

Supplemental Digital Content is available in the text
Keywords: bone marrow stromal cells, exosomes, miRNA, multiple myeloma
Abstract
Interaction with bone marrow stromal cells (BMSCs) has been suggested as an important mechanism for the progression of multiple myeloma (MM) cells, while exosomes are crucial mediators for cell-to-cell communication. The study was to investigate the miRNA profile changes in exosomes released by BMSCs of MM patients and explore their possible function roles.
The microarray datasets of exosomal miRNAs in BMSCs were downloaded from the Gene Expression Omnibus database (GSE110271: 6 MM patients, 2 healthy donors; GSE78865: 4 donors and 2 MM patients; GSE39571: 7 MM patients and 4 controls). The differentially expressed miRNAs (DEMs) were identified using the LIMMA method. The target genes of DEMs were predicted by the miRwalk 2.0 database and the hub genes were screened by constructing the protein–protein interaction (PPI) network, module analysis and overlapping with the differentially expressed genes (DEGs) after overexpression or knockout of miRNAs.
Three downregulated DEMs were found to distinguish MM from normal and MM-MGUS controls in the GSE39571 dataset; one downregulated and one upregulated DEMs (hsa-miR-10a) could differentiate MM from normal and MM-MGUS controls in the GSE110271-GSE78865 merged dataset. Furthermore, 11 downregulated (hsa-miR-16) and 1 upregulated DEMs were shared between GSE39571 and merged dataset when comparing MM with normal samples. The target genes were predicted for these 17 DEMs. PPI with module analysis showed IGF1R and CCND1 were hub genes and regulated by hsa-miR-16. Furthermore, EPHA8 was identified as a DEG that was downregulated in MM cells when the use of has-miR-10a mimics; while IGF1R, CCND1, CUL3, and ELAVL1 were also screened as DEGs that were upregulated in MM cells when silencing of hsa-miR-16.
BMSCs-derived exosomal miR-10a and miR-16 may be involved in MM progression by regulating EPHA8 or IGF1R/CCND1/CUL3/ELAVL1, respectively. These exosomal miRNAs or genes may represent potential biomarkers for diagnosis of MM and prediction of progression and targets for developing therapeutic drugs.
1. Introduction
Multiple myeloma (MM) is a common hematological cancer, with an estimated incidence of 4.7 to 5.0/100,000 person-years that accounts for about 13% of all hematological neoplasms.[1] MM is characterized by accumulation of monoclonal plasma cells in bone marrow (BM), which frequently leads to osteolysis, anemia, renal impairment, and immune dysfunction, causing the death or disability of patients.[1] Despite advances have been made in therapies (i.e., chemotherapy, plasmapheresis, autologous stem cell transplantation), MM remains an incurable disease and the 5-year survival rate is lower than 50%.[1,2] Thus, there is an urgent need to further understand the molecular mechanisms of MM to develop novel therapeutic strategies.
Accumulating evidence suggests that interaction with BM mesenchymal stromal cells (BMSCs) may be an important mechanism for the progression of MM cells.[3–5] The main cause is that MSCs secrete various molecules to MM cells. For example, Zhang et al found that gap junction gene Cx43 was upregulated in BMSCs of patients with MM compared with BMSCs of normal donors. Co-culture with BMSCs promoted the migration of MM cells, while this process can be inhibited by the use of gap junction inhibitor 18α-glycyrrhetinic acid.[6] Corre et al identified that the concentration of interleukin (IL)-6 was higher in MM BMSCs than that of normal BMSCs.[7] Co-culture with MM BMSCs increased proliferation of MM cells, while the addition of an anti-IL-6 monoclonal antibody[7] or siRNA[8] in MSCs inhibited MM cell growth in vitro and in vivo. The study of Zhao et al showed that BMSCs co-culture could upregulate the expression of key stemness genes in MM cells by Bruton tyrosine kinase signal pathway and increase their clonogenicity.[9] Shen et al demonstrated that microRNA (miRNA)-202 was weakly expressed in BMSCs of MM patients. Overexpression of miR-202 in BMSCs and then co-culture with MM cells significantly inhibited MM cell survival and growth.[10] These findings imply that genes secreted by BMSCs may be potential targets for the treatment of MM. However, there is a question unanswered that how these BMSCs genes are transferred to MM.
Recently, cell-to-cell communication mediated by exosomes (also known as microvesicles), naturally occurring 40- to 100-nm membrane vesicles of endocytic origin that carry messenger RNAs (mRNAs), miRNAs, and proteins, are gaining much attention. There has a study to show that PSMA3 and PSMA3-AS1 in MSCs could be packaged into exosomes and transferred to MM cells, thus promoting proteasome inhibitor resistance. Intravenous administration of siPSMA3-AS1 increased the chemotherapy sensitivity in mice.[11] Roccaro et al identified that miR-15a was downregulated in MM BMSC-derived exosomes compared with normal BMSCs. Overexpression of miR-15a in exosomes isolated from BMSCs significantly inhibited MM cell proliferation.[12] The results of Umezu et al showed that exosomal miR-10a expression was high in MM-BMSCs. The proliferation of MM cells was enhanced when miR-10a derived from MM-BMSCs was transferred.[13] MM MSCs was also proved to induce a rapid and continued activation of mitogen-activated protein kinases and translation initiation in MM cells, which promoted cell viability, proliferation and migration.[14] However, the exosomal genes released by BMSCs remain rarely studied in MM. This study was to further investigate the miRNA profile changes in exosomes released by BMSCs of MM patients and explored their possible function roles.
2. Materials and methods
2.1. Microarray data of exosomal miRNAs
The microarray datasets of exosomal miRNAs in BMSCs were downloaded from the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/) under accession number GSE110271,[13] GSE78865,[15] and GSE39571.[12] The GSE110271 dataset[13] analyzed the miRNA profile changes in BMSCs derived from 6 MM patients, 2 healthy donors, and 3 monoclonal gammopathy of undetermined significance (MGUS) patients. The GSE78865 dataset[13,15] included human BMSCs samples derived from 4 donors and 2 MM patients. GSE110271 and GSE78865 datasets were provided by the same author (Umezu T) and done using the same microarray platform (GPL13987, Taqman Array Human MicroRNA A Card v2.0) and they were merged for the following analysis. In the GSE39571 dataset,[12] exosomes were isolated from cell culture supernatant of BMSCs in 7 MM patients, 2 MGUS patients, and 4 normal controls and then miRNA profiling was determined by using Applied Biosystems Human TaqMan Low Density Array (platform, GPL15838TLDA). MGUS is generally defined as asymptomatic premalignant clonal plasma cell disorder and should be identified from MM. Thus, we also included the samples from these patients. Patient consent and ethical approval were not necessary since this is a study to analyze the data from the public database.
2.2. Differential analysis for exosomal miRNAs
The series matrix data were downloaded and quantile normalized using the Linear Models for Microarray Data (LIMMA) package (version 3.38.3; https://bioconductor.org/packages/release/bioc/html/limma.html) in R (version 3.5.2; http://www.R-project.org/).[16] The differentially expressed miRNAs (DEMs) between MM and healthy donors as well as between MM and MGUS were identified using empirical Bayes moderated t-statistics from the LIMMA package in R. |log2fold change (FC)| > 1 and P < .05 were set as the threshold to define DEMs. Heat map was created using the “pheatmap” package (version: 1.0.8; https://cran.r-project.org/web/packages/pheatmap) based on the Euclidean distance. Moreover, Venn diagram (http://bioinformatics.psb.ugent.be/webtools/Venn/) was also drawn to compare the difference in different groups of the same dataset or the difference between two datasets.
2.3. Target gene prediction for exosomal miRNAs
The target genes of crucial exosomal DEMs were predicted using the miRwalk database (version 2.0; http://www.zmf.umm.uni-heidelberg.de/apps/zmf/mirwalk2),[17] which is a comprehensive archive for miRNA-target interactions predicted by 12 algorithms (miRWalk, MicroT4, miRanda, miRBridge, miRDB, miRMap, miRNAMap, PICTAR2, PITA, RNA22, RNAhybrid, and Targetscan). Only the genes that were predicted in ≥8 databases were considered to be targets of exosomal DEMs.
2.4. Function enrichment analysis of target genes
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses for target genes of exosomal DEMs were performed using the clusterProfiler tool (version 3.2.11; http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html).[18] False discovery rate (FDR) < 0.05 was considered to be statistically significant.
2.5. Protein–protein interaction (PPI) network
PPI pairs between target genes of crucial exosomal DEMs were collected from STRING (Search Tool for the Retrieval of Interacting Genes; version 10.0; http://string.db.org/) database.[19] Only interactions with combined score >0.4 were selected to construct the PPI network. Eight topology measures of the nodes (protein) in the PPI network were calculated using the CytoNCA plugin in the Cytoscape software (http://apps.cytoscape.org/apps/cytonca)[20] to screen hub genes, including subgragh centrality (SC), degree centrality (DC), eigenvector centrality (EC), betweenness centrality (BC), closeness centrality (CC), information centrality (IC), network centrality (NC) and local average connectivity (LAC). Moreover, several function-related modules were also extracted from the PPI network using the Molecular Complex Detection (MCODE; version, 1.4.2; http://apps.cytoscape.org/apps/mcode) plugin of the Cytoscape software. Modules with MCODE score > 4 and a node number > 6 were considered to be significant.
2.6. Validation of target genes of exosomal miRNAs in MM cells
The mRNA expression profile changes before or after miRNA treatment in MM cells were analyzed using the microarray data deposited in GEO, including the GSE118282 dataset[13] which analyzed the difference between MM cell lines RPMI8226, KMS-11 and U266) transfected with miR-10a mimic (n = 3) and scrambled control (n = 3), and the GSE24522 dataset which analyzed the difference between two MM cell lines (RPMI and MMS1) undergoing miR-16 knockout (with three repeat in each cell line, total n = 6) and scrambled control (with three repeat in each cell line, total n = 6). The differentially expressed genes (DEGs) between miRNA treatment and controls were identified by the LIMMA method or using GEO2R, which were then overlapped with the target genes of exosomal miRNAs.
3. Results
3.1. Differentially expressed exosomal miRNAs
Among the 644 miRNAs included in GSE39571 dataset, 68 of them were found to be differentially expressed in BMSCs of MM patients compared with the normal control, including 66 downregulated and 2 upregulated miRNAs (Table S1, http://links.lww.com/MD/F431); while in comparison with MGUS patients, 13 of them were found to be DEMs in BMSCs of MM patients, including 8 downregulated and 5 upregulated ones (Table S1, http://links.lww.com/MD/F431). Among the 376 miRNAs included in the merged dataset of GSE78865 and GSE110271, 97 and 160 miRNAs were respectively identified as downregulated and upregulated DEMs in BMSCs of MM patients compared with the normal control (Table S1, http://links.lww.com/MD/F431); while 1 and 16 miRNAs were screened as downregulated and upregulated DEMs between MM and MGUS patients, respectively (Table S1, http://links.lww.com/MD/F431). The heat maps of these DEMs in two datasets are shown in Figure 1.
Figure 1.
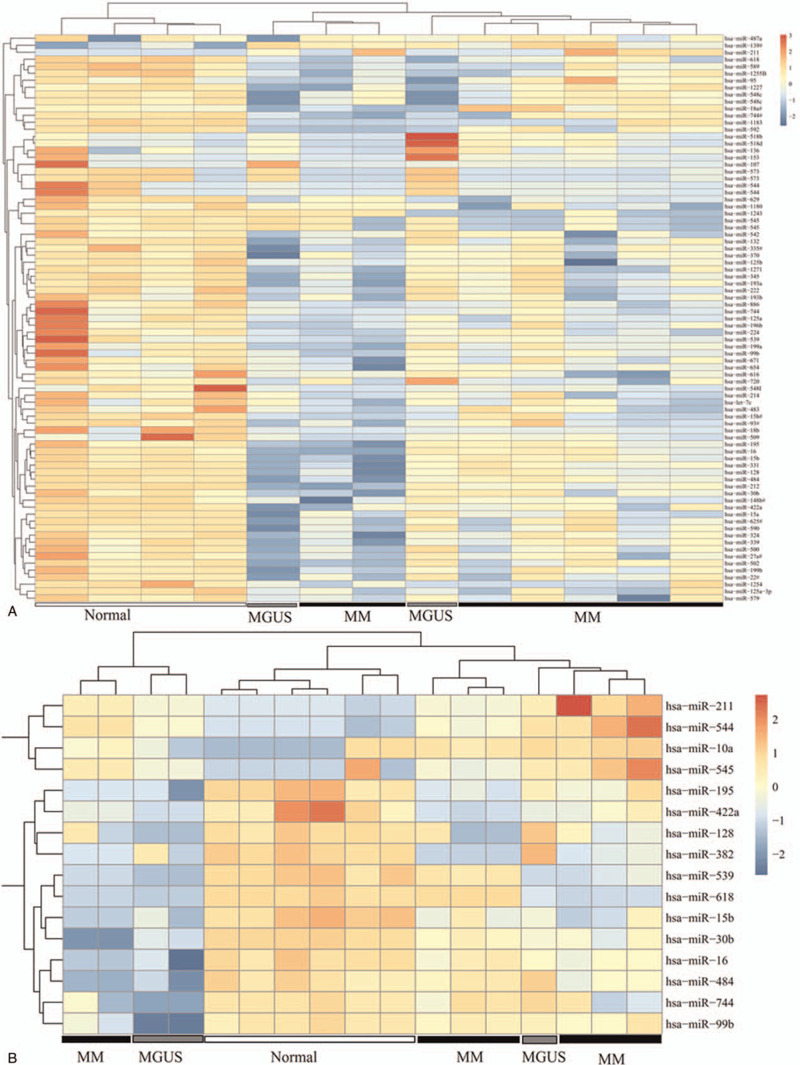
Hierarchical clustering and heat map analysis of differentially expressed miRNAs. (A) GSE39571; (B) merged GSE110271-GSE78865 dataset. The color to red, high expression; the color to light blue, low expression. MGUS = monoclonal gammopathy of undetermined significance, MM = multiple myeloma.
In order to screen crucial DEMs for MM, we first compared the MM-normal and MM-MGUS comparison groups in the GSE39571 and merged datasets. As a result, three downregulated miRNAs (hsa-miR-573, hsa-miR-544, and hsa-miR-545) were found to be common in MM-normal and MM-MGUS groups of the GSE39571 dataset (Fig. 2A; Table 1); one downregulated (hsa-miR-382) and one upregulated miRNA (hsa-miR-10a) were found to be shared in MM-normal and MM-MGUS groups of the merged dataset (Fig. 2B; Table 1). These findings indicated that these five miRNAs can distinguish MM from normal and MGUS controls. Then, we also compared the DEMs in two datasets in order to screen their overlap. The results showed that there were 1 commonly upregulated (hsa-miR-211) and 11 commonly downregulated (hsa-miR-618, hsa-miR-422a, hsa-miR-744, hsa-miR-128, hsa-miR-484, hsa-miR-539, hsa-miR-99b, hsa-miR-195, hsa-miR-30b, hsa-miR-16, and hsa-miR-15b) DEMs between the GSE39571 and merged datasets when comparing MM with normal samples (Fig. 2C; Table 1). No common DEMs were observed between the GSE39571 and merged datasets when comparing the MM with MGUS samples (Fig. 2D).
Figure 2.
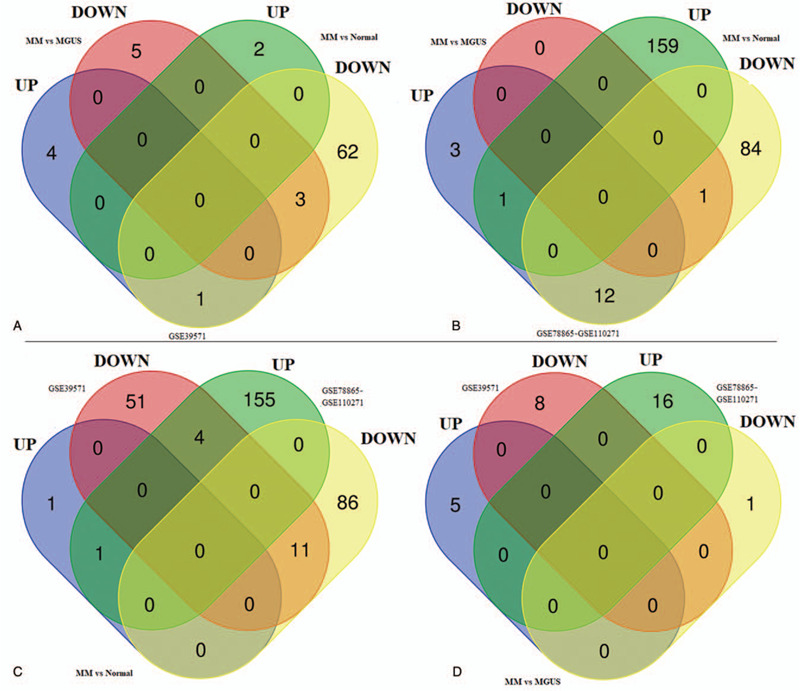
Venn diagram. (A) the shared differentially expressed miRNAs between MM-normal and MM-MGUS comparisons in GSE39571 dataset; (B) the shared differentially expressed miRNAs between MM-normal and MM-MGUS comparison in merged GSE110271-GSE78865 dataset; (C) common differentially expressed miRNAs between GSE39571 and merged dataset when comparing the MM with normal samples; (D) common differentially expressed miRNAs between GSE39571 and merged dataset when comparing the MM with MGUS samples. MGUS = monoclonal gammopathy of undetermined significance, MM = multiple myeloma.
Table 1.
Expression of exosomal miRNAs in BMSCs of patients with MM compared with normal control.
| GSE39571 | GSE78865-GSE110271 | |||||
| miRNA | logFC | P | FDR | logFC | P | FDR |
| hsa-miR-573 | −13.48 | 2.34E−03 | 0.24 | |||
| hsa-miR-544 | −3.51 | 4.28E−02 | 0.42 | |||
| hsa-miR-545 | −5.17 | 3.83E−03 | 0.24 | |||
| hsa-miR-382 | −8.38 | 2.44E−10 | 2.12E−08 | |||
| hsa-miR-10a | 5.97 | 6.67E−03 | 1.20E−02 | |||
| hsa-miR-618 | −3.20 | 8.15E−04 | 0.21 | −15.97 | 5.67E−03 | 1.03E−02 |
| hsa-miR-422a | −1.46 | 1.13E−02 | 0.31 | −3.45 | 5.25E−04 | 1.49E−03 |
| hsa-miR-744 | −1.16 | 1.28E−02 | 0.31 | −2.46 | 2.46E−02 | 3.88E−02 |
| hsa-miR-128 | −1.55 | 2.23E−02 | 0.37 | −4.19 | 1.10E−03 | 2.27E−03 |
| hsa-miR-484 | −1.21 | 3.39E−02 | 0.42 | −1.84 | 1.91E−02 | 3.05E−02 |
| hsa-miR-539 | −1.43 | 3.77E−02 | 0.42 | −4.14 | 6.67E−04 | 1.49E−03 |
| hsa-miR-99b | −1.10 | 3.88E−02 | 0.42 | −1.80 | 1.87E−02 | 3.00E−02 |
| hsa-miR-195 | −1.46 | 4.19E−02 | 0.42 | −2.32 | 4.48E−04 | 1.49E−03 |
| hsa-miR-30b | −1.18 | 4.56E−02 | 0.42 | −5.41 | 1.35E−03 | 2.71E−03 |
| hsa-miR-16 | −1.83 | 4.57E−02 | 0.42 | −2.43 | 2.38E−03 | 4.68E−03 |
| hsa-miR-15b | −1.51 | 4.96E−02 | 0.42 | −7.21 | 2.09E−05 | 3.60E−04 |
| hsa-miR-211 | 6.23 | 4.67E−02 | 0.42 | 2.64 | 2.54E−03 | 4.94E−03 |
BMSCs = bone marrow mesenchymal stromal cells, FC = fold change, FDR = false discovery rate, MM = multiple myeloma.
3.2. Target gene prediction of exosomal miRNAs
Using the miRwalk 2.0 database, 1371 genes were predicted to be regulated by 3 miRNAs (hsa-miR-573, hsa-miR-544, and hsa-miR-545) in the GSE39571 dataset (Table S2, http://links.lww.com/MD/F432); 341 and 236 genes were predicted to be regulated by hsa-miR-382 and hsa-miR-10a in the merged dataset, respectively (Table S2, http://links.lww.com/MD/F432); 1047 and 773 genes were predicted to be respectively modulated by 11 commonly upregulated miRNAs and 1 downregulated miRNA in two datasets (Table S2, http://links.lww.com/MD/F432).
3.3. Function enrichment analysis of target genes
The above target genes of exosomal DEMs were uploaded to the DAVID database to predict their function. The results showed that 12 significant GO biological process terms were enriched, such as GO:0000122∼negative regulation of transcription from RNA polymerase II promoter (CCND1, cyclin D1; CUL3, cullin 3), GO:0048013∼ephrin receptor signaling pathway (EPHA8, Eph tyrosine kinase receptor A8) and GO:0007165∼signal transduction (IGF1R, insulin like growth factor 1 receptor) (Table 2; Fig. 3A). Furthermore, 25 KEGG pathways were also enriched, such as hsa05205:Proteoglycans in cancer (CCND1, IGF1R), hsa04151:PI3K-Akt signaling pathway (IGF1R, CCND1), hsa04068:FoxO signaling pathway (IGF1R, CCND1), hsa04510:Focal adhesion (IGF1R, CCND1), hsa04360:Axon guidance (EPHA8), hsa04310:Wnt signaling pathway (CCND1), hsa04120:Ubiquitin mediated proteolysis (CUL3), hsa04152:AMPK signaling pathway (IGF1R, CCND1; ELAVL1, ELAV like RNA binding protein 1), et al (Table 2; Fig. 3B).
Table 2.
Function enrichment analysis for target genes of crucial miRNAs.
| Term | Genes | FDR | Count |
| GO:0045944∼positive regulation of transcription from RNA polymerase II promoter | CTNNB1, MAPK3, VEGFA, MAPK14, PIK3R1 | 3.89E−12 | 239 |
| GO:0000122∼negative regulation of transcription from RNA polymerase II promoter | CTNNB1, VEGFA, CCND1, CUL3 | 2.75E−07 | 172 |
| GO:0045893∼positive regulation of transcription, DNA-templated | CTNNB1, MAPK1, MAPK3, CDH1, IGF1 | 4.13E−07 | 132 |
| GO:0006468∼protein phosphorylation | MAPK1, MAPK3, MAPK8, CCND1, PIK3R1 | 1.79E−05 | 115 |
| GO:0048013∼ephrin receptor signaling pathway | CDC42, RHOA, EPHA8 | 4.66E−05 | 35 |
| GO:0007411∼axon guidance | BDNF, MAPK1, MAPK3, NRAS, EPHA8 | 2.09E−04 | 51 |
| GO:0006366∼transcription from RNA polymerase II promoter | MEF2C, AKNA, MEF2A, RNMT, LMO4, STAT5B, TBP, REST, FOXO3, FOXO4… | 2.15E−04 | 122 |
| GO:0035556∼intracellular signal transduction | MAPK14, MAP3K4, CRKL | 1.49E−03 | 98 |
| GO:0018105∼peptidyl-serine phosphorylation | MAPK1, MAPK14, MAPK3, MAPK8 | 6.29E−03 | 40 |
| GO:0061024∼membrane organization | SGCZ, YWHAZ, CCDC88A, PRKCI, CHP1, LPCAT2, YWHAE, YWHAG, YWHAH, YWHAQ… | 6.38E−03 | 17 |
| GO:0030182∼neuron differentiation | MEF2C, WNT5A, EDN3, COPS2, CDK5R1, WNT3A, MYEF2, HOXD1, ITM2C, WNT2… | 7.77E−03 | 33 |
| GO:0007165∼signal transduction | MAPK1, MAPK14, IGF1R, PIK3R1… | 2.59E−02 | 226 |
| hsa05205:Proteoglycans in cancer | CTNNB1, CDC42, RHOA, MAPK1, CCND1, VEGFA, MAPK3, IGF1R, PIK3R1, IGF1, NRAS, MAPK14 | 2.79E−07 | 68 |
| hsa04722:Neurotrophin signaling pathway | CDC42, BDNF, RHOA, PIK3R1, MAPK1, NRAS, MAPK14, MAPK3, MAPK8 | 1.79E−05 | 45 |
| hsa04010:MAPK signaling pathway | CDC42, BDNF, MAPK1, MAPK3, MAPK8, NRAS, MAPK14, MAP3K4 | 2.91E−05 | 75 |
| hsa05200:Pathways in cancer | CTNNB1, MAPK1, VEGFA, MAPK3, MAPK8, CDC42, RHOA, CCND1, CDH1, IGF1R, PIK3R1, IGF1, NRAS, LAMC1 | 6.93E−05 | 103 |
| hsa04360:Axon guidance | CDC42, RHOA, MAPK1, NRAS, EPHA8, MAPK3 | 1.25E−04 | 45 |
| hsa04022:cGMP-PKG signaling pathway | RHOA, MAPK1, MAPK3 | 1.65E−04 | 52 |
| hsa04151:PI3K-Akt signaling pathway | MAPK1, CCND1, VEGFA, MAPK3, IGF1R, PIK3R1, IGF1, NRAS, LAMC1 | 1.68E−04 | 92 |
| hsa04068:FoxO signaling pathway | IGF1R, PIK3R1, IGF1, MAPK1, NRAS, CCND1, MAPK14, MAPK3, MAPK8 | 7.11E−04 | 45 |
| hsa04510:Focal adhesion | CTNNB1, CDC42, RHOA, MAPK1, CCND1, VEGFA, MAPK3, MAPK8, IGF1R, PIK3R1, IGF1, LAMC1 | 7.61E−04 | 61 |
| hsa04014:Ras signaling pathway | CDC42, RHOA, MAPK1, MAPK3, VEGFA, MAPK8, IGF1R, PIK3R1, IGF1, NRAS | 9.62E−04 | 65 |
| hsa04152:AMPK signaling pathway | IGF1R, PIK3R1, CCND1, ELAVL1 | 1.11E−03 | 42 |
| hsa04310:Wnt signaling pathway | CTNNB1, RHOA, CCND1, MAPK8 | 1.77E−03 | 45 |
| hsa05220:Chronic myeloid leukemia | PIK3R1, NRAS, MAPK1, CCND1, MAPK3, CRKL | 2.02E−03 | 29 |
| hsa04015:Rap1 signaling pathway | CTNNB1, CDC42, RHOA, MAPK1, MAPK3, VEGFA, CDH1, IGF1R, PIK3R1, IGF1, NRAS, MAPK14, CRKL | 3.48E−03 | 60 |
| hsa04611:Platelet activation | RHOA, PIK3R1, MAPK1, MAPK14, MAPK3 | 5.52E−03 | 42 |
| hsa04666:Fc gamma R-mediated phagocytosis | CDC42, PIK3R1, MAPK1, MAPK3, CRKL | 6.84E−03 | 31 |
| hsa05214:Glioma | IGF1R, PIK3R1, IGF1, NRAS, MAPK1, CCND1, MAPK3 | 9.25E−03 | 26 |
| hsa05211:Renal cell carcinoma | CDC42, PIK3R1, NRAS, MAPK1, VEGFA, MAPK3, CRKL | 1.27E−02 | 26 |
| hsa04550:Signaling pathways regulating pluripotency of stem cells | CTNNB1, IGF1R, PIK3R1, IGF1, MAPK1, NRAS, MAPK14, MAPK3 | 1.72E−02 | 43 |
| hsa04520:Adherens junction | CDH1, CTNNB1, CDC42, IGF1R, RHOA, MAPK1, MAPK3 | 1.73E−02 | 27 |
| hsa04066:HIF-1 signaling pathway | IGF1R, PIK3R1, IGF1, MAPK1, VEGFA, MAPK3 | 1.76E−02 | 33 |
| hsa04120:Ubiquitin mediated proteolysis | UBE2Z, XIAP, UBE2G1, BTRC, UBA6, SAE1, UBE3C, UBE2R2, PRPF19, CUL3… | 2.32E−02 | 42 |
| hsa04114:Oocyte meiosis | IGF1R, IGF1, MAPK1, MAPK3 | 2.78E−02 | 36 |
| hsa04668:TNF signaling pathway | PIK3R1, MAPK1, MAPK14, MAPK3, MAPK8 | 3.03E−02 | 35 |
| hsa04012:ErbB signaling pathway | PIK3R1, MAPK1, NRAS, MAPK3, MAPK8, CRKL | 4.44E−02 | 30 |
FDR = false discovery rate, GO = Gene ontology, KEGG = Kyoto encyclopedia of genes and genomes. Only the hub genes enriched in GO or pathways were listed.
Figure 3.
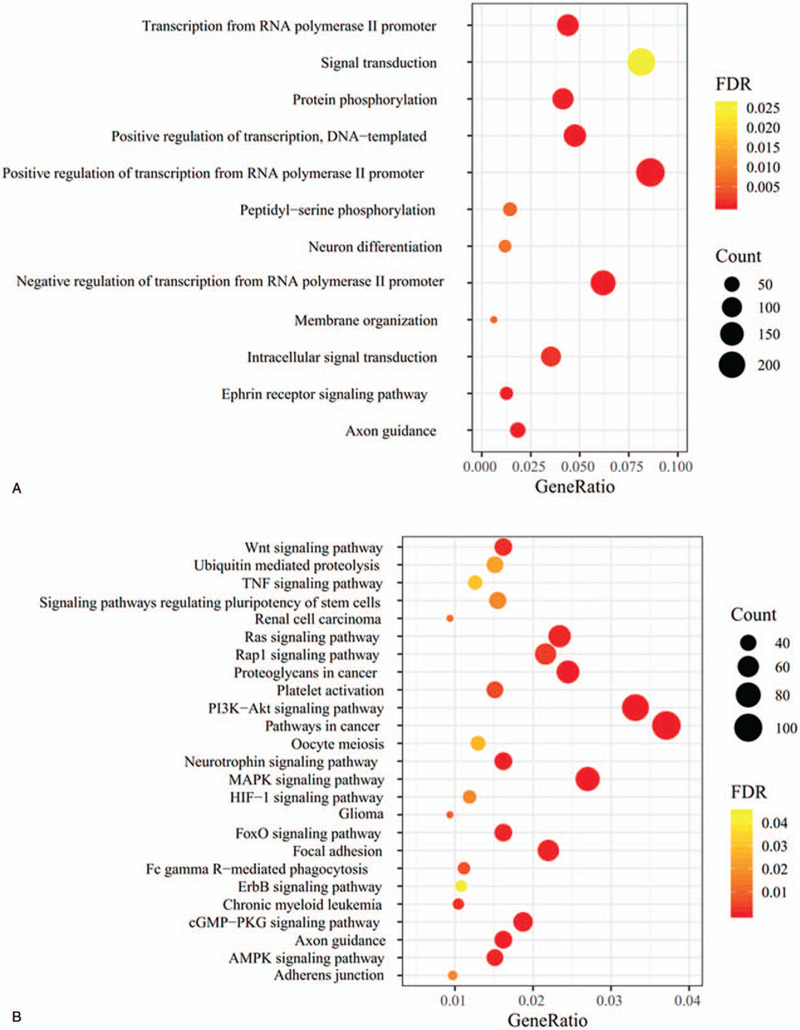
Function enrichment analysis for target genes of differentially expressed miRNAs. (A) GO terms; (B) KEGG pathways. GO = gene ontology, KEGG = Kyoto Encyclopedia of Genes and Genomes.
3.4. Selection of crucial target genes of exosomal DEMs by PPI network
The PPI data of the 420 genes enriched in the above 25 KEGG pathways were collected from the STRING database, including 6346 interaction pairs among 419 nodes (such as CUL3-CCND1, EPHA8-SEMA5A [semaphorin 5A], ELAVL1-IGF1R/CCND1) (Table S3, http://links.lww.com/MD/F433). CCND1 may be a hub gene in the PPI network because it ranked the top 20 in all eight topological measures (Table 3). Furthermore, IGF1R was also important because it ranked the top 20 in seven topological measures except LAC (Table 3). In addition, six significant modules (Fig. 4; Table 4) were extracted from this PPI network. Both of the above hub genes were enriched in significant modules: hub gene IGF1R was included in module 2; hub gene CCND1 was included in module 3; their interacted CUL3 and ELAVL1 were respectively enriched in module 1 and 4. EPHA8 was also enriched in module 3. Thus, the miRNA–mRNA regulatory network was constructed using the 96 genes of the significant modules and their corresponding 10 miRNAs (Fig. 5), such as hsa-miR-16-5p-CCND1/IGF1R/CUL3/ELAVL1 and hsa-miR-10a-5p-EPHA8.
Table 3.
Topology measures for nodes of the PPI network.
| SC | DC | EC | IC | LAC | BC | CC | NC | ||||||||
| MAPK3 | 1.37E+21 | MAPK3 | 171 | MAPK3 | 0.19 | MAPK3 | 14.89 | UBE2Q1 | 30 | MAPK3 | 11987.87 | MAPK3 | 0.62 | MAPK3 | 117.18 |
| MAPK1 | 1.06E+21 | MAPK1 | 148 | MAPK1 | 0.17 | MAPK1 | 14.69 | UBE3C | 29.88 | MAPK1 | 9465.96 | MAPK1 | 0.59 | MAPK1 | 94.16 |
| VEGFA | 1.04E+21 | RHOA | 135 | VEGFA | 0.17 | RHOA | 14.55 | UBE2J1 | 29.88 | CTNNB1 | 9329.19 | CTNNB1 | 0.59 | RHOA | 86.82 |
| RHOA | 9.10E+20 | CTNNB1 | 134 | RHOA | 0.15 | CTNNB1 | 14.54 | IGF1 | 29.46 | RHOA | 7752.85 | VEGFA | 0.59 | VEGFA | 82.56 |
| CTNNB1 | 9.07E+20 | VEGFA | 133 | CTNNB1 | 0.15 | VEGFA | 14.53 | VEGFA | 29.05 | VEGFA | 6987.20 | RHOA | 0.58 | CTNNB1 | 79.70 |
| CDC42 | 8.33E+20 | CDC42 | 121 | CDC42 | 0.15 | CDC42 | 14.38 | UBE2Z | 28.97 | MAPK8 | 6486.17 | MAPK8 | 0.57 | CDC42 | 72.93 |
| MAPK8 | 8.15E+20 | MAPK8 | 120 | MAPK8 | 0.15 | MAPK8 | 14.36 | MAPK3 | 28.76 | CDC42 | 5282.59 | CDC42 | 0.57 | MAPK8 | 69.27 |
| PIK3R1 | 7.49E+20 | CDH1 | 109 | PIK3R1 | 0.14 | CDH1 | 14.20 | KLHL13 | 28.48 | CDH1 | 4037.51 | CDH1 | 0.57 | CDH1 | 61.09 |
| CDH1 | 7.42E+20 | PIK3R1 | 103 | CDH1 | 0.14 | PIK3R1 | 14.09 | UBE4A | 28.24 | PRKCA | 3062.30 | NRAS | 0.55 | PIK3R1 | 57.98 |
| IGF1 | 7.35E+20 | NRAS | 100 | IGF1 | 0.14 | NRAS | 14.04 | UBE2H | 28.24 | RAC1 | 2845.44 | PIK3R1 | 0.55 | IGF1 | 56.48 |
| CCND1 | 6.48E+20 | IGF1 | 97 | CCND1 | 0.13 | IGF1 | 13.98 | UBE2R2 | 28.24 | NRAS | 2794.93 | MAPK14 | 0.55 | NRAS | 53.36 |
| NRAS | 6.40E+20 | CCND1 | 94 | NRAS | 0.13 | CCND1 | 13.91 | FGF2 | 28.16 | SIRT1 | 2703.91 | IGF1 | 0.55 | CCND1 | 53.28 |
| IGF1R | 6.34E+20 | MAPK14 | 94 | IGF1R | 0.13 | MAPK14 | 13.91 | SHC1 | 27.95 | MAPK14 | 2595.51 | CCND1 | 0.55 | FGF2 | 50.07 |
| FGF2 | 6.08E+20 | RAC1 | 91 | FGF2 | 0.13 | RAC1 | 13.85 | TRIM37 | 27.94 | BDNF | 2517.18 | FGF2 | 0.55 | MAPK14 | 49.67 |
| MAPK14 | 5.63E+20 | IGF1R | 88 | MAPK14 | 0.12 | IGF1R | 13.78 | PIK3R1 | 27.61 | PIK3R1 | 2473.35 | IGF1R | 0.54 | RAC1 | 47.68 |
| KDR | 5.56E+20 | BDNF | 88 | KDR | 0.12 | BDNF | 13.78 | CCND1 | 27.49 | CCND1 | 2427.10 | MAP2K1 | 0.54 | BDNF | 47.07 |
| BDNF | 5.29E+20 | FGF2 | 87 | BDNF | 0.12 | FGF2 | 13.75 | UBE2W | 27.49 | IGF1R | 2356.76 | PRKCA | 0.54 | KDR | 44.41 |
| MAP2K1 | 5.19E+20 | KDR | 87 | MAP2K1 | 0.12 | KDR | 13.75 | UBE2G1 | 27.39 | IGF1 | 2353.36 | KDR | 0.54 | IGF1R | 43.30 |
| PTPN11 | 5.18E+20 | MAP2K1 | 82 | PTPN11 | 0.12 | MAP2K1 | 13.623 | CUL5 | 27.37 | SOCS1 | 2345.63 | RAC1 | 0.54 | MAP2K1 | 42.79 |
| SHC1 | 5.15E+20 | PRKCA | 81 | SHC1 | 0.12 | PRKCA | 13.60 | RHOA | 27.13 | PTPN11 | 2292.86 | BDNF | 0.54 | PTPN11 | 41.90 |
BC = betweenness centrality, CC = closeness centrality, DC = degree centrality, EC = eigenvector centrality, IC = information centrality, LAC = local average connectivity, NC = network centrality, PPI = Protein-protein interaction, SC = subgragh centrality.
Figure 4.
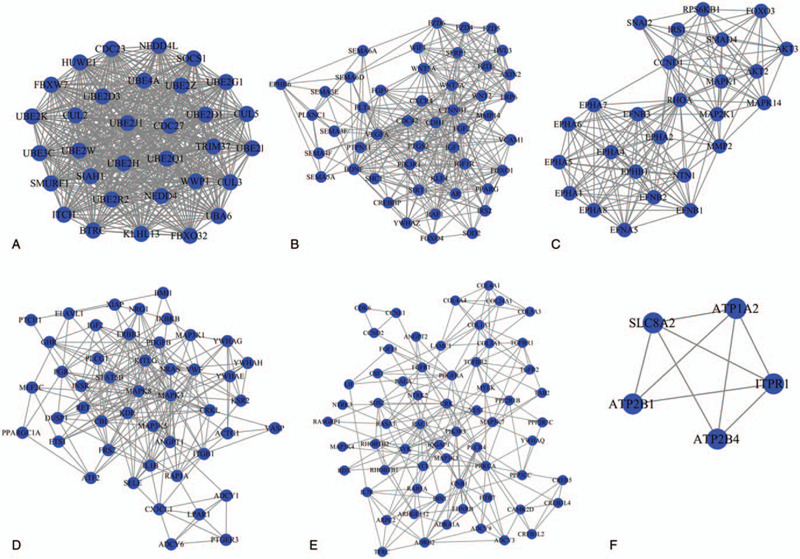
Significant modules extracted from the protein–protein interaction network. (A) Module 1; (B) module 2; (C) module 3; (D) module 4; (E) module 5; (F) module 6.
Table 4.
significant module screened from the PPI network.
| Cluster | Score (Density∗#Nodes) | Nodes | Edges | Node IDs |
| 1 | 32 | 32 | 496 | BTRC, WWP1, UBE2B, SMURF1, UBE2H, UBE2D1, UBE2W, CUL3, KLHL13, SOCS1, UBE2G1, UBE2R2, CUL2, HUWE1, UBE4A, UBE2D3, SIAH1, FBXO32, UBE2K, UBA6, UBE2J1, UBE2Z, FBXW7, NEDD4, ITCH, UBE3C, CUL5, UBE2Q1, CDC23, CDC27, TRIM37, NEDD4L |
| 2 | 16.085 | 48 | 378 | FOXO1, IRS2, WNT3A, FZD4, FZD6, WNT2, CDC42, BDNF, SIRT1, AXIN2, SOD2, VEGFA, FGF2, SEMA3F, PTGS2, VCAM1, PLXNC1, SEMA3E, AR, WNT5A, WIF1, FZD5, FGF9, RAF1, SEMA4F, KLF4, YWHAZ, CTNNB1, DVL3, PIK3R1, SEMA6D, CREBBP, IGF1R, LRP6, IGF1, SHC1, MMP14, SEMA6A, CXCR4, SEMA5A, CDH1, FOXO4, FLT1, FZD3, EPHB6, SFRP1, PPARG, PTPN11 |
| 3 | 13.6 | 26 | 170 | MAPK14, NTN1, EPHA6, FOXO3, AKT3, EPHA5, EPHB1, SNAI2, MAP2K1, RHOA, EFNA5, MAPK1, EPHA7, EPHA2, EFNB2, EPHA4, CCND1, EFNB3, RPS6KB1, MMP2, EPHA8, EFNB1, IRS1, AKT2, SMAD4, EPHA1 |
| 4 | 9.913 | 47 | 228 | VWF, KITLG, ATF2, KDR, BMI1, FRS2, XIAP, GHR, PGR, ELAVL1, YWHAE, LPAR1, KSR2, NRAS, VASP, ADCY1, DUSP1, RAP1A, PTGER3, MAP3K1, MEF2C, MAPK3, PLCG1, PTCH1, ANGPT1, IL1B, CRKL, ITGB1, INSR, ETS1, SELE, CBL, MAP3K5, MAPK8, IGF2, IKBKB, STAT5B, YWHAG, ADCY6, YWHAH, ACTG1, PPARGC1A, ERBB3, RET, CX3CL1, NRG1, PDGFB |
| 5 | 7.129 | 63 | 221 | CCNE1, RHOBTB1, HTR7, CAMK2D, SOS2, RHOBTB2, PLCB4, TGFB2, CCND2, RDX, COL5A3, MAP3K3, ARPC2, ADCY9, YWHAQ, RAC1, MYLK, CDK6, VCL, MAP3K7, FGF18, IL7R, GNB1, LIF, CREB3L4, COL4A4, RALA, PPP2R1B, PPP2R5C, COL24A1, EDNRB, CREB3L2, RAB5A, TFRC, NTRK3, NTRK2, RASGRP1, FGFR1, COL3A1, RASA1, ANGPT2, ADCY3, COL1A1, SYK, PRKCA, YES1, ARHGEF12, LAMC1, MAP3K4, IQGAP1, COL4A1, ADRA1A, PDGFRA, CRK, CREB5, PIK3R3, PPP3CC, ADRB2, TAB2, TGFBR2, TGFBR1, BIN1, CSF1 |
| 6 | 4.5 | 5 | 9 | ATP1A2, ATP2B1, ITPR1, ATP2B4, SLC8A2 |
Figure 5.
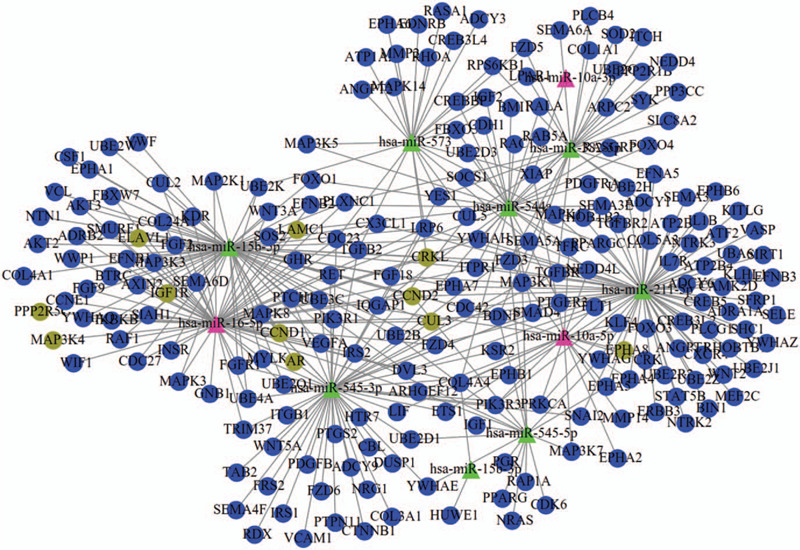
miRNA–mRNA regulatory network. Triangle, miRNAs; circular, target genes. Pink was miRNAs that had the target genes (brown green) to be demonstrated in validation datasets; green were miRNAs that had the target genes (blue) to be only predicted by miRwalk database.
3.5. Validation of the expression of target genes
Among these 10 miRNAs, 2 (hsa-miR-16 and hsa-miR-10a; both had the adjusted P-value < .05 in the analysis of GSE78865-GSE110271 dataset) were previously investigated to explore their downstream genes in MM cells using microarray data. Thus, we also analyzed the GSE118282 dataset to obtain the differentially expressed targets of hsa-miR-10a mimic treatment. Unfortunately, the results of GSE118282 dataset seemed to be poor and only one gene (PQLC2) was found to be significantly downregulated using the LIMMA method according to P-value < .05; while 73 were identified to be significantly downregulated using the GEO2R method. Among them, only EPHA8 (Table 5) was overlapped with the target genes of hsa-miR-10a in Figure 5. Thus, exosomal hsa-miR-10a in BMSCs may be involved in the progression of MM by downregulating EPHA8 in MM cells.
Table 5.
Overlapped differentially expressed genes regulated by miR-10a and miR-16.
| GSE118282 | GSE24522 | ||||||
| logFC | P | FDR | logFC | P | FDR | ||
| miR-10a | EPHA8(GEO2R) | −0.37 | 4.61E−02 | 1.00 | |||
| miR-16 | IGF1R(LIMMA) | 0.87 | 9.34E−04 | 0.08 | |||
| IGF1R(GEO2R) | 0.87 | 1.88E−05 | 2.97E−02 | ||||
| MAP3K4(LIMMA) | 0.28 | 5.02E−03 | 0.14 | ||||
| MAP3K4(GEO2R) | 0.32 | 5.96E−03 | 0.19 | ||||
| CUL3(LIMMA) | 0.27 | 7.92E−03 | 0.17 | ||||
| CUL3(GEO2R) | 0.46 | 2.75E−05 | 3.38E-02 | ||||
| CCND1(LIMMA) | 0.27 | 4.44E−02 | 0.33 | ||||
| CCND1 (GEO2R) | 0.50 | 7.01E−03 | 0.20 | ||||
| CRKL(LIMMA) | 0.24 | 2.73E−02 | 0.27 | ||||
| CRKL(GEO2R) | 0.34 | 3.65E−02 | 0.36 | ||||
| ELAVL1(LIMMA) | 0.23 | 4.36E−03 | 0.13 | ||||
| ELAVL1(GEO2R) | 0.32 | 1.05E−02 | 0.23 | ||||
| CCND2(LIMMA) | 0.23 | 9.90E−03 | 0.18 | ||||
| CCND2(GEO2R) | 0.34 | 1.80E−02 | 0.28 | ||||
| LAMC1 | 0.16 | 3.85E−02 | 0.31 | ||||
| AR(LIMMA) | 0.16 | 3.51E−02 | 0.30 | ||||
| AR(GEO2R) | 0.32 | 8.51E−03 | 0.21 | ||||
| PPP2R5C(LIMMA) | 0.15 | 3.25E−02 | 0.29 | ||||
| PPP2R5C(GEO2R) | 0.42 | 2.46E−02 | 0.31 | ||||
FC = fold change, FDR = false discovery rate.
GSE24522 explored the effects of hsa-miR-16 silencing on MM cells. The expression of a total of 3174 genes was found to be changed with P < .05 using the LIMMA method. Among them, 15 (including 10 upregulated) were overlapped with the target genes of hsa-miR-16 predicted by the miRwalk database (Table 5). Nine of 10 upregulated genes were also confirmed to be differentially expressed using the GEO2R method. Therefore, downregulated exosomal hsa-miR-16 in BMSCs may lead to the progression of MM by upregulating these 9 targets, especially IGF1R and CUL3 (which had the adjusted P-value < .05) in MM cells.
4. Discussion
Using three microarray datasets and a series of bioinformatic analyses, we identified upregulation of exosomal hsa-miR-10a and downregulation of exosomal hsa-miR-16 in BMSCs may be underlying mechanisms to result in the progression of MM cells. Upregulated hsa-miR-10a may be transferred to MM cells and inhibit the expression of EPHA8 and influence the ephrin receptor signaling pathway and axon guidance by interacting with downstream SEMA3F; while downregulated exosomal hsa-miR-16 lead to the high expression of CUL3 and ELAVL1, which may promote MM cell proliferation and metastasis by interacting with CCND1 and IGF1R/CCND1 and then activating AMPK and PI3K-Akt pathways.
Extensive studies have demonstrated that hsa-miR-10a may be an oncogenic miRNA for hematological cancers. Dumas et al reported that miR-10a was the most significantly upregulated miRNA in atypical myeloproliferative neoplasms.[21] Zhi et al detected that the serum level of miR-10a-5p was significantly higher in patients with acute myeloid leukemia than that of the controls.[22] miR-10a-5p expression can be used for diagnosis (with the area under the receiver operator characteristic curve ranging from 0.8129 to 0.9531) and prediction of poor overall survival for acute myeloid leukemia patients.[22,23] Transfection of miR-10a mimics promoted proliferation of leukemia cells,[24] while inhibition of miR-10a expression increased leukaemic cell death, cell cycle arrest and reduced clonogenic capacity.[25] Thus, we speculated that hsa-miR-10a transferred to MM cells by exosomes may also promote deterioration of MM, which was confirmed by the study of Umezu et al.[13] Furthermore, it is consensus that miRNAs play important roles in various physiological and pathological processes by negatively regulating gene expression at the post-transcriptional and/or translational level by binding with their 3′-untranslated region. Thus, the target genes of miR-10a were also explored previously to explain its function mechanisms. Tao et al used the luciferase activity assays to identify phosphatase and tensin homolog (PTEN) as a direct target of miR-10a in non-small cell lung cancer. Downregulation of PTEN by siRNA or miR-10a promoted the migration and invasion of SPC-A-1 cells.[26] Xiong et al confirmed that transcription factor activating protein 2 gamma (TFAP2C) was a target of miR-10a-5p. High miR-10a-5p expression levels and low TFAP2C expression levels were independent adverse prognostic factors in patients with human pancreatic ductal adenocarcinoma.[27] Targeting of PTEN, annexin A7, cyclin-dependent kinase 6 (CDK6) and transferrin receptor protein 1[13] by miR-10a were proved as important mechanisms for proliferation of MM cells. However, the downstream mechanisms of miR-10a in MM remain rarely reported. In this study, by miRwalk2 database prediction, PPI screening and expression profile validation after miR-10a overexpression, we inferred that EphA8, a receptor of ephrin, may be a potential target gene of miR-10a. Although this interaction between miR-10a and EphA8 was previously reported in glioma cells,[28] it was firstly demonstrated for MM. There was also no direct evidence to study the roles of EphA8 in MM. Nevertheless, the research on its family genes may indirectly indicate the function of EphA8 in MM. For example, Ding et al found that high expressed EphA4 promoted proliferation and cell adhesion-mediated drug resistance of MM cells via interacting with cell cycle gene CDK5 and then activating AKT pathway.[29] La Rocca et al found that EphA3 was overexpressed in MM-derived cell lines compared to healthy controls. Silencing of EphA3 by using siRNA and antibody significantly inhibited in vivo tumor growth in mouse xenograft models.[30] More interestingly, we predicted EphA8-mediated signaling pathway may be involved in MM progression by regulating the expression of an axon guidance molecule SEMA5A. SEMA5A was also identified to be highly expressed in newly diagnosed MM patients and led to their decreased survival time.[31] Ectopic expression of SEMA5A significantly enhanced tumorigenesis, growth and metastasis in vivo as well as proliferation and invasiveness in vitro for pancreatic cancer.[32] Our results also showed EphA8 was enriched in axon guidance pathway. Therefore, exosomes-mediated miR-10a-EphA8-SEMA5A axis may represent a novel, credible mechanism for explaining the progression of MM.
Previous research had verified the tumor suppressor roles of hsa-miR-16 in MM. miR-16 was found to be significantly decreased in MM specimens,[33] peripheral blood samples[34] and BM aspirates[35] compared with normal control. Overexpression of miR-16 significantly inhibited MM cell proliferation and angiogenesis in vitro and in vivo.[36,37] Experimental mechanism analyses showed that downregulated miR-16 may contribute to the progression of MM by upregulating calcineurin binding protein 1,[33] WT1[36] or vascular endothelial growth factor A.[37] miR-16 was also predicted to regulate IGF1R, CCND2 and CCND1 to participate in MM[34]; while these regulatory relationships had been validated in other cancers, such as osteosarcoma.[38,39] In line with these studies, we also identified these interaction axes by the miRwalk2 prediction and more importantly, our microarray analysis directly demonstrated IGF1R, CCND2, and CCND1 were upregulated when silencing of miR-16 in MM cells. Also, IGF1R, CCND2, and CCND1 also had been implied to be upregulated in MM[40–42]; amplification of them was a significantly unfavorable parameter with regard to shorter overall survival[40,42,43]; mechanism studies showed upon binding by IGF-1, IGF1R promoted the growth and migration of MM cells by activating the PI3K-Akt signaling pathway,[44] which subsequently upregulated cell cycle genes CCND2 and CCND1.[45,46] This signaling pathway was also enriched in our study by IGF1R and CCND1. In addition to these known target genes, our study also suggested CUL3 and ELAVL1 as underlying novel targets of miR-16 in MM. E3 ubiquitin ligase CUL3[47,48] had been correlated with other cancers, with high expressed cells showing higher proliferative, migration and invasive rates. However, no studies focused on its roles in MM and their interactions with miR-16 were also not reported. In our study, we predicted CUL3 may interact with CCND1. Its mechanism may be similar to CCNE1: Cul3 mediated N-terminal ubiquitinylation of cyclin D by recognition of the degron on cyclin D, which led to increased activity of cyclin D.[49] There was only one study to report the roles of ELAVL1, an RNA binding protein, in MM: it induced MM cell proliferation by interacting with FUT4 and then stabilizing its mRNA.[50] Thus, further mechanism investigation is still needed. In this study, we found ELAVL1 was enriched in AMPK pathway and interacted with IGF1R and CCND1. This theory can be indirectly explained by the fact that: Wang et al demonstrated AMPK activation decreased both the cytoplasmic ELAVL1 levels and associative transcripts encoding proliferative genes[51]; while activation of AMPK was reported to induce apoptosis of MM cells.[52] Accordingly, miR-16-IGF1R/CCND1, miR-16-CUL3-CCND1, and miR-16-ELAVL1-IGF1R/CCND1 interaction axes may represent potentially believable mechanisms for MM progression.
There were some limitations in this study. First, the sample size was not large, although 3 datasets were included in our study. Second, several diseases (severe burns, traumas, major bleeding, major surgery, sepsis, acute GvHD, and macrophage activation syndromes) are associated with hyper-inflammatory and hyper-proliferative pathways, which were also identified in our results (Table 2, such as TNF signaling pathway). Thus, in addition to healthy donor, these diseases should be selected as the controls in future high-throughput experiment (microarray or sequencing) to identify MM-specific genes. Third, although our study preliminarily demonstrated the crucial exosomal miRNAs may function by regulating their target genes in MM cells, the exosomal delivery mechanisms remained not directly investigated by co-culture experiments. Fourth, the expression and regulatory relationships between miR-10a and EphA8 as well as between miR-16 and CUL3/ELAVL1 need further experiment validation (such as PCR, Dual luciferase reporter assay, overexpression or knockout). Fifth, the diagnostic and prognostic values of these two exosomal miRNAs for MM need confirmation in a cohort of patients in our hospital.[53]
In conclusion, BMSCs-derived exosomal miR-10a and miR-16 may be involved in MM progression by regulating EPHA8-SEMA5A or CUL3-CCND1/ELAVL1-IGF1R, respectively. These exosomal miRNAs or genes may represent potential biomarkers for diagnosis of MM and prediction of progression and targets for developing therapeutic drugs. (Figure S1, http://links.lww.com/MD/F429, Figure S2, http://links.lww.com/MD/F430).
Author contributions
Conceptualization: Ye Peng, Xiaolu Song.
Data curation: Ye Peng, Xiaolu Song.
Formal analysis: Ye Peng, Xiaolu Song.
Investigation: Jianping Lan.
Methodology: Jianping Lan.
Resources: Xiaogang Wang.
Software: Xiaogang Wang.
Validation: Manling Wang.
Visualization: Manling Wang.
Writing – original draft: Ye Peng, Xiaolu Song.
Writing – review & editing: Ye Peng.
Glossary
Abbreviations: BC = betweenness centrality, BMSCs = bone marrow mesenchymal stromal cells, CC = closeness centrality, CCND1 = cyclin D1, CDK = cyclin-dependent kinase, CUL3 = cullin 3, DC = degree centrality, DEGs = differentially expressed genes, DEMs = differentially expressed miRNAs, EC = eigenvector centrality, ELAVL1 = ELAV like RNA binding protein 1, EPHA8 = Eph tyrosine kinase receptor A8, FC = fold change, FDR = false discovery rate, GEO = Gene Expression Omnibus, GO = Gene ontology, IC = information centrality, IGF1R = insulin like growth factor 1 receptor, IL-6 = interleukin-6, KEGG = Kyoto Encyclopedia of Genes and Genomes, LAC = local average connectivity, LIMMA = Linear Models for Microarray Data, MCODE = Molecular Complex Detection, MGUS = monoclonal gammopathy of undetermined significance, miRNA = microRNA, MM = multiple myeloma, mRNAs = messenger RNAs, NC = network centrality, PPI = protein–protein interaction, PTEN = phosphatase and tensin homolog, SC = subgragh centrality, SEMA5A = semaphorin 5A, STRING = Search Tool for the Retrieval of Interacting Genes.
References
- [1].Andres M, Feller A, Arndt V. Trends of incidence, mortality, and survival of multiple myeloma in Switzerland between 1994 and 2013. Cancer Epidemiol 2018;53:105–10. [DOI] [PubMed] [Google Scholar]
- [2].Costa LJ, Brill IK, Omel J, et al. Recent trends in multiple myeloma incidence and survival by age, race, and ethnicity in the United States. Blood Adv 2017;1:282–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Xu S, De Veirman K, De Becker A, et al. Mesenchymal stem cells in multiple myeloma: a therapeutical tool or target? Leukemia 2018;32:1500–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Tsukamoto S, Løvendorf MB, Park J, et al. Inhibition of microRNA-138 enhances bone formation in multiple myeloma bone marrow niche. Leukemia 2018;32:1739–50. [DOI] [PubMed] [Google Scholar]
- [5].Reagan MR, Mishima Y, Glavey SV, et al. Investigating osteogenic differentiation in multiple myeloma using a novel 3D bone marrow niche model. Blood 2014;124:3250–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Shen X, Guo Y, Yu J, et al. miRNA-202 in bone marrow stromal cells affects the growth and adhesion of multiple myeloma cells by regulating B cell-activating factor. Clin Exp Med 2016;16:307–16. [DOI] [PubMed] [Google Scholar]
- [7].Zhang X, Sun Y, Wang Z, et al. Up-regulation of connexin-43 expression in bone marrow mesenchymal stem cells plays a crucial role in adhesion and migration of multiple myeloma cells. Leuk Lymphoma 2015;56:211–8. [DOI] [PubMed] [Google Scholar]
- [8].Corre J, Mahtouk K, Attal M, et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia 2007;21:1079–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Teoh HK, Pei PC, Abdullah M, et al. Small interfering RNA silencing of interleukin-6 in mesenchymal stromal cells inhibits multiple myeloma cell growth. Leuk Res 2016;40:44–53. [DOI] [PubMed] [Google Scholar]
- [10].Zhao P, Chen Y, Yue Z, et al. Bone marrow mesenchymal stem cells regulate stemness of multiple myeloma cell lines via BTK signaling pathway. Leuk Res 2017;57:20–6. [DOI] [PubMed] [Google Scholar]
- [11].Xu H, Han H, Song S, et al. Exosome-transmitted PSMA3 and PSMA3-AS1 promote proteasome inhibitor resistance in multiple myeloma. Clin Cancer Res 2019;25:1923–35. [DOI] [PubMed] [Google Scholar]
- [12].Roccaro AM, Sacco A, Maiso P, et al. BM mesenchymal stromal cell-derived exosomes facilitate multiple myeloma progression. J Clin Invest 2013;123:1542–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Umezu T, Imanishi S, Yoshizawa S, et al. Induction of multiple myeloma bone marrow stromal cell apoptosis by inhibiting extracellular vesicle miR-10a secretion. Blood Adv 2019;3:3228–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Dabbah M, Attar-Schneider O, Tartakover MS, et al. Microvesicles derived from normal and multiple myeloma bone marrow mesenchymal stem cells differentially modulate myeloma cells’ phenotype and translation initiation. Carcinogenesis 2017;38:708–16. [DOI] [PubMed] [Google Scholar]
- [15].Umezu T, Imanishi S, Azuma K, et al. Replenishing exosomes from older bone marrow stromal cells with miR-340 inhibits myeloma-related angiogenesis. Blood Adv 2017;1:812–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Diboun I, Wernisch L, Orengo CA, et al. Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genomics 2006;7:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dweep H, Gretz N. miRWalk2.0: a comprehensive atlas of microRNA-target interactions. Nat Methods 2015;12:697. [DOI] [PubMed] [Google Scholar]
- [18].Yu G, Wang LG, Han Y, et al. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43(Database issue):D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tang Y, Li M, Wang J, et al. CytoNCA: a cytoscape plugin for centrality analysis and evaluation of protein interaction networks. Biosystems 2015;127:67–72. [DOI] [PubMed] [Google Scholar]
- [21].Dumas PY, Mansier O, Prouzet-Mauleon V, et al. MiR-10a and HOXB4 are overexpressed in atypical myeloproliferative neoplasms. BMC Cancer 2018;18:1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhi Y, Xie X, Wang R, et al. Serum level of miR-10-5p as a prognostic biomarker for acute myeloid leukemia. Int J Hematol 2015;102:296–303. [DOI] [PubMed] [Google Scholar]
- [23].Zhi F, Cao X, Xie X, et al. Identification of circulating microRNAs as potential biomarkers for detecting acute myeloid leukemia. PLoS One 2013;8:e56718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bi L, Sun L, Jin Z, et al. MicroRNA-10a/b are regulators of myeloid differentiation and acute myeloid leukemia. Oncol Lett 2018;15:5611–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Bryant A, Palma CA, Jayaswal V, et al. miR-10a is aberrantly overexpressed in Nucleophosmin1 mutated acute myeloid leukaemia and its suppression induces cell death. Mol Cancer 2012;11:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Yu T, Liu L, Li J, et al. MiRNA-10a is upregulated in NSCLC and may promote cancer by targeting PTEN. Oncotarget 2015;6:30239–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xiong G, Huang H, Feng M, et al. MiR-10a-5p targets TFAP2C to promote gemcitabine resistance in pancreatic ductal adenocarcinoma. J Exp Clin Cancer Res 2018;37:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yan Y, Wang Q, Yan XL, et al. miR-10a controls glioma migration and invasion through regulating epithelial–mesenchymal transition via EphA8. FEBS Lett 2016;589:756–65. [DOI] [PubMed] [Google Scholar]
- [29].Ding L, Shen Y, Ni J, et al. EphA4 promotes cell proliferation and cell adhesion-mediated drug resistance via the AKT pathway in multiple myeloma. Tumour Biol 2017;39: doi: 10.1177/1010428317694298. [DOI] [PubMed] [Google Scholar]
- [30].La Rocca F, Airoldi I, Di Carlo E, et al. EphA3 targeting reduces in vitro adhesion and invasion and in vivo growth and angiogenesis of multiple myeloma cells. Cell Oncol 2017;40:483–96. [DOI] [PubMed] [Google Scholar]
- [31].Huang LJ, Shen Y, Bai J, et al. High expression levels of long noncoding RNA small nucleolar RNA host gene 18 and Semaphorin 5A indicate poor prognosis in multiple myeloma. Acta Haematol 2020;143:279–88. [DOI] [PubMed] [Google Scholar]
- [32].Sadanandam A, Varney ML, Singh S, et al. High gene expression of semaphorin 5A in pancreatic cancer is associated with tumor growth, invasion and metastasis. Int J Cancer 2010;127:1373–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Zhang L, Zhou L, Shi M, et al. Downregulation of miRNA-15a and miRNA-16 promote tumor proliferation in multiple myeloma by increasing CABIN1 expression. Oncol Lett 2018;15:1287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Yyusnita N, Chang KM, Zakiah I, et al. MicroRNA (miRNA) expression profiling of peripheral blood samples in multiple myeloma patients using microarray. Malays J Pathol 2012;34:133–43. [PubMed] [Google Scholar]
- [35].Wang W, Corrigan-Cummins M, Barber EA, et al. Aberrant levels of miRNAs in bone marrow microenvironment and peripheral blood of myeloma patients and disease progression. J Mol Diagn 2015;17:669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Gao SM, Xing CY, Chen CQ, et al. miR-15a and miR-16-1 inhibit the proliferation of leukemic cells by down-regulating WT1 protein level. J Exp Clin Cancer Res 2011;30:110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sun CY, She XM, Qin Y, et al. miR-15a and miR-16 affect the angiogenesis of multiple myeloma by targeting VEGF. Carcinogenesis 2013;34:426–35. [DOI] [PubMed] [Google Scholar]
- [38].Chen L, Wang Q, Wang GD, et al. miR-16 inhibits cell proliferation by targeting IGF1R and the Raf1-MEK1/2-ERK1/2 pathway in osteosarcoma. FEBS Lett 2013;587:1366–72. [DOI] [PubMed] [Google Scholar]
- [39].Cai CK, Zhao GY, Tian LY, et al. miR-15a and miR-16-1 downregulate CCND1 and induce apoptosis and cell cycle arrest in osteosarcoma. Oncol Rep 2012;28:1764–70. [DOI] [PubMed] [Google Scholar]
- [40].Hoechtlen-Vollmar W, Menzel G, Bartl R, et al. Amplification of cyclin D1 gene in multiple myeloma: clinical and prognostic relevance. Br J Haematol 2015;109:30–8. [DOI] [PubMed] [Google Scholar]
- [41].Misiewicz-Krzeminska I, Sarasquete ME, Vicente-Dueñas C, et al. Post-transcriptional modifications contribute to the upregulation of Cyclin D2 in multiple myeloma. Clin Cancer Res 2016;22:207–17. [DOI] [PubMed] [Google Scholar]
- [42].Chng WJ, Gualberto A, Fonseca R. IGF-1R is overexpressed in poor-prognostic subtypes of multiple myeloma. Leukemia 2006;20:174–6. [DOI] [PubMed] [Google Scholar]
- [43].Ngo BT, Felthaus J, Hein M, et al. Monitoring bortezomib therapy in multiple myeloma: screening of cyclin D1, D2, and D3 via reliable real-time polymerase chain reaction and association with clinico-pathological features and outcome. Leuk Lymphoma 2010;51:1632–42. [DOI] [PubMed] [Google Scholar]
- [44].Menu E, van Valckenborgh E, van Camp B, et al. The role of the insulin-like growth factor 1 receptor axis in multiple myeloma. Arch Physiol Biochem 2009;115:49–57. [DOI] [PubMed] [Google Scholar]
- [45].Mao X, Cao B, Wood TE, et al. A small-molecule inhibitor of D-cyclin transactivation displays preclinical efficacy in myeloma and leukemia via phosphoinositide 3-kinase pathway. Blood 2011;117:1986–97. [DOI] [PubMed] [Google Scholar]
- [46].Glassford J, Kassen D, Quinn J, et al. Inhibition of cell cycle progression by dual phosphatidylinositol-3-kinase and mTOR blockade in cyclin D2 positive multiple myeloma bearing IgH translocations. Blood Cancer J 2012;2:e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Laura G, Luque-Garcia JL, Pilar GP, et al. A quantitative proteomic analysis uncovers the relevance of CUL3 in bladder cancer aggressiveness. PLoS One 2013;8:e53328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zeng R, Tan G, Li W, et al. Increased expression of Cullin 3 in nasopharyngeal carcinoma and knockdown inhibits proliferation and invasion. Oncol Res 2018;26:111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Davidge B, Rebola KGO, Agbor LN, et al. Cul3 regulates cyclin E1 protein abundance via a degron located within the N-terminal region of cyclin E. J Cell Sci 2019;132:jcs233049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Chen R, Zhang X, Wang C. LncRNA HOXB-AS1 promotes cell growth in multiple myeloma via FUT4 mRNA stability by ELAVL1. J Cell Biochem 2020;121:4043–51. [DOI] [PubMed] [Google Scholar]
- [51].Wang W, Yang X, López de Silanes I, et al. Increased AMP:ATP ratio and AMP-activated protein kinase activity during cellular senescence linked to reduced HuR function. J Biol Chem 2003;278:27016–23. [DOI] [PubMed] [Google Scholar]
- [52].Mei H, Xiang Y, Mei H, et al. Pterostilbene inhibits nutrient metabolism and induces apoptosis through AMPK activation in multiple myeloma cells. Int J Mol Med 2018;42:2676–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Manier S, Liu CJ, Avet-Loiseau H, et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood 2017;129:2429–36. [DOI] [PMC free article] [PubMed] [Google Scholar]