

Abstract
The relevance of hemodynamic derangements on the incidence of recurrent acute kidney injury (AKI) and chronic kidney disease (CKD) in patients with cirrhosis is largely unknown. Consecutive patients with cirrhosis with a complete record of baseline hemodynamics were followed for identifying risk factors for the development of recurrent AKI and CKD by using negative binomial regression and competing risk analysis, respectively. Consecutive patients with cirrhosis (n = 2013, age 50.1 ± 11.8 years, 80% male, Child A:B:C percentage 13.7:52.9:33.4, and mean Child‐Turcotte‐Pugh score 8.6 ± 1.8) were enrolled, 893 (44.3%) of whom received beta‐blockers, with 44.2% responders. Prior AKI was noted in 12.4% at enrollment. At a median follow‐up of 379 (interquartile range: 68‐869) days, AKI developed at a rate of 0.37 episodes per person‐year, and 26% patients developed CKD. A lower mean number of AKI episodes (0.05 ± 0.25 vs. 0.42 ± 0.868; P < 0.001), CKD (subdistribution hazard ratio 0.74 [0.54‐1.02]), and mortality (hazard ratio 0.21 [0.06‐0.73]) were observed in beta‐blocker responders. Albuminuria was an independent risk factor for recurrent AKI, CKD, and mortality (P < 0.05). Lower systemic vascular resistance index predicted hemodynamic response (odds ratio 2.04 [1.29‐3.22]), cumulative AKI episodes (ratio of means 0.10 [0.08‐0.14]), and development of CKD (subdistribution hazard ratio 0.70 [0.58‐0.83]). Higher hepatic venous pressure gradient (≥17 mm Hg) predicted AKI episodes (ratio of means 1.76 [1.32‐2.35]) but not CKD. Conclusion: High portal pressure and severe vasodilatation predispose patients with cirrhosis to repeated AKI episodes and development of CKD. Response to beta‐blockers and therapies targeting the vasodilatory state could prevent frequent AKI and the risk of CKD development. Albuminuria could serve as an early marker of renal dysfunction in patients with cirrhosis.
Abbreviations
- ATN
acute tubular necrosis
- BB
beta‐blocker
- BMI
body mass index
- CRDI
Cardiac Index
- CTP
Child‐Turcotte‐Pugh
- eGFR
estimated glomerular filtration rate
- HR
hazard ratio
- HRS
hepatorenal syndrome
- HVPG
hepatic venous pressure gradient
- INR
international normalized ratio
- MAP
mean arterial pressure
- MELD
Model for End‐Stage Liver Disease
- NB
negative binomial
- PVRI
Pulmonary Vascular Resistance Index
- RM
ratio of means
- SVRI
Systemic Vascular Resistance Index
The prognostic significance of acute kidney injury (AKI) is well‐known in patients with cirrhosis. AKI is seen in almost 20% of hospitalized patients with cirrhosis and associated with significant morbidity and mortality.( 1 ) Patients with cirrhosis are predisposed to develop multiple AKI episodes. It has been shown that AKI is a predisposing factor for subsequent AKI and in almost one‐third of these patients’ progress to chronic kidney disease (CKD).( 2 ) This is governed by the number of AKI episodes alongside the severity of liver disease and the status of kidney reserve as determined by cystatin C.( 2 ) Severity, cause, and duration of AKI also determine the risk of CKD development.( 2 , 3 )
Patients with cirrhosis characteristically have increased intrahepatic resistance and high portal pressures consequent to the development of fibrosis and regenerative nodules. Moreover, there is systemic and splanchnic vasodilatation secondary to an increase in the production of nitric oxide and other vasodilators (e.g., calcitonin gene‐related peptide, copeptin, adrenomedullin,) in the vascular endothelium and a concomitant decreased sensitivity to the vasoconstrictors (e.g., angiotensin II, noradrenaline, endothelin‐1, vasopressin).( 1 , 2 ) This systemic vasodilatory state and hemodynamic alterations are known risk factors for AKI; however, currently, this has not been studied in the context of CKD development.
Nonselective beta‐blockers (BBs) have been the backbone for the management of portal hypertension in patients with cirrhosis. However, recent data have suggested harm when these drugs are used in patients with advanced decompensated cirrhosis.( 4 , 5 ) A concept of “window hypothesis” for BBs has been suggested, in which these drugs are not effective when used in either very early or in patients with very advanced cirrhosis.( 6 , 7 ) On the contrary, studies have shown that the use of BBs is associated with improved survival in patients with advanced cirrhosis with refractory ascites awaiting liver transplantation. In the context of AKI, studies have shown that BBs are associated with increased incidence of AKI in patients with ascites and patients with spontaneous bacterial peritonitis.( 8 , 9 ) Therefore, as of now, the controversy still exists regarding the use of BBs in patients with cirrhosis.( 4 , 5 ) Furthermore, no studies have explored the role of BBs in modulating the hemodynamic response concerning the risk of CKD development.
Therefore, we undertook the current study to evaluate in a large cohort of patients with cirrhosis, the correlation of altered portal, systemic and pulmonary hemodynamics, and their influence on recurrent AKI and CKD development.
Patients and Methods
The study is a retrospective evaluation of a prospective cohort of patients which was conducted from August 2012 to July 2016 at the Institute of Liver and Biliary Sciences. Consecutive patients with cirrhosis visiting the outpatient department or admitted as in‐patients in the Hepatology department were screened for enrollment in the current study. Patients with cirrhosis who underwent hemodynamic assessment (i.e., hepatic venous pressure gradient [HVPG] as a part of clinical evaluation with no AKI at enrollment) were followed for the development of new AKI episodes or CKD for a median period of about 1 year (379 days [interquartile range (IQR): 68‐869]). Patients with at least three follow‐up visits 3 months apart were included in the current study. Patients aged less than 18 or more than 70 years, with pre‐existing CKD, with AKI at enrollment, following liver or kidney transplant, transjugular intrahepatic portosystemic shunt, obstructive uropathy, with hepatocellular carcinoma, advanced co‐existing cardiopulmonary diseases, portal vein or hepatic vein thrombosis, pregnancy, without availability of baseline serum creatinine, and patients with missing information on any of the considered variables in this study were excluded. CKD was defined as persistent reduction in estimated glomerular filtration rate (eGFR) < 60 mL/min/1.73 m2 by modification of diet in end‐stage renal disease (MDRD6) equation for more than 3 months (see Supporting Information).( 2 , 3 )
Study Population
The patients were enrolled from the time of measurement of HVPG, at which time the baseline serum creatinine was recorded. These patients were evaluated for the development of new AKI, prior AKI in the last 3 months, the number of episodes of AKI, and development of CKD at last follow‐up visit. HVPG was performed in these patients as part of different clinical protocols. For patients with multiple AKI episodes, the peak episode and the worst cause were considered for analysis. The number of hospital admissions after enrollment were recorded for each patient (see Supporting Information).
Hemodynamic Studies
The hemodynamic studies were done after a 6‐hour or overnight fast. Under aseptic conditions, under local anesthesia, a venous introducer was placed in the right femoral or the internal jugular vein using the Seldinger technique.( 10 ) For measuring HVPG, a 7Fr balloon‐tipped Swan‐Ganz catheter was introduced into the main right hepatic vein under fluoroscopic guidance. Free hepatic venous pressure (FHVP) and wedged hepatic venous pressure (WHVP) were measured using pressure transducers and hemodynamic monitor. Measurements were performed in triplicate, and a mean value was taken. HVPG was obtained as the difference between WHVP and FHVP. Subsequently, the Swan–Ganz catheter was advanced into the pulmonary artery under fluoroscopic control for the measurement of cardiopulmonary pressures and cardiac index (CRDI) by the Fick’s principle as oxygen consumption (mL min‐1) ÷ arteriovenous oxygen difference (mL l‐1). Aortic pressure was measured simultaneously, and the mean arterial pressure (MAP) was calculated. Samples were taken from the right atrium, pulmonary artery, and aorta. Systemic Vascular Resistance Index (SVRI) (dyns/cm5/m2) was calculated as (MAP [mm Hg] right atrial pressure [mm Hg]) 80 ÷ CRDI (L/min/m2). Pulmonary Vascular Resistance Index (PVRI) (dyns /cm5/m2) was calculated as (mean pulmonary artery pressure pulmonary capillary wedge pressure) 80 ÷ CRDI. Heart rate was also obtained from continuous electrocardiogram monitoring. The hemodynamic studies were done after an overnight fast. For all patients, the HVPG and values of other hemodynamic parameters were recorded at enrollment.
Urine Analysis for Albuminuria
The urine data were analyzed using the dipstick method for presence of albuminuria, which was graded semi‐quantitatively with values reported as absent, trace, 1+, 2+, 3+, or 4+ (corresponding to albumin levels of undetectable or <10 mg/dL, 10‐29 mg/dL, 30‐99 mg/dL, 100‐299 mg/dL, 300‐999 mg/dL, and 1,000 mg/dL or greater, respectively). At enrollment, albuminuria was assessed for all patients as a risk factor for recurrent AKI, development of CKD, and mortality.
Beta‐Blockers
In the absence of contraindications, 893 patients were initiated on oral propranolol or carvedilol. Carvedilol was administered orally at a dose of 3.125 mg twice daily. The treatment was increased every week, to 6.25 mg twice daily, and, if required, further expanded up to a maximum dose of 12.5 mg twice daily with a caution that the systolic blood pressure did not fall below 100 mm Hg and heart rate <55 bpm. Similarly, propranolol was initiated at 20 mg twice daily. This dose was increased until the heart rate had fallen by 25% or below 55/minute, or systolic blood pressure was below 90 mm Hg or to the maximal tolerable dose. Patients were asked to coordinate with the primary care physician to monitor the heart rate and blood pressure and report any adverse effects. The second hemodynamic evaluation was done in 495 patients at a median of 12 weeks (range 8‐16). Patients were considered as responders if, at the second hemodynamic evaluation, the recorded HVPG was either less than 12 mm Hg or had decreased by at least 20% from the baseline value, and nonresponders if these criteria were not met. Beta‐blockers were discontinued during an episode of AKI, spontaneous bacterial peritonitis or any other decompensating event, and reinitiated within a week after episode’s resolution.
Statistical Methods
For continuous variables, descriptive statistics were presented in the form of mean ± SD. For categorical variables such as sex, descriptive statistics were presented in the form of frequencies and percentages. Bivariate association between categorical variables was assessed using either chi‐square tests or Fisher’s exact test, whichever is applicable. Independent sample t tests were performed to test the difference in mean values between the two groups. Analysis of variance was used to test the difference in mean values of more than two groups. Time to CKD development or death was recorded for all patients, alongside the number of AKI episodes during the follow‐up period. Univariate and multivariate competing‐risk analysis with death as the competing event was performed for identifying risk factors for CKD (as patients who underwent liver transplant or transjugular intrahepatic portosystemic shunt were excluded). Univariate and multivariate Cox regression analyses was used to identify predictors of mortality. Kaplan‐Meier survival estimation procedure was followed to provide nonparametric survival estimates by various durations since the baseline time point. Log‐rank tests were used to compare survival curves across different categories of a categorical variable.
Negative binomial (NB) regression analysis was used to identify factors that influenced the number of AKI episodes. Before using NB regression, we tested whether the conservative Poisson regression model could be used for the objective, as mentioned previously. NB regression avoids the strict and restrictive assumption that the variance is equal to the mean made by the Poisson model. We found the NB regression model was more suitable than the Poisson regression model, as judged by the chi‐square test for dispersion. NB regression model is similar to the linear regression model with two main exceptions. First, while the dependent variable in the linear regression model is continuous, the dependent (Y) variable in NB regression is a count variable. Second, while linear models assume a normal distribution for the outcome variable, the NB regression model assumes negative binomial distribution for the outcome variable. Apart from this, the NB model also has characteristics of the logistic regression model. Like in the logistic regression model, exponents of parameters have simple interpretations in NB regression. Unlike in logistic regression analysis, parameters are interpreted in terms of the ratio of means (RM) and 95% confidence intervals. NB regression model was fitted by using the SAS University Edition, using the PROC GENMOD procedure. Log of follow‐up time was used as the offset variable in the PROC GENMOD procedure.
For this study, the optimal cutoff points for HVPG, SVRI, PVRI, and CRDI for determining time to AKI were obtained based on the Contal and O’Quigley method.( 11 ) This technique uses a log‐rank test statistic in its procedure to estimate the optimal cutoff point. The optimal cutoff point based on this method is 17 mm Hg for HVPG, 2008 dyns/cm5/m2 for SVRI, 68 dyns /cm5/m2 for PVRI, and 4 L/min/m2 for CRDI. Based on these cutoff points, patients with HVPG ≤ 17 mm Hg were considered as having low HVPG, and patients with HVPG > 17 mm Hg were considered as having high HVPG. Similarly, patients with SVRI ≤ 2,008 dyns/cm5/m2, PVRI ≤ 68 dyns/cm5/m2, and CRDI ≤ 4 L/min/m2 were considered as having low SVRI, low PVRI, and low CRDI, respectively. Their counterparts with SVRI > 2,008 dyns/cm5/m2, PVRI > 68 dyns/cm5/m2, and CRDI > 4 L/min/m2 were considered as having high SVRI, high PVRI, and high CRDI, respectively. In this study, these cutoff point–based groups were used in all of the analyses except for descriptive analysis.
Variables found significant at P < 0.05 in univariate analysis were considered for multivariate analysis. Highly correlated variables with the main variables of interest were not considered for multivariate analysis. All tests were two‐tailed, and a P value of less than 0.05 was deemed to be significant. Data were analyzed using SPSS software version 22 and SAS University Edition.
Results
A total of 2,013 patients were enrolled in the study (Supporting Fig. S1). These patients’ mean age was 50.1 ± 11.8 years, 1,251 (79.5%) were males, with alcohol as the predominant etiology of liver disease in 772 (38.4%). One‐hundred and seventy‐one (8.5%) of the study participants had diabetes, and 24 (1.2%) had hypertension. The mean Model for End‐Stage Liver Disease (MELD) score of these patients was 14.2 ± 6.7, and Child‐Turcott Pugh (CTP) score was 8.6 ± 1.8. The mean HVPG at baseline was 16.2 ± 5.8, and the eGFR was 93.1 ± 56.0 mL/min. The mean SVRI, PVRI, and CDRI were 2,292.5 ± 810.6 dyns/cm5/m2, 127.4 ± 88.1 dyns/cm5/m2, and 3.71 ± 1.35 L/min/m2, respectively. Prior AKI was noted in 249 (12.4%) of patients at enrollment (Table 1).
FIG. 1.
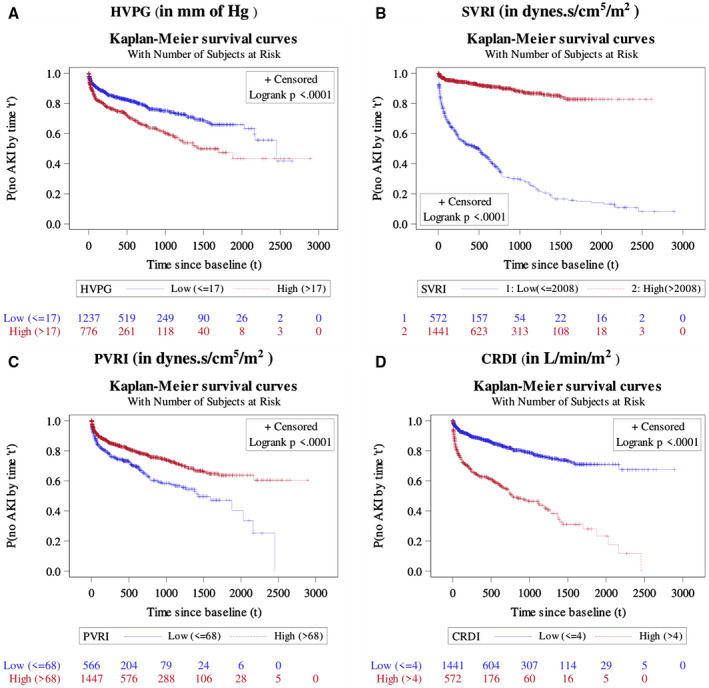
Probability of no AKI by various durations since enrollment, as estimated by Kaplan‐Meier survival method, among different categories of HVPG (A), SVRI (B), PVRI (C), and CRDI (D). Note: Log‐rank test was used to test whether the pattern of incidence of mortality vary across deifferent categories of a considered variable. The result of it was shown in the form of P value.
TABLE 1.
Baseline Characteristics of Study Cohort Based on Their Status of AKI on Follow‐up
| Characteristic | Total (n = 2013) | No AKI During the Follow‐up Period (n = 1,573) | Had AKI During the Follow‐up Period (n = 440) | P Value |
|---|---|---|---|---|
| Age (years) | 50 ± 11 | 49 ± 11 | 50 ± 11 | 0.36 |
| Gender (males) | 1,619 (80.4) | 1,251 (79.5) | 368 (83.6) | 0.05 |
| Etiology | <0.0001 | |||
| Alcohol | 772 (38.4) | 578 (36.8) | 194 (44.1) | |
| Viral | 604 (30.0) | 453 (28.8) | 151 (34.3) | |
| Others | 637 (31.6) | 542 (34.5) | 95 (21.6) | |
| Diabetes mellitus | 171 (8.5) | 108 (6.9) | 63 (14.3) | <0.0001 |
| Hypertension | 24 (1.2) | 12 (0.8) | 12 (2.7) | <0.0018 |
| BMI category (kg/m2) | 0.86 | |||
| <18.5 | 98 (4.9) | 74 (4.7) | 24 (5.5) | |
| 18.5‐25 | 844 (41.9) | 662 (42.1) | 182 (41.4) | |
| 25‐30 | 698 (34.7) | 549 (34.9) | 149 (33.9) | |
| ≥30 | 373 (18.5) | 288 (18.3) | 85 (19.3) | |
| MELD | 14 ± 6 | 13 ± 6 | 16 ± 6 | <0.0001 |
| CTP | 8 ± 2 | 8 ± 2 | 9 ± 2 | <0.0001 |
| Ascites | <0.0001 | |||
| No ascites | 306 (15.2) | 251 (15.9) | 55 (12.5) | |
| Grade 1 | 980 (48.7) | 805 (51.2) | 175 (39.8) | |
| Grade 2 and 3 | 727 (36.1) | 517 (32.9) | 210 (47.7) | |
| eGFR (mL/min) | 93±55 | 94±57 | 86±47 | 0.003 |
| MAP (mm Hg) | 75 ± 10 | 75 ± 10 | 76 ± 11 | 0.16 |
| Esophageal varices | 0.24 | |||
| No varices | 16 (0.8) | 12 (0.8) | 4 (0.9) | |
| Esophageal varices | 1,962 (97.5) | 1,537 (97.7) | 425 (96.6) | |
| Isolated gastric varix | 8 (0.4) | 7 (0.5) | 1 (0.2) | |
| Esophageal and gastric varices | 27 (1.3) | 17 (1.1) | 10 (2.3) | |
| Grade of esophageal varices (n = 1,992) | 0.59 | |||
| Small | 828 (41.1) | 638 (40.6) | 190 (43.2) | |
| Large | 1,164 (57.8) | 918 (58.4) | 246 (55.9) | |
| Prior variceal bleed | 963 (47.8) | 701 (44.6) | 262 (59.6) | <0.0001 |
| Beta‐blockers | 893 (44.4) | 679 (43.2) | 214 (48.6) | 0.04 |
| HVPG (mm Hg) | 16±5 | 15.±5 | 17±6 | <0.0001 |
| SVRI (dyns/cm5/m2) | 2,292 ± 810 | 2,498 ± 709 | 1,555 ± 716 | <0.0001 |
| PVRI (dyns/cm5/m2) | 127 ± 88 | 132 ± 90 | 109 ± 78 | <0.0001 |
| CRDI (L/min/m2) | 3.7 ± 1.4 | 3.6 ± 1.3 | 4.1 ± 1.3 | <0.0001 |
| Leucocyte counts (×103/m3) | 7.5 ± 5.1 | 7.2 ± 4.8 | 8.5 ± 5.9 | <0.0001 |
| Serum bilirubin (mg/dL) | 7.2 ± 9.8 | 6.3 ± 9.1 | 10.1 ± 11.7 | <0.0001 |
| Serum albumin (g/dL) | 2.9 ± 0.7 | 2.9 ± 0.7 | 2.6 ± 0.5 | <0.0001 |
| INR | 1.6 ± 0.5 | 1.54 ± 0.40 | 1.77 ± 0.56 | <0.0001 |
| Serum creatinine at enrollment (mg/dL) | 0.57 ± 0.28 | 0.57 ± 0.25 | 0.58 ± 0.38 | 0.74 |
| Serum sodium (mEq/L) | 134 ± 5.8 | 135 ± 5.5 | 132 ± 6.5 | <0.0001 |
| Prior AKI | 249 (12.4) | 135 (8.6) | 114 (25.9) | <0.001 |
| Urine for albumin | ||||
| Absent | 1,267 (62.9) | 1,064 (67.6) | 203 (46.1) | <0.001 |
| Trace or 1+ | 662 (32.9) | 471 (29.9) | 191 (43.4) | |
| 2+ | 61 (3) | 33 (2.1) | 28 (6.4) | |
| 3+ or 4+ | 23 (1.1) | 5 (0.3) | 18 (4.1) |
Abbreviations: BMI, body mass index; INR, international normalized ratio.
Incidence of AKI
A total of 440 (21.9%) patients had at least one episode of AKI on follow‐up. Furthermore, the overall incidence rate (or density) of AKI episodes was 0.37 AKI episodes per person‐year follow‐up, with an incidence rate of 0.05 episodes per person‐year follow‐up among patients with compensated cirrhosis CTP class A at enrollment, 0.30 episodes among patients with cirrhosis CTP class B, and 1.04 episodes per person‐year follow‐up among patients with CTP class C. There were a total of 787 episodes of AKI in 440 patients, yielding a mean of 1.79 episodes of AKI per patient (range one to eight episodes) at a median of 322 (41‐728) days. Of these patients, 214 (48.6%) had one, 132 (30%) had two, 78 (17.7%) had three, 9 (2.05%) had four, and 7 (1.6%) had five or more episodes. The peak AKI stage reached was stage 1 in 121 (28%), stage 2 in 77 (17%), and stage 3 in 242 (55%). The cause was prerenal volume‐responsive AKI in 92 (21%), hepatorenal syndrome (HRS) in 259 (59%) and acute tubular necrosis (ATN) and/or miscellaneous forms of AKI (ATN) in 89 (20%) patients. On follow‐up, median 379 (IQR 68‐869) days, 518 (26%) patients developed CKD. A significantly higher proportion of patients with AKI died on follow‐up, as compared with those who did not develop AKI (190 [43.2%] vs. 18 [1.1%]; P < 0.001). Table 1 lists the baseline characteristics of patients who developed AKI and who did not during the follow‐up. Patients who developed AKI were significantly more vasodilated, had higher mean CRDI (4.1 ± 1.3 vs. 3.6 ± 1.3 L/min/m2; P < 0.001), lower SVRI (1,555.4 ± 716.1 vs. 2,498.6 ± 709.4 dyns/cm5/m2; P < 0.001), lower PVRI (109.5 ± 78.0 vs. 132.5 ± 90.1 dyns /cm5/m2; P < 0.001) and higher HVPG (17.7 ± 6.4 vs. 15.7 ± 5.5 mm Hg; P < 0.001) (Fig. 1). In addition, they had lower eGFR (86.9 ± 47.9 vs. 94.9 ± 57.9 mL/min/m2; P < 0.001), higher MELD (16.2 ± 6.9 vs. 13.6 ± 6.5; P < 0.001) and CTP scores (9.4 ± 1.7 vs. 8.4 ± 1.8; P < 0.001) (Supporting Figs. 2 and 3).
FIG. 2.
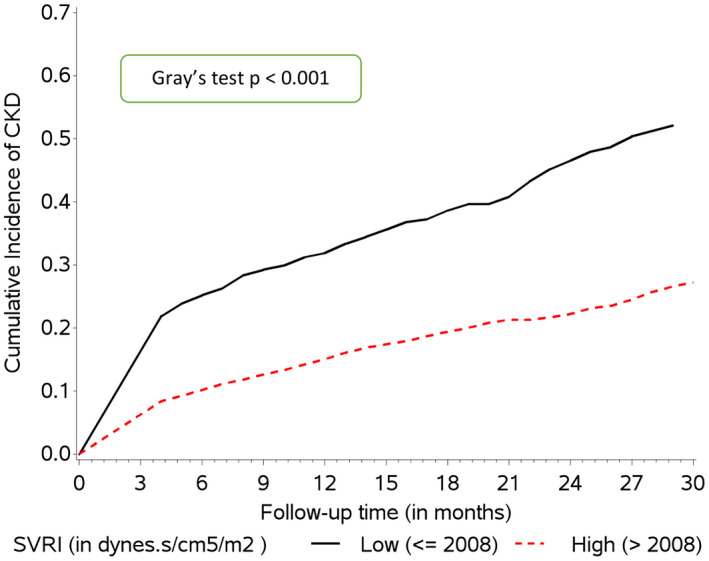
Probability of development of CKD by various durations since enrollment, as estimated by Fine and Gray competing risk survival analysis, among different categories of SVRI.
FIG. 3.
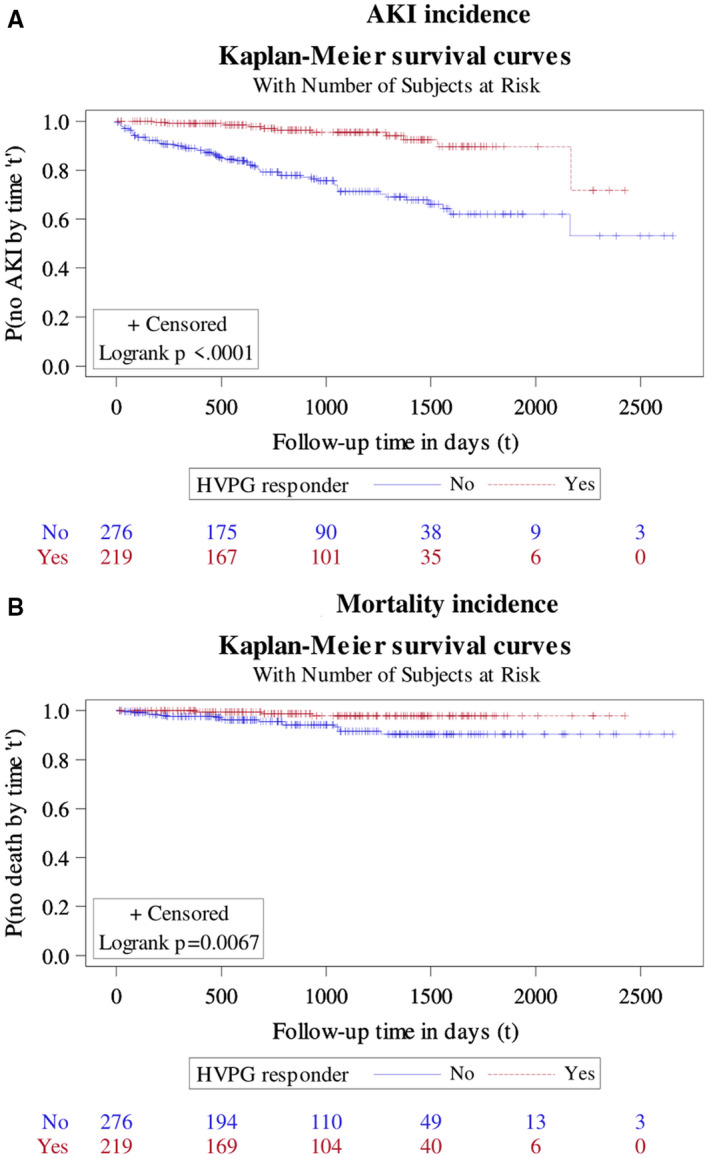
Probability of no AKI and no mortality by various durations since enrollment, as estimated by Kaplan‐Meier survival method, stratified based on HVPG‐related responders versus nonresponders. (A) AKI incidence. (B) Mortality incidence.
Albuminuria as a Risk Factor for AKI
At enrollment, 1,267 (63%) had no albumin, 662 (33%) had trace or 1+ albumin, 61 (3.0%) had 2+ albumin, and 23 (1%) had 3+ or 4+ albumin in the microscopic urine analysis. A significant difference was noted in the albuminuria in patients who developed AKI on follow‐up versus those who did not (Table 1). A significant difference was also noted in the presence of albuminuria in patients with prior AKI. These patients had a higher proportion of patients with albuminuria (P < 0.001), trace or 1+ (103 [41%] vs. 559 [32%]), 2+ albumin (16 [6%] vs. 45 [3%]), and 3+ or 4+ albumin (11 [4%] vs. 12 [0.7%]), respectively. At the same time, a significantly lower proportion of patients with prior AKI had absent albumin in the urine as compared to those without prior AKI (119 [49%] vs. 1,148 [65%]), respectively.
Risk Factors for the Number of AKI Episodes—Negative Binomial Regression
Univariate and multivariate negative binomial regression analyses to determine factors affecting the number of AKI episodes are given in Table 2. On multivariate analysis, higher HVPG (>17 mm Hg) (RM 1.76, 95% CI 1.32‐2.35) and lower SVRI (<2,008 dyns/cm5/m2) (RM 0.10, 95% CI 0.08‐0.14) were significant predictors of number of AKI episodes, alongside viral etiology (RM 1.56, 95% CI 1.08‐2.26), presence of diabetes (RM 1.77, 95% CI 1.13‐2.76), higher MELD‐Na at enrollment (RM 1.08, 95% CI 1.01‐1.10), and higher leucocyte counts (RM 1.35, 1.03‐1.76). The use of BBs was associated with a significantly lower mean number of AKI episodes on follow‐up (RM 0.56, 95% CI 0.42‐0.74). It was interesting to find prior AKI (RM 2.04, 1.41‐2.95) and increasing concentration of albumin in the urine as an independent risk factor of repeated AKI episodes (Table 2).
TABLE 2.
Univariate and Multivariate Analysis for Risk Factors of Number of AKI Episodes—Negative Binomial Regression Analysis
| Factor | Crude RM | 95% CI for RM | Adjusted RM | 95% CI for RM |
|---|---|---|---|---|
| Age (years) | 0.99 | 0.98, 1.01 | ||
| Gender (female) | 0.61* | 0.38, 0.96 | ||
| Etiology (ref = others) | 1 | 1,1 | 1 | 1, 1 |
| Alcohol | 3.01 † | 1.97, 4.62 | 1.05 | 0.73,1.52 |
| Viral | 2.69 † | 1.71, 4.25 | 1.56* | 1.08, 2.26 |
| Diabetes | 1.74 | 0.98, 3.07 | 1.77* | 1.13, 2.76 |
| Hypertension | 1.26 | 0.25, 6.33 | ||
| BMI (kg/m2) (ref = 18.5‐25) | ||||
| <18.5 | 1.06 | 0.45, 2.49 | ||
| 25‐30 | 0.74 | 0.50, 1.10 | ||
| ≥30 | 0.71 | 0.43, 1.15 | ||
| MELD | 1.16 † | 1.13, 1.19 | ||
| CTP | 1.96 † | 1.79, 2.14 | ||
| Ascites (ref = no ascites) Grade 1 | 1.56 | 0.91, 2.66 | ||
| Grade 2 and 3 | 4.30 † | 2.50, 7.41 | ||
| eGFR ‡ (mL/min) | 0.70* | 0.49, 0.99 | ||
| MAP (mm Hg) | 0.99 | 0.97, 1.01 | ||
| Beta‐blockers | 0.59 † | 0.41, 0.83 | 0.56 † | 0.42, 0.74 |
| HVPG (>17 mm Hg) | 2.48 † | 1.75, 3.52 | 1.76 † | 1.32, 2.35 |
| SVRI (>2,008 dyns/cm5/m2) | 0.06 † | 0.04, 0.08 | 0.10 † | 0.08, 0.14 |
| PVRI (>68 dyns/cm5/m2) | 0.58 † | 0.40, 0.84 | ||
| CRDI (>4 L/min/m2) | 6.07 † | 4.31, 8.56 | ||
| Leucocyte counts ‡ (×103/m3) | 2.94 † | 2.25, 3.86 | 1.35* | 1.03, 1.76 |
| Serum bilirubin ‡ (mg/dL) | 2.57 † | 2.27, 2.90 | ||
| Serum albumin (g/dL) | 0.20 † | 0.15, 0.26 | ||
| INR (seconds) | 6.88 † | 4.58, 10.34 | ||
| Serum creatinine at enrollment (mg/dL) | 0.84 | 0.51, 1.38 | ||
| Serum sodium (mEq/L) | 0.90 † | 0.87, 0.92 | ||
| MELD‐Na | 1.15 † | 1.13, 1.17 | 1.08 † | 1.05, 1.10 |
| Prior AKI | 4.00 † | 2.49, 6.42 | 2.04 † | 1.41, 2.95 |
| Urine for albumin | ||||
| Absent | 1 | 1, 1 | 1 | 1, 1 |
| Trace or 1+ | 2.93 † | 2.04, 4.21 | 2.13* | 1.08, 4.21 |
| 2+ | 4.30 † | 1.80, 10.25 | 5.42 † | 2.07, 14.22 |
| 3+ or 4+ | 5.07* | 1.40, 18.28 | 1.38* | 1.02, 1.86 |
MELD‐Na was calculated using bilirubin, INR, creatinine and sodium. Therefore, these components were not considered for the multivariate regression model, as MELD‐Na was included in the model. Also, because CTP is highly associated with MELD‐Na, CTP was not considered for the multivariate analysis, to avoid multicollinearity. PVRI, CRDI, and MAP were not included in the multivariate analysis, as they are highly associated with SVRI. Similarly, eGFR contains albumin, age, gender and creatinine, so eGFR was not included in the model. HVPG, SVRI, PVRI, and CVRI were considered as category variables in the regression analysis. Of them, factors significant in the multivariate analysis were only reported. Urine analysis for albuminuria was graded semi‐quantitatively, with values reported as absent or trace (±), 1+, 2+, and 3+, or 4+ (corresponding to albumin levels of undetectable or <10 mg/dL, 10‐29 mg/dL, 30‐99 mg/dL, 100‐299 mg/dL, 300‐999 mg/dL, and 1,000 mg/dL. Variables considered in multivariate analysis were etiology, diabetes mellitus, ascites use of BBs, HVPG, SVRI, log‐transformed leucocyte count, MELD‐Na, prior AKI, and urine albumin.
Significant at 5% level of significance.
Significant at 1% level of significance.
Corresponding log‐transformed variables were used in the regression analysis.
Risk Factors for CKD Development—Competing Risk Survival Analysis
Patients with CKD had higher frequency of AKI episodes subdistribution hazard ratio (SHR 1.37, 95% CI 1.27‐1.48), higher peak severity of AKI, stage 0 (as reference; SHR 1), stage 1 (SHR 2.26, 95% CI 1.73‐2.94), stage 2 (SHR 1.8, 95% CI 1.26‐2.56), and stage 3 (SHR 2.12, 1.71‐2.63). These patients also had more frequent hospitalizations (SHR 1.05, 1.03‐1.07). The mean number of AKI episodes after development of CKD was higher as compared with before development of CKD. Patients with prior AKI at enrollment had higher risk of CKD development (SHR 2.31, 95% CI 1.90‐2.81). Albuminuria was also a significant risk factor for CKD development (Table 3). On multivariate analysis, adjusting for collinearity, prior AKI (SHR 1.72, 95% CI 1.40‐2.11), presence of diabetes (SHR 1.79, 95% CI 1.44‐2.21), hypertension (SHR 1.74, 1.04‐2.91), higher MELD‐Na (SHR 1.05, 95% CI 1.03‐1.06), higher age (SHR 1.03, 95% CI 1.02‐1.04), lower SVRI (2,008 dyns/cm5/m2) (SHR 0.70, 95% CI 0.58‐0.83), lower serum albumin (SHR 0.84, 0.72‐0.97), presence of albuminuria (>100 mg/dL) (2+; SHR 1.64, 1.13‐2.39), and 3+ or 4+ albuminuria (SHR 2.74, 95% CI 1.83‐4.11) as compared with absent albumin (as reference category) were independent predictors of CKD. Importantly, HVPG and BB use were not identified as risk factors for CKD development (Fig. 2).
TABLE 3.
Univariate and Multivariate Competing Risk Survival Analysis Regression Results for Identifying Factors Associated With CKD Development Since Enrollment
| Factor | Crude SHR | 95% CI for SHR | Adjusted SHR | 95% CI for SHR |
|---|---|---|---|---|
| Age (years) | 1.03* | 1.02, 1.04 | 1.03* | 1.03, 1.04 |
| Gender (female) | 0.82 | 0.65, 1.03 | ||
| Etiology (ref = others) | ||||
| Alcohol | 1.53* | 1.24, 1.88 | ||
| Viral | 1.42* | 1.14, 1.77 | ||
| Diabetes | 3.64* | 3.07, 4.32 | 1.79* | 1.44, 2.21 |
| Hypertension | 2.91* | 1.73, 4.91 | 1.74 † | 1.04, 2.91 |
| BMI (kg/m2 ) (Ref = 18.5‐25) | ||||
| <18.5 | 0.78 | 0.49, 1.26 | ||
| 25‐30 | 1.16 | 0.96, 1.40 | ||
| ≥30 | 1.24 | 0.99, 1.54 | ||
| MELD | 1.05* | 1.04, 1.06 | ||
| CTP | 1.08* | 1.03, 1.13 | ||
| Ascites (ref = no ascites) | ||||
| Grade 1 | 1.05 | 0.83, 1.34 | ||
| Grade 2 and 3 | 1.38 † | 1.08, 1.77 | ||
| eGFR ‡ (mL/min) | 0.34* | 0.30, 0.38 | ||
| MAP (mm Hg) | 1.00 | 0.99, 1.01 | ||
| Beta‐blockers | 1.05 | 0.89, 1.24 | ||
| HVPG (>17 mm Hg) | 1.06 | 0.90, 1.26 | ||
| SVRI (>2,008 dyns/cm5/m2) | 0.53* | 0.45, 0.63 | 0.70* | 0.58, 0.83 |
| PVRI (>68 dyns/cm5/m2) | 0.95 | 0.79, 1.14 | ||
| CRDI (>4 L/min/m2) | 1.25 † | 1.04, 1.49 | ||
| Leucocyte counts ‡ (×103/m3) | 1.16 † | 1.01, 1.34 | ||
| Serum bilirubin ‡ (mg/dL) | 0.10 | 0.93, 1.07 | ||
| Serum albumin (g/dL) | 0.66* | 0.58, 0.74 | 0.84 † | 0.72, 0.97 |
| INR (seconds) | 1.18 | 0.10, 1.39 | ||
| Serum creatinine at enrollment (mg/dL) | 3.55* | 2.68, 4.69 | ||
| Serum sodium (mEq/L) | 0.97* | 0.96, 0.99 | ||
| Episodes of AKI (n) | 1.37* | 1.27, 1.48 | ||
| Prior AKI | 2.31* | 1.90, 2.81 | 1.72* | 1.40, 2.11 |
| MELD‐Na | 1.07* | 1.04, 1.06 | 1.05* | 1.03, 1.06 |
| Urine for albumin | ||||
| Absent or trace | 1.00 | 1.00, 1.00 | 1.00 | 1.00, 1.00 |
| 1+ | 1.84 | 1.51‐2.25 | 0.88 | 0.73, 1.06 |
| 2+ | 3.11* | 2.09‐4.62 | 1.64* | 1.13, 2.39 |
| 3+ or 4+ | 8.12* | 5.00‐13.18 | 2.74* | 1.83, 4.11 |
HVPG, SVRI, PVRI, and CVRI were considered as category variables in the regression analysis. Of them, only those factors significant in the multivariate analysis were reported. Urine analysis for albuminuria was graded semi‐quantitatively, with values reported as absent or trace (±), 1+, 2+, and 3+ or 4+ (corresponding to albumin levels of undetectable or <10 mg/dL, 10‐29 mg/dL, 30‐99 mg/dL, 100‐299 mg/dL, 300‐999 mg/dL, and 1,000 mg/dL. MELD‐Na was calculated using bilirubin, INR, creatinine, and sodium. Therefore, these components were not considered for the multivariate regression model, as MELD‐Na was included in the model. Also, because CTP is highly associated with MELD‐Na, CTP was not considered for the multivariate analysis, to avoid multicollinearity. PVRI, CRDI, and MAP were not included in the multivariate analysis, as they are highly associated with SVRI. Similarly, because eGFR contains albumin, age, gender and creatinine, it was not included in the model. Variables considered in multivariate analysis were age, etiology, diabetes mellitus, hypertension, ascites category, SVRI, albumin, MELD‐Na, prior AKI, and urine albumin.
Significant at 1% level of significance.
Significant at 5% level of significance.
Corresponding log‐transformed variables were used in the regression analysis.
Impact of BBs on HVPG Response and AKI
Of the total cohort of 2,013 patients, 893 patients received BBs. Of these, 697 (78%) had received carvedilol, and the remaining 196 (22%) received propranolol. The mean dose of carvedilol was 12.5 ± 2 mg/day, and for propranolol it was 66 ± 20.1 mg/day. A comparison of baseline characteristics of patients who received versus who did not receive BBs is found in Supporting Table S1. Patients on BBs had higher HVPG and prior variceal bleed. A second HVPG was performed in 495 (55.4%) patients. For this reason, the number of responders (defined as a reduction in HVPG of >20% from baseline or ≤12 mm Hg in HVPG) was 219 (44.2%), and the rest were nonresponders. A comparison of baseline characteristics of responders as compared with nonresponders is found in Supporting Table S2. A lower incidence of AKI (HR 0.18, 95% CI 0.10‐0.34) (Fig. 3) as well as a lower mean number of AKI episodes (0.05 ± 0.25 vs. 0.42 ± 0.868; P < 0.001) were observed in responders as compared with nonresponders. Using negative binomial regression analysis, a lower mean number of AKI episodes by 92% was observed for HVPG responders as compared with nonresponders (95% CI for the RM, 0.04‐0.17). Furthermore, in patients who developed AKI (n = 76), responders had less severe AKI (stage 1, 9 [80.0%] vs. 24 [38.1%]; stage 2, 1 [10%] vs. 28 [44.4%]; and stage 3, 1 [10%] vs. 13 [17.5%]; P = 0.046) and lower development of HRS‐AKI (2 [14%] vs. 55 [60%]) and ATN (1 [7%] vs. 5 [6%]; P = 0.003) as compared with nonresponders, respectively. Urine microscopy showed significant differences between patients with HRS and ATN. Patients with ATN had granular casts and higher urine sodium concentration. These patients also had significantly higher proportion of patients with tubule epithelial cells and presence of albuminuria in the urine analysis as compared to patients with HRS (Supporting Table S3) Responders also showed significantly lower mortality (HR 0.21, 95% CI 0.06‐0.73). The development of CKD, even though not statistically significant, was also lower among responders (RM 0.74, 95% CI 0.54‐1.01; P = 0.06). Higher MELD‐Na signifying less severe liver disease, BBs, and higher systemic vascular resistance index were predictors of the hemodynamic response on univariate and multivariate logistic regression analysis (Table 4).
TABLE 4.
Univariate and Multivariate Binary Logistic Regression Results for Identifying Factors Associated With Hemodynamic Response
| Factor | Crude Odds Ratio | 95% CI for Odds Ratio | Adjusted Odds Ratio | 95% CI for HR |
|---|---|---|---|---|
| Age (years) | 1.01 | 0.99, 1.03 | ||
| Gender (female) | 1.15 | 0.72, 1.83 | ||
| Etiology (ref = others) | ||||
| Alcohol | 0.72 | 0.47, 1.10 | ||
| Viral | 0.76 | 0.49, 1.17 | ||
| Diabetes | 0.88 | 0.41, 1.89 | ||
| Hypertension | 0.42 | 0.04, 4.04 | ||
| BMI (kg/m2) (ref = 18.5‐25) | ||||
| <18.5 | 0.49 | 0.15, 1.59 | ||
| 25‐30 | 1.23 | 0.82, 1.85 | ||
| ≥30 | 1.03 | 0.63, 1.68 | ||
| MELD | 0.98 | 0.95, 1.01 | ||
| CTP | 0.84* | 0.75, 0.94 | ||
| Ascites (ref = no ascites) | ||||
| Grade 1 | 0.79 | 0.47, 1.32 | ||
| Grade 2 and 3 | 0.51 † | 0.28, 0.91 | ||
| eGFR (mL/min) | 1.34 | 0.94, 1.90 | ||
| MAP | 1.02 | 1.00, 1.03 | ||
| Beta‐blocker | 3.10* | 1.39, 6.92 | 3.35* | 1.49, 7.54 |
| HVPG (>17 mm Hg) | 1.21 | 0.85, 1.74 | ||
| SVRI (>2,008 dyns/cm5/m2) | 2.11* | 1.35, 3.29 | 2.04* | 1.29, 3.22 |
| PVRI (>68 dyns/cm5/m2) | 0.99 | 0.66, 1.47 | ||
| CRDI (>4 L/min/m2) | 1.17 | 0.76, 1.79 | ||
| Leucocyte counts ‡ (×103/m3) | 1.15 | 0.81, 1.63 | ||
| Serum bilirubin ‡ (mg/dL) | 0.93 | 0.77, 1.11 | ||
| Serum albumin (g/dL) | 1.80* | 1.34, 2.41 | ||
| INR (seconds) | 0.57 † | 0.35, 0.94 | ||
| Serum creatinine at enrollment (mg/dL) | 1.52 | 0.71, 3.24 | ||
| Serum sodium (mEq/L) | 1.06* | 1.02, 1.10 | ||
| MELD‐Na | 0.96* | 0.94, 0.99 | 0.97 † | 0.94, 1.00 |
MELD‐Na was calculated using bilirubin, INR, creatinine, and sodium. Therefore, these components were not considered for the multivariate regression model, as MELD‐Na was included in the model. Also, because CTP is highly associated with MELD‐Na, CTP was not considered for the multivariate analysis, to avoid multicollinearity. PVRI, CRDI, and MAP were not included in the multivariate analysis, as they are highly associated with SVRI. HVPG, SVRI, PVRI, and CVRI were considered as category variables in the regression analysis. Variables considered in multivariate analysis were use of BBs, SVRI, and MELD‐Na.
Significant at 1% level of significance.
Significant at 5% level of significance.
Corresponding log‐transformed variables were used in the regression analysis.
Predictors of Mortality—Multivariate Cox Regression Analysis
Lower SVRI (HR 0.17, 95% CI 0.12‐0.23) was associated with increased risk of mortality (Fig. 4) alongside higher MELD‐Na score (HR 1.06, 95% CI 1.03‐1.08), lower serum albumin (HR 0.59, 95% CI 0.45‐0.77), and higher albumin concentration in the urine (>10 mg/dL) was associated with increased mortality. It was interesting to see a protective effect of BBs (HR 0.54, 95% CI 0.40‐0.72) on mortality (Supporting Table S4).
FIG. 4.
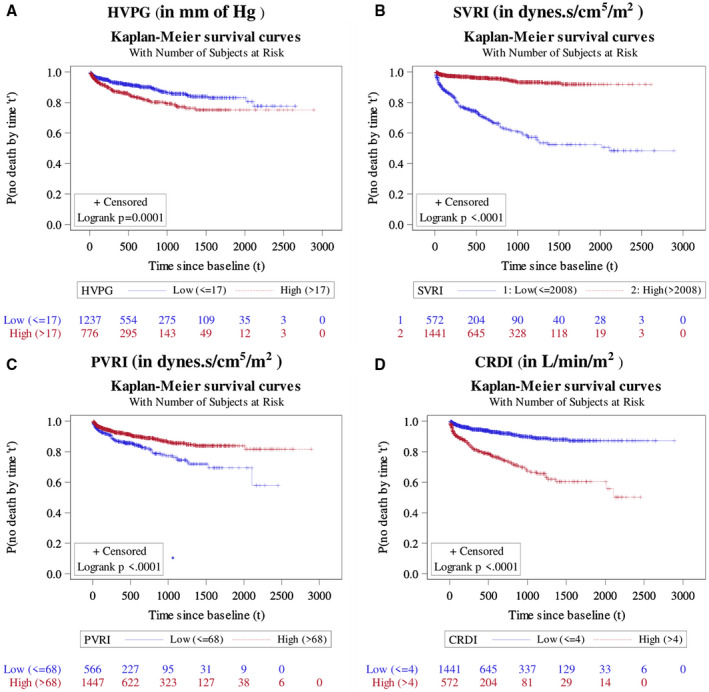
Probability of survival up to various durations since enrollment, as estimated by Kaplan‐Meier survival method, among different categories of HVPG (A), SVRI (B), PVRI (C), and CRDI (D). Note: Log‐rank test was used to test whether the pattern of incidence of mortality vary across deifferent categories of a considered variable. The result of it was shown in the form of P value.
Discussion
In this large cohort of patients with cirrhosis, we demonstrate the association of hepatic and systemic hemodynamics in the development of recurrent AKI and CKD. We also showed that patients who develop repeated AKI episodes and CKD were more severely vasodilated with lower SVRI and higher CDRI. Patients with CKD had frequent AKI. Among all of the hemodynamic parameters, lower SVRI predicted repeated AKI episodes and CKD development. High portal pressures were associated with risk of AKI but not CKD. The hemodynamic response further correlated with lower development and frequency of AKI episodes, lesser severity of AKI, lower CKD risk, and improved survival. The severity of liver disease and degree of systemic vasodilatation is associated with a higher rate of hemodynamic nonresponse, while the use of BBs improved hemodynamic response. We also report on albuminuria as a risk factor for repeated AKI, CKD, and mortality. Stratification of patients with cirrhosis based on their HVPG or SVRI could easily predict the risk of future episode(s) of AKI and CKD. These data could help stratify patients with high HVPG and low SVRI for a targeted portal pressure reduction. Improvement of the systemic vasodilatory state with additional pharmacotherapy could be an effective strategy for preventing recurrent AKI and CKD in patients with cirrhosis.
The incidence rate of AKI episodes in our study was 0.45 per person‐year follow‐up. Furthermore, we noted a higher incidence of AKI with the increase in severity of liver disease, suggesting a more deranged hemodynamic state and high portal pressures that contribute to ascites and AKI in these patients.( 12 ) It was also interesting to see that these patients were more hyperdynamic at baseline. We demonstrate a crucial contribution of the systemic and pulmonary hemodynamics in determining the risk of repeated AKI in patients with cirrhosis. As previously shown, patients with high HVPG were more systemically vasodilated and developed more AKI episodes on follow‐up.( 13 , 14 , 15 , 16 , 17 ) However, we used the revised definition of AKI in our study. We evaluated hemodynamic alterations in driving repeated episodes of AKI in patients with cirrhosis for the first time. Our study included patients with compensated and decompensated cirrhosis, contrary to prior studies that enrolled patients with compensated cirrhosis. Apart from HVPG, we evaluated the systemic, cardiac, and pulmonary hemodynamic parameters in predicting the risk of cumulative episodes of AKI. The impact of frequent AKI episodes on CKD development has already been reported.( 2 , 3 ) Similar to the study by Bassegoda et al., development of CKD was associated with a higher number of hospitalizations and more frequent AKI episodes.( 3 ) The impact of hemodynamic alterations in CKD development has never been reported in patients with cirrhosis. The degree of systemic vasodilation was identified as an independent risk factor for CKD. We also observed that SVRI was a more important predictor than HVPG in predicting hemodynamic response and CKD development. Drugs like carvedilol have an anti‐alpha adrenergic activity and a potential to worsen vasodilatation. They may predispose to CKD in patients with lower SVRI and should be administered carefully in these patients. Orally administered vasoconstrictor drugs like midodrine may prevent CKD by counteracting systemic vasodilatation. Midodrine has the potential to improve the systemic vasodilatory state and has shown benefits in the context of recurrent ascites and could prevent development of CKD in these patients.( 18 ) In the large multicentric Italian trial, the benefits of long‐term albumin administration in patients with decompensated cirrhosis, and uncomplicated ascites were reported.( 19 ) Albumin can improve systemic inflammation and cardiocirculatory function in patients with decompensated cirrhosis.( 20 ) The impact of long‐term albumin administration in improving systemic vasodilation and preventing incident CKD also merits investigation. Stratifying patients with cirrhosis based on their SVRI and providing targeted therapy to prevent repeated AKI episodes or CKD development should be evaluated in future trials.
It was interesting to observe the significance of albuminuria as a risk factor of recurrent AKI and CKD. Studies in the critically ill have suggested that albuminuria is associated with nonrecovery of AKI.( 21 ) Even a minor leakage of albumin in the urine could be a marker of early chronic renal impairment.( 22 ) We observed a stepwise increase in the risk of AKI and CKD with increase in the urine albumin levels. Levels as low as 10 mg/dL were independently associated with repeated AKI, and levels above 100 mg/dL were associated with CKD development. Emerging data have suggested an association of albuminuria with tubulointerstitial inflammation and progressive renal damage.( 22 ) We previously reported predominance of tubulointerstitial damage in renal biopsies from patients with cirrhosis.( 2 ) The loss of glomerular filtration barrier causes higher exposure of renal tubules to increased albumin concentrations. In the proximal tubules, albumin uses the megalin and cubulin receptors for internalization, from where it is transported to the dendritic cells, eliciting an inflammatory response. Higher uptake of albumin is directly related to cytotoxic effects on the renal tubules.( 23 ) The significance of albuminuria is established for diabetic nephropathy.( 23 ) It may be worthwhile to screen patients with cirrhosis routinely for urine albumin, to identify CKD development risk. The lack of detailed information on follow‐up is, however, a limitation of our study. Only a proportion (i.e., 1% of the enrolled cohort) had significant albuminuria of more than 300 mg/dL at enrollment, and urine analysis was not used for defining CKD in our study.
Repeat HVPG assessment was available in almost two‐thirds of the patients enrolled in the current study. Most of these patients were on carvedilol (mean dose of 12.5 ± 2 mg/day) and remaining on propranalol (mean dose of 66 ± 20.1 mg/day). Almost 50% of these patients were deemed to have a hemodynamic response. Responders had a lower incidence of cumulative AKI and CKD compared with nonresponders. Prior studies have shown a correlation of hemodynamic response with decreased incidence of liver‐related complications.( 12 , 13 , 14 ) Among the patients who developed AKI, it was less aggressive, and of lower severity. The severity of AKI is a proven risk factor for CKD.( 2 , 3 ) Interestingly, HVPG at baseline did not predict nonresponse to therapy. Together the data suggest that HVPG is a driver of clinical complications; therefore, additional pharmacotherapy should be considered in HVPG nonresponders. In these patients, drugs such as statins or placement of transjugular intrahepatic portosystemic shunt (TIPS) may lower portal pressure and prevent AKI recurrence and other complications driven by high portal pressures. The data showed that hyperdynamic circulation drives repeated AKI and CKD development in patients with cirrhosis. However, at the same time, we observed that response in HVPG significantly reduced this risk, signifying HVPG as a modifiable factor by targeted treatment for preventing the development of subsequent AKI episodes, which lead to CKD.
The severity of liver disease and comorbid diseases like diabetes and hypertension were associated with recurrent AKI episodes and CKD development in patients with cirrhosis.
Our study’s strength is the large cohort of patients with cirrhosis included for evaluating portal and systemic hemodynamics in CKD development. Moreover, in a significant number of patients, a second HVPG was also available to identify responders and the influence of hemodynamic response on clinical events. Furthermore, the data also support the stratification of patients based on the HVPG. We found patients with HVPG above 17 mm Hg had the highest risk of repeated AKI. Alternatively, measuring SVRI was found to be more relevant in the context of AKI development and CKD. One may also argue the utility of HVPG in clinical practice, which is invasive, requires expertise, and is not available in most centers and cannot be used routinely. Measuring SVRI, a simple noninvasive parameter,( 24 ) could be done at any center by Doppler echocardiography. SVRI is a more relevant parameter and should be targeted to prevent AKI and CKD development in patients with cirrhosis. We propose the incorporation of SVRI in the therapeutic algorithms for patients with decompensated cirrhosis who develop AKI, for preventing future episodes of AKI and CKD development. However, the main limitation of our study is the retrospective study design. In fact, previous studies have shown that unless AKI is specifically and prospectively investigated, AKI episodes may be missed. To handle the problem of missing data, we excluded all patients who had missing data on any of the essential variables. We did not perform biomarkers for assessment of renal function, which is also a limitation. We also excluded patients who underwent TIPS and liver transplantation, as inclusion of these patients would have confounded the results. There could also be a possibility of bias associated with the use of BBs. Similarly, multiple reasons could have influenced the second hemodynamic assessment, which was available for only a subset of patients. Therefore, the potential of selection bias in the use of BBs and second hemodynamic assessment remains a major limitation of the current study.
In conclusion, the results of our study demonstrate that the degree of systemic vasodilatation drives the development of CKD and frequent AKI in patients with cirrhosis. Portal pressure, which is a modifiable factor, should be targeted to lower the frequency and severity of AKI, the development of incident CKD, and overall patient outcomes. Albuminuria is an independent risk factor for recurrent AKI and CKD development in patients with cirrhosis and may be a potential marker of early renal dysfunction. Patients with HVPG above 17 mm Hg are at highest risk of developing frequent AKI episodes, which predisposes them to permanent renal damage if the insult remains unabated. Stratification of patients for the development of CKD can be done based on SVRI, which may guide targeted treatment. Therapeutic strategies for improving the hemodynamic response should be incorporated in nonresponders to prevent portal hypertension–driven complications, including repeated AKI and CKD.
Supporting information
Fig S1
Supplementary Material
A part of this work was presented as an oral paper at AASLD 2017, Washington DC.
Potential conflict of interest: Nothing to report.
References
- 1. Møller S, Krag A, Bendtsen F. Kidney injury in cirrhosis: pathophysiological and therapeutic aspects of hepatorenal syndromes. Liver Int 2014;34:1153‐1163. [DOI] [PubMed] [Google Scholar]
- 2. Maiwall R, Pasupuleti SSR, Bihari C, Rastogi A, Singh PK, Naik V, et al. Incidence, risk factors, and outcomes of transition of acute kidney injury to chronic kidney disease in cirrhosis: a prospective cohort study. Hepatology 2020;71:1009‐1022. [DOI] [PubMed] [Google Scholar]
- 3. Bassegoda O, Huelin P, Ariza X, Solé C, Juanola A, Gratacós‐Ginès J, et al. Development of chronic kidney disease after acute kidney injury in patients with cirrhosis is common and impairs clinical outcomes. J Hepatol 2020;72:1132‐1139. [DOI] [PubMed] [Google Scholar]
- 4. Sersté T, Melot C, Francoz C, Durand F, Rautou PE, Valla D, et al. Deleterious effects of beta‐blockers on survival in patients with cirrhosis and refractory ascites. Hepatology 2010;52:1017‐1022. [DOI] [PubMed] [Google Scholar]
- 5. Robins A, Bowden A, Watson W, Smith F, Gelson W, Griffiths W. Beta‐blockers in cirrhosis patients with refractory ascites. Hepatology 2014;59:2054‐2055. [DOI] [PubMed] [Google Scholar]
- 6. Moctezuma‐Velazquez C, Kalainy S, Abraldes JG. Beta‐blockers in patients with advanced liver disease: Has the dust settled? Liver Transpl 2017;23:1058‐1069. [DOI] [PubMed] [Google Scholar]
- 7. Krag A, Wiest R, Albillos A, Gluud LL. The window hypothesis: the hemodynamic and non‐hemodynamic effects of β‐blockers improve the survival of patients with cirrhosis during a window in the disease. Gut 2012;61:967‐969. [DOI] [PubMed] [Google Scholar]
- 8. Kim SG, Larson JJ, Lee JS, Therneau TM, Kim WR. Beneficial and harmful effects of nonselective beta blockade on acute kidney injury in liver transplant candidates. Liver Transpl 2017;23:733‐740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mandorfer M, Bota S, Schwabl P, Bucsics T, Pfisterer N, Kruzik M, et al. Nonselective β blockers increase risk for hepatorenal syndrome and death in patients with cirrhosis and spontaneous bacterial peritonitis. Gastroenterology 2014;146:1680‐1690.e1. [DOI] [PubMed] [Google Scholar]
- 10. Bosch J, Abraldes JG, Berzigotti A, García‐Pagan JC. The clinical use of HVPG measurements in chronic liver disease. Nat Rev Gastroenterol Hepatol 2009;6:573‐582. [DOI] [PubMed] [Google Scholar]
- 11. Contal C, O'Quigley J. An application of changepoint methods in studying the effect of age on survival in breast cancer. Comput Stat Data Anal 1999;30:253‐270. [Google Scholar]
- 12. Maiwall R, Kumar A, Bhardwaj A, Kumar G, Bhadoria AS, Sarin SK. Cystatin C predicts acute kidney injury and mortality in cirrhotics: a prospective cohort study. Liver Int 2018;38:654‐664. [DOI] [PubMed] [Google Scholar]
- 13. Merkel C, Montagnese S. Hepatic venous pressure gradient measurement in hepatology. Dig Liver Dis 2011;43:762‐767. [DOI] [PubMed] [Google Scholar]
- 14. Bosch J, Abraldes JG, Berzigotti A, García‐Pagan JC. The clinical use of HVPG measurements in chronic liver disease. Nat Rev Gastroenterol Hepatol 2009;6:573‐582. [DOI] [PubMed] [Google Scholar]
- 15. Ripoll C, Groszmann R, Garcia‐Tsao G, Grace N, Burroughs A, Planas R, et al; Portal Hypertension Collaborative Group . Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology 2007;133:481‐488. [DOI] [PubMed] [Google Scholar]
- 16. Hernández‐Gea V, Aracil C, Colomo A, Garupera I, Poca M, Torras X, et al. Development of ascites in compensated cirrhosis with severe portal hypertension treated with β‐blockers. Am J Gastroenterol 2012;107:418‐427. [DOI] [PubMed] [Google Scholar]
- 17. Villanueva C, Albillos A, Genescà J, Garcia‐Pagan JC, Calleja JL, Aracil C, et al. β blockers to prevent decompensation of cirrhosis in patients with clinically significant portal hypertension (PREDESCI): a randomized, double‐blind, placebo‐controlled, multicentre trial. Lancet 2019;20:1597‐1608. [DOI] [PubMed] [Google Scholar]
- 18. Singh V, Dhungana SP, Singh B, Vijayverghia R, Nain CK, Sharma N, et al. Midodrine in patients with cirrhosis and refractory or recurrent ascites: a randomized pilot study. J Hepatol 2012;56:348‐354. [DOI] [PubMed] [Google Scholar]
- 19. Caraceni P, Riggio O, Angeli P, Alessandria C, Neri S, Foschi FG, et al. Long‐term albumin administration in decompensated cirrhosis (ANSWER): an open‐label randomised trial [published correction appears in Lancet 2018;392:386]. Lancet 2018;391:2417‐2429. [DOI] [PubMed] [Google Scholar]
- 20. Fernández J, Clària J, Amorós A, Aguilar F, Castro M, Casulleras M, et al. Systemic and portal hemodynamics and systemic inflammation in patients with decompensated cirrhosis. Gastroenterology 2019;157:149‐162. [DOI] [PubMed] [Google Scholar]
- 21. Neyra JA, Li X, Yessayan L, Adams‐Huet B, Yee J, Toto RD. Dipstick albuminuria and acute kidney injury recovery in critically ill septic patients. Nephrology 2016;21:512‐518. [DOI] [PubMed] [Google Scholar]
- 22. Lambers Heerspink HJ, Gansevoort RT. Albuminuria is an appropriate therapeutic target in patients with CKD: the pro view. Clin J Am Soc Nephrol 2015;10:1079‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gekle M. Renal tubule albumin transport. Annu Rev Physiol 2005;67:573‐594. [DOI] [PubMed] [Google Scholar]
- 24. Porter TR, Shillcutt SK, Adams MS, Desjardins GKE, Olson JJ, Troughton RW. Guidelines for the use of echocardiography as a monitor for therapeutic intervention in adults: a report from the American Society of Echocardiography. J Am Soc Echocardiogr 2015;28:40‐56. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Supplementary Material