

Abstract
Objectives
Clinical presentation of primary Sjögren’s syndrome (pSS) varies considerably. A shortage of evidence-based objective markers hinders efficient drug development and most clinical trials have failed to reach primary endpoints.
Methods
We performed a multicentre study to identify patient subgroups based on clinical, immunological and genetic features. Targeted DNA sequencing of 1853 autoimmune-related loci was performed. After quality control, 918 patients with pSS, 1264 controls and 107 045 single nucleotide variants remained for analysis. Replication was performed in 177 patients with pSS and 7672 controls.
Results
We found strong signals of association with pSS in the HLA region. Principal component analysis of clinical data distinguished two patient subgroups defined by the presence of SSA/SSB antibodies. We observed an unprecedented high risk of pSS for an association in the HLA-DQA1 locus of odds ratio 6.10 (95% CI: 4.93, 7.54, P=2.2×10−62) in the SSA/SSB-positive subgroup, while absent in the antibody negative group. Three independent signals within the MHC were observed. The two most significant variants in MHC class I and II respectively, identified patients with a higher risk of hypergammaglobulinaemia, leukopenia, anaemia, purpura, major salivary gland swelling and lymphadenopathy. Replication confirmed the association with both MHC class I and II signals confined to SSA/SSB antibody positive pSS.
Conclusion
Two subgroups of patients with pSS with distinct clinical manifestations can be defined by the presence or absence of SSA/SSB antibodies and genetic markers in the HLA locus. These subgroups should be considered in clinical follow-up, drug development and trial outcomes, for the benefit of both subgroups.
Keywords: Sjögren’s syndrome, autoimmunity, gene polymorphism, autoantibodies
Rheumatology key messages
Clinical data analysis provides evidence for two subgroups of primary Sjögren’s syndrome best defined by SSA/SSB antibodies.
Signals within the HLA region are unique to patients with SSA/SSB autoantibodies.
Genetic markers of the HLA locus and SSA/SSB autoantibodies define two distinct subgroups of primary Sjögren’s syndrome.
Introduction
Primary Sjögren’s syndrome (pSS) is a systemic autoimmune disease that predominantly affects women [1, 2]. Patients are classified as having pSS when fulfilling internationally accepted criteria, but the clinical presentation varies considerably [3, 4]. While sicca symptoms, with dryness of the eyes and mouth, pain, and fatigue are common, some patients present with extra-glandular manifestations such as arthritis, purpura or interstitial lung disease. Additionally, immune-variables differ substantially among the patients. The typical disease-related autoantibodies against SSA and SSB are found in 70% and 45% of patients, respectively, and in some a mild leukopenia or hypergammaglobulinaemia may be detected [5]. Given the heterogeneity in clinical presentation of what is currently referred to as pSS, selecting patients and evaluating outcomes in clinical trials has proven difficult. A recent study suggested patient-reported outcome measures to classify patients with pSS into different subtypes [6]. Variation in clinical manifestations or outcome based on presence or absence of particular biomarkers has also been highlighted [1, 7]. However, patient-reported symptoms and some biomarkers may vary over time and these approaches do not take into account possible underlying genetic predisposition for different clinical subgroups.
During the past decade, genetic association studies have revealed several loci linked to pSS (reviewed in [8, 9]). The most prominent associations are with variants in the HLA region, but associations have also been found with single-nucleotide polymorphisms (SNPs) in or around other genes with immunological functions [10–13]. However, the impact of many of these polymorphisms in pSS pathogenesis has not been studied, nor how differences in the clinical presentation relate to genetic variants.
Using a large set of cases of well-characterized patients with pSS, the aim of this study was to investigate if genetic heterogeneity and variation in clinical phenotypes represent different disease subtypes that may require distinction for both diagnosis and treatment. We sequenced >1800 autoimmunity-related gene loci in nearly 1000 patients from Sweden and Norway and analysed clinical features of the patients focusing on immunological manifestations intersected with genetic associations.
Methods
Patients and controls
A total of 982 patients with pSS from Sweden and Norway, and 1342 healthy blood donors and population controls were included in the study (Supplementary Table S1, available at Rheumatology online). All patients fulfilled the American European Consensus Group (AECG) criteria for pSS [4] (Table 1 and Supplementary Table S2, available at Rheumatology online). For replication, an independent set of 177 patients with pSS from Sweden and Norway and 7672 controls (n = 918 from the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) and n = 6754 from the Swedish Twin Registry (STR) were included (Supplementary Table S1, available at Rheumatology online) [14, 15]). The study was approved by the local ethics committees and patients gave written informed consent.
Table 1.
Clinical characteristics of patients with primary Sjögren’s syndrome
| All patients | Anti-SSA/SSB positive | Anti-SSA/SSB negative | P-valuea | ||
|---|---|---|---|---|---|
| (n = 982) | (n = 717) | (n = 265) | |||
| Females, % | 93.1 | 92.1 | 95.8 | 0.037 | |
| Age, mean (s.d.), years | |||||
| At symptom onset | 46.2 (14.7) | 45.1 (15.1) | 49.0 (13.3) | 5.0 × 10−4 | |
| At diagnosis | 52.6 (13.7) | 51.3 (14.0) | 56.1 (12.1) | <1 × 10−4 | |
| Laboratory findings, % | |||||
| ANA | 74.8 | 85.8 | 44.9 | <1 × 10−4 | |
| Anti-SSA | 70.5 | 96.5 | 0.0 | ||
| Anti-SSB | 42.8 | 58.8 | 0.0 | ||
| Anti-SSA and/or anti-SSB | 73.0 | 100 | 0.0 | ||
| Anaemia Hb <120 g/l | 22.3 | 26.5 | 10.9 | <1 × 10−4 | |
| Leukopenia <4.0 × 109/l | 30.5 | 36.8 | 13.9 | <1 × 10−4 | |
| Thrombocytopenia <100 × 109/l | 3.7 | 4.3 | 2.0 | 0.15 | |
| P-IgG >15 g/l | 48.8 | 59.6 | 14.5 | <1 × 10−4 | |
| Minor salivary gland biopsies | |||||
| Focus score, mean (s.d.) | 2.4 (2.4) | 2.5 (2.6) | 2.2 (1.9) | 0.04 | |
| Germinal centre formations, % | 21.8 | 25.5 | 14.2 | 0.013 | |
| Extraglandular manifestations, % | |||||
| Raynaud | 29.1 | 29.1 | 29.0 | 0.98 | |
| Arthritis | 19.4 | 20.6 | 16.1 | 0.12 | |
| Purpura | 10.7 | 13.8 | 2.8 | <1 × 10−4 | |
| Major salivary gland swelling | 30.0 | 32.8 | 22.6 | 3.9 × 10−3 | |
| Lymphadenopathy | 9.1 | 10.5 | 5.3 | 0.014 | |
| Hypothyreoidism | 22.5 | 21.3 | 25.4 | 0.21 | |
| Myositis | 0.9 | 1.1 | 0.5 | 0.38 | |
| Interstitial lung disease | 6.8 | 7.6 | 4.3 | 0.12 | |
| Interstitial nephritis | 3.0 | 3.3 | 2.2 | 0.46 | |
| Lymphoma | 4.6 | 5.5 | 2.3 | 0.036 | |
| Age at lymphoma onset, mean (s.d.), years | 57.1 (13.7) | 54.5 (12.4) | 73.8 (9.0) | 8.8 × 10−4 | |
P-value for the comparison between anti-SSA/SSB (anti-SSA and/or anti-SSB) positive and anti-SSA/SSB negative patients. Continuous variables compared with Student’s unpaired t-test, frequencies with χ2 test.
Targeted sequencing, genotyping and quality control
A sequence capture array was designed to target 1853 genes including their coding and regulatory regions, covering 32.2 Mbp (see Supplementary Materials and methods, Supplementary Table S3 and Supplementary Fig. S1, available at Rheumatology online). Genes were selected based on their known role in immunological processes, inflammation and autoimmune diseases. Sequencing libraries were prepared from genomic DNA, hybridized (Roche NimbleGen, Basel, Switzerland) and then sequenced with 100-bp paired-end reads using an Illumina HiSeq 2500 (Illumina, Inc., San Diego, CA, USA). Samples with a mean target coverage of <10× were excluded. A set of bi-allelic single nucleotide variants (SNVs) was generated with call rate 90% for SNVs and 80% for samples. Population outliers and related samples were excluded and 918 patients with pSS and 1264 controls remained for analysis.
The replication set was genotyped on Illumina OmniExpressExome (cases and PIVUS) and Illumina OmniExpress (STR). Quality control was performed and additional variants imputed based on the Haplotype reference consortium r1.1 (Supplementary Materials and methods, available at Rheumatology online) [16].
Statistical analysis
Single variant association analysis in the main set of cases and controls for variants with a minor allele frequency (MAF) of ≥1% (n = 107 045) was performed in PLINK using a logistic regression model [17]. Experiment-wide Bonferroni corrected significance was set to P < 8.7×10−7 after removal of highly correlated SNVs [57 768 SNVs remaining with linkage disequilibrium (LD) r2<0.8] and a suggestive significance threshold of P<1×10−5 was applied. For clinical data, continuous variables were analysed using Student’s unpaired t-test and frequencies were compared with χ2 unless there were <5 observations, in which case Fisher’s exact test was applied (Statistica version 13.4.0.14, TIBCO Software Inc., Palo Alto, CA, USA). Correlations between clinical variables were assessed using Spearman’s correlation in GraphPad Prism 6 (GraphPad Software Inc., La Jolla, CA, USA) and results plotted in Morpheus (https://software.broadinstitute.org/morpheus/). Principal component analysis of clinical data and plotting of results was carried out in R [18]. Logistic regression analyses of genotypes and clinical variables were performed using a generalized linear model and plotted using R. Association analysis for variants selected for replication was performed in PLINK using a logistic regression model with the first three population stratification principal components as covariates (Supplementary Materials and methods, available at Rheumatology online).
Results
Targeted sequencing suggests novel loci and confirms known genetic variants associated with primary Sjögren’s syndrome
To map the genetic variability in pSS, targeted sequencing of coding regions of 1853 immune-related genes, including their upstream and downstream regulatory regions, was performed on samples from patients with pSS and controls. Our analysis revealed strong signals of association for pSS in the HLA region, with the top variant in the HLA-DQA1 locus [rs6933289; odds ratio (OR) 3.88; 95% CI: 3.22, 4.66; P = 1.4 × 10−46]. Suggestive associations were also found with variants in the interferon regulatory factor 5-transportin 3 (IRF5-TNPO3) locus (OR 1.39; 95% CI: 1.22, 1.61; P = 1.8 × 10−6), as well as in two novel loci not previously associated with pSS containing the glutamic-oxaloacetic transaminase 1 (GOT1) (OR 1.43; 95% CI: 1.23, 1.64; P = 1.1 × 10−6) and mitogen-activated protein kinase 2 (MAP2K2) (OR 1.82; 95% CI: 1.43, 2.33; P = 1.7 × 10−6) genes (Table 2, Fig. 1A, Supplementary Fig. S2 and Supplementary Table S4, available at Rheumatology online).
Table 2.
Allelic association analysis in patients with primary Sjögren’s syndrome compared with healthy controls
| Variant | Position | Gene/region | P-value | Conditional P-value | OR (95% CI) | RAF, cases/controls | Minor/major alleles | snpEff annotation [19] |
|---|---|---|---|---|---|---|---|---|
| All cases vs controlsa | ||||||||
| rs6933289 | chr6:32604551 | HLA-DQA1 | 1.4 × 10−46 | — | 3.88 (3.22, 4.66) | 0.31/0.13 | T/C | Upstream |
| rs3099839 | chr6:31430065 | HCP5 | 1.6 × 10−43 | 2.4 × 10−6b | 3.75 (3.11, 4.52) | 0.30/0.11 | T/C | Upstream |
| rs4919321 | chr10:101230461 | GOT1 | 1.1 × 10−6 | — | 1.43 (1.23, 1.64) | 0.73/0.67 | C/G | Intergenic |
| rs6630 | chr19:4090422 | MAP2K2 | 1.7 × 10−6 | — | 1.82 (1.43, 2.33) | 0.94/0.88 | T/G | 3′ UTR |
| rs11761199 | chr7:128581835 | IRF5 | 1.8 × 10−6 | — | 1.39 (1.22, 1.61) | 0.55/0.47 | A/G | Intronic |
| rs7197 | chr6:32412580 | HLA-DRA | 9.2 × 10−5 | 1.6 × 10−13c | 1.34 (1.16, 1.55) | 0.27/0.23 | T/C | 3′ UTR |
| SSA/SSB antibody positive cases vs controlsa | ||||||||
| rs6933289 | chr6:32604551 | HLA-DQA1 | 2.2 × 10−62 | — | 6.10 (4.93, 7.54) | 0.37/0.13 | T/C | Upstream |
| rs2523607 | chr6:31322790 | HLA-B | 5.3 × 10−58 | 6.8 × 10−07b | 5.27 (4.30, 6.45) | 0.36/0.12 | A/T | Upstream |
| rs7197 | chr6:32412580 | HLA-DRA | 5.2 × 10−8 | 2.6 × 10−25c | 1.56 (1.33, 1.84) | 0.30/0.23 | T/C | 3′ UTR |
| rs3823536 | chr7:128579666 | IRF5 | 7.4 × 10−8 | — | 1.52 (1.30, 1.79) | 0.57/0.47 | G/A | Upstream |
| rs4919321 | chr10:101230461 | GOT1 | 2.4 × 10−6 | — | 1.47 (1.25, 1.72) | 0.73/0.67 | C/G | Intergenic |
Independent signals with uncorrected or conditional P-values exceeding a suggestive significance threshold of P < 1 × 10−5 are shown. The risk allele is marked in bold.
All cases n = 918, SSA and/or SSB antibody positive cases n = 663, controls n = 1264.
Analysis passing suggestive significance threshold with rs6933289 and rs7197 as covariates.
Analysis passing suggestive significance threshold with rs6933289 as covariate. Chr, chromosome, OR, odds ratio; RAF, risk allele frequency; UTR, untranslated region.
Fig. 1.

Genetic association and subgroup analysis of primary Sjögren’s syndrome patients vs controls
(A) Single variant association analysis between 918 pSS cases and 1264 healthy controls. Logistic regression with minor allele frequency ≥0.01 and three principal components as covariates. A total of 107 045 variants included after quality control. Red line indicates the experiment wide Bonferroni cutoff (P = 8.7 × 10−7); blue line represents the suggestive significance threshold (P = 1 × 10−5). (B) PCA of clinical data collected for 982 pSS cases. (C) Single variant association analysis between anti-SSA/SSB positive patients (dark blue in PCA plot) vs controls. (D) Single variant association analysis between anti-SSA and SSB negative patients (light blue in PCA plot) vs controls. PCA: principal component analysis.
Clinical features distinguish distinct patient subgroups identifiable by unique HLA associations
To identify patient subgroups, we used the clinical information available for all 982 patients and performed a principal component analysis (PCA). Interestingly, this approach distinguished two clinically distinct subgroups of patients. Regression analysis revealed that the clinical variable best corresponding to the first principal component (explaining 16.2% of the overall variability in the clinical data) was SSA and/or SSB (hereafter SSA/SSB) autoantibody status (Fig. 1B and Supplementary Fig. S3A, available at Rheumatology online). This was confirmed using k-means clustering on the PCA results to reveal two groups that predicted SSA/SSB positivity with 99.3% accuracy (Supplementary Fig. S3B, available at Rheumatology online). The clinical variables best associated with the second principal component (explaining 8.35% of the variability in the clinical data) were the two interrelated variables age at symptom onset and age at diagnosis (Supplementary Fig. S3A, C and D, available at Rheumatology online).
Since subgroups within the patients were best defined by the presence of SSA/SSB autoantibodies, we stratified the patients based on SSA/SSB autoantibody status and further explored differences in their clinical presentation. Patients positive for SSA/SSB were younger at disease onset and diagnosis, and presented more frequently with anaemia, leukopenia, and hypergammaglobulinaemia. Further, SSA/SSB antibody positive patients displayed an increased prevalence of purpura, major salivary gland swelling, lymphadenopathy and lymphoma, showing an overall more severe disease phenotype compared with patients negative for both SSA and SSB autoantibodies (Table 1).
As the two identified pSS patient subgroups differed in their clinical presentation, we explored genetic associations separately for each of the two groups. When only including patients positive for SSA/SSB antibodies in the analysis and comparing them with controls, the association with variants in the HLA region was distinctly stronger compared with the analysis of the whole set of cases, with an OR as high as 6.1 for the top associated variant in the HLA-DQA1 region (rs6933289; OR 6.10; 95% CI: 4.93, 7.54; P = 2.2 × 10−62). The association with variants in the IRF5-TNPO3 locus was also more prominent (OR 1.52; 95% CI: 1.30, 1.79; P = 7.4 × 10−8), while the association with GOT1 remained unchanged (OR 1.47; 95% CI: 1.25, 1.72; P = 2.4 × 10−6) and MAP2K2 did not pass the suggestive significance threshold of P < 1 × 10−5 (OR 1.79; 95% CI: 1.35, 2.33; P = 4.2 × 10−5) (Table 2, Fig. 1C, Supplementary Fig. S4 and Supplementary Table S5, available at Rheumatology online).
In contrast, when comparing SSA and SSB autoantibody negative patients with controls, surprisingly no association with HLA was observed. In fact, no variant associated with SSA/SSB autoantibody negative pSS exceeded the suggestive significance threshold of P < 1 × 10−5, but two signals near kinesin family member 1B (KIF1B) on chromosome 1 (rs149524751; OR 4.55; 95% CI: 2.32, 8.92; P = 1.0 × 10−5) and caspase 8 (CASP8) on chromosome 2 (rs17860432; OR 2.75; 95% CI: 1.74, 4.31; P = 1.2 × 10−5) nearly reached the suggestive cutoff (Fig. 1D, Supplementary Fig. S5 and Supplementary Table S6, available at Rheumatology online).
A case–case analysis between patients positive for SSA/SSB autoantibodies and patients negative for both autoantibodies identified rs9273058, a variant downstream of HLA-DQA1 (OR 0.28; 95% CI: 0.22, 0.35; P = 6.1 × 10−26) as the most significantly associated variant (Supplementary Fig. S6 and Supplementary Table S7, available at Rheumatology online). The HLA-DQA1 variant most strongly associated with SSA/SSB positive pSS (rs6933289) remained significantly associated in the case–case comparison of the two patients subgroups (OR 4.03; 95% CI: 2.97, 5.47; P = 3.0 × 10−19), despite the lower power. This demonstrates that the association with the HLA region in pSS is unique to the SSA/SSB autoantibody positive patient subgroup, providing evidence for distinct genetic aetiologies in the two groups.
Independent genetic signals within the HLA region are unique to patients with SSA/SSB autoantibodies
Considering that the associations with variation in the HLA domain differ between the two pSS subgroups, we examined the HLA region in more detail. When conditioning on the top variant in the HLA-DQA1 locus (rs6933289) in the analysis of all patients with pSS vs controls and SSA/SSB positive cases vs controls, independent signals with the highest peak in HLA-DRA (rs7179) remained (for all patients vs controls, OR 1.85; 95% CI: 1.57, 2.17; P = 1.6 × 10−13; and for SSA/SSB positive cases vs controls, OR 2.85; 95% CI: 2.34, 3.47; P = 2.6 × 10−25), passing the experiment-wide Bonferroni correction cutoff of P < 8.7 × 10−7 (Table 2, Supplementary Tables S8 and S9, available at Rheumatology online). Further, conditioning on the top SNVs from both independent signals revealed a third association signal in the HLA-B/MICA/HCP5 locus exceeding the suggestive significance threshold of P < 1 × 10−5 (rs3099839 in HCP5 for all cases vs controls, OR 2.07; 95% CI: 1.53, 2.80; P = 2.4 × 10−6; and rs2523607 in HLA-B for SSA/SSB positive cases vs controls, OR 2.14; 95% CI: 1.59, 2.89; P = 6.8 × 10−7). These signals are in high LD (r2 = 0.91) (Table 2, Fig. 2A–D, Supplementary Tables S10 and S11, available at Rheumatology online). Each of these three independent HLA signals were more significant in the SSA/SSB positive cases compared with the analysis of all patients (Fig. 2C). The LD between the independent signals and the positions of the nearest genes is depicted in Fig. 2D and E. Together, these data suggest several independent HLA associations in pSS, which all are unique to patients with SSA/SSB autoantibodies.
Fig. 2.

HLA associations with primary Sjögren’s syndrome
Stepwise adjustment for the top associated variants. (A, B) Logistic regression analysis of all patients vs controls (A), or anti-SSA/SSB positive patients vs controls (B). Second panel after conditioning on rs6933289, bottom panel after conditioning on rs6933289 and rs7197, with rs3099839 top remaining variant in all cases vs controls, rs2523607 top remaining variant in anti-SSA/SSB positive vs controls. (C) Unadjusted P-values for all cases vs controls (blue), SSA/SSB positive vs controls (red), and SSA and SSB negative vs controls (grey). (D) Linkage disequilibrium (r2) between the variants. (E) Gene regions of the independent HLA variants: HLA-DQA1, HLA-DRA and the HLA-B/MICA/HCP5 locus.
Genetic variants identify patients with extraglandular manifestations
To examine the interplay between the three independently associated HLA genetic variants and clinical presentation, we performed a non-parametric correlation analysis between risk allele counts and clinical variables (Fig. 3A and Supplementary Table S12, available at Rheumatology online). This analysis revealed significant correlations between HLA-DQA1 (rs6933289) and HCP5 (rs3099839) and a number of clinical variables including younger age at symptom onset and diagnosis, ANA, SSA and SSB autoantibodies, hypergammaglobulinaemia, leukopenia, anaemia, purpura, major salivary gland swelling and lymphadenopathy (−0.10 < ρ > 0.10, P < 0.05). These correlations mirror the grouping according to SSA/SSB antibody status previously identified in the clinical data.
Fig. 3.

Correlations between clinical phenotypes and associated variants
(A) Non-parametric correlations between associated genetic variants and clinical variables. The purple scale represents P-values for the correlation (darker represents more significant), and the blue–red scale represents the correlation coefficient, with darker blue representing stronger negative correlation, and deeper red representing stronger positive correlation. (B) Logistic regression of different clinical variables with risk allele count for the top associated independent genetic variants. Red lines represent significant positive associations, blue lines significant negative associations. Whiskers indicate 95% CI. OR: odds ratio.
To further examine the associations between genotypes and phenotypes, we also performed a logistic regression analysis between the top associated variants and the clinical variables (Fig. 3B). The HLA-DQA1 and HCP5 risk variants significantly increased the OR of the clinical variables previously shown to co-occur with SSA/SSB autoantibodies, emphasizing the phenotype identified in the SSA/SSB positive patient subgroup. In contrast, the HLA-DRA risk variant rs7197 was only associated with an increased OR for SSA and leukopenia, and with a significantly reduced OR for hypothyroidism. In all, we conclude that carrying the risk alleles at HLA-DQA1 and HCP5 predicts distinct clinical manifestations of pSS and the presence of SSA/SSB autoantibodies.
Replication analysis in additional Scandinavian patients and controls
To replicate the MHC class I association at HLA-B/HCP5, and the suggestive associations with GOT1 and MAP2K2, an independent set of 177 Scandinavian pSS cases (n = 153 SSA/SSB antibody positive) and 7672 controls were included. We confirmed the association between HLA and pSS being confined to the SSA/SSB autoantibody positive subgroup. The MHC class I association HLA-B (rs2523607)/HCP5 (rs3099839) replicated in all patients vs controls (rs3099839; OR 3.47; 95% CI: 2.74, 4.41; P = 1.6 × 10−24) and SSA/SSB positive patients vs controls (rs2523607; OR 3.83; 95% CI: 2.98, 4.92; P = 5.3 × 10−26). There were no significant associations with GOT1 (rs49193219) or MAP2K2 (rs6630), in the analyses of all patients with pSS or SSA/SSB antibody positive patients vs controls. The results are presented in Supplementary Table S13, available at Rheumatology online.
Discussion
pSS is a heterogeneous disease with an apparent need for biomarkers to identify patient subgroups for monitoring, prognosis, treatment and inclusion in clinical trials. Using a combination of genetic information and analysis of extensive clinical information for nearly a thousand Scandinavian patients, we detected clear signs of grouping in the patient data, evident in both clinical manifestations and genetic associations. We found that pSS with associated variants in the HLA region is unique to a subgroup of patients, best identified by the presence of the hallmark SSA/SSB autoantibodies, and that the top variant is associated with a six times increased risk of pSS in this group. Further, to the best of our knowledge, this is the first time a direct correlation between HLA risk variants and specific clinical features in patients with pSS has been described.
Notably, we detected clear grouping of the patients, with SSA/SSB antibodies and age at disease onset and diagnosis being the factors explaining most of the variability in the data. Earlier reports confirm the validity of the observation as patients positive for SSA/SSB antibodies are known to have an earlier disease onset and present with more systemic extraglandular manifestations, such as leukopenia, hypergammaglobulinaemia, purpura, and major salivary gland swelling [1]. Furthermore, in a recent epidemiological study we found that patients positive for SSA/SSB antibodies are particularly at increased risk for cardiovascular disease, highlighting the importance of subgroup stratification in clinical monitoring and risk assessment [20].
Combining high resolution variant information, together with the observation of clinical clustering of patients, we could demonstrate an unprecedented high odds ratio of 6.10 for developing pSS compared with the general population in carriers of the associated variant rs6933289 in the HLA-DQA1 region, when restricting the analysis to the identified SSA/SSB antibody positive subgroup. Notably, there was no risk of SSA/SSB antibody negative pSS associated with the same genetic variant. Previous studies in Caucasians have described OR of different genetic variants of the HLA locus in the range of 2–3.5, depicted in Fig. 4 [10, 13].
Fig. 4.
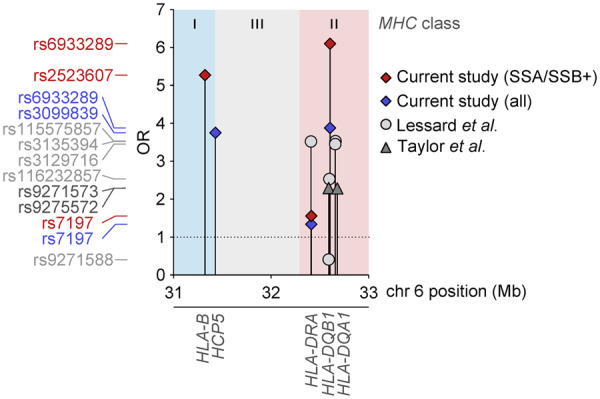
Comparison of effect sizes for associated variants in the HLA region
Top variants from Lessard et al. [10] are shown as light grey circles and variants from Taylor et al. [13] are depicted as dark grey triangles. Red diamonds represent SSA and/or SSB positive primary Sjögren’s syndrome associations and blue diamonds represent the full primary Sjögren’s syndrome associations from the current study. Position on chromosome 6 is shown on the x-axis and ORs on the y-axis. The nearest genes are labelled below. OR: odds ratio.
Different strategies in subgrouping patients with pSS can be applied. A recent study defined four different subgroups based on patient-reported outcome measures of levels of pain, dryness, fatigue, depression and anxiety [6]. The subgroups differed in laboratory parameters and gene expression as well as response in clinical trials in retrospective analysis of pain, dryness and fatigue scores, and salivary flow measurements. Our approach of letting clinical data guide subgrouping of pSS clearly identifies distinct groups also at the genetic level. We believe these two subgroups defined by the presence or absence of SSA/SSB antibodies, demarking autoimmune disease processes, need to be considered before, or in addition to, applying additional markers for further subgrouping. The genetic differences are likely to relate to different pathogenic mechanisms, and this knowledge will be valuable both for continued efforts in understanding the mechanisms driving development of the respective subtypes and for designing treatment strategies for each group. Further, with an OR higher than 6 detectable by a single variant, the precision approaches clinical usability, at least in the population studied (Caucasian).
The hitherto few genome-wide association studies performed in pSS have not stratified patients according to autoantibodies or clinical manifestations [10, 11, 13]. The association of HLA variants with the presence of SSA/SSB autoantibodies has previously been described in several smaller studies including patients of different ethnicities [21–23]. However, none of these studies described associations between HLA and clinical features, either because the question was not addressed or possibly because of lack of power [21]. Here we extend our knowledge by establishing a direct link between specific HLA risk variants and clinical manifestations. This implicates that HLA risk variants not only drive the autoantibody response, but also predict the various disease manifestations seen in patients with pSS.
In our study we confirmed the association between variants in IRF5 and pSS, previously established in multiple ethnicities (reviewed in [8, 9]). Additionally, we found associations with GOT1 and MAP2K2 passing our suggestive significance threshold. The variants near GOT1 are located in the intergenic region between GOT1 and NKX2-3, a gene encoding a NKX2 homeobox protein, necessary for marginal-zone B cell development and implicated in lymphomagenesis [24]. This is intriguing given the increased risk for B cell lymphoma in pSS. Gene variants in GOT1-NKX2-3 have previously been associated with inflammatory bowel disease [25]. MAP2K2 encodes a protein kinase not previously associated with autoimmune diseases. However, we were not able to confirm these associations in a small independent Scandinavian pSS set of cases. To clarify the role of GOT1 and MAP2K2 variants in pSS susceptibility, these results need confirmation in additional samples.
The mechanisms that explain the high risk of developing pSS with SSA/SSB autoantibodies in carriers of specific HLA variants remain to be understood. While the HLA proteins have their main role in antigen presentation, many additional genes are encoded within the locus and understanding which genes, or combination of genes and their respective variants, drive the autoimmune reaction will require detailed mechanistic investigations. The SSA and SSB antigens are RNA-binding proteins which together with SSA/SSB autoantibodies form immune complexes that induce type I IFN production, eliciting an immune response [26, 27]. The upregulated expression of IFN-induced genes, i.e. the IFN signature, is mainly seen in SSA/SSB positive patients, which is also supported by observations from studies on epigenetic regulation [28, 29]. These autoantibodies appear before pSS is clinically apparent [30]. Notably, an association with HLA class II alleles in patients with different systemic autoimmune diseases and an IFN signature has been described [31]. Consequently, activation of the type I IFN system as an aetiopathogenic mechanism for clinical disease in SSA/SSB antibody positive patients has been proposed, and variants of IRF5 may amplify this process, while it is likely that other immune mechanisms operate in the antibody negative subgroup of patients with pSS [27, 32, 33]. Further studies into the pathogenic mechanisms behind antibody negative pSS are warranted, to discover potential therapeutic targets for this patient subgroup.
Limitations of our study include the low frequency of certain clinical manifestations and missing data, possibly precluding some additional associations with SSA/SSB antibodies or HLA risk variants. Unfortunately, data on the severity of dryness, pain, fatigue, anxiety or depression, which are common manifestations in patients with pSS, were not available. Antibody data were retrieved from the medical records and analysed according to routine clinical immunology methods that may have varied over time, but SSA/SSB antibody status in pSS is stable through the disease course [34]. The targeted sequencing approach precludes identification of novel risk genes not included in the panel. Future studies should aim at whole genome sequencing to fully elucidate the genetic background to pSS and its subphenotypes. Strengths of our study are the large number of patients included, the homogeneous genetic background, meticulous evaluation of diagnostic criteria and detailed clinical data collected in a similar manner by the participating clinicians.
In conclusion, we define two subgroups of patients with pSS based on HLA association, SSA/SSB antibodies and clinical manifestations, and demonstrate a direct correlation between HLA risk variants, age of onset and several clinical features. The SSA/SSB antibody positive subgroup clearly presents with a systemic autoimmune disease. In contrast, the patients negative for these antibodies and lacking the HLA-associated genetic features could be defined as having an organ-specific disease with less obvious autoimmune features and potentially a different underlying pathogenesis, not least in terms of the predisposing genetic makeup. These differences need to be considered not only during clinical follow-up, but also in drug development and when designing clinical trials and determining trial outcomes, for the benefit of all patients with pSS.
Supplementary Material
Acknowledgements
This study was supported by the Swedish Research Council for Medicine and Health (Dnr 2016–01982 to G.N., 2018–02399 to L.R.), the Swedish Rheumatism Association, the King Gustav V’s 80-year Foundation, a Wallenberg Scholar Award (to K.L.T.) and an AstraZeneca-Science for Life Laboratory Research Collaboration grant (DISSECT). DNA sequencing was performed at the SNP&SEQ Technology Platform in Uppsala, part of the National Genomics Infrastructure (NGI) Sweden, and supported by Science for Life Laboratory, the Swedish Research Council (VR-RFI), Uppsala University and the Knut and Alice Wallenberg Foundation. We acknowledge The Swedish Twin Registry for access to data. The Swedish Twin Registry is managed by Karolinska Institutet and receives funding through the Swedish Research Council under the grant no 2017–00641. PIVUS was supported by Wellcome Trust Grants (WT098017, WT064890, WT090532), Uppsala University, Uppsala University Hospital, the Swedish Research Council and the Swedish Heart-Lung Foundation. The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) through Uppsala Multidisciplinary Center for Advanced Computational Science (UPPMAX) under projects SNIC SENS 2017142 and 2017107. We thank Rezvan Kiani Dehkordi, Karolina Tandre, Käth Nilsson, Marianne Eidsheim, Kjerstin Jacobsen, Ingeborg Kvivik and Kjetil Bårdsen for collecting patient blood samples. DNA sequencing data can be obtained from the authors on a collaborative basis. K.L.T., L.R., J.K.S., L.H.R., J.R.S.M., M.W.H. and G.N. designed the study. E.T., M.K., H.F.d’E., S.M.B., K.B.N., S.J.A.J., D.H., K.S., M.V.J., E.B., L.A.A., J.L.J., Ø.P., S.R.D., T.M., P.E., R.O., R.J. and G.N. contributed to the acquisition of patients and control samples and clinical data. A.M. and L.L. provided genotype control data. G.E.T., L.H.R., M.B., J.K.S., P.P. and J.I.K. analysed the data. G.E.T., M.W.H. and G.N. drafted the manuscript and all authors approved of the final version of the manuscript.
The DISSECT consortium: 1. Andrei Alexsson, Pascal Pucholt, Carin Backlin, Eva Baecklund, Gunnel Nordmark, Johanna K. Sandling, Juliana Imgenberg-Kreuz, Lars Rönnblom, Lilian Vasaitis, Maija-Leena Eloranta, Department of Medical Sciences, Rheumatology, Uppsala University, Sweden. 2. Ann-Christine Syvänen, Department of Medical Sciences, Molecular Medicine and Science for Life Laboratory, Uppsala University, Sweden. 3. Argyri Mathioudaki, Fabiana H. G. Farias, Jennifer Meadows, Jessika Nordin, Lina Hultin-Rosenberg, Matteo Bianchi, Science for Life Laboratory, Department of Medical Biochemistry and Microbiology, Uppsala University, Sweden. 4. Kerstin Lindblad-Toh, Science for Life Laboratory, Department of Medical Biochemistry and Microbiology, Uppsala University, Sweden and Broad Institute of MIT and Harvard, Boston, MA, USA. 5. Albin Björk, Guðný Ella Thorlacius, Ingrid E. Lundberg, Jorge I. Ramírez Sepúlveda, Marie Wahren-Herlenius, Marika Kvarnström, Department of Medicine, Rheumatology Unit, Karolinska Institutet, Stockholm, Sweden. 6. Daniel Eriksson, Department of Medicine, Karolinska Institutet and Department of Endocrinology, Metabolism and Diabetes, Karolinska University Hospital, Sweden. 7. Helena Forsblad-d’Elia, Department of Public Health and Clinical Medicine/Rheumatology, Umeå University, Sweden and Department of Rheumatology and Inflammation Research, Institute of Medicine, Sahlgrenska Academy at University of Gothenburg, Sweden. 8. Per Eriksson, Christopher Sjöwall, Department of Clinical and Experimental Medicine, Rheumatology/Division of Neuro and Inflammation Sciences, Linköping University, Sweden. 9. Elke Theander, Thomas Mandl, Department of Rheumatology, Skåne University Hospital Malmö/Lund University, Sweden. 10. Solbritt Rantapää-Dahlqvist, Department of Public Health and Clinical Medicine/Rheumatology, Umeå University, Sweden. 11. Sara Magnusson Bucher, Department of Rheumatology, Faculty of Medicine and Health, Örebro University, Sweden. 12. Daniel Hammenfors, Karl A. Brokstad, Kathrine Skarstein, Roland Jonsson, Silke Appel, Broegelmann Research Laboratory, Department of Clinical Science, University of Bergen, and Department of Rheumatology, Haukeland University Hospital, Bergen, Norway. 13. Malin V. Jonsson, Section for Oral and Maxillofacial Radiology, Department of Clinical Dentistry, University of Bergen, Norway. 14. Johan G. Brun, Department of Rheumatology, Haukeland University Hospital, University of Bergen, Norway. 15. Katrine Brække Norheim, Roald Omdal, Svein Joar Auglænd Johnsen, Clinical Immunology unit, Department of Internal Medicine, Stavanger University Hospital, Norway. 16. Øyvind Palm, Oslo University Hospital, Norway, 17. Janicke Liaaen Jensen, Lara Adnan Aqrawi, University of Oslo, Norway.
The ImmunoArray consortium: Principal InvestigatorKerstin Lindblad-Toh, Science for Life Laboratory, Department of Medical Biochemistry and Microbiology, Uppsala University, Sweden & Broad Institute of MIT and Harvard, Cambridge, MA, USA, Laboratory Protocol Principal InvestigatorGerli Rosengren Pielberg, Science for Life Laboratory, Department of Medical Biochemistry and Microbiology, Uppsala University, Sweden, Laboratory ProtocolEva Murén, Åsa Karlsson, Science for Life Laboratory, Department of Medical Biochemistry and Microbiology, Uppsala University, Sweden, ContributorsGöran Andersson, Department of Animal Breeding and Genetics, Swedish University of Agricultural Sciences, Uppsala, Sweden. Kerstin M. Ahlgren, Department of Surgical Sciences, Uppsala University, Sweden. Anna Lobell, Office for Medicine and Pharmacy, Uppsala University, Sweden. Lars Rönnblom, Maija-Leena Eloranta, Science for Life Laboratory, Department of Medical Sciences, Rheumatology, Uppsala University, Sweden. Peter Söderkvist, Department of Clinical and Experimental Medicine, Linköping University, Sweden. Olle Kämpe, Department of Medicine, Center for Molecular Medicine, Karolinska Institutet, Stockholm, Sweden and Department of Endocrinology, Metabolism and Diabetes Karolinska University Hospital, Stockholm, Sweden and Science for Life Laboratory, Department of Medical Sciences, Uppsala University, Sweden. Nils Landegren, Department of Medicine, Center for Molecular Medicine, Karolinska Institutet, Stockholm, Sweden and Science for Life Laboratory, Department of Medical Sciences, Uppsala University, Sweden.
Funding: The Swedish Research Council for Medicine and Health, the Swedish Rheumatism Association, King Gustav V’s 80-year Foundation, the Knut and Alice Wallenberg Foundation, AstraZeneca-Science for Life Laboratory Research Collaboration grant (DISSECT).
Disclosure statement: E.T. is Medical Advisor at Janssen Cilag and T.M. is a Medical Advisor at Novartis. The other authors have declared no conflicts of interest.
Supplementary data
Supplementary data are available at Rheumatology online.
Contributor Information
the DISSECT consortium:
Andrei Alexsson, Pascal Pucholt, Carin Backlin, Eva Baecklund, Gunnel Nordmark, Johanna K Sandling, Juliana Imgenberg-Kreuz, Lars Rönnblom, Lilian Vasaitis, Maija-Leena Eloranta, Ann-Christine Syvänen, Argyri Mathioudaki, Fabiana H G Farias, Jennifer Meadows, Jessika Nordin, Lina Hultin-Rosenberg, Matteo Bianchi, Kerstin Lindblad-Toh, Albin Björk, Guðný Ella Thorlacius, Ingrid E Lundberg, Jorge I Ramírez Sepúlveda, Marie Wahren-Herlenius, Marika Kvarnström, Daniel Eriksson, Helena Forsblad-d’Elia, Per Eriksson, Christopher Sjöwall, Elke Theander, Thomas Mandl, Solbritt Rantapää-Dahlqvist, Sara Magnusson Bucher, Daniel Hammenfors, Karl A Brokstad, Kathrine Skarstein, Roland Jonsson, Silke Appel, Malin V Jonsson, Johan G Brun, Katrine Brække Norheim, Roald Omdal, Svein Joar Auglænd Johnsen, Øyvind Palm, Janicke Liaaen Jensen, and Lara Adnan Aqrawi
the ImmunoArray consortium:
Kerstin Lindblad-Toh, Gerli Rosengren Pielberg, Eva Murén, Åsa Karlsson, Göran Andersson, Kerstin M Ahlgren, Anna Lobell, Lars Rönnblom, Maija-Leena Eloranta, Peter Söderkvist, Olle Kämpe, and Nils Landegren
References
- 1. Brito-Zeron P, Acar-Denizli N, Ng WF. et al. How immunological profile drives clinical phenotype of primary Sjögren’s syndrome at diagnosis: analysis of 10,500 patients (Sjögren Big Data Project). Clin Exp Rheumatol 2018;36(Suppl 112):102–12. [PubMed] [Google Scholar]
- 2. Kvarnstrom M, Ottosson V, Nordmark B. et al. Incident cases of primary Sjögren’s syndrome during a 5-year period in Stockholm County: a descriptive study of the patients and their characteristics. Scand J Rheumatol 2015;44:135–42. [DOI] [PubMed] [Google Scholar]
- 3. Shiboski CH, Shiboski SC, Seror R. et al. 2016 American College of Rheumatology/European League Against Rheumatism classification criteria for primary Sjögren’s syndrome: a consensus and data-driven methodology involving three international patient cohorts. Ann Rheum Dis 2017;76:9–16. [DOI] [PubMed] [Google Scholar]
- 4. Vitali C, Bombardieri S, Jonsson R. et al. Classification criteria for Sjögren’s syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 2002;61:554–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mariette X, Criswell LA.. Primary Sjögren’s syndrome. N Engl J Med 2018;378:931–9. [DOI] [PubMed] [Google Scholar]
- 6. Tarn JR, Howard-Tripp N, Lendrem DW. et al. Symptom-based stratification of patients with primary Sjögren’s syndrome: multi-dimensional characterisation of international observational cohorts and reanalyses of randomised clinical trials. Lancet Rheumatol 2019;1:e85–e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baldini C, Ferro F, Elefante E. et al. Biomarkers for Sjogren’s syndrome. Biomark Med 2018;12:275–86. [DOI] [PubMed] [Google Scholar]
- 8. Imgenberg-Kreuz J, Rasmussen A, Sivils K. et al. Genetics and epigenetics in primary Sjögren’s syndrome. Rheumatology (Oxford) 2019. (in press) doi: 10.1093/rheumatology/key330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thorlacius GE, Wahren-Herlenius M, Ronnblom L.. An update on the role of type I interferons in systemic lupus erythematosus and Sjögren’s syndrome. Curr Opin Rheumatol 2018;30:471–81. [DOI] [PubMed] [Google Scholar]
- 10. Lessard CJ, Li H, Adrianto I. et al. Variants at multiple loci implicated in both innate and adaptive immune responses are associated with Sjögren’s syndrome. Nat Genet 2013;45:1284–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Li Y, Zhang K, Chen H. et al. A genome-wide association study in Han Chinese identifies a susceptibility locus for primary Sjögren’s syndrome at 7q11.23. Nat Genet 2013;45:1361–5. [DOI] [PubMed] [Google Scholar]
- 12. Nordmark G, Kristjansdottir G, Theander E. et al. Additive effects of the major risk alleles of IRF5 and STAT4 in primary Sjögren’s syndrome. Genes Immun 2009;10:68–76. [DOI] [PubMed] [Google Scholar]
- 13. Taylor KE, Wong Q, Levine DM. et al. Genome-wide association analysis reveals genetic heterogeneity of Sjögren’s syndrome according to ancestry. Arthritis Rheumatol 2017;69:1294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lind L, Fors N, Hall J. et al. A comparison of three different methods to determine arterial compliance in the elderly: the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) study. J Hypertens 2006;24:1075–82. [DOI] [PubMed] [Google Scholar]
- 15. Magnusson PK, Almqvist C, Rahman I. et al. The Swedish Twin Registry: establishment of a biobank and other recent developments. Twin Res Hum Genet 2013;16:317–29. [DOI] [PubMed] [Google Scholar]
- 16. McCarthy S, Das S, Kretzschmar W. et al. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016;48:1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Purcell S, Neale B, Todd-Brown K. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ihaka R, Gentleman R.. R: A Language for data analysis and graphics. J Comput Graph Stat 1996;5:299–314. [Google Scholar]
- 19. Cingolani P, Platts A, Wang le L. et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mofors J, Holmqvist M, Westermark L. et al. Concomitant Ro/SSA and La/SSB antibodies are biomarkers for the risk of venous thromboembolism and cerebral infarction in primary Sjögren’s syndrome. J Intern Med 2019;286:458–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gottenberg JE, Busson M, Loiseau P. et al. In primary Sjögren’s syndrome, HLA class II is associated exclusively with autoantibody production and spreading of the autoimmune response. Arthritis Rheum 2003;48:2240–5. [DOI] [PubMed] [Google Scholar]
- 22. Miyagawa S, Shinohara K, Nakajima M. et al. Polymorphisms of HLA class II genes and autoimmune responses to Ro/SS-A-La/SS-B among Japanese subjects. Arthritis Rheum 1998;41:927–34. [DOI] [PubMed] [Google Scholar]
- 23. Tzioufas AG, Wassmuth R, Dafni UG. et al. Clinical, immunological, and immunogenetic aspects of autoantibody production against Ro/SSA, La/SSB and their linear epitopes in primary Sjögren’s syndrome (pSS): a European multicentre study. Ann Rheum Dis 2002;61:398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robles EF, Mena-Varas M, Barrio L. et al. Homeobox NKX2-3 promotes marginal-zone lymphomagenesis by activating B-cell receptor signalling and shaping lymphocyte dynamics. Nat Commun 2016;7:11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lu X, Tang L, Li K. et al. Contribution of NKX2-3 polymorphisms to inflammatory bowel diseases: a meta-analysis of 35358 subjects. Sci Rep 2015;4:3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bodewes ILA, Al-Ali S, van Helden-Meeuwsen CG. et al. Systemic interferon type I and type II signatures in primary Sjögren’s syndrome reveal differences in biological disease activity. Rheumatology (Oxford) 2018;57:921–30. [DOI] [PubMed] [Google Scholar]
- 27. Båve U, Nordmark G, Lövgren T. et al. Activation of the type I interferon system in primary Sjögren’s syndrome: a possible etiopathogenic mechanism. Arthritis Rheum 2005;52:1185–95. [DOI] [PubMed] [Google Scholar]
- 28. Brkic Z, Maria NI, van Helden-Meeuwsen CG. et al. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjögren’s syndrome and association with disease activity and BAFF gene expression. Ann Rheum Dis 2013;72:728–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Imgenberg-Kreuz J, Sandling JK, Almlof JC. et al. Genome-wide DNA methylation analysis in multiple tissues in primary Sjögren’s syndrome reveals regulatory effects at interferon-induced genes. Ann Rheum Dis 2016;75:2029–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jonsson R, Theander E, Sjostrom B. et al. Autoantibodies present before symptom onset in primary Sjögren syndrome. JAMA 2013;310:1854–5. [DOI] [PubMed] [Google Scholar]
- 31. Barturen G, Babaei S, Català-Moll F. et al. Integrative analysis reveals a molecular stratification of systemic autoimmune diseases. medRxiv 2020.02.21.20021618 [preprint]. [DOI] [PubMed] [Google Scholar]
- 32. Bodewes ILA, Versnel MA.. Interferon activation in primary Sjögren’s syndrome: recent insights and future perspective as novel treatment target. Expert Rev Clin Immunol 2018;14:817–29. [DOI] [PubMed] [Google Scholar]
- 33. Nordmark G, Eloranta ML, Rönnblom L.. Primary Sjögren’s syndrome and the type I interferon system. Curr Pharm Biotechnol 2012;13:2054–62. [DOI] [PubMed] [Google Scholar]
- 34. Davidson BK, Kelly CA, Griffiths ID.. Primary Sjögren’s syndrome in the North East of England: a long-term follow-up study. Rheumatology (Oxford) 1999;38:245–53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.