D, spheroidal cultures and may yield new insights on disease mechanisms and progression and support new programs of drug discovery.( 33 )
HCs account for ~78% of liver volume( 34 ) and are responsible for most of the vital liver functions, such as protein synthesis, metabolic regulation, and detoxification.( 35 ) We used collagen type I, the major matrix component of liver tissue,( 21 , 36 ) to enhance the growth and preserve the differentiated state of HCs.( 14 ) More importantly, culturing the HCs in a 3D collagen matrix allows closer simulation to the spatial distribution of cells in vivo because it facilitates cell‐to‐cell and cell‐to‐matrix interactions in all directions. For example, HCs cultured in a 3D collagen gel showed superior activity compared to HCs cultured as a monolayer on a collagen gel‐coated 2D surface.( 22 ) In addition, we used dynamic flow conditions to control the supply of oxygen, nutrients, and other soluble factors to the culture,( 37 ) thus enhancing cell survival and activity, as measured by albumin and urea secretion over 14 days (Fig. 2). Interestingly, lactate production tendency in our liver chips matched well the reported extraordinary capacity of HCs to tolerate hypoxia and satisfy energy requirements by anaerobic glycolysis (Fig. 2E). We observed a reduction in lactate production at later culture time points, indicating that the dynamic flow condition can result in improved oxygen delivery and reverse the potentially anaerobic metabolic status of the cells leading to decreased lactate production.( 38 ) It is worth mentioning that media free from serum proteins and growth factors was used starting from day 4 of the experiment to prepare the chips for disease induction and drug testing. During this phase of the experiment, it is important to keep the cells under chemically defined serum and growth factor‐free conditions because the presence of serum proteins in the media may cause uncontrolled drug binding that may affect the experimental results. Furthermore, some of the added growth factors to the media can potentially cause activation of cellular signaling pathways involved in disease progression, thus leading to misinterpretation of the data.
We selected PA and OA, the most common long‐chain FFAs that accumulate in the liver of patients with NASH, as lipotoxic stressors in the culture system and used them at concentrations typically present in the plasma of patients with NASH.( 33 ) Further, we used LPS to trigger disease progression because recent clinical data indicate that patients with NAFLD and NASH have high serum LPS levels.( 39 ) HCs in culture accumulated lipids in their cytoplasm in a time‐dependent manner in response to lipotoxic stress (Fig. 3; Supporting Fig. S3), and a number of these HCs eventually died of apoptosis, evidenced by increased levels of cleaved caspase‐3 and LDH activity and mirroring closely a key feature of the transition between NAFL and NASH (Fig. 4).( 27 ) Lipotoxic sublethal injury of HCs in human NASH leads to hepatocellular ballooning and causes the release of extracellular vesicles that initiate HSC activation through different processes mediated by Fas and TNF‐related apoptosis‐inducing ligand pathways( 40 ) and consistent with this mechanism of paracrine signaling. To the best of our knowledge, this is the first in vitro model platform that has shown systematic morphologic changes of HCs due to lethal and sublethal injury during NASH progression, indicating that our NASH‐on‐a‐chip system can be useful to study NASH pathogenesis.
It is widely known that TIMP‐1 is up‐regulated in liver tissue during hepatic fibrogenesis and that it promotes fibrosis by subsequent accumulation of ECM proteins in the injured liver. Although TIMP‐1 is mainly secreted by HSCs and KCs in the liver, it is also reported to be expressed by HCs under diseased conditions, such as CCl4‐induced liver fibrosis.( 41 ) This is in agreement with our findings as we observed elevated TIMP‐1 expression in HSCs, KCs, and HCs under a NASH condition. Increased expression of TIMP‐1 and COL1A1 and the increased number of α‐SMA‐positive HSCs in the FFA + LPS‐treated chips versus the FFA‐treated chips (Fig. 5) suggest that LPS can potentially drive the phenotype toward NASH even though clinically the levels of LPS are similar in NAFL and NASH.
It has been reported that the Hh signaling pathway is widely involved in the development of fibrosis in patients with NASH, where both ballooned and apoptotic HCs produce and release SHh ligands.( 42 ) Binding of SHh ligands to the SHh receptor on the surface of HSCs leads to activation of smoothened (Smo) nuclear accumulation of GLI2 and subsequent transcription of GLI1 and its target genes leading to activation and differentiation of HSCs.( 28 ) Interestingly, this finding is mirrored in our NASH model (Fig. 6), highlighting another advantage of our NASH model that allows crosstalk between different types of liver cells by either direct cell to cell contact and/or factors released in the matrix.
While treatment with FFA alone resulted in up‐regulation of MCP1 and TGF‐β, the addition of LPS resulted in activation of MCP1, MIP1α, TNF‐α, and OPN in a time‐dependent manner, confirming that distinct pathways are involved in and can discriminate different stages of the disease leading to HSC activation and proliferation. Hepatocellular lipotoxic stress and apoptotic cell death further act as the main triggers for stimulation of innate immune cells to elicit inflammatory response,( 43 ) which in turn leads to further hepatocellular damage and death.( 44 ) KCs contribute to tissue inflammation through secretion of MCP1 and TNF‐α,( 27 ) and the results of our culture system (Figs. 7 and 8) indicate that the engineered NASH‐on‐chips platform captures the key proinflammatory and profibrotic features of the progression from NAFL to NASH.
A major advantage of using a human disease‐on‐a‐chip model is that it can be used to study new chemical entities in a preclinical setting.( 45 ) We observed that elafibranor could inhibit disease progression and fibrosis, as observed in clinical trials.( 46 ) Interestingly, elafibranor failed to inhibit the late induction of TGF‐β but inhibited the induction of other molecular markers, including the early increase in OPN induced by the combination of FFA + LPS. This is consistent with previous reports where PPAR has been implicated in signaling pathways downstream of TGF‐β.( 47 ) These results indicate that monotherapies, such as elafibranor, may impact certain steps of NASH progression, but the complexity of NASH suggests that combined therapies may ultimately be more successful in disease control. The NASH‐on‐a‐chip culture system appears ideal for studying the prospective synergy of drugs that target distinct pathogenetic components of the disease, such as HC death, inflammation, and fibrosis, leading to the development of novel combination therapies.
In conclusion, we have developed a 3D microfluidic culture system that captures the essential morphologic features of NAFL and NASH and the progression from NAFL to NASH. Of further note is the endothelialized inlet microfluidic channel and outlet microfluidic channel in our chip‐based device that mimic a blood vessel for drug diffusion and allow easy collection of culture medium for protein analysis for NASH molecular markers. We also envisage further and major developments of the culture system, namely, (1) the design of the 3D architecture of liver tissue by integrating 3D printing, (2) the use of more complex ECM instead of the collagen type I used in the current study, and (3) the integration of immune cells beyond the tissue‐resident KCs included in the present design.( 48 ) Overall, we believe that the platform described can have a potential contribution in the medical field by providing a noninvasive, human cell‐based, in vitro model of NASH that can make significant contributions to the understanding of NASH pathogenesis and development of novel NASH therapies.
Supporting information
Fig S1
Fig S2
Fig S3
Supplementary Material
Supported by the National Institutes of Health (CA236702, CA214411 to H.L.J., S.S.; AR073135 to H.L.J.; CA229772 to S.S.), American Lung Association Cancer Discovery Award (LCD‐618834 to S.S.), Department of Defense (PC180355 to H.L.J.), and AIL Trentino and Italian Ministry of Education, Universities, and Research Departments of Excellence Initiative (to E.G.).
Potential conflict of interest: Nothing to report.
Contributor Information
Shiladitya Sengupta, Email: ssengupta2@bwh.harvard.edu.
Hae Lin Jang, Email: hjang@bwh.harvard.edu.
References
- 1. Eslam M, Sanyal AJ, George J. Toward more accurate nomenclature for fatty liver diseases. Gastroenterology 2019;157:590‐593. [DOI] [PubMed] [Google Scholar]
- 2. Byrne CD, Targher G. NAFLD: a multisystem disease. J Hepatol 2015;62(Suppl.):S47‐S64. [DOI] [PubMed] [Google Scholar]
- 3. Angulo P. Nonalcoholic fatty liver disease. N Engl J Med 2002;346:1221‐1231. [DOI] [PubMed] [Google Scholar]
- 4. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686‐690. [DOI] [PubMed] [Google Scholar]
- 5. Schumacher JD, Guo GL. Mechanistic review of drug‐induced steatohepatitis. Toxicol Appl Pharmacol 2015;289:40‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cassidy S, Syed BA. Nonalcoholic steatohepatitis (NASH) drugs market. Nat Rev Drug Discov 2016;15:745‐746. [DOI] [PubMed] [Google Scholar]
- 8. Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology 2016;151:513‐525.e0. [DOI] [PubMed] [Google Scholar]
- 9. Janorkar AV, Harris LM, Murphey BS, Sowell BL. Use of three‐dimensional spheroids of hepatocyte‐derived reporter cells to study the effects of intracellular fat accumulation and subsequent cytokine exposure. Biotechnol Bioeng 2011;108:1171‐1180. [DOI] [PubMed] [Google Scholar]
- 10. Ijssennagger N, Janssen AW, Milona A, Pittol JMR, Hollman DA, Mokry M, et al. Gene expression profiling in human precision cut liver slices in response to the FXR agonist obeticholic acid. J Hepatol 2016;64:1158‐1166. [DOI] [PubMed] [Google Scholar]
- 11. Gomez‐Lechon MJ, Donato MT, Martínez‐Romero A, Jiménez N, Castell JV, O’Connor J‐E. A human hepatocellular in vitro model to investigate steatosis. Chem Biol Interact 2007;165:106‐116. [DOI] [PubMed] [Google Scholar]
- 12. Boeckmans J, Buyl K, Natale A, Vandenbempt V, Branson S, De Boe V, et al. Elafibranor restricts lipogenic and inflammatory responses in a human skin stem cell‐derived model of NASH. Pharmacol Res 2019;144:377‐389. [DOI] [PubMed] [Google Scholar]
- 13. Suurmond CE, Lasli S, van den Dolder FW, Ung A, Kim HJ, Bandaru P, et al. In vitro human liver model of nonalcoholic steatohepatitis by coculturing hepatocytes, endothelial cells, and Kupffer cells. Adv Healthc Mater 2019;8:e1901379. [DOI] [PubMed] [Google Scholar]
- 14. Shulman M, Nahmias Y. Long‐term culture and coculture of primary rat and human hepatocytes. Methods Mol Biol 2013;945:287‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ingber DE. Reverse engineering human pathophysiology with organs‐on‐chips. Cell 2016;164:1105‐1109. [DOI] [PubMed] [Google Scholar]
- 16. Bovard D, Iskandar A, Luettich K, Hoeng J, Peitsch MC. Organs‐on‐a‐chip: a new paradigm for toxicological assessment and preclinical drug development. Toxicol Res Appl 2017. 10.1177/2397847317726351. [DOI] [Google Scholar]
- 17. Gori M, Simonelli MC, Giannitelli SM, Businaro L, Trombetta M, Rainer A. Investigating nonalcoholic fatty liver disease in a liver‐on‐a‐chip microfluidic device. PLoS One 2016;11:e0159729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Malarkey DE, Johnson K, Ryan L, Boorman G, Maronpot RR. New insights into functional aspects of liver morphology. Toxicol Pathol 2005;33:27‐34. [DOI] [PubMed] [Google Scholar]
- 19. Bale SS, Geerts S, Jindal R, Yarmush ML. Isolation and co‐culture of rat parenchymal and non‐parenchymal liver cells to evaluate cellular interactions and response. Sci Rep 2016;6:25329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, et al. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003;125:437‐443. [DOI] [PubMed] [Google Scholar]
- 21. Esch MB, Prot JM, Wang YI, Miller P, Llamas‐Vidales JR, Naughton BA, et al. Multi‐cellular 3D human primary liver cell culture elevates metabolic activity under fluidic flow. Lab Chip 2015;15:2269‐2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang YJ, Liu HL, Guo HT, Wen HW, Liu J. Primary hepatocyte culture in collagen gel mixture and collagen sandwich. World J Gastroenterol 2004;10:699‐702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lackner C. Hepatocellular ballooning in nonalcoholic steatohepatitis: the pathologist’s perspective. Expert Rev Gastroenterol Hepatol 2011;5:223‐231. [DOI] [PubMed] [Google Scholar]
- 24. Caldwell S, Ikura Y, Dias D, Isomoto K, Yabu A, Moskaluk C, et al. Hepatocellular ballooning in NASH. J Hepatol 2010;53:719‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al.; Nonalcoholic Steatohepatitis Clinical Research Network . Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 26. Friedman SL. Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 2008;88:125‐172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ibrahim SH, Hirsova P, Gores GJ. Non‐alcoholic steatohepatitis pathogenesis: sublethal hepatocyte injury as a driver of liver inflammation. Gut 2018;67:963‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chung SI, Moon H, Ju H‐L, Cho KJ, Kim DY, Han K‐H, et al. Hepatic expression of Sonic Hedgehog induces liver fibrosis and promotes hepatocarcinogenesis in a transgenic mouse model. J Hepatol 2016;64:618‐627. [DOI] [PubMed] [Google Scholar]
- 29. Urtasun R, Lopategi A, George J, Leung TM, Lu Y, Wang X, et al. Osteopontin, an oxidant stress sensitive cytokine, up‐regulates collagen‐I via integrin αVβ3 engagement and PI3K/pAkt/NFκB signaling. Hepatology 2012;55:594‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Westerouen Van Meeteren MJ, Drenth JPH, Tjwa E. Elafibranor: a potential drug for the treatment of nonalcoholic steatohepatitis (NASH). Expert Opin Investig Drugs 2020;29:117‐123. [DOI] [PubMed] [Google Scholar]
- 31. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, et al.; GOLDEN‐505 Investigator Study Group . Elafibranor, an agonist of the peroxisome proliferator‐activated receptor‐α and ‐δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology 2016;150:1147‐1159.e5. [DOI] [PubMed] [Google Scholar]
- 32. Bhatia SN, Underhill GH, Zaret KS, Fox IJ. Cell and tissue engineering for liver disease. Sci Transl Med 2014;6:245sr242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Feaver RE, Cole BK, Lawson MJ, Hoang SA, Marukian S, Blackman BR, et al. Development of an in vitro human liver system for interrogating nonalcoholic steatohepatitis. JCI Insight 2016;1:e90954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blouin A, Bolender RP, Weibel ER. Distribution of organelles and membranes between hepatocytes and nonhepatocytes in the rat liver parenchyma. A stereological study. J Cell Biol 1977;72:441‐455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou Z, Xu M‐J, Gao B. Hepatocytes: a key cell type for innate immunity. Cell Mol Immunol 2016;13:301‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aycock RS, Seyer JM. Collagens of normal and cirrhotic human liver. Connect Tissue Res 1989;23:19‐31. [DOI] [PubMed] [Google Scholar]
- 37. Gori M, Simonelli MC, Giannitelli SM, Businaro L, Trombetta M, Rainer A. Investigating nonalcoholic fatty liver disease in a liver‐on‐a‐chip microfluidic device. PLoS One 2016;11:e0159729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bader A, Frühauf N, Tiedge M, Drinkgern M, De Bartolo L, Borlak J, et al. Enhanced oxygen delivery reverses anaerobic metabolic states in prolonged sandwich rat hepatocyte culture. Exp Cell Res 1999;246:221‐232. [DOI] [PubMed] [Google Scholar]
- 39. Carpino G, Del Ben M, Pastori D, Carnevale R, Baratta F, Overi D, et al. Increased liver localization of lipopolysaccharides in human and experimental NAFLD. Hepatology 2020;72:470‐485. [DOI] [PubMed] [Google Scholar]
- 40. Gandhi CR. Hepatic stellate cell activation and pro‐fibrogenic signals. J Hepatol 2017;67:1104‐1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang H, Lafdil F, Wang L, Yin S, Feng D, Gao B. Tissue inhibitor of metalloproteinase 1 (TIMP‐1) deficiency exacerbates carbon tetrachloride‐induced liver injury and fibrosis in mice: involvement of hepatocyte STAT3 in TIMP‐1 production. Cell Biosci 2011;1:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hirsova P, Gores GJ. Ballooned hepatocytes, undead cells, sonic hedgehog, and vitamin E: therapeutic implications for nonalcoholic steatohepatitis. Hepatology 2015;61:15‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hirsova P, Gores GJ. Death receptor‐mediated cell death and proinflammatory signaling in nonalcoholic steatohepatitis. Cell Mol Gastroenterol Hepatol 2015;1:17‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Heymann F, Tacke F. Immunology in the liver—from homeostasis to disease. Nat Rev Gastroenterol Hepatol 2016;13:88‐110. [DOI] [PubMed] [Google Scholar]
- 45. Griffith LG, Swartz MA. Capturing complex 3D tissue physiology in vitro. Nat Rev Mol Cell Biol 2006;7:211‐224. [DOI] [PubMed] [Google Scholar]
- 46. Connolly JJ, Ooka K, Lim JK. Future pharmacotherapy for non‐alcoholic steatohepatitis (NASH): review of phase 2 and 3 trials. J Clin Transl Hepatol 2018;6:264‐275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Németh Á, Mózes MM, Calvier L, Hansmann G, Kökény G. The PPARγ agonist pioglitazone prevents TGF‐β induced renal fibrosis by repressing EGR‐1 and STAT3. BMC Nephrol 2019;20:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Narayanan S, Surette FA, Hahn YS. The immune landscape in nonalcoholic steatohepatitis. Immune Netw 2016;16:147‐158. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Fig S3
Supplementary Material