

Abstract
Therapy for intracerebral hemorrhage (ICH) remains elusive, in part dependent on the severity of the hemorrhage itself as well as multiple deleterious effects of blood and its breakdown products such as hemin and free iron. While oxidative injury and genomic damage have been seen following ICH, the details of this injury and implications remain unclear. Here, we discovered that, while free iron produced mostly reactive oxygen species (ROS)-related single-strand DNA breaks, hemin unexpectedly induced rapid and persistent nuclear and mitochondrial double-strand breaks (DSBs) in neuronal and endothelial cell genomes and in mouse brains following experimental ICH comparable to that seen with γ radiation and DNA-complexing chemotherapies. Potentially as a result of persistent DSBs and the DNA damage response, hemin also resulted in senescence phenotype in cultured neurons and endothelial cells. Subsequent resistance to ferroptosis reported in other senescent cell types was also observed here in neurons. While antioxidant therapy prevented senescence, cells became sensitized to ferroptosis. To address both senescence and resistance to ferroptosis, we synthesized a modified, catalytic, and rapidly internalized carbon nanomaterial, poly(ethylene glycol)-conjugated hydrophilic carbon clusters (PEG-HCC) by covalently bonding the iron chelator, deferoxamine (DEF). This multifunctional nanoparticle, DEF-HCC-PEG, protected cells from both senescence and ferroptosis and restored nuclear and mitochondrial genome integrity in vitro and in vivo. We thus describe a potential molecular mechanism of hemin/iron-induced toxicity in ICH that involves a rapid induction of DSBs, senescence, and the consequent resistance to ferroptosis and provide a mechanistic-based combinatorial therapeutic strategy.
Keywords: intracerebral hemorrhage, hemin, genome damage, senescence, ferroptosis, nanomaterial
Graphical Abstract
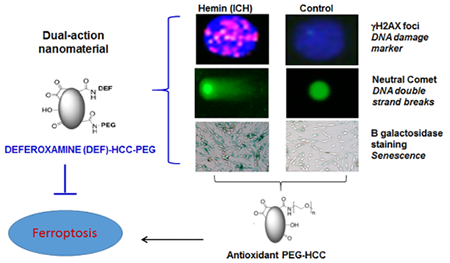
Although hemorrhagic stroke is less frequent than ischemic stroke, hemorrhages comprise 40% of all stroke morbidity and mortality,1 and therapy remains elusive despite some recent hints of promise.2 A number of factors have been identified as mechanisms of injury in intracerebral hemorrhage (ICH).3 ICH releases blood cells and their breakdown products into the brain parenchyma. This release subsequently damages neurons and neural tissues.4–6 In addition to the initial injury and mass effects, processes associated with brain injury during ICH include red blood cell (RBC) lysis, which results in the release of free hemoglobin (Hb).7,8 Free Hb is unstable and undergoes unchecked auto-oxidation to release hemin and globin chains. While the amino acids of globin chains are recycled for protein synthesis, the fate of hemin may be converted into biliverdin, which can act as an antioxidant, or into redox-active iron, which can be reused for Hb synthesis9 or as free hemin. Excess free Hb is toxic to neurons and the endothelium.10–12 Though the precise time course of RBC lysis after clinical or experimental ICH is variable, as much as 10 mM hemin is liberated from Hb in the hematomal region.13 Hb breakdown can form toxic products and generate reactive oxygen species (ROS) such as direct heme-derived superoxide (O2•−), H2O2, and their products via secondary oxidative reactions of H2O2 with Hb and ferrous and ferric Hb, thereby producing Fe(IV)-ferrylHb and oxyferrylHb. Furthermore, these products can undergo additional oxidative reactions and are pro-inflammatory.9,14,15 Finally, free Hb produces Hb–Hb dimers that readily enter cells and are oxidized to methemoglobin, which then reacts with H2O2 to produce genotoxic oxyferryl Hb.15–17 The amount of hemin liberated from hematoma during ICH may vary depending on hematoma size,18 ultimately affecting the severity and nature of hemin-induced neurotoxicity.
ICH and cerebral microbleeds have been linked to both acute and chronic neurological dysfunctions, long-term disability, and increased predisposition to various neurodegenerative disorders.6,19–25 Importantly, the major challenge in the clinical management of ICH is the lack of precise mechanistic insights into the pathways underlying neuronal and vasculature toxicity. While inflammation, ROS, and redox iron are thought to play critical roles in ICH-induced neurotoxicity, efforts at exploring antioxidant-based therapeutic strategies have not successfully transitioned to clinics.26–32 Importantly, we and others recently reported that catabolites of Hb, including iron, contribute to oxidative DNA damage not only by direct DNA damage induction but also by oxidizing repair proteins such as NEIL1, thereby inhibiting DNA repair.33–35 Thus, prevention of iron-mediated genome toxicity required both iron chelation and reduction of reversibly oxidized cysteine residues in DNA repair proteins33,34 in this case using two separate therapies. Ultimately, these studies highlight the need for revisiting the mechanisms underlying iron-induced toxicity and ROS formation in conditions like ICH.
Genome damage and instability have been linked to hemin and iron, two catabolic derivatives of Hb in ICH. In vitro studies have suggested that hemin has DNA strand scission activity in plasmid DNA.36,37 However, the role of hemin in nuclear or mitochondrial genome damage and their implications in neuronal and vascular pathology after ICH are unknown. Previous studies have suggested that heme oxygenase (HO)-mediated degradation of hemin into free iron and biliverdin prevents cellular senescence.38–40 However, the complexity and diverse implications of hemin/iron-mediated genome damage in cellular senescence versus iron-mediated ferroptosis have not been investigated. Additionally, the extent of hemin/iron-induced genotoxic stress in mitochondria versus nuclei is unclear. To address these outstanding questions, we used neuron-like cells differentiated from a neuroblastoma SH-SY5Y line, induced pluripotent stem cell (iPSC)-derived neural progenitor stem cells (NPSCs), mouse brain endothelial cell line bEnd.3, and an in vivo mouse model of experimental ICH. Our studies revealed several phenomena, including the generation of DNA double-strand breaks (DSBs), activation of DNA damage response (DDR) signaling, and significantly increased senescence in hemin-treated cells. Consistent with reports in other cell types,41 the senescent cells were resistant to iron-mediated ferroptosis. To explore the role of ROS in genome damage, we exposed hemin-treated cells to poly-(ethylene glycol)-conjugated hydrophilic carbon clusters (PEG-HCCs), a catalytic, broadly acting antioxidant recently generated in our laboratory.42 PEG-HCCs are a highly oxidized and functionalized graphene-based material, 3 × 40 nm, that are high-capacity catalytic antioxidants and rapidly internalized. The PEG-HCCs demonstrate therapeutic efficacy after systemic injection in models of ischemic stroke and traumatic brain injury.42–44 Here we show that PEG-HCCs inhibited hemin-induced senescence, but sensitized cells to iron-mediated toxicity and ferroptosis.
In ICH, iron toxicity is treated with chelating agents such as deferoxamine (DEF, also known as deferoxamine). While DEF improves outcomes in ICH animal models, dose-limited side effects occurred in human ICH trials, and the recent iDEF trial showed that while DEF was safe with a trend toward improvement in outcome, the effect was small and did not meet the threshold for significance.45 Furthermore, DEF has the limitation of poor and variable cellular uptake, toxicity, and a short shelf life because of its propensity to oxidation.46,47 To overcome the limitations of DEF and PEG-HCCs individually, we developed a multifunctional nanoparticle by exploiting the similar amine binding moiety between DEF and PEG-NH2 at the nonchelating site of DEF. This allowed us to covalently bond DEF and PEG-NH2 to the HCC carboxyl units forming amide linkages, thereby generating DEF-HCC-PEG. DEF-HCC-PEG more effectively than the individual therapies individually or together prevented both hemin- and iron-mediated toxicity and reduced both senescence and ferroptosis. Together, our studies report a mechanism of hemin- and iron-mediated neurotoxicity that is linked to genomic DSB damage and interactions between senescence and ferroptosis. We also established a combinatorial strategy to mitigate neurotoxicity in ICH.
RESULTS AND DISCUSSION
Hemin Induces DNA DSBs in the Nuclear Genome.
Previous studies have shown that hemorrhagic stroke is associated with extensive genome damage.37,48–50 However, the effect of hemin, the predominant Hb catabolite, on genome integrity and neuronal toxicity is unclear. To test this, we first used the MTT assay to evaluate hemin-induced cytotoxicity at various doses (0–25 μM) in neuron-like cells differentiated from human neuroblastoma SH-SY5Y cells. Hemin caused 50% cell mortality (IC50) at 5 μM (Figure 1a). Notably, even at 100 μM, iron (FeSO4) reduced cell viability by only 30%. Indeed, the IC50 of FeSO4 or Fe-nitrilotriacetate (NTA) was substantially (~4-fold) higher than that of hemin (Figure S1a,b). Despite likely differences in cellular absorption rates, hemin’s higher IC50, even at a 10-fold lower concentration compared to iron, suggested that hemin, which is an iron-porphyrin complex (at 1:1 ratio), was significantly more cytotoxic than iron alone (Figure 1a). Furthermore, hemin’s IC50 was modestly higher for undifferentiated SH-SY5Y cells than differentiated neuron-like cells, and it was ~2-fold higher (~10 μM; data not shown) for mouse endothelial cells (bEnd.3) than for neurons. Thus, cultured neurons appear to be more vulnerable to hemin-induced toxicity than other cell types including cycling cells.
Figure 1.
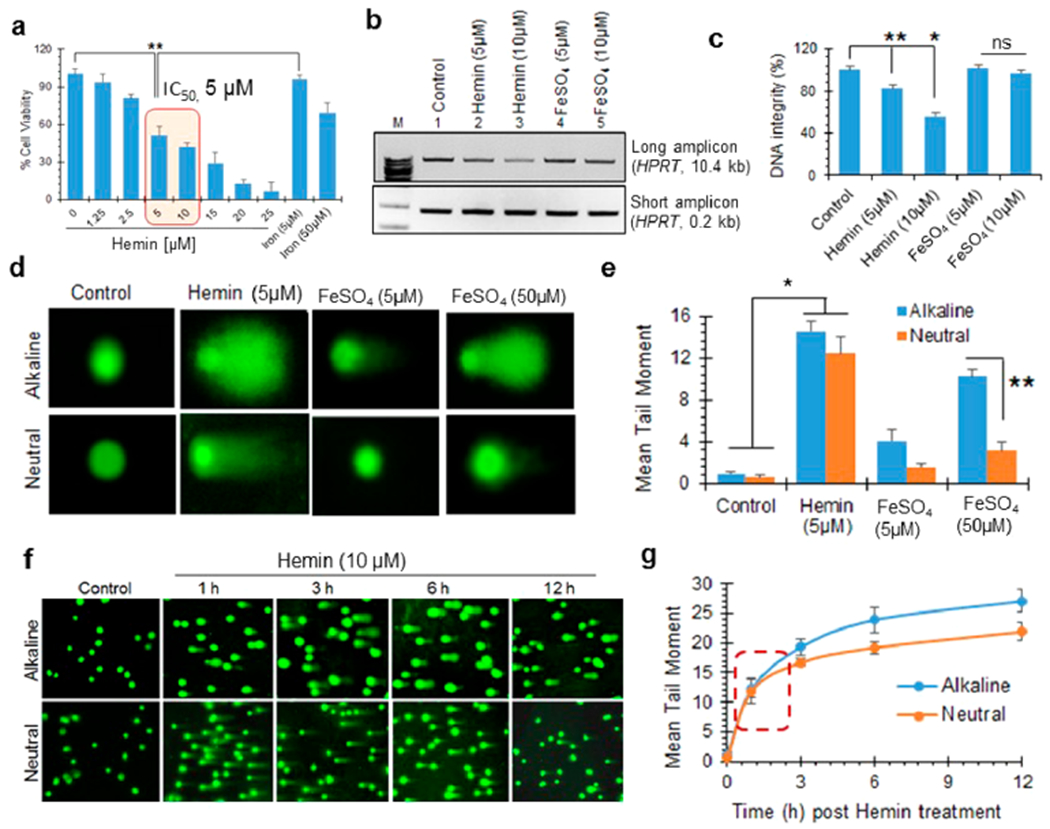
Hemin induces DSBs in the nuclear genome. (a) Cell viability analysis by MTT assay in differentiated neuron-like SH-SY5Y cells treated with increasing doses (0–25 μM) of hemin. The IC50 was determined at 5 μM. Iron salt (FeSO4) at 5 and 50 μM was also used for comparison. (b and c) Quantitation of hemin-induced DNA strand breaks using LA-PCR analysis. Genomic DNA extracted from differentiated SH-SY5Y cells treated with hemin and iron for 12 h was subjected to LA-PCR. The products were separated on a 1% agarose gel (b). A 10.4 kb region in the HPRT gene was used as long amplicon (LA), while a 0.2 kb region was used as a short amplicon control. (c) Picogreen-based quantitation of amplified DNA expressed as percent (%) DNA integrity. (d and e) Comet assay (alkaline and neutral) performed on differentiated neurons 12 h after hemin (5 μM) or FeSO4 (5 and 50 μM) treatment. Quantitation of mean comet tail moment showed marked increases in both alkaline and neutral comet tails after hemin treatment compared to control cells. In contrast, iron predominantly caused alkaline comet tails, even at 50 μM. (f and g) Time-dependent kinetics of alkaline versus neutral comet tails in hemin (10 μM)-treated cells. The comparable increases in alkaline and neutral comet tails at early time points suggested direct induction of DSBs. Results are represented as mean ± SEM from samples assayed in triplicate (for each experiment) and three independent experiments. *p < 0.01, **p < 0.05, ns: nonsignificant.
To evaluate the effect of hemin on genome integrity, we first extracted the total genomic DNA from control-, hemin-, and iron-treated neurons. We then used long-amplicon PCR (LA-PCR) to quantitate DNA strand breaks51–53 and assess DNA integrity. In this assay, we PCR-amplified specific, large (~10 kb) segments of genomic DNA. The presence of strand breaks in the amplicon would cause reduced amplification of the fragment. Hemin treatment significantly reduced HPRT amplification in a dose-dependent manner (5 and 10 μM) compared to FeSO4 at comparable concentrations (Figure 1b). Genomic DNA from untreated cells and short amplicon products served as a control. Quantification of the PCR products by Picogreen revealed that 5 μM hemin decreased genome integrity by ~50% (Figure 1c). These results revealed accumulation of strand breaks in the genome after hemin treatment.
To further characterize the nature of these strand breaks, we next performed alkaline and neutral comet assays in hemin-treated neurons. Although alkaline comets measure all alkali-labile genomic sites, including both single- and double-strand breaks, neutral comet tails exclusively reflect DSBs.53 Hemin treatment (5 μM) significantly increased both alkaline and neutral comet tails (Figure 1d,e). Again, hemin-induced genome damage was significantly higher than that generated by FeSO4, even at a 10-fold higher concentration (50 μM). Interestingly, quantification of the mean comet tail moment revealed a comparable level of damage in alkaline and neutral comet analyses (Figure 1e), suggesting that hemin predominantly induced DSBs in the nuclear genome. This was unexpected, because previous studies had suggested that hemin causes genome damage indirectly via ROS or other oxidative mechanisms.54,55 ROS primarily induce oxidized DNA base lesions, alkali-labile abasic sites, and single-strand breaks.56,57 To address this further, we examined the kinetics of alkaline and neutral comet tails in hemin-treated neurons. The cells predominantly accumulated DSBs at 1 h post-hemin treatment (Figure 1f,g). At longer times, the increase in alkaline comet tails was moderately higher than neutral tails, which was likely due to ROS-mediated additional non-DSB damage. Thus, hemin directly induces DSBs in the nuclear genome.
Hemin Induces Robust DDR Signaling.
We next used immunoblotting (IB) and immunofluorescence (IF) to analyze the activation of DDR factors in hemin-treated cells. IB of control and hemin-treated differentiated SH-SY5Y cells showed that hemin markedly increased the levels of γH2AX, p-53BP1, and p-ATM (S1981) (Figure 2a,b). γH2AX and p-53BP1 are markers of DSBs,58 whereas p-ATM is an early activator of DDR signaling after DSB induction.59 Consistent with the IB data, a significant increase in the nuclear foci formation of γH2AX, p-53BP1 (Figure 2c,d), and p-ATM (Figure 2e,f) was observed in IF analysis. Ionizing radiation (IR) was used as a positive control for DSB-induced ATM activation. Notably, the number of p-ATM foci induced by 5 μM hemin was comparable to that induced by 2 Gy of IR.
Figure 2.
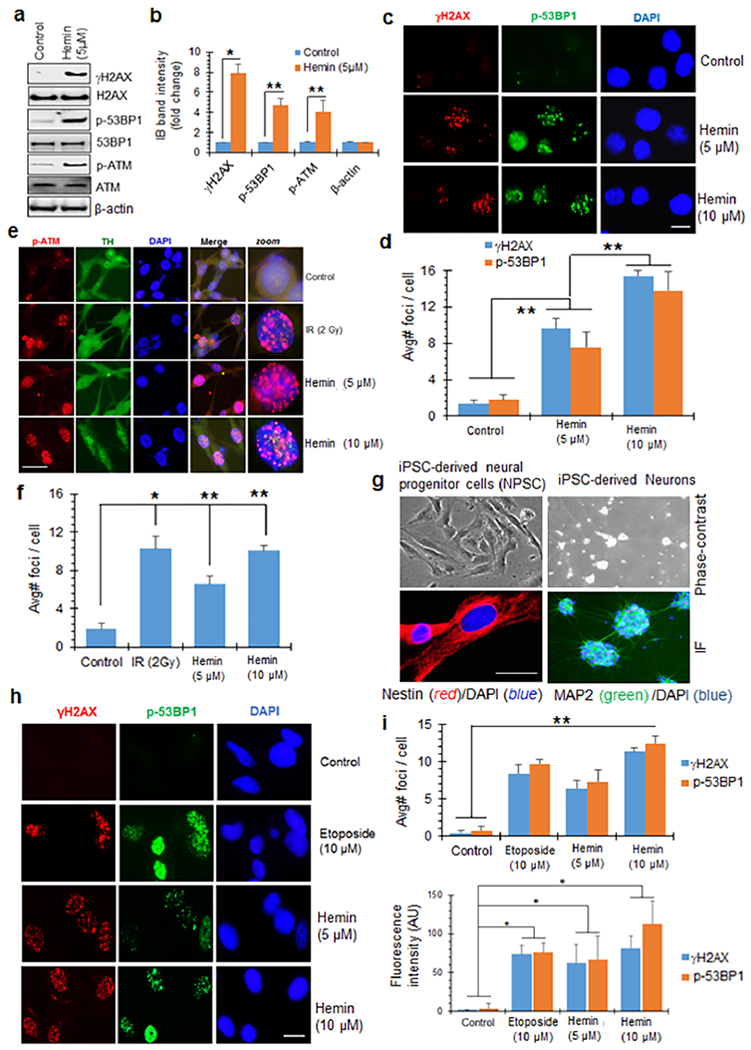
Hemin induces robust DNA damage response (DDR) signaling. (a and b) Immunoblot analysis (IB) of γH2AX, p-53BP1, and p-ATM in differentiated SH-SY5Y cells treated with hemin (5 μM). Quantitation of IB band intensity shows ~4–8-fold higher levels of γH2AX, p-ATM, and p-53BP1 in differentiated SH-SY5Y cells than in control cells. β-Actin served as loading control. (c and d) Immunofluorescence (IF) analysis of γH2AX and p-53BP1 in neurons treated with 5 or 10 μM hemin for 12 h. Nuclei were stained with DAPI. The average number of nuclear foci per cell was quantified from at least 25 cells and mean ± SEM represented in the histogram from three independent experiments. (e and f) IF analysis of p-ATM in hemin-treated neurons. Ionizing radiation (IR, 2 Gy) was used as a positive control for inducing DSBs. Nuclei were again stained with DAPI, and neuronal differentiation of SH-SY5Y cells was confirmed by tyrosine hydroxylase (TH). Quantitation of the average number of foci per cell from 25 cells is shown in the histogram as mean ± SEM from three independent experiments. (g) IF staining of human neural progenitor cells (NPCs) and matured neurons derived from an iPSC line. The NPCs were stained with the neural stem cell marker Nestin, and the neurons were stained with neuronal marker MAP2. Nuclei were stained with DAPI. Corresponding phase contrast images are shown. (h–j) IF analysis of γH2AX and p-53BP1 in NPSCs treated with hemin (5 or 10 μM, 12 h) or with 10 μM etoposide as a positive control. Quantitation of the average number of foci/nucleus (i) and mean fluorescence intensity/field (j) shown in histograms. *p < 0.01, **p < 0.05. Scale bars, 5 μm.
To further test hemin-induced DDR activation in human primary cells, we generated NPSCs from a human iPSC line (Figure 2g).52,53 DDR activation observed in NPSCs was comparable to that in differentiated SH-SY5Y cells (Figure 2h–j). The chemotherapeutic drug topoisomerase II inhibitor, etoposide, was used as a positive control for DSB induction. Again, the number of DDR foci induced by 10 μM hemin was comparable to that induced by 10 μM etoposide.
Early Kinetics of Hemin-Induced Activation of DDR Signaling.
We next analyzed the early response kinetics of hemin-induced DDR activation in cultured cells (Figure 3a,b). In cultured neurons, γH2AX and p-53BP1 foci were observed as early as 5 min post-hemin treatment, and the number of foci increased further at 15, 30, and 60 min post-treatment. As before, 10 μM etoposide was used as a positive control for DSB induction. The rapid induction of DSB markers revealed the generation of DNA DSBs within 5 min post-hemin treatment.
Figure 3.
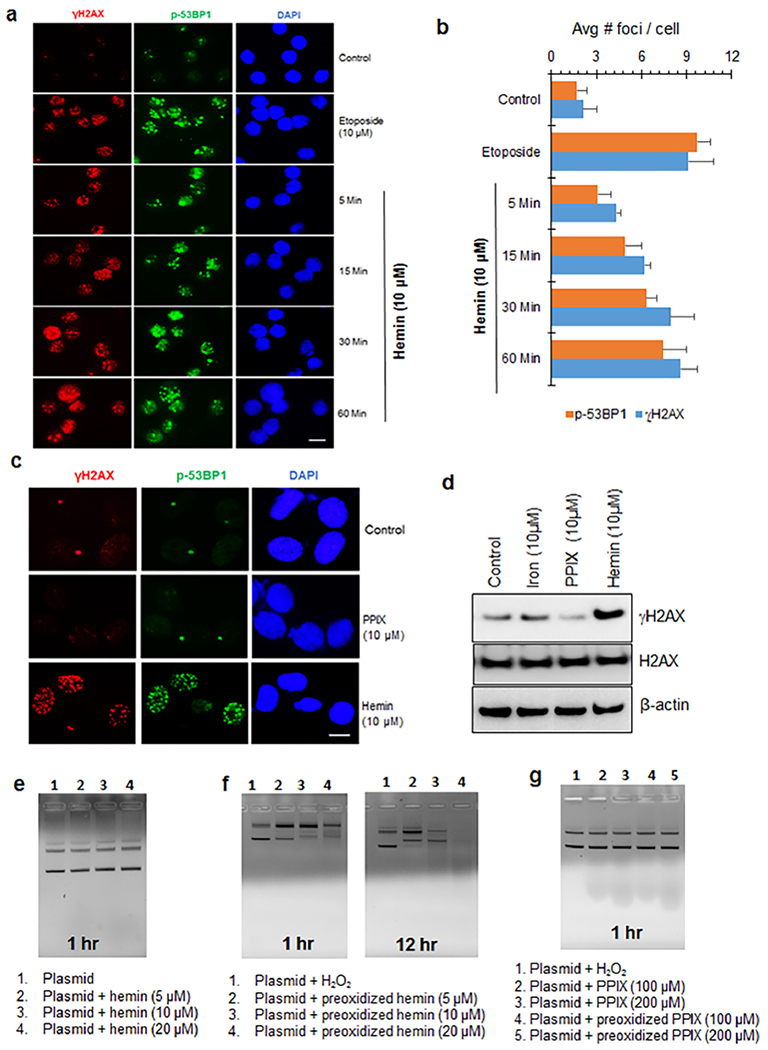
Early kinetics of hemin-induced DDR activation suggests direct DSB induction. (a and b) Kinetics of γH2AX and p-53BP1 foci formation in differentiated SH-SY5Y cells treated with hemin. The cells were treated with 10 μM hemin for 0, 5, 15, 30, and 60 min, respectively, and then foci were analyzed by IF. Etoposide (10 μM) was used as a positive control. Scale bar, 10 μm. Quantitation of the average number of foci/nucleus is shown in the histogram. (c) IF analysis of γH2AX and p-53BP1 foci in hemin-treated BJ-hTERT fibroblasts. Protoporphyrin IX (PPIX, 10 μM, 12 h) did not induce DDR foci. Scale bar, 5 μm. (d) IB analysis of γH2AX levels in differentiated SH-SY5Y cells treated with FeSO4 (10 μM), PPIX (10 μM), or hemin (10 μM) for 12 h. (e–g) Agarose gel analysis of plasmid DNA in the presence of native or preoxidized hemin at different concentrations. Hemin and protoporphyrin were preoxidized with 1 μM H2O2 for 30 min and then incubated with pUC19 plasmid DNA for 1 and 12 h, respectively. All experiments are performed in three independent repeats. Scale bar, 5 μm.
We next investigated whether hemin induces DSB damage in all cell types. Figure 3c shows hemin-induced activation of DSB markers γH2AX and p-53BP1 in human primary fibroblast cells (BJ-hTERT), similar to the pattern observed in cultured neurons and endothelial cells, suggesting a phenomenon independent of cell type. Notably, the structural analogue of hemin that lacks iron, protoporphyrin IX (10 μM), or iron (10 μM) alone did not induce DSB damage at a similar concentration (10 μM) (Figure 3c). The lack of DSB induction by protoporphyrin IX was confirmed by immunoblotting in SH-SY5Y cells (Figure 3d). These data underscore the specific genotoxic role of the iron-porphyrin complex in inducing DSBs.
We then examined the effect of hemin and protoporphyrin IX preoxidized by hydrogen peroxide (1 μM for 30 min) on plasmid DNA in vitro. Compared to the native hemin, the preoxidized hemin induced DNA strand breaks in a dose- and time-dependent manner (Figure 3e,f). Furthermore, protoporphyrin and preoxidized protoporphyrin, even at a 10-fold higher concentration, did not damage DNA (Figure 3g). Thus, the iron-porphyrin complex mediates hemin-induced genotoxicity. Furthermore, hemin oxidation is required for its ability to induce DSBs in DNA.
Hierarchy in Subcellular ROS Accumulation and Mitochondrial DNA Damage after Hemin Treatment.
To evaluate how ROS formation contributed to hemin-mediated nuclear genome damage, we used a plasmid-based ROS sensor, p-HyPer. This sensor localizes to the cytosol (p-HyPer-Cyto), mitochondria (p-HyPer-Mito), and nucleus (p-HyPer-Nuclear) of cultured neurons and elicits green fluorescence upon ROS formation.60 Treatment with the p-HyPer vector and with 5 μM hemin significantly induced green fluorescence in mitochondria as early as 5 min, which gradually increased at 10, 15, 30, and 60 min post-treatment (Figure 4a,b). ROS similarly accumulated in the cytosol, albeit at a reduced intensity compared to that in the mitochondria. However, the nucleus accumulated ROS at a significantly slower rate, and fluorescence was only detectable at 30 min post-treatment. These data suggested that the early DSB induction in the nuclear genome likely involved hemin binding directly to chromatin rather than being mediated by global ROS stress. This could eventually contribute to enhanced genome damage at later time points.
Figure 4.
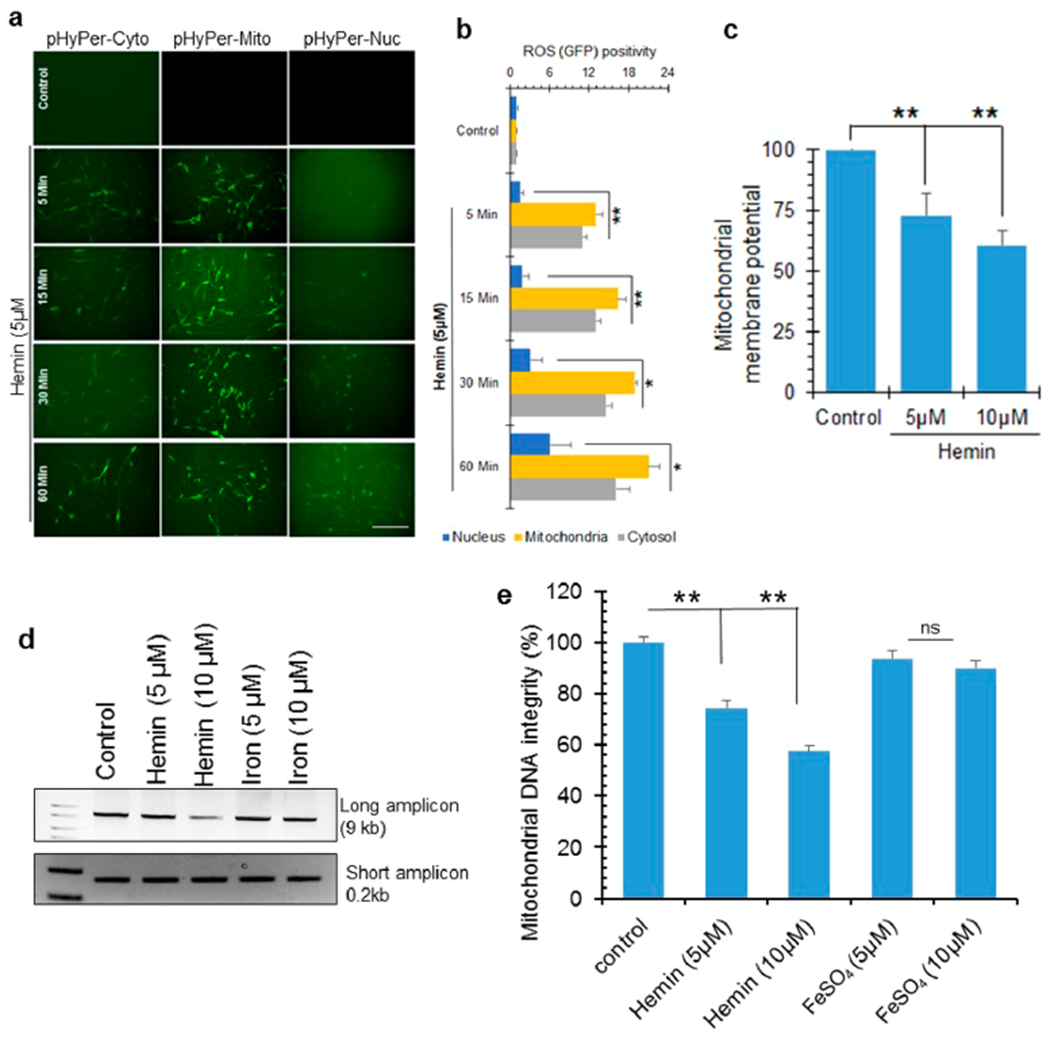
ROS formation and mitochondrial DNA damage in hemin-treated cells. (a and b) Cellular ROS levels in differentiated SH-SY5Y cells in the presence or absence of hemin (5 μM) was assessed using the H2O2 sensor pHyPer. This sensor emits green fluorescence in the presence of ROS, allowing detection by IF. pHyper expression vectors with targeting sequences for the cytosol, mitochondria, or nuclei were stably transfected in SH-SY5Y cells. Hemin-induced ROS accumulation in mitochondria occurred very early at 5 min and at a significantly higher level than the cytosol or nucleus. Nuclear ROS accumulation had slower kinetics. The histogram represents quantitation of GFP fluorescence intensity from 10 different fields. Scale bar, 20 μm. (c) Loss of mitochondrial membrane potential in differentiated neurons treated with hemin (5 or 10 μM, 1 h) compared to control cells. (d and e) Analysis of DNA integrity measured by LA-PCR of mitochondrial DNA extracted from differentiated neurons treated with hemin or iron (5 or 10 μM) for 12 h. Picogreen-based quantitation of amplified DNA expressed as percent (%) of DNA integrity. *p < 0.01, **p < 0.05. Three independent experiments were performed, and the results are represented as mean ± SEM. Scale bar, 10 μm.
Based on the robust and early accumulation of mitochondrial ROS, we next evaluated the impact of hemin on mitochondrial DNA integrity and membrane potential. LA-PCR analysis of mitochondrial DNA from hemin-treated cells revealed an ~5-fold increase in mitochondrial DNA damage at 12 h after hemin treatment relative to the untreated cells (Figure 4d,e). Furthermore, compared to the level of nuclear genome damage at the same time point, mitochondrial genome damage was significantly higher (Figure S2c–f). Hemin also reduced mitochondrial membrane potential (Figure 4c), indicating that hemin is also involved in mitochondrial oxidative stress. Finally, we tested whether hemin affected PGC-1α, a key factor in mitochondrial biogenesis. IB analysis revealed that hemin significantly reduced PGC-1α levels compared to control cells (Figure S2a,b). Thus, hemin likely affects the integrity of both the nuclear and mitochondrial genomes and may involve both ROS and the direct binding of hemin to chromatin.
DNA Damage in an ICH Mouse Model.
Genome damage and DDR activation in an ICH mouse model was examined. To induce ICH, C57BL/6 mice were injected using a Hamilton syringe with intracerebral hemolyzed blood in the left cerebral hemisphere, while the right hemisphere served as a control (Figure 5a). We used hemolyzed blood to model the acute effects of blood degradation products and to delineate the molecular mechanisms, as it hastens the degradation. At 12 h after ICH induction, the mice were euthanized and both brain hemispheres were snap-frozen for IB analysis and DNA isolation. Brain tissue around the visible blood products on the ICH hemisphere and comparable location on the contralateral hemisphere were carefully dissected for analysis. We found that the peri-ICH region of the left hemisphere had significantly higher (3- to 4-fold) expression of γH2AX compared with the contralateral (control) hemisphere (Figure 5b,c). Six mice each in both the control and experimental group were analyzed for deriving mean ± SEM in quantitation histograms, and representative data from two mice are shown in the immunoblotting and LA-PCR (Figure 5). LA-PCR analysis showed that ICH significantly reduced DNA integrity in both nuclear (Figure 5d,e) and mitochondrial (Figure 5f,g) DNA compared to the control. Furthermore, ICH significantly reduced PGC-1α expression relative to the control region, consistent with mitochondrial toxicity (Figure S3a,b).
Figure 5.
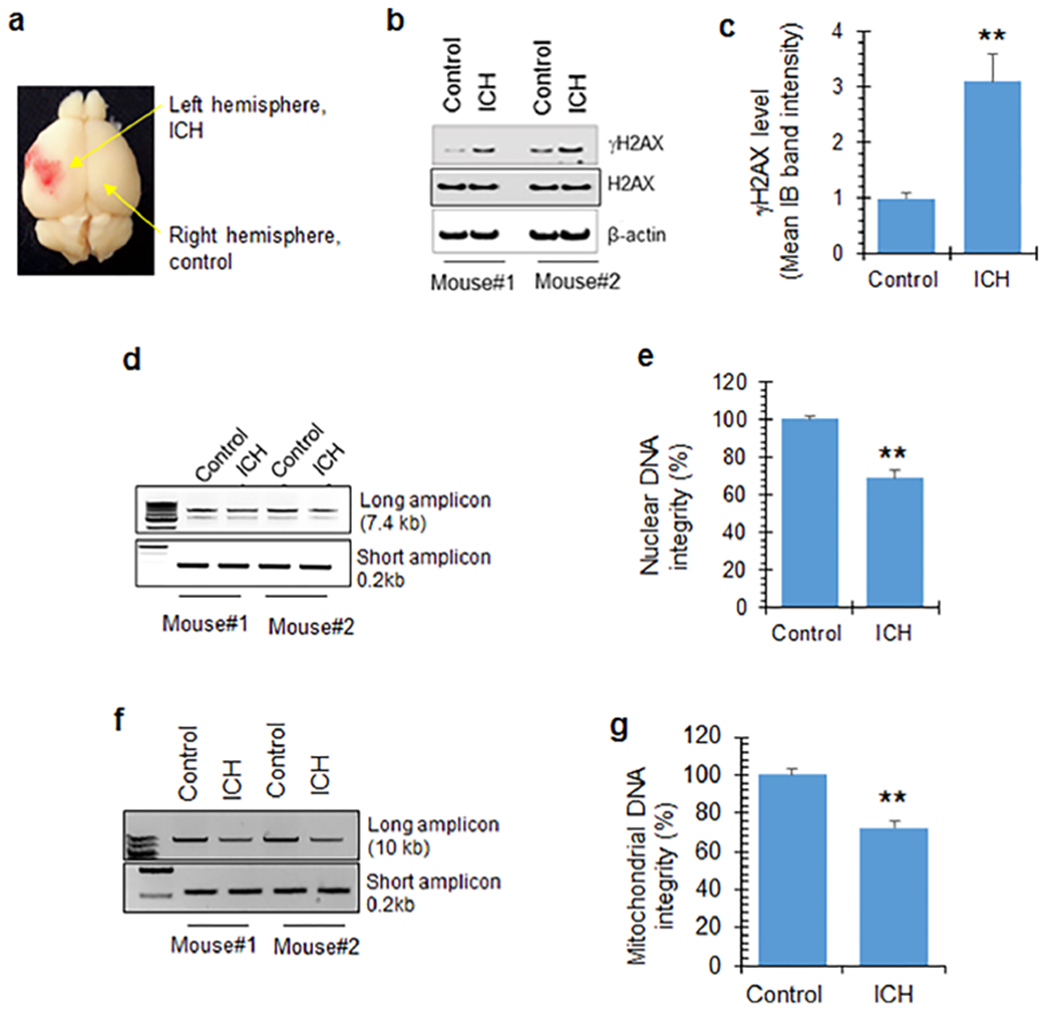
DNA damage in an ICH mouse model. (a) Schematic demonstrating ICH induction using hemolyzed blood in mouse brains. The left hemisphere was injected with intracerebral hemolyzed blood, while the right hemisphere served as the control. (b and c) IB of γH2AX levels in brain extracts from two representative mice. Quantitation of IB bands from 6 mice shows increased γH2AX expression in the left hemisphere (ICH) relative to the right (control). Data represented as mean ± SEM. (d and e) LA-PCR analysis of genomic DNA extracted from mouse brains. NanoG (7.4 kb) was used as a long amplicon (LA), while a 0.2 kb segment within that same gene was used as a short amplicon. Picogreen-based quantitation of amplified DNA was expressed as percent (%) of DNA integrity. (f and g) LA-PCR analysis of mitochondrial DNA damage. Mitochondrial DNA (10 kb) was used as a LA, while 0.2 kb within the LA region was used as a short amplicon. Picogreen-based quantitation of amplified DNA expressed as percent (%) of DNA integrity. The quantitative histograms represent data from 6 mice each in control and experimental groups, expressed as mean ± SEM, and representative data from 2 mice are shown in the immunoblotting and agarose gel separation of LA-PCR products. **p < 0.05.
PEG-HCCs Prevent Hemin-Induced Genome Damage and Only Moderately Reduced Cell Death.
We have developed nanoantioxidants that are highly oxidized 3 × 40 nm carbon nanoparticles,42,43 termed poly(ethylene glycol)-functionalized hydrophilic carbon clusters (Figure 6a). The synthesis of these particles is described in detail elsewhere.61 PEG-HCCs are catalytic superoxide dismutase mimetics that convert superoxide (O2•−) into H2O2 and are effective scavengers of hydroxyl radicals. They have been shown to reduce lesion size and improve functional outcome in acute nonhemorrhagic traumatic brain injury and ischemic stroke.37,43,62 Here, we tested how these broadly active antioxidants altered the effects of hemin by adding PEG-HCCs to cell cultures 30 min after hemin treatment. In hemin-treated bEnd.3 cells, PEG-HCCs significantly reduced ROS accumulation (Figure 6b). We then evaluated how PEG-HCCs affected hemin-induced genome damage in neurons. PEG-HCCs significantly reduced γH2AX and p-ATM levels compared to hemin treatment alone (Figure 6c,d). Consistently, LA-PCR analysis showed that PEG-HCCs prevented hemin-induced nuclear and mitochondrial DNA damage (Figure S4a,b). Intriguingly, although PEG-HCCs effectively prevented both ROS accumulation and genome damage, this treatment only partially (30–40%) prevented hemin-induced total cell death (Figure 6e). These results suggest that additional pathways likely contribute to hemin-induced cell death or neurotoxicity.
Figure 6.
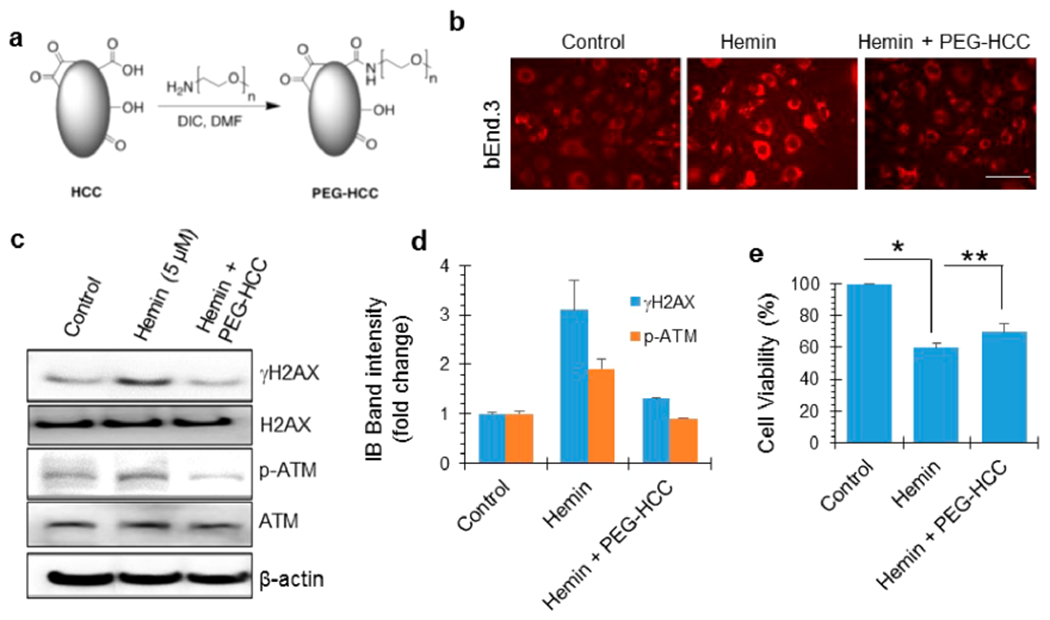
Antioxidant-based nanoparticle PEGylated hydrophilic carbon clusters (PEG-HCCs) prevent hemin-induced genome damage but not cytotoxicity. (a) Chemical structure of PEG-HCCs generated by a DCC coupling reaction. (b) Validation of antioxidant activity of the PEG-HCC preparation in bEnd.3 cells (mouse brain endothelial cells). PEG-HCC effectively sequestered hemin-induced ROS levels. (c and d) Effect of PEG-HCC on the hemin-induced increase in γH2AX and p-ATM levels in bEnd.3 cells, as analyzed by IB. Quantitation of IB band intensity. (e) Analysis of cell viability in b.End3 cells treated with hemin and PEG-HCC. PEG-HCC partially prevented hemin-induced cell death. Results are represented as mean ± SEM from three independent experiments. *p < 0.01, **p < 0.05. Scale bar, 10 μm.
Hemin Activates Senescence and Inhibits Ferroptosis.
Recent studies have suggested that iron-induced ferroptosis contributes to ICH and other neurodegenerative diseases.10,63,64 Furthermore, unrepaired DNA damage, particularly DSBs, activates senescence.65,66 Senescence in normal situations contributes to the removal of dysfunctional cells by inducing tissue remodeling during development and tissue damage. However, in pathological conditions, accumulation of senescent cells causes functional decline. This is consistent with studies reporting a major role for senescence in age-related diseases.67,68 Here, we investigated the implications of hemin and iron toxicity on cellular senescence versus ferroptosis. Our observation of persistent DDR foci, even at 12 h after hemin exposure, suggested possible induction of senescence. To evaluate this, we examined β-galactosidase staining as a marker for cellular senescence in hemin-treated SH-SY5Y cells (Figure 7a,b).69,70 Hemin treatment caused a significant increase in β-galactosidase positive cells (~40% at 5 μM hemin) compared to untreated control cells. Similar increased expression of β-galactosidase positive cells was seen in cultured b.End3 cells following 5 days of treatment with 50 μM hemin (Figure S5). Furthermore, PCR analysis in SH-SY5Y cells revealed that hemin increased the expression of a number of senescence-associated factors, including ankyrin repeat domain 1 (ANKRD1), endothelin 1 (EDN1), cyclin-dependent kinase inhibitor (CDKN1A/p21), and poliovirus receptor-related protein 4 (PVRL4) (Figure 7c). To confirm the involvement of hemin-induced DNA damage and ROS stress in senescence, we then tested the effect of PEG-HCCs, which effectively reduced the level of senescence markers (Figure 7d,e). These observations implicate hemin-induced genome damage in neuronal senescence and its prevention by antioxidants.
Figure 7.
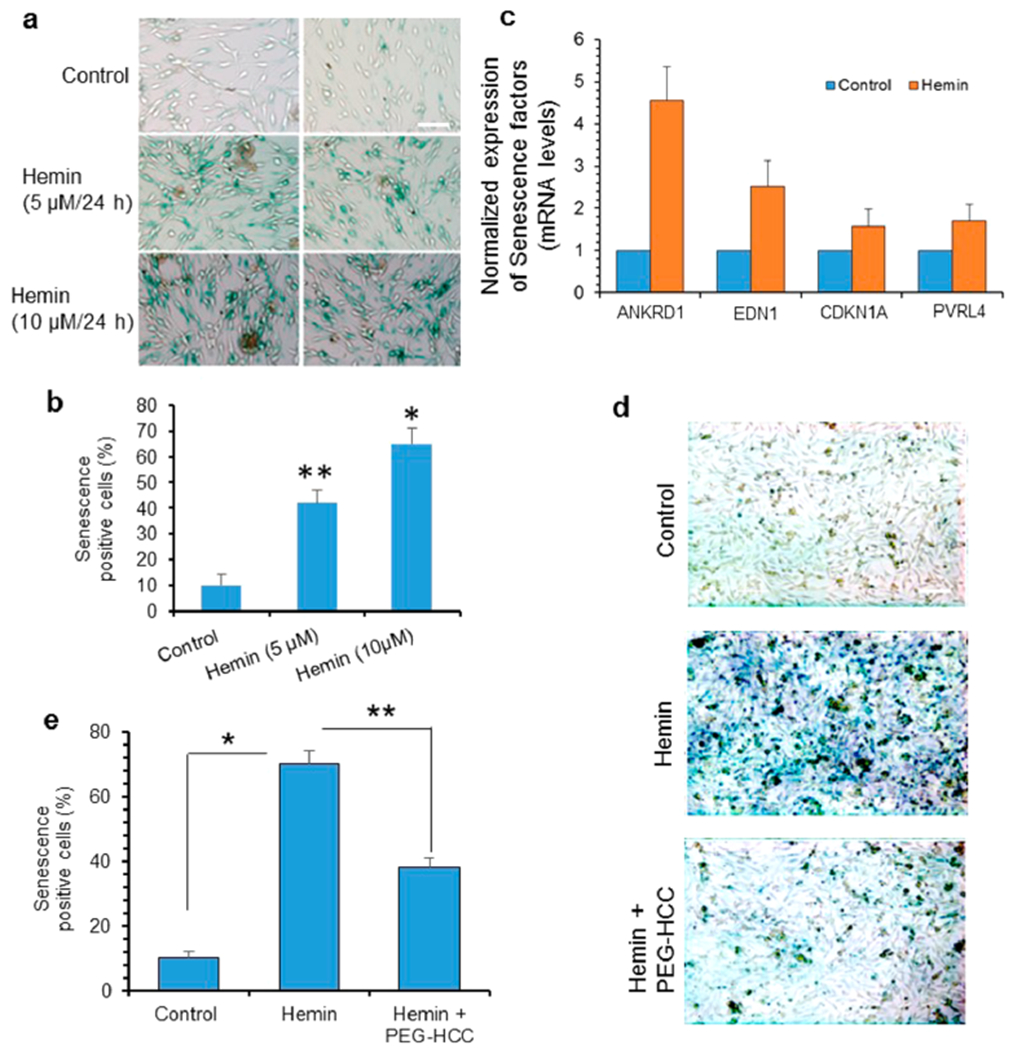
Hemin causes senescence in neurons; PEG-HCC reduces hemin-induced senescence. (a and b) Representative light microscopy images of staining for β-galactosidase, a marker of cellular senescence, in differentiated neurons treated with hemin (5 or 10 μM, 24 h). Quantitation of β-galactosidase-positive cells is shown in the histogram. (c) Real-time mRNA quantitation of senescence-associated genes indicated senescence-associated secretory phenotypes. (d and e) Effect of PEG-HCC on hemin-induced senescence. Neurons were treated with hemin (10 μM) with or without PEG-HCC for 24 h. PEG-HCC reduced hemin-induced senescence. Results are represented as mean ± SEM from samples assayed in triplicate (for each experiment) and 3 independent experiments. *p < 0.01, **p < 0.05. Scale bar, 10 μm.
Alternatively, ferroptosis has been implicated in various iron overload conditions71,72 including in animal models of ICH.10 While ferroptosis is critically involved in Fe-mediated cell death,73,74 its role in hemin-induced cytotoxicity is unknown. Furthermore, because senescent cells are reportedly resistant to cell death or ferroptosis,41 we tested the effects of hemin-induced senescence on ferroptosis. We measured levels of two key markers of ferroptosis, glutathione peroxidase 4 (GPx4) and lipid peroxidation product malondialdehyde (MDA),73,75,76 in cells treated with hemin (10 μM), iron (100 μM), or hemin (10 μM) for 30 min and then supplemented with iron (90 μM). The iron-treated cells were found to have significantly higher MDA levels compared to hemin treatment alone, as later shown in Figure 8g. Furthermore, incubation with both hemin and free iron salt reduced the magnitude of ferroptosis-related markers after addition of iron compared to iron alone (Figure 8f, GPx4 levels, and 8g, MDA levels). Thus, hemin-induced senescent cells appeared to have resistance to ferroptosis. The presence of ferroptosis was confirmed using erastin (5 μM) as a positive control and using a known ferroptosis inhibitor, ferrostatin 1, demonstrating a reduction in GPx4 (Figure S6).
Figure 8.
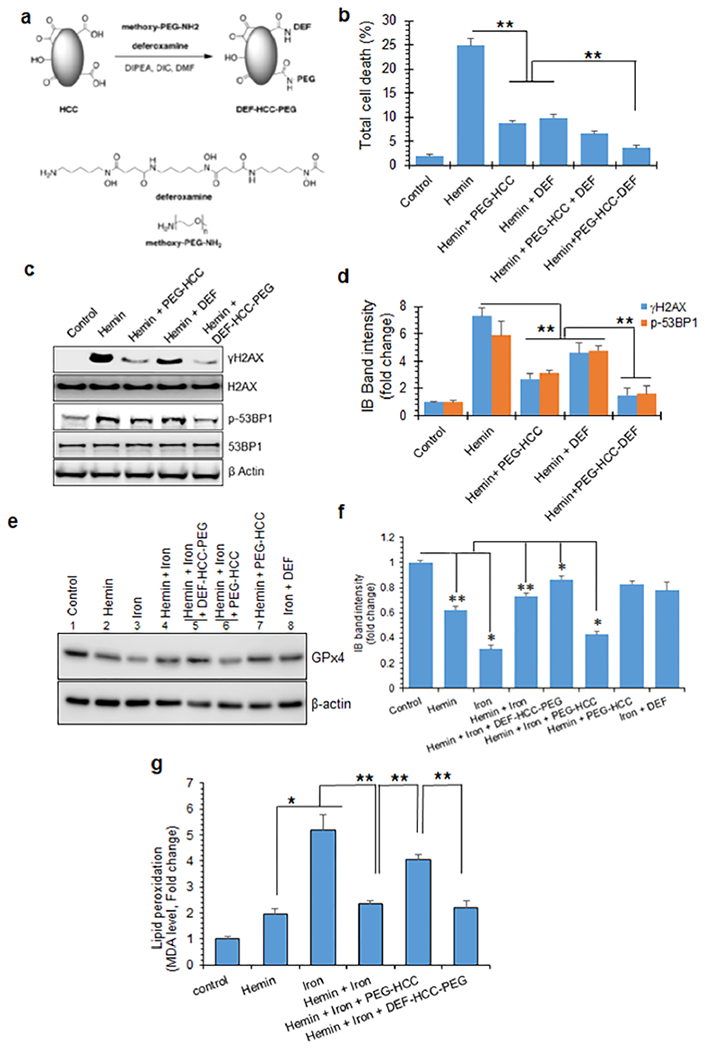
Modified nanoformulation of PEG-HCC and the iron chelator deferoxamine (DEF-HCC-PEG) mitigates genome damage and ferroptosis in cultured neurons. (a) Synthesis and chemical structure of a modified nanococktail, DEF-HCC-PEG. (b) Effect of PEG-HCC and DEF alone or of DEF-HCC-PEG on the viability of hemin-treated cultured neurons. Neurons were generated from human iPS cells and treated with hemin (5 μM) in the absence or presence of DEF-HCC-PEG or PEG-HCC. Hemin-treated cells were also treated with PEG-HCC or DEF alone as controls. Cell death was quantitated by MTT assay. (c and d) IB of γH2AX and p-53BP1 levels to determine the protective efficacy of DEF-HCC-PEG against hemin-mediated genome damage. Histogram shows quantitation of IB band intensity. (e and f) IB analysis of GPx4 expression as a measure of ferroptosis in iPSC-derived neurons treated with hemin, iron, PEG-HCC, DEF, or DEF-HCC-PEG. Histogram shows quantitative analysis of GPx4 expression in neurons treated with hemin, iron, and DEF-HCC-PEG. (g) Effect of hemin on iron-mediated ferroptosis. Increase in MDA levels as an indicator of lipid peroxidation (ferroptosis) was analyzed in iron-treated cells in the presence or absence of hemin, PEG-HCC, or DEF-HCC-PEG. PEG-HCC reduced hemin-induced senescence but sensitized cells to iron-mediated ferroptosis. DEF-HCC-PEG effectively prevented ferroptosis in cultured neurons. Data indicate mean ± SEM from three independent experiments. *p < 0.01, **p < 0.05.
Combinatorial Antioxidant–Iron Chelator Therapy: Multifunctional Nanoparticle DEF-HCC-PEG Ameliorates Hemin-Induced Neurotoxicity.
Having established that hemin inhibited iron-mediated cell death, we investigated the effects of intervention with our prototype antioxidant nanoparticle, PEG-HCCs. While PEG-HCCs reduced the presence of senescence marker when treated following exposure to hemin as shown above, PEG-HCCs in hemin-treated cells caused an increase in sensitivity to iron-mediated ferroptosis marker expression (Figure 8f,g). This effect suggested that an antioxidant alone may not be sufficient and may require combining iron sequestration with an antioxidant to effectively address both senescence and ferroptosis. To test this possibility, we first used the MTT assay to evaluate the effect of the iron chelator DEF on cell death in cultured neurons treated with hemin alone or in combination with PEG-HCCs (Figure 8b). DEF (100 μM) demonstrated a protective effect comparable to that of PEG-HCC, while incubating hemin-treated cells with both PEG-HCCs and DEF further increased cell survival by 10–20%. The DEF concentration used was guided by published literature.73
While deferoxamine is a prototype intervention to assess the contribution of ferroptosis,77,78 it has several weaknesses when used as a therapy.79,80 Among the most important of these is inconsistent cellular uptake, requiring large doses and continuous infusion, risking toxicity. To address some of these limitation, we generated a multifunctional nanoparticle, DEF-HCC-PEG, by conjugating PEG-HCC with DEF into a single nanoparticle (Figure 8a), given the excellent cellular uptake.44 PEG-NH2 and DEF have a terminal amine that was covalently attached via an amide linkage to HCC to achieve an approximate loading of 58 ± 15 DEF molecules per DEF-HCC-PEG. We tested the effectiveness of DEF-HCC-PEG on hemin-treated neurons and in an experimental ICH mouse model. The concentration and dose of DEF-HCC-PEG were guided by our prior work with PEG-HCCs.43,81 The DEF-HCC-PEG treatment restored viability of hemin-treated cells at a significantly higher efficiency compared to PEG-HCC or DEF treatment alone (Figure 8b). DEF-HCC-PEG treatment also strongly reduced the levels of DNA damage markers γH2AX and p-53BP1 compared to DEF or PEG-HCC treatment (Figure 8c,d). DEF-HCC-PEG also prevented hemin-induced strand breaks in plasmid DNA in vitro, supporting the above results (Figure S7).
We then evaluated whether DEF-HCC-PEG treatment could also prevent the increased sensitivity to ferroptosis we observed after hemin and PEG-HCCs. To test this, we treated differentiated neurons derived from a human iPSC line with both hemin (5 μM) and FeSO4 (100 μM) and then treated with DEF-HCC-PEG or PEG-HCC. We performed IB on neuronal extracts to measure ferroptosis markers GPx4 and MDA levels in hemin-treated cells. We observed that DEF-HCC-PEG significantly prevented GPx4 depletion compared to PEG-HCC or DEF treatment alone (Figure 8e,f). Lower levels of GPx4 after PEG-HCC treatment were consistent with its inability to prevent ferroptosis. We also observed similar significant decrease by DEF-HCC-PEG in ferroptosis-associated lipid peroxidation as measured by MDA levels, confirming ferroptosis prevention (Figure 8g). Notably, FeSO4 (100 μM) increased MDA levels by 5- to 6-fold compared to hemin, indicating robust ferroptosis. Alternatively, increase in MDA levels was less for treatment with both hemin and iron, compared to iron alone, suggesting that these senescent cells were resistant to ferroptosis. Similarly, PEG-HCCs increased MDA levels, consistent with sensitization of hemin-treated cells to iron-mediated toxicity.
We tested the efficacy of DEF-HCC-PEG in preventing genome damage and senescence in vivo in mouse brains, following injection of hemolyzed blood into the left hemisphere of the brain. Thirty minutes after ICH induction, we injected DEF-HCC-PEG intraperitoneally (ip). For in vivo dosing, we employed a similar dose as in our prior work in other injury models, based on extrapolation from in vitro concentrations that achieve maximal efficacy then adjusted for mouse blood volume. Our prior work82 showed that following ip injection, PEGylated HCCs are taken up systemically, whose levels peak at approximately 12 h postinjection. We performed LA-PCR analysis of genomic DNA isolated from the right (control) and left hemispheres (ICH) sampled after 12 h. DEF-HCC-PEG treatment significantly restored both nuclear (Figure 9a,b) and mitochondrial genome integrity (Figure 9c,d). DNA integrity in two untreated (untreated were treated with ip saline as a control) ICH mice brains (mouse #1 and #2) and two DEF-HCC-PEG-treated ICH brains (mouse #3 and #4) is shown in the representative gels. ICH increased levels of the senescence factors ANIKRD1, EDN1, p21, and PVRL4 expression by qRT-PCR quantitation of respective mRNA levels, and DEF-HCC-PEG-treated ICH mice tissue showed substantial reduction of senescence markers after treatment (Figure 9e). Taken together, these results demonstrated that the multifunctional nanoparticle, DEF-HCC-PEG, was effect at inhibiting both hemin- and iron-induced neurotoxicity both in cultured cells and in experimental ICH mice.
Figure 9.
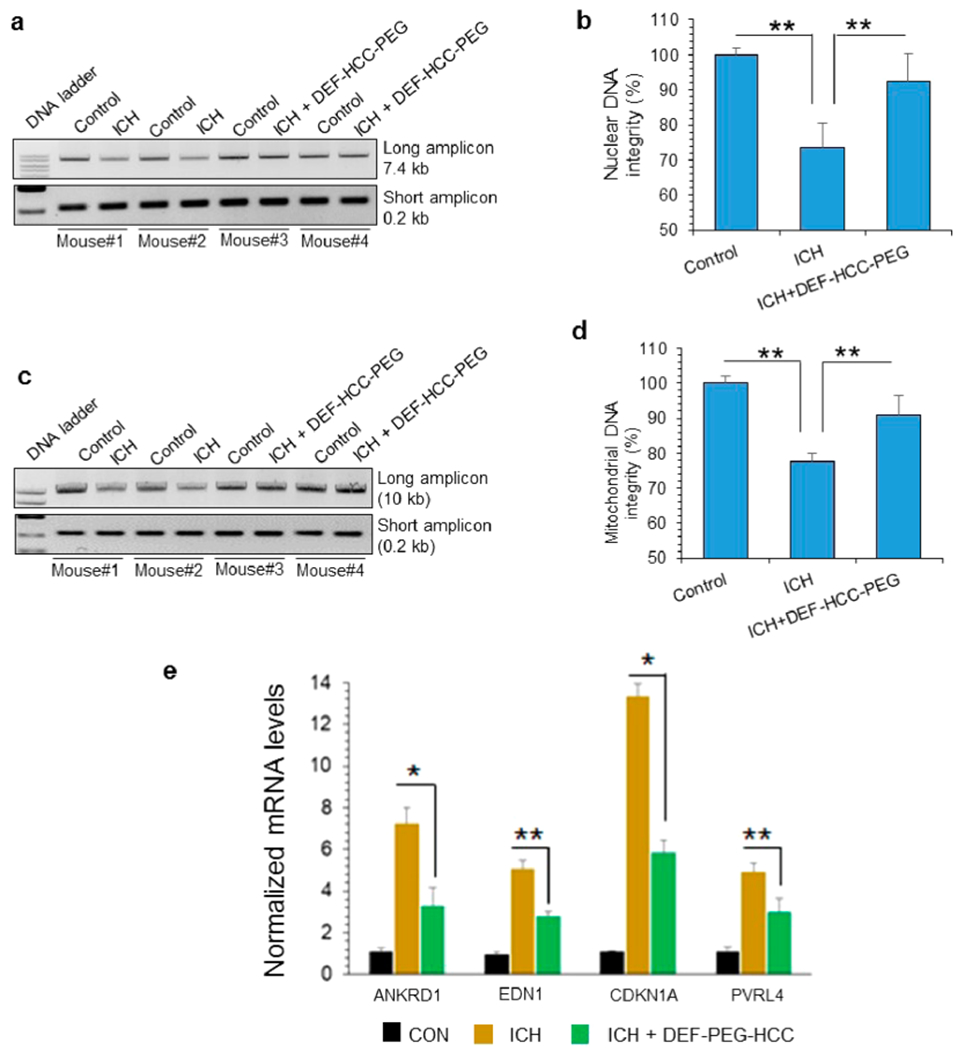
DEF-HCC-PEG alleviates genome damage and senescence in experimental ICH model. (a–d) LA-PCR of total DNA extracted from 6 untreated and 6 DEF-HCC-PEG-treated ICH mouse brains. Untreated mice were treated with saline. Left hemisphere served as non-ICH control in each mouse brain, while right hemisphere was induced with ICH as described in Methods. Two untreated (mouse #1 and #2) and two DEF-HCC-PEG-treated ICH mouse brains (mouse #3 and #4) are represented in the gel image. (a and b) Nuclear (7.4 kb) and (c and d) mitochondrial DNA (10 kb) were used as long amplicons (LA), while 0.2 kb within the LA region was used as a short amplicon. Picogreen-based quantitation of amplified DNA from 6 mice, expressed as percent (%) of DNA integrity in the histograms. (e) qRT-PCR analysis to measure expression of senescence-associated factors’ mRNA levels from sham control, ICH-induced, and ICH-induced along with DEF-HCC-PEG-treated mice (n = 6). *p < 0.01, **p < 0.05.
CONCLUSION
In this study, we observed both ROS- and non-ROS-mediated interactions among hemin- and iron-mediated toxicity, leading to specific types of genome damage, a persistent DDR in both neurons and endothelial cells, and activation of signals associated with senescence and ferroptosis. However, anti-ROS, while suppressing senescence, increased sensitivity to the iron-mediated cell death pathway ferroptosis. Addressing ROS or iron individually was not as effective as a combination approach. This interaction could explain disappointing results in clinical trials of either antioxidants or deferoxamine alone.2
Unexpectedly, we observed that Hb-derived hemin, a blood breakdown product, rapidly generated DSBs in the nuclear genome of multiple cultured cell types in vitro. These included neurons, endothelial cells, immortalized BJ-hTERT fibroblasts, and iPSC-derived neural stem cells. The rapid induction of DSBs by hemin resembled the effects of IR exposure or antitumor drugs such as bleomycin or etoposide.83,84 Our studies established the cell-type-independent nature of hemin-induced DSB generation, as we observed a similar pattern in four different cell lines including endothelial, neuronal, and primary fibroblast lines. Because hemin is a complex that contains iron and protoporphyrin IX, we tested the ability of iron and protoporphyrin IX to induce genome damage individually and observed that neither induced comparable levels of DSBs, even at 10-fold higher concentrations. This suggested genotoxicity of the iron-porphyrin complex. Our in vitro data using preoxidized hemin and naked plasmid DNA suggested that the hemin-induced DSBs in genomic DNA are dependent on the oxidation of hemin. We also ruled out that global ROS induced DSBs by demonstrating delayed nuclear ROS accumulation in hemin-treated neurons compared to mitochondrial ROS, which was generated much earlier. Furthermore, the fast kinetics of DSB induction in cultured neurons by 5 μM hemin, comparable to the effect of 2 Gy IR, suggested that DSBs are directly induced by hemin and not secondarily from multiple closely spaced single-strand breaks in the genome. Consistent with this scenario, previous studies suggested nuclear localization of heme, possibly resulting from translocation by a carrier protein.85
Our proof-of-principle in vivo data showed a significant increase in DNA DSBs in ICH-treated mouse brains coupled with DDR activation. Notably, we used hemolyzed blood to model the acute effects of blood degradation products, as it hastens the degradation. While, we use whole blood or collagenase to model chronic ICH effects including phenotypic changes, in this study, aimed at delineating the molecular mechanisms, hemolyzed blood injection was employed.
Previous studies suggested that hemin may exist as a dimer in aqueous solutions.86,87 It is possible that while oxidized hemin monomers nick DNA, dimeric hemin generates breaks on both DNA strands, causing DSB formation. Consistently, studies have shown that hemin can bind DNA87 and may preferentially bind G-quadruplex-forming DNA sequences.88–90 These sequences are generally enriched in gene promoter and termination regions in the mammalian genome.91 Thus, hemin-induced DSB damage may be euchromatin- or gene region-specific, a potential topic for future studies.
Pro-oxidant iron salts can generate ROS via the Fenton reaction, which causes oxidative DNA damage, including oxidized bases and single-strand breaks. It is important to note that ROS is not known to produce DSBs directly, although ROS-induced single-strand breaks in close proximity can lead to secondary DSBs eventually.53,92 Consistently, our alkaline comet assay indicated single-strand breaks in the cellular genome predominated after FeSO4 treatment. On the contrary, the presence of both alkaline and neutral comet tails indicated DSBs in the genomes of hemin-treated cells. We observed primarily alkaline comet tails with iron, but both alkaline and neutral comet tails in hemin-treated cells. Furthermore, our recent data demonstrate that ROS and pro-oxidant metal species like iron not only induce genome damage but also inhibit key DNA repair pathways and significantly affect damage repair signaling in neurons.33,34,56,57 Free iron promotes oxidation of cysteine residues in DNA repair enzymes. It also replaces Zn from their Zn-finger domains. We showed that defective repair due to free iron can be prevented with a combinatorial treatment including an iron chelator and an antioxidant.42,44,64,65 In view of their subdued signaling processes, senescent cells can be resistant to various stimuli and toxic substances.93,94
How hemin-induced senescence and iron-mediated ferroptosis interact in ICH is unknown; however, there is likely synergistic pathological activity between iron and hemin.95,96 To address this, we evaluated both senescence and ferroptosis markers in cultured neurons exposed to hemin or iron alone, or in combination. While hemin induced robust senescence and moderate ferroptosis, free iron primarily induced ferroptosis. When combined, hemin suppressed iron-mediated ferroptosis. We evaluated lipid peroxidation (MDA level) and GPx4 (glutathione peroxidase 4) expression levels, which are two critical parameters associated with ferroptosis.76,97 Further support for involvement of ferroptosis comes from the effects of ferrostatin and deferoxamine, two agents employed to elucidate a potential contribution of ferroptosis. This convergence of evidence supports our conclusion of the involvement of ferroptosis. This finding supports work in other cell types, that senescent neurons are resistant to iron toxicity and ferroptosis,41 and iron accumulates in senescent cells, indicating failure of ferroptosis. This constellation of findings indicates why single therapies, e.g., antioxidants or iron chelators alone, do not sufficiently prevent neurotoxicity after ICH. Thus, our data have significant implications for ICH treatment strategies.
To pursue potential therapies, we initially evaluated the protective efficacy of the catalytic antioxidant carbon nano-particle, PEG-HCC, previously developed by us and shown to have efficacy in other brain injury models not involving primary hemorrhage.43 In our model illustrated schematically in Figure 10, while PEG-HCCs prevented senescence, they also increased sensitivity to ferroptosis-induced cell death. These data suggest that the antioxidant PEG-HCC is not sufficient to ameliorate the dual toxic effects of hemin and iron. While PEG-HCC reduces oxidative damage, genome instability, and senescence post hemin, its beneficial effect of senescence reversal was counteracted by the sensitivity of cells to iron and induction of ferroptosis. To overcome these limitations of antioxidants and the poor uptake of DEF, we modified PEG-HCCs by attaching the chelating agent DEF at its nonchelating terminal amine, to develop a targeted therapy for both the conse uences of free iron/hemin and ROS. The DEF-HCC-PEG was significantly more effective in preventing both senescence and ferroptosis in cultured neurons than PEG-HCCs, DEF alone, or combined but as separate drugs. Importantly, DEF-HCC-PEG also showed good efficacy in reducing DNA damage in vivo when adminstred ip after infusion of hemolyzed blood into the subcortical mouse brain. The enhanced efficacy is likely due to the ability of the PEG-HCC to enhance the uptake of the DEF, given that DEF alone has uncertain and variable uptake.47 The in vivo proof-of-principle efficacy data for DEF-HCC-PEG treatment (Figure 9) performed in 6 mice were highly consistent with our extensive in vitro results from multiple cell lines, which showed a clear difference between the untreated and treated groups. Thus, our combinatorial approach using DEF-HCC-PEG was significantly more effective in mitigating multifaceted pathological events in these models. It is important to note that while antioxidants or iron chelators have been explored individually in ICH trials with limited success, such a combination was not tested earlier. Our study thus provides a strong rationale for exploring such a mechanism-based combinatorial approach to treating ICH. It is important to mention that no acute toxicity was observed with the DEF-HCC-PEG treatment in mice, although we did not perform a thorough toxicology study using these materials. A full dose/response study to assess the translational potential of our particles together with additional toxicology assessment is needed to establish the safety and long-term effects of the material, which is being pursued in our ongoing study. Another key question is the distribution and uptake of DEF in DEF-HCC-PEG. We have elsewhere shown that PEG-HCCs are rapidly taken up and distributed intracellularly including to colocalize with mitochondria.98 We found similar rapid uptake of the DEF-HCC-PEG particles (results not shown), suggesting that ultimately lower systemic concentrations can be used. Indeed, based on DEF content of the DEF-HCC-PEG, we calculate 200–300-fold lower overall DEF has been administered than traditional doses for chelation used in patients.
Figure 10.
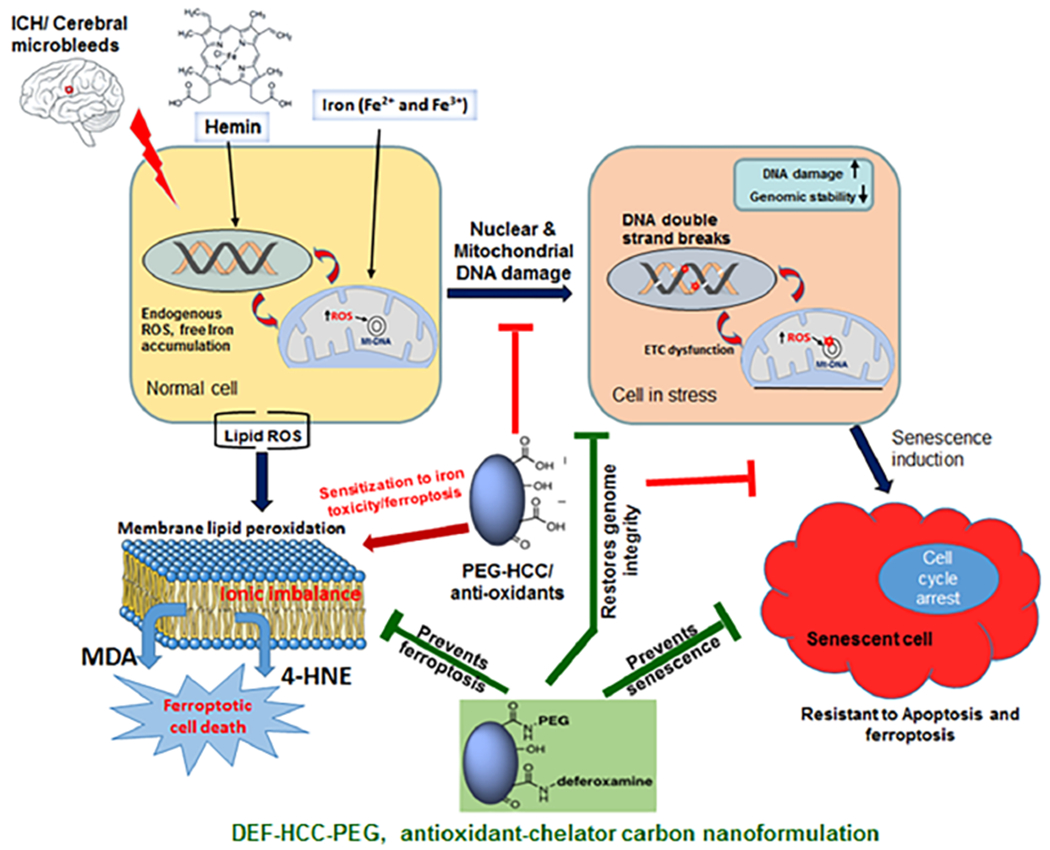
Model illustrating hemorrhage/hemin-induced genome instability, resulting interaction between senescence and ferroptosis, and a mechanism-driven combinatorial antioxidant plus chelator therapy for ICH. Hemin rapidly induces lethal DSBs in addition to ROS injury, which activate neuronal senescence. Prevention of oxidative injury by antioxidants then sensitizes cells to iron toxicity and ferroptosis. A nanoparticle of the iron chelator deferoxamine (DEF) and antioxidant PEG-HCC effectively prevents genome damage, senescence, and ferroptosis, thus mitigating ICH-induced neuronal toxicity in preclinical models.
The distinct nature of cellular toxicity and death caused by hemin versus iron is an important finding in our study. Unlike iron, which predominantly induces oxidative (non-DSB) DNA damage, hemin directly induced DSBs nearly exclusively; this finding was not previously reported. Because of acute induction of DSB damage, hemin largely induces cellular senescence together with a small fraction of cell death. Previous studies by Rajiv Ratan and colleagues showed that such cell death may involve necroptosis.10 Although, cells are exposed to both hemin and free iron under ICH conditions, hemin-mediated senescence obscures the ability of iron to induce ferroptosis at least initially. Our previous studies showed that PEG-HCCs primarily scavenge superoxide radicals.42,44 Here, we demonstrate that PEG-HCCs are able to reduce hemin-induced genome damage as well as senescence; however, this leads to sensitization of cells to iron toxicity. While ferroptosis involves oxidation/peroxidation of membrane lipids, it is specifically triggered by iron and not by reactive oxygen species. Thus, while antioxidants may have minor impact in reducing ferroptosis, iron chelation is required to significantly inhibit ferroptosis. In our study, antioxidant PEG-HCC reduced hemin-induced DNA DSB formation as well as senescence phenotype; nonetheless, this led to an increase in ferroptosis in the presence of iron. It is important to note that senescent cells are resistant to iron toxicity. The combinatorial nanozyme of PEG-HCC and iron chelator DEF, PEG-HCC-DEF, significant reduced both senescence and ferroptosis caused by hemin and iron.
Taken together, these results showed that our combinatorial strategy using a multifunctional nanoparticle of an antioxidant and an iron chelator effectively protected neurons against the toxicity of blood breakdown products in hemorrhagic stroke.
METHODS
Antibodies, Kits, and Cell Culture Reagents.
Antibodies against the following proteins were used in our study: γH2AX (CST9718; 1:1000), p-53BP1 (CST2675; 1:1000), p-ATM (ab81292; 1:1000), PGC1α (CST2178S; 1:1000), glutathione peroxidase 4 (ab185689; 1:1000), and β-actin (GTX109639; 1:4000). Antibodies were purchased from Abcam (Cambridge, UK), Cell Signaling Technology (Danvers, MA, USA), Sigma-Aldrich (St. Louis, MO, USA), Thermo Fisher Scientific (Waltham, MA, USA), GeneTex (Zeeland, MI, USA), and Santa Cruz Biotechnology (Dallas, TX, USA). The beta-galactosidase staining kit (CST#9860) was purchased from Cell Signaling Technology, the lipid peroxidation assay kit (KA1381) was purchased from Abnova (Taipei, Taiwan), the SYBR Green Master Mix was purchased from Qiagen (Hilden, Germany), and the plasmid (PGL4.23-GW, #60323) was purchased from Addgene (Watertown, MA, USA). Hemin (#51280) and all-trans retinoic acid (#R2625) were purchased from Sigma. The RNAeasy extraction kit (#74106) and DNeasy blood and tissue kit (#69504) were purchased from Qiagen. The Quant-iT Picogreen dsDNA assay kit and Verso cDNA synthesis kit (#AB1453B) were purchased from Thermo Fisher (#P11496). The TMRE mitochondrial membrane potential assay kit (ab113852) was purchased from Abcam. Cell imaging slides (#PEZGS0816) were purchased from Millicell EZ (EMD Millipore, Darmstadt, Germany). Cell culture reagents such as Dulbecco’s modified Eagle medium (DMEM), trypsin-EDTA, fetal bovine serum (FBS), and penicillin/streptomycin were purchased from Gibco BRL (Thermo Fisher Scientific). SH-SY5Y and bEnd.3 cells were purchased from ATCC (Manassas, VA, USA).
Cell Culture and Differentiation.
Human iPSC Cell Culture and Neuronal Differentiation.
Human iPSCs (KYOU-DXR0109B (201B7)) were purchased from ATCC and were grown using cell matrix basement membrane gel (ACS-3035) and pluripotent stem cell SFM XF/FF cell culture media (ACS-3002) at 37 °C and 5% CO2. NPSCs were derived from iPSCs using PSC neural induction medium according to the manufacturer’s instructions. NPCs were generated by replacing E8M with neural induction medium about 24 h after passing iPSCs, and then these cells were maintained for days. Derived NPSCs (P0) were then passaged onto six-well plates coated with Geltrex (Thermo Fisher), and the lineage was expanded in stemPRO neural stem cell SFM media (A1050901). Finally, the efficiency of neural induction was determined at passage 3 by IF staining with Nestin.52,53 Neurons were generated from iPSC cells as described previously (Figure S1e).53
SH-SY5Y Cell Culture.
The SH-SY5Y human neuroblastoma cell line was obtained from ATCC CRL-2266. Cells were cultured and maintained in DMEM/F12 (1:1) supplemented with 10% FBS (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Corning Inc., Corning, NY, USA) in a humidified chamber at 5% CO2 and 37 °C. To induce differentiation, cells were treated with differentiation medium containing 1% FBS and 10 μM all-trans retinoic acid (RA) for 4 days. Cells were replenished with freshly prepared differentiation medium containing RA every 24 h for up to 4 days (Figure S1d).
bEnd.3 Cell Culture.
Mouse brain endothelial cells (bEnd.3; ATCC CRL# 2299) were cultured in DMEM supplemented (50:50) with 4 mM glutamine, 10% FBS (Gibco), and 1 % penicillin/streptomycin (10 000 U/mL; Corning) in a humidified chamber at 5% CO2 and 37 °C. Cells were subcultured at a ratio of 1:6 with supplementation of fresh media once in 3 days.99
Hemin Preparation.
Hemin was purchased from Sigma (cat.# 51280). Hemin purity was verified by HPLC-MS/MS with some modifications.100 Briefly, high-resolution mass spectrometry (HRMS) of hemin (Figure S1e) was performed on a Thermo Scientific Q Exactive hybrid quadrupole-Orbitrap mass spectrometer. Peak detection was done at 395 nm. Samples were introduced into the HRMS via reversed-phase HPLC on an Accucore Vanquish C18+ column (2.1 × 100 mm, 1.5 μm) eluting with a linear gradient over 6 min. A gradient solvent system consisting of solvent A [water/acetonitrile (95:5)] and solvent B [water/acetonitrile (5:95)] with 0.1% formic acid was used as the mobile phase. The gradient elution was as follows: 80% A to 0% A in 5 min, 0% A in 0.5 min, 0% A to 80% A in 0.01 min, and 80% A in 0.49 min, followed by a 2 min postrun for column reconditioning.
For treatment in cell culture, a stock solution of 1 mM hemin was dissolved in dimethyl sulfoxide (DMSO), and then a serial dilution was made using serum-free DMEM/F12 to 0.1 mM. The working solution was vortexed and immediately used to obtain a desired concentration of hemin in tissue culture plates to induce neurotoxicity. A freshly prepared stock solution was used each time.
Cell Viability Assay.
About 2 × 104 cells were seeded onto 96-well plates and cultured for 24 h. Then, the cells were treated with differentiation medium containing 10 μM RA for 4 days. Differentiated SH-SY5Y cells were treated with different concentrations of hemin for 12 h. Cell viability was determined using the MTT assay. MTT solution (10 μL, 5 mg/mL) was added to each well and incubated for 2 h. Then, the supernatant was removed, and 100 μL of DMSO was added to each well. The plates were gently mixed by shaking, and then the absorbance was measured at 570 nm on a microplate reader. All cell viability assays were performed in triplicate (for each experiment) and three independent biological repeats. Results are represented as mean ± SEM.
Long-Amplicon Polymerase Chain Reaction.
Differentiated SH-SY5Y cells grown on 60 mm dishes that were exposed to 10 μM hemin for various incubation periods were harvested and washed with phosphate-buffered saline (PBS). Genomic DNA was isolated using a DNeasy blood and tissue kit according to the manufacturer’s instructions. Genome integrity was assessed using LongAmpTaq DNA polymerase101 to amplify the nuclear and mitochondrial genome damage.51 PCR amplification was performed using the primer sequences listed below, with a short region of the respective genes also amplified and used as a control to normalize the amplification obtained from the large fragment.102 The primer sequences used for LA-PCR amplification have been given in Supplemenatry Table 1.
PCR products were electrophoretically separated on 0.8% and 2% agarose gels for long- and short-amplicon products, respectively. These products were then stained with ethidium bromide, and images were acquired using the Gel Documentation System xR+ with Image Lab software (Bio-Rad Laboratories, Hercules, CA, USA). The relative DNA amplification percentage was quantified by band intensities using ImageJ software (NIH, Bethesda, MD, USA). The Quant-iTPicogreen dsDNA assay kit was used to quantify PCR products.
IF Analysis.
SH-SY5Y cells were seeded onto cell imaging slides until they reached 70% confluence. Cells were then treated with differentiation media for 4 d. Differentiated SH-SY5Y cells were exposed to 10 μM hemin for various incubation periods (5, 10, 15, 30, or 60 min), and the cells were washed twice with ice-cold PBS. Cells were then fixed using ice cold methanol for 5 min at 4 °C. Prior to antibody incubation, cells were permeabilized using 0.2% Triton X-100 in 1× PBS for 10 min. After blocking with 3% bovine serum albumin (BSA)/PBS for 1 h at room temperature (RT), cells were incubated with primary antibody (1:200 in 1% BSA/PBS) against γH2AX, p-53BP1, and p-ATM overnight at 4 °C in a humidified chamber. After washing three times in PBS for 10 min each, cells were incubated with fluorescent Alexa Fluor 488 (green) or 588 (red) (Life Technologies, Carlsbad, CA, USA) conjugated secondary antibodies (1:700, 1.5% BSA/PBs) for 1 h at RT. After washing with PBS three times for 10 min each, cells were counterstained with DAPI-containing (4,6-diamidino-2-phenylidole) mounting media. Images were immediately acquired using an AXIO Observer inverted microscope (Carl Zeiss, Oberkochen, Germany).
β-Gal Staining for Senescence.
SH-SY5Y cells were seeded onto six-well plates until they attained 70% confluence. Cells were then exposed to 10 μM hemin for 1 h, followed by PEG-HCC or PEG-HCC-DEF treatment for 24 h. Then, the cells were washed with ice cold PBS, and the senescence assay was performed according to the manufacturer’s protocol (Cell Signaling, CST#9860).
Single-Cell Gel Electrophoresis (Comet) Assay.
The alkaline and neutral comet assays were performed on differentiated SH-SY5Y cells at 12 h after hemin treatment. For the kinetics studies, assays were performed from 1 to 12 h, respectively, according to the manufacturer’s protocol (Trevigen, Gaithersburg, MD, USA). Briefly, treated cells were mixed with low-melting grade agarose and placed on comet assay slides provided by the manufacturer (Trevigen). Slides were immersed in neutral and alkaline electrophoresis buffer, respectively, after immersion in lysis solution. For the alkaline comet assay, slides were subjected to electrophoresis at 21 V for 30 min, while for the neutral comet assay, slides were subjected to electrophoresis at 21 V for 45 min. Slides were then fixed with 70% ethanol and stained with SYBR Green for visualization using a fluorescence microscope (Carl Zeiss).
Detection of ROS by pHyper-Cytoplasmic-, Mitochondrial-, and Nuclear-Specific Sensors.
Cellular ROS levels in hemin-treated differentiated SH-SY5Y cells were assessed using the hydrogen peroxide sensor pHyPer (Evrogen, Axxora Inc., Farmingdale, NY, USA).103 Vectors included pHyPer-Cyto (without targeting signal), pHyPer-dMito (mitochondria-targeting signal fused to the HyPer N-terminus), and pHyPer-Nuc (nuclear localization signal fused to the HyPer C-terminus). The assay was performed as described by Hajas et al.60 Cells were transfected with the vectors using Lipofectamine 2000 as per the manufacturer’s instructions (Life Technologies). Seventy-two hours after transfection, cells were incubated in medium containing 1% FBS for 4 h and challenged with hemin (5 μM) or H2O2 (10 μM). At 0, 5, 10, 30, and 60 min postexposure, cells were fixed in formalin (3.7%), dried, and mounted onto microscope slides. Images were acquired using an AXIO Observer inverted microscope (Carl Zeiss).
Measurement of Mitochondrial Membrane Potential (ΔΨm).
SH-SY5Y cells were seeded onto six-well plates until they attained 70% confluence. Cells were then exposed to 5 and 10 μM hemin for 1 h. Next, mitochondrial membrane potential was determined using a TMRE mitochondrial membrane potential assay kit according to the manufacturer’s protocol.
Synthesis of PEG-HCC and DEF-HCC-PEG Nanoparticles.
HCCs (51 mg, 4.3 mmol of carbon) were suspended in DMF (50 mL). The mixture was subjected to bath sonication for 45 min to completely dissolve the HCCs. Methoxy-PEG-NH2 (1.01 g, 0.202 mmol) with an average molecular weight of 5 kDa was added, and the reaction mixture was sonicated for 20 min. N,N′-Diisopropylcarbodiimide (DIC) (0.50 mL, 0.41 g, 3.2 mmol) was added, and the resulting solution was stirred at RT for 72 h. This solution was then diluted in water and purified by tangential flow filtration (Spectrum Laboratories Krosflo, Research IIi TFF System) with a 50 kDa poly(ether sulfone) (PES) dialysis column to yield 42 mL of PEG-HCCs with a carbon-core concentration of 1.05 mg/mL.
For DEF-HCC-PEG, HCCs (20 mg, 1.7 mmol of carbon) were suspended in DMF (20 mL), and the mixture was placed in a bath sonicator for 45 min. DEF (25 mg, 0.045 mmol), N,N-diisopropylethylamine (DIPEA) (0.040 mL, 30 mg, 0.23 mmol), and methoxy-PEG-NH2 (200 mg, 0.0400 mmol) with an average molecular weight of 5 kDa were then added. The solution was again placed in a sonication bath. After 30 min, DIC (0.20 mL, 0.16 g, 1.3 mmol) was added, and the reaction was stirred at RT for 72 h. The resulting solution was diluted in water and purified by tangential flow filtration (Spectrum Laboratories Krosflo, Research IIi TFF System) with a 50 kDa PES dialysis column to yield 25 mL of DEF PEG-HCCs with a carbon core concentration of 0.72 mg/mL.
Characterization of Nanoparticle-Calculating DEF and PEG Loading of DEF-HCC-PEG.
In order to determine the DEF loading per DEF-HCC-PEG nanoparticle, the nanomaterial was characterized via thermogravimetric analysis (TGA). The acquired data were then compared to the TGA of HCC1 and PEG-HCCs (see Supporting Information for methods and Figure S8).
Lipid Peroxidation Assay.
Lipid peroxides in the hemin-treated cells and mouse brain tissues were assayed in terms of malondialdehyde. MDA levels were measured spectrophotometrically using thiobarbituric acid reactive substances (TBARS). The assay was performed using a TBARS kit according to the manufacturer’s protocol.
Animal Experiments and Treatment Regimen.
Animal experiments were performed after obtaining approval from the Baylor College of Medicine and Michael E. DeBakey VA Medical Research and Development Animal Ethical Clearance Committees (IACUC—AN4510). C57BL/6 mice (Jackson Laboratories, Bar Harbor, ME, USA) were allowed to acclimatize for at least 5 d following arrival at the vivarium. Mice received standard conditions of food, water, lighting, and temperature in environmentally controlled cages. Each animal underwent a tail vein puncture to obtain 20 μL of blood to be utilized for the experimental ICH. This is a modification of the method of Nakamura et al.104 To maximize exposure to blood elements, blood was lysed by freeze–thawing prior to injection. For the procedure, mice were anesthetized by isoflurane (1.5–3%) inhalation. Following skin cleansing and an infusion of a 1:20 dilution of bupivacaine and lidocaine 50/50 sq, the animals were affixed in a stereotaxic frame (Kopf) and the calvarium was exposed. Next, a 1 mm burr hole was made in the calvarium at the appropriate coordinates, and the lysed blood was injected under stereotaxic control at coordinates corresponding to the striatum. Following these procedures, the skin was closed over the various incisions with 6-0 proline. Animals were kept warm on a warming pad until sternally recumbent and then returned to their cages. Postoperative sc buprenorphine (0.6 mg/kg) was given every 12 h, with ketoprofen 2.5–5 mg/kg sq daily. The sterile-filtered carbon nanoparticle preparations, DEF-HCC-PEG (2 mg/kg), were administered in saline solution ip or saline solution alone as a control at 30 min after ICH. Mice received supportive postoperative care and were monitored twice daily. At 12–24 h, mice were sacrificed under deep anesthesia, and their brains were removed and dissected into peri-hemorrhage and contralateral control regions. Peri-hemorrhage tissue was identified based on the visible blood following injection and careful dissection of the adjacent tissue. Comparable regions from the contralateral hemisphere were used as control.
Tissue Processing and Protein Isolation.
Brain tissues were processed under liquid nitrogen, ground into fine powder, and stored at −80 °C until use. Proteins were extracted from tissues using RIPA buffer, including protease and phosphatase inhibitors (20 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1 mM EDTA, 0.5 EGTA; 1% sodium deoxycholate; 1% Triton-X-100; and 0.1% SDS). Tissue lysates were sonicated 8–10 times, and then debris was removed by centrifugation at 13 000 rpm for 15 min at 4 °C.
IB Analysis.
Protein concentrations in cell or tissue extracts were determined using the Bradford assay (Bio-Rad). Protein mixed with loading buffer was resolved on NuPAGE Bis-Tris gels (4–12%), and then proteins were electrotransferred onto nitrocellulose (0.45 μm) membranes. Membranes were then blocked at RT in 3% BSA/TBST, followed by incubation with primary antibodies in 1% BSA overnight at 4 °C on a shaker. The next day, membranes were washed three times in TBST for 7 min each at RT. Subsequently, membranes were probed with appropriate secondary antibodies in 1% BSA/TBST for 1 h at RT. After washing three times in TBST, membranes were finally developed using ECL substrate (Thermo Fisher) and scanned using the gel documentation system (Odyssey FC). Band intensity was calculated using ImageJ (NIH).
RNA Extraction and Quantitative Real-Time PCR.
Total RNA was extracted from cells or tissues using a Qiagen RNAeasy extraction kit as per the manufacturer’s protocol. DNA-free RNA was reverse-transcribed into cDNA using the verso cDNA synthesis kit (cat.# AB1453B, Thermo Fisher). PCR was performed using a real-time PCR thermocycler (Applied Biosystems, 7500) and the SYBR Green method. Amplification of Actin/GAPDH/18S was used to normalize expression of the target gene. Optimal primer concentrations and annealing temperatures were determined by preliminary experiments. Gene expression analysis was performed using the 2−ΔΔCt method.
Statistical Analysis.
A minimum of three independent biological experiments was always performed, in addition to the triplicate measurements for each assay. Data and statistical analyses were performed using Microsoft Excel or GraphPad Prism 6 software. Group comparisons were analyzed for significant differences using two-way ANOVA or Student’s t test. The p values are indicated in the associated figure legends, with p < 0.05 considered statistically significant. Results are represented as mean ± SEM from samples assayed in triplicate (for each experiment) and three independent experiments.
Study Approval.
Animal experiments were performed after obtaining approval from the Baylor College of Medicine and Michael E. DeBakey VA Medical Research and Development Animal Ethical Clearance Committees (IACUC–AN4510).
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by grants from the National Institute of Neurological Disorders and Stroke (NINDS) of the National Institutes of Health (NIH) under the award numbers R01NS088645 (M.L.H.), NIH R01NS094535 (T.A.K. and J.M.T.), R01NS094535 (T.A.K., J.M.T., and M.L.H.), and Welch Foundation Grant BE-0048 (T.A.K.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsnano.9b05821.
List of gene targets and primer sequences used for LA-PCR experiment and additional experimental data for dose and time kinetics of iron (FeSO4 and Fe-NTA)-induced cell death analysis and hemin purity analysis chromatogram; PGC-1α levels and DNA integrity in hemin-treated neurons; PGC-1α levels in ICH mice; DNA strand breaks quantitation in hemin-treated b.End3 cells and its amelioration by PEG-HCC; inhibition of hemin-induced senescence by PEG-HCC in b.End3 cells; GPx4 levels in hemin-treated iPSC-derived neurons together with erastin/ferrostatin controls; in vitro DNA damage by hemin and iron and characterization of nanoparticles via thermogravimetric analysis (PDF)
Complete contact information is available at: https://pubs.acs.org/10.1021/acsnano.9b05821
The authors declare the following competing financial interest(s): T.A.K. and J.M.T. are inventors on patents granted and under review related to the therapeutic potential of oxidized carbon nanomaterials.
Contributor Information
Prakash Dharmalingam, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, Texas 77030, United States.
Girish Talakatta, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, Texas 77030, United States.
Joy Mitra, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, Texas 77030, United States.
Haibo Wang, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, Texas 77030, United States.
Paul J. Derry, Institute of Biosciences and Technology, Texas A&M Health Science Center, Houston, Texas 77030, United States
Lizanne Greer Nilewski, Department of Chemistry, Rice University, Houston, Texas 77005, United States.
Emily A. McHugh, Department of Chemistry, Rice University, Houston, Texas 77005, United States
Roderic H. Fabian, Department of Neurology, Baylor College of Medicine, Michael E. DeBakey VA Medical Center, Houston, Texas 77030, United States
Kimberly Mendoza, Department of Chemistry, Rice University, Houston, Texas 77005, United States.
Velmarini Vasquez, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, Texas 77030, United States.
Pavana M. Hegde, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, Texas 77030, United States
Eugenia Kakadiaris, Department of Chemistry, Rice University, Houston, Texas 77005, United States.
Trenton Roy, Department of Chemistry, Rice University, Houston, Texas 77005, United States.
Istvan Boldogh, Department of Microbiology and Immunology, University of Texas Medical Branch, Galveston, Texas 77555, United States.
Venkatesh L. Hegde, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, Texas 77030, United States
Sankar Mitra, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, Texas 77030, United States; Weill Medical College of Cornell University, New York, New York 10065, United States.
James M. Tour, Departments of Chemistry, Computer Science, Materials Science and NanoEngineering Smalley-Curl Institute and the NanoCarbon Center, Rice University, Houston, Texas 77005, United States.
Thomas A. Kent, Institute of Biosciences and Technology, Texas A&M Health Science Center, Houston, Texas 77030, United States; Department of Chemistry, Rice University, Houston, Texas 77005, United States; Stanley H. Appel Department of Neurology, Houston Methodist Hospital and Research Institute, Houston, Texas 77030, United States
Muralidhar L. Hegde, Department of Radiation Oncology, Houston Methodist Research Institute, Houston, Texas 77030, United States; Weill Medical College of Cornell University, New York, New York 10065, United States; Center for Neuroregeneration, Department of Neurosurgery, Houston Methodist Neurological Institute, Houston, Texas 77030, United States.
REFERENCES
- (1).Chen-Roetling J; Kamalapathy P; Cao Y; Song W; Schipper HM; Regan RF Astrocyte Heme Oxygenase-1 Reduces Mortality and Improves Outcome after Collagenase-Induced Intracerebral Hemorrhage. Neurobiol. Dis 2017, 102, 140–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Selim M; Foster LD; Moy CS; Xi G; Hill MD; Morgenstern LB; Greenberg SM; James ML; Singh V; Clark WM; Norton C; Palesch YY; Yeatts SD Deferoxamine Mesylate in Patients with Intracerebral Haemorrhage (I-Def): A Multicentre, Randomised, Placebo-Controlled, Double-Blind Phase 2 Trial. Lancet Neurol. 2019, 18, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Lok J; Leung W; Murphy S; Butler W; Noviski N; Lo EH Intracranial Hemorrhage: Mechanisms of Secondary Brain Injury. Acta Neurochir. Suppl 2011, 111, 63–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sidyakin AA; Kaysheva AL; Kopylov AT; Lobanov AV; Morozov SG Proteomic Analysis of Cerebral Cortex Extracts from Sus Scrofa with Induced Hemorrhagic Stroke. J. Mol. Neurosci 2018, 65, 28–34. [DOI] [PubMed] [Google Scholar]
- (5).Xi G; Strahle J; Hua Y; Keep RF Progress in Translational Research on Intracerebral Hemorrhage: Is There an End in Sight? Prog. Neurobiol 2014, 115, 45–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Vijayan M; Reddy PH Stroke, Vascular Dementia, and Alzheimer’s Disease: Molecular Links. J. Alzheimer’s Dis 2016, 54, 427–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Augustynek B; Kudin AP; Bednarczyk P; Szewczyk A; Kunz WS Hemin Inhibits the Large Conductance Potassium Channel in Brain Mitochondria: A Putative Novel Mechanism of Neurodegeneration. Exp. Neurol 2014, 257, 70–75. [DOI] [PubMed] [Google Scholar]
- (8).Dang G; Yang Y; Wu G; Hua Y; Keep RF; Xi G Early Erythrolysis in the Hematoma after Experimental Intracerebral Hemorrhage. Transl. Transl. Stroke Res 2017, 8, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Robinson SR; Dang TN; Dringen R; Bishop GM Hemin Toxicity: A Preventable Source of Brain Damage Following Hemorrhagic Stroke. Redox Rep. 2009, 14, 228–235. [DOI] [PubMed] [Google Scholar]
- (10).Zille M; Karuppagounder SS; Chen Y; Gough PJ; Bertin J; Finger J; Milner TA; Jonas EA; Ratan RR Neuronal Death after Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke 2017, 48, 1033–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jeney V; Balla J; Yachie A; Varga Z; Vercellotti GM; Eaton JW; Balla G Pro-Oxidant and Cytotoxic Effects of Circulating Heme. Blood 2002, 100, 879–887. [DOI] [PubMed] [Google Scholar]
- (12).Balla G; Jacob HS; Eaton JW; Belcher JD; Vercellotti GM Hemin: A Possible Physiological Mediator of Low Density Lipoprotein Oxidation and Endothelial Injury. Arterioscler. Thromb 1991, 11, 1700–1711. [DOI] [PubMed] [Google Scholar]
- (13).Chen-Roetling J; Regan RF Targeting the Nrf2-Heme Oxygenase-1 Axis after Intracerebral Hemorrhage. Curr. Pharm. Des 2017, 23, 2226–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rifkind JM; Nagababu E; Ramasamy S; Ravi LB Hemoglobin Redox Reactions and Oxidative Stress. Redox Rep. 2003, 8, 234–237. [DOI] [PubMed] [Google Scholar]
- (15).Rifkind JM; Mohanty JG; Nagababu E The Pathophysiology of Extracellular Hemoglobin Associated with Enhanced Oxidative Reactions. Front. Physiol 2015, 5, 500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Umbreit J Methemoglobin–It’s Not Just Blue: A Concise Review. Am. J. Hematol 2007, 82, 134–144. [DOI] [PubMed] [Google Scholar]
- (17).Chakane S; Markad V; Kodam K; Bulow L The Penultimate Tyrosine Residues Are Critical for the Genotoxic Effect of Human Hemoglobin. Adv. Exp. Med. Biol 2017, 977, 351–357. [DOI] [PubMed] [Google Scholar]
- (18).Qureshi AI; Mohammad YM; Yahia AM; Suarez JI; Siddiqui AM; Kirmani JF; Suri MF; Kolb J; Zaidat OO A Prospective Multicenter Study to Evaluate the Feasibility and Safety of Aggressive Antihypertensive Treatment in Patients with Acute Intracerebral Hemorrhage. J. Intensive Care Med 2005, 20, 34–42. [DOI] [PubMed] [Google Scholar]
- (19).Akoudad S; Wolters FJ; Viswanathan A; de Bruijn RF; van der Lugt A; Hofman A; Koudstaal PJ; Ikram MA; Vernooij MW Association of Cerebral Microbleeds with Cognitive Decline and Dementia. JAMA Neurol 2016, 73, 934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Vijayan M; Kumar S; Yin X; Zafer D; Chanana V; Cengiz P; Reddy PH Identification of Novel Circulatory Microrna Signatures Linked to Patients with Ischemic Stroke. Hum. Mol. Genet 2018, 27, 2318–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ghosh S; Canugovi C; Yoon JS; Wilson DM 3rd; Croteau DL; Mattson MP; Bohr VA Partial Loss of the DNA Repair Scaffolding Protein, Xrcc1, Results in Increased Brain Damage and Reduced Recovery from Ischemic Stroke in Mice. Neurobiol. Aging 2015, 36, 2319–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yates PA; Villemagne VL; Ellis KA; Desmond PM; Masters CL; Rowe CC Cerebral Microbleeds: A Review of Clinical, Genetic, and Neuroimaging Associations. Front. Neurol 2014, 4, 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kitago T; Ratan RR Rehabilitation Following Hemorrhagic Stroke: Building the Case for Stroke-Subtype Specific Recovery Therapies. F1000Research 2017, 6, 2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Theriault BC; Woo SK; Karimy JK; Keledjian K; Stokum JA; Sarkar A; Coksaygan T; Ivanova S; Gerzanich V; Simard JM Cerebral Microbleeds in a Neonatal Rat Model. PLoS One 2017, 12, No. e0171163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Aburatani H; Hippo Y; Ishida T; Takashima R; Matsuba C; Kodama T; Takao M; Yasui A; Yamamoto K; Asano M Cloning and Characterization of Mammalian 8-Hydroxyguanine-Specific DNA Glycosylase/Apurinic, Apyrimidinic Lyase, a Functional Mutm Homologue. Cancer Res. 1997, 57, 2151–2156. [PubMed] [Google Scholar]
- (26).Margaill I; Plotkine M; Lerouet D Antioxidant Strategies in the Treatment of Stroke. Free Radical Biol. Med 2005, 39, 429–443. [DOI] [PubMed] [Google Scholar]
- (27).Firuzi O; Miri R; Tavakkoli M; Saso L Antioxidant Therapy: Current Status and Future Prospects. Curr. Med. Chem 2011, 18, 3871–3888. [DOI] [PubMed] [Google Scholar]
- (28).Shirley R; Ord EN; Work LM Oxidative Stress and the Use of Antioxidants in Stroke. Antioxidants 2014, 3, 472–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Duan X; Wen Z; Shen H; Shen M; Chen G Intracerebral Hemorrhage, Oxidative Stress, and Antioxidant Therapy. Oxid. Med. Cell. Longevity 2016, 2016, 1203285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Fouda AY; Newsome AS; Spellicy S; Waller JL; Zhi W; Hess DC; Ergul A; Edwards DJ; Fagan SC; Switzer JA Minocycline in Acute Cerebral Hemorrhage: An Early Phase Randomized Trial. Stroke 2017, 48, 2885–2887. [DOI] [PubMed] [Google Scholar]
- (31).Perry G; Taddeo MA; Nunomura A; Zhu X; Zenteno-Savin T; Drew KL; Shimohama S; Avila J; Castellani RJ; Smith MA Comparative Biology and Pathology of Oxidative Stress in Alzheimer and Other Neurodegenerative Diseases: Beyond Damage and Response. Comp. Biochem. Physiol., Part C: Toxicol. Pharmacol 2002, 133, 507–513. [DOI] [PubMed] [Google Scholar]
- (32).Zhao BQ; Tejima E; Lo EH Neurovascular Proteases in Brain Injury, Hemorrhage and Remodeling after Stroke. Stroke 2007, 38, 748–752. [DOI] [PubMed] [Google Scholar]
- (33).Hegde ML; Hegde PM; Holthauzen LM; Hazra TK; Rao KS; Mitra S Specific Inhibition of Neil-Initiated Repair of Oxidized Base Damage in Human Genome by Copper and Iron: Potential Etiological Linkage to Neurodegenerative Diseases. J. Biol. Chem 2010, 285, 28812–28825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hegde ML; Hegde PM; Jagannatha Rao KS; Mitra S Oxidative Genome Damage and Its Repair in Neurodegenerative Diseases: Function of Transition Metals as a Double-Edged Sword. J. Alzheimer’s Dis 2011, 24, 183–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Li H; Swiercz R; Englander EW Elevated Metals Compromise Repair of Oxidative DNA Damage via the Base Excision Repair Pathway: Implications of Pathologic Iron Overload in the Brain on Integrity of Neuronal DNA. J. Neurochem 2009, 110, 1774–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Aft RL; Mueller GC Hemin-Mediated DNA Strand Scission. J. Biol. Chem 1983, 258, 12069–12072. [PubMed] [Google Scholar]
- (37).Chakane S; Matos T; Kettisen K; Bulow L Fetal Hemoglobin Is Much Less Prone to DNA Cleavage Compared to the Adult Protein. Redox Biol 2017, 12, 114–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Even B; Fayad-Kobeissi S; Gagliolo JM; Motterlini R; Boczkowski J; Foresti R; Dagouassat M Heme Oxygenase-1 Induction Attenuates Senescence in Chronic Obstructive Pulmonary Disease Lung Fibroblasts by Protecting against Mitochondria Dysfunction. Aging Cell 2018, 17, No. e12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Shan H; Li T; Zhang L; Yang R; Li Y; Zhang M; Dong Y; Zhou Y; Xu C; Yang B; Liang H; Gao X; Shan H Heme Oxygenase-1 Prevents Heart against Myocardial Infarction by Attenuating Ischemic Injury-Induced Cardiomyocytes Senescence. EBioMedicine 2019, 39, 59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hedblom A; Hejazi SM; Canesin G; Choudhury R; Hanafy KA; Csizmadia E; Persson JL; Wegiel B Heme Detoxification by Heme Oxygenase-1 Reinstates Proliferative and Immune Balances Upon Genotoxic Tissue Injury. Cell Death Dis 2019, 10, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Masaldan S; Clatworthy SAS; Gamell C; Meggyesy PM; Rigopoulos AT; Haupt S; Haupt Y; Denoyer D; Adlard PA; Bush AI; Cater MA Iron Accumulation in Senescent Cells Is Coupled with Impaired Ferritinophagy and Inhibition of Ferroptosis. Redox Biol 2018, 14, 100–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Samuel EL; Marcano DC; Berka V; Bitner BR; Wu G; Potter A; Fabian RH; Pautler RG; Kent TA; Tsai AL; Tour JM Highly Efficient Conversion of Superoxide to Oxygen Using Hydrophilic Carbon Clusters. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 2343–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Fabian RH; Derry PJ; Rea HC; Dalmeida WV; Nilewski LG; Sikkema WKA; Mandava P; Tsai AL; Mendoza K; Berka V; Tour JM; Kent TA Efficacy of Novel Carbon Nanoparticle Antioxidant Therapy in a Severe Model of Reversible Middle Cerebral Artery Stroke in Acutely Hyperglycemic Rats. Front. Neurol 2018, 9, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Bitner BR; Marcano DC; Berlin JM; Fabian RH; Cherian L; Culver JC; Dickinson ME; Robertson CS; Pautler RG; Kent TA; Tour JM Antioxidant Carbon Particles Improve Cerebrovascular Dysfunction Following Traumatic Brain Injury. ACS Nano 2012, 6, 8007–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Veltkamp R; Purrucker J Management of Spontaneous Intracerebral Hemorrhage. Curr. Neurol. Neurosci. Rep 2017, 17 (10), 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hegde ML; Bharathi P; Suram A; Venugopal C; Jagannathan R; Poddar P; Srinivas P; Sambamurti K; Rao KJ; Scancar J; Messori L; Zecca L; Zatta P Challenges Associated with Metal Chelation Therapy in Alzheimer’s Disease. J. Alzheimer’s Dis 2009, 17, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hegde ML; Bharathi P; Suram A; Venugopal C; Jagannathan R; Poddar P; Srinivas P; Sambamurti K; Rao KJ; Scancar J; Messori L; Zecca L; Zatta P Challenges Associated with Metal Chelation Therapy in Alzheimer’s Disease. J. Alzheimer’s Dis 2009, 17, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Jorgensen A; Staalsoe JM; Simonsen AH; Hasselbalch SG; Hogh P; Weimann A; Poulsen HE; Olsen NV Progressive DNA and RNA Damage from Oxidation after Aneurysmal Subarachnoid Haemorrhage in Humans. Free Radical Res 2018, 52, 51–56. [DOI] [PubMed] [Google Scholar]
- (49).Gu Y; Hua Y; He Y; Wang L; Hu H; Keep RF; Xi G Iron Accumulation and DNA Damage in a Pig Model of Intracerebral Hemorrhage. Acta Neurochir. Suppl 2011, 111, 123–128. [DOI] [PubMed] [Google Scholar]
- (50).Tripathi AK; Singh N Prion Protein-Hemin Interaction Upregulates Hemoglobin Synthesis: Implications for Cerebral Hemorrhage and Sporadic Creutzfeldt-Jakob Disease. J. Alzheimer’s Dis 2016, 51, 107–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Furda AM; Bess AS; Meyer JN; Van Houten B Analysis of DNA Damage and Repair in Nuclear and Mitochondrial DNA of Animal Cells Using Quantitative Pcr. Methods Mol. Biol 2012, 920, 111–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Vasquez V; Mitra J; Hegde PM; Pandey A; Sengupta S; Mitra S; Rao KS; Hegde ML Chromatin-Bound Oxidized Alpha-Synuclein Causes Strand Breaks in Neuronal Genomes in In Vitro Models of Parkinson’s Disease. J. Alzheimer’s Dis 2017, 60, S133–S150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Wang H; Guo W; Mitra J; Hegde PM; Vandoorne T; Eckelmann BJ; Mitra S; Tomkinson AE; Van Den Bosch L; Hegde ML Mutant FUS Causes DNA Ligation Defects to Inhibit Oxidative Damage Repair in Amyotrophic Lateral Sclerosis. Nat. Commun 2018, 9, 3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Glei M; Klenow S; Sauer J; Wegewitz U; Richter K; Pool-Zobel BL Hemoglobin and Hemin Induce DNA Damage in Human Colon Tumor Cells Ht29 Clone 19a and in Primary Human Colonocytes. Mutat. Res., Fundam. Mol. Mech. Mutagen 2006, 594, 162–171. [DOI] [PubMed] [Google Scholar]
- (55).Ishikawa S; Tamaki S; Ohata M; Arihara K; Itoh M Heme Induces DNA Damage and Hyperproliferation of Colonic Epithelial Cells via Hydrogen Peroxide Produced by Heme Oxygenase: A Possible Mechanism of Heme-Induced Colon Cancer. Mol. Nutr. Food Res 2010, 54, 1182–1191. [DOI] [PubMed] [Google Scholar]
- (56).Mitra J; Guerrero EN; Hegde PM; Wang H; Boldogh I; Rao KS; Mitra S; Hegde ML New Perspectives on Oxidized Genome Damage and Repair Inhibition by Pro-Oxidant Metals in Neurological Diseases. Biomolecules 2014, 4, 678–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Mitra J; Vasquez V; Hegde PM; Boldogh I; Mitra S; Kent TA; Rao KS; Hegde ML Revisiting Metal Toxicity in Neurodegenerative Diseases and Stroke: Therapeutic Potential. Neurol. Res. Ther 2014, 1, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Sahan AZ; Hazra TK; Das S The Pivotal Role of DNA Repair in Infection Mediated-Inflammation and Cancer. Front. Microbiol 2018, 9, 663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Wang H; Adhikari S; Butler BE; Pandita TK; Mitra S; Hegde ML A Perspective on Chromosomal Double Strand Break Markers in Mammalian Cells. Jacobs J. Radiat. Oncol 2014, 1, 003. [PMC free article] [PubMed] [Google Scholar]
- (60).Hajas G; Bacsi A; Aguilera-Aguirre L; Hegde ML; Tapas KH; Sur S; Radak Z; Ba X; Boldogh I 8-Oxoguanine DNA Glycosylase-1 Links DNA Repair to Cellular Signaling via the Activation of the Small Gtpase Rac1. Free Radical Biol. Med 2013, 61, 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Berlin JM; Pham TT; Sano D; Mohamedali KA; Marcano DC; Myers JN; Tour JM Noncovalent Functionalization of Carbon Nanovectors with an Antibody Enables Targeted Drug Delivery. ACS Nano 2011, 5, 6643–6650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Jalilov AS; Nilewski LG; Berka V; Zhang C; Yakovenko AA; Wu G; Kent TA; Tsai AL; Tour JM Perylene Diimide as a Precise Graphene-Like Superoxide Dismutase Mimetic. ACS Nano 2017, 11, 2024–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Li Q; Han X; Lan X; Gao Y; Wan J; Durham F; Cheng T; Yang J; Wang Z; Jiang C; Ying M; Koehler RC; Stockwell BR; Wang J Inhibition of Neuronal Ferroptosis Protects Hemorrhagic Brain. JCI Insight 2017, 2, No. e90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Stockwell BR; Friedmann Angeli JP; Bayir H; Bush AI; Conrad M; Dixon SJ; Fulda S; Gascon S; Hatzios SK; Kagan VE; Noel K; Jiang X; Linkermann A; Murphy ME; Overholtzer M; Oyagi A; Pagnussat GC; Park J; Ran Q; Rosenfeld CS; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Bielak-Zmijewska A; Mosieniak G; Sikora E Is DNA Damage Indispensable for Stress-Induced Senescence? Mech. Ageing Dev 2018, 170, 13–21. [DOI] [PubMed] [Google Scholar]
- (66).Her J; Bunting SF How Cells Ensure Correct Repair of DNA Double-Strand Breaks. J. Biol. Chem 2018, 293, 10502–10511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).McHugh D; Gil J Senescence and Aging: Causes, Consequences, and Therapeutic Avenues. J. Cell Biol 2018, 217, 65–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Munoz-Espin D; Serrano M Cellular Senescence: From Physiology to Pathology. Nat. Rev. Mol. Cell Biol 2014, 15, 482–496. [DOI] [PubMed] [Google Scholar]
- (69).Dong CM; Wang XL; Wang GM; Zhang WJ; Zhu L; Gao S; Yang DJ; Qin Y; Liang QJ; Chen YL; Deng HT; Ning K; Liang AB; Gao ZL; Xu J A Stress-Induced Cellular Aging Model with Postnatal Neural Stem Cells. Cell Death Dis 2017, 8, No. e3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Baker DJ; Petersen RC Cellular Senescence in Brain Aging and Neurodegenerative Diseases: Evidence and Perspectives. J. Clin. Invest 2018, 128, 1208–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Fang S; Yu X; Ding H; Han J; Feng J Effects of Intracellular Iron Overload on Cell Death and Identification of Potent Cell Death Inhibitors. Biochem. Biophys. Res. Commun 2018, 503, 297–303. [DOI] [PubMed] [Google Scholar]
- (72).Tang M; Chen Z; Wu D; Chen L Ferritinophagy/Ferroptosis: Iron-Related Newcomers in Human Diseases. J. Cell. Physiol 2018, 233, 9179–9190. [DOI] [PubMed] [Google Scholar]
- (73).Dixon SJ; Lemberg KM; Lamprecht MR; Skouta R; Zaitsev EM; Gleason CE; Patel DN; Bauer AJ; Cantley AM; Yang WS; Morrison B 3rd; Stockwell BR Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Masaldan S; Bush AI; Devos D; Rolland AS; Moreau C Striking While the Iron Is Hot: Iron Metabolism and Ferroptosis in Neurodegeneration. Free Radical Biol. Med 2019, 133, 221–233. [DOI] [PubMed] [Google Scholar]
- (75).Hassannia B; Wiernicki B; Ingold I; Qu F; Van Herck S; Tyurina YY; Bayir H; Abhari BA; Angeli JPF; Choi SM; Meul E; Heyninck K; Declerck K; Chirumamilla CS; Lahtela-Kakkonen M; Van Camp G; Krysko DV; Ekert PG; Fulda S; De Geest BG; et al. Nano-Targeted Induction of Dual Ferroptotic Mechanisms Eradicates High-Risk Neuroblastoma. J. Clin. Invest 2018, 128, 3341–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Imai H; Matsuoka M; Kumagai T; Sakamoto T; Koumura T Lipid Peroxidation-Dependent Cell Death Regulated by Gpx4 and Ferroptosis. Curr. Top. Microbiol. Immunol 2016, 403, 143–170. [DOI] [PubMed] [Google Scholar]
- (77).Yao X; Zhang Y; Hao J; Duan HQ; Zhao CX; Sun C; Li B; Fan BY; Wang X; Li WX; Fu XH; Hu Y; Liu C; Kong XH; Feng SQ Deferoxamine Promotes Recovery of Traumatic Spinal Cord Injury by Inhibiting Ferroptosis. Neural Regener. Res 2019, 14, 532–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Wu C; Zhao W; Yu J; Li S; Lin L; Chen X Induction of Ferroptosis and Mitochondrial Dysfunction by Oxidative Stress in Pc12 Cells. Sci. Rep 2018, 8, 574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Mobarra N; Shanaki M; Ehteram H; Nasiri H; Sahmani M; Saeidi M; Goudarzi M; Pourkarim H; Azad M A Review on Iron Chelators in Treatment of Iron Overload Syndromes. Int. J. Hematol. Oncol. Stem Cell Res 2016, 10, 239–247. [PMC free article] [PubMed] [Google Scholar]
- (80).Di Nicola M; Barteselli G; Dell’Arti L; Ratiglia R; Viola F Functional and Structural Abnormalities in Deferoxamine Retinopathy: A Review of the Literature. BioMed Res. Int 2015, 2015, 249617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Mendoza K; Derry PJ; Cherian L; Garcia R; Nilewski L; Goodman JC; Mbye L; Robertson CS; Tour JM; Kent T Improved Outcome with a Catalytic Carbon Nano-Antioxidant in Experimental Traumatic Brain Injury Complicated by Hypotension. J. Neurotrauma 2019, 36, 2139–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Huq R; Samuel EL; Sikkema WK; Nilewski LG; Lee T; Tanner MR; Khan FS; Porter PC; Tajhya RB; Patel RS; Inoue T; Pautler RG; Corry DB; Tour JM; Beeton C Preferential Uptake of Antioxidant Carbon Nanoparticles by T Lymphocytes for Immunomodulation. Sci. Rep 2016, 6, 33808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Chen J; Ghorai MK; Kenney G; Stubbe J Mechanistic Studies on Bleomycin-Mediated DNA Damage: Multiple Binding Modes Can Result in Double-Stranded DNA Cleavage. Nucleic Acids Res 2008, 36, 3781–3790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Smith BL; Bauer GB; Povirk LF DNA Damage Induced by Bleomycin, Neocarzinostatin, and Melphalan in a Precisely Positioned Nucleosome. Asymmetry in Protection at the Periphery of Nucleosome-Bound DNA. J. Biol. Chem 1994, 269, 30587–30594. [PubMed] [Google Scholar]
- (85).Tudor C; Lerner-Marmarosh N; Engelborghs Y; Gibbs PE; Maines MD Biliverdin Reductase Is a Transporter of Haem into the Nucleus and Is Essential for Regulation of Ho-1 Gene Expression by Haematin. Biochem. J 2008, 413, 405–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Golnak R; Xiao J; Atak K; Khan M; Suljoti E; Aziz EF Local Energy Gap Opening Induced by Hemin Dimerization in Aqueous Solution. J. Phys. Chem. B 2015, 119, 3058–3062. [DOI] [PubMed] [Google Scholar]
- (87).Sen D; Poon LC Rna and DNA Complexes with Hemin [Fe(Iii) Heme] Are Efficient Peroxidases and Peroxygenases: How Do They Do It and What Does It Mean? Crit. Rev. Biochem. Mol. Biol 2011, 46, 478–492. [DOI] [PubMed] [Google Scholar]
- (88).Li W; Li Y; Liu Z; Lin B; Yi H; Xu F; Nie Z; Yao S Insight into G-Quadruplex-Hemin Dnazyme/Rnazyme: Adjacent Adenine as the Intramolecular Species for Remarkable Enhancement of Enzymatic Activity. Nucleic Acids Res 2016, 44, 7373–7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Golub E; Freeman R; Willner I A Hemin/G-Quadruplex Acts as an Nadh Oxidase and Nadh Peroxidase Mimicking Dnazyme. Angew. Chem., Int. Ed 2011, 50, 11710–11714. [DOI] [PubMed] [Google Scholar]
- (90).Travascio P; Li Y; Sen D DNA-Enhanced Peroxidase Activity of a DNA-Aptamer-Hemin Complex. Chem. Biol 1998, 5, 505–517. [DOI] [PubMed] [Google Scholar]
- (91).Hansel-Hertsch R; Di Antonio M; Balasubramanian S DNA G-Quadruplexes in the Human Genome: Detection, Functions and Therapeutic Potential. Nat. Rev. Mol. Cell Biol 2017, 18, 279–284. [DOI] [PubMed] [Google Scholar]
- (92).Yang N; Galick H; Wallace SS Attempted Base Excision Repair of Ionizing Radiation Damage in Human Lymphoblastoid Cells Produces Lethal and Mutagenic Double Strand Breaks. DNA Repair 2004, 3, 1323–1334. [DOI] [PubMed] [Google Scholar]
- (93).Park SC Survive or Thrive: Tradeoff Strategy for Cellular Senescence. Exp. Mol. Med 2017, 49, No. e342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Achuthan S; Santhoshkumar TR; Prabhakar J; Nair SA; Pillai MR Drug-Induced Senescence Generates Chemoresistant Stemlike Cells with Low Reactive Oxygen Species. J. Biol. Chem 2011, 286, 37813–37829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Owen JE; Bishop GM; Robinson SR Uptake and Toxicity of Hemin and Iron in Cultured Mouse Astrocytes. Neurochem. Res 2016, 41, 298–306. [DOI] [PubMed] [Google Scholar]
- (96).Goldstein L; Teng ZP; Zeserson E; Patel M; Regan RF Hemin Induces an Iron-Dependent, Oxidative Injury to Human Neuron-Like Cells. J. Neurosci. Res 2003, 73, 113–121. [DOI] [PubMed] [Google Scholar]
- (97).Derry PJ; Hegde ML; Jackson GR; Kayed R; Tour JM; Kent TA; Tsa AL Revisiting the Intersection of Amyloid, Pathologically Modified Tau and Iron in Alzheimer’s Disease from a Ferroptosis Perspective. Prog. Neurobiol 2020, 184, 101716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (98).Derry PJ; Nilewski LG; Sikkema WKA; Mendoza K; Jalilov A; Berka V; McHugh EA; Tsai AL; Tour JM; Kent TA Catalytic Oxidation and Reduction Reactions of Hydrophilic Carbon Clusters with Nadh and Cytochrome C: Features of an Electron Transport Nanozyme. Nanoscale 2019, 11, 10791–10807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Marcano DC; Bitner BR; Berlin JM; Jarjour J; Lee JM; Jacob A; Fabian RH; Kent TA; Tour JM Design of Poly(Ethylene Glycol)-Functionalized Hydrophilic Carbon Clusters for Targeted Therapy of Cerebrovascular Dysfunction in Mild Traumatic Brain Injury. J. Neurotrauma 2013, 30, 789–796. [DOI] [PubMed] [Google Scholar]
- (100).Fyrestam J; Ostman C Determination of Heme in Microorganisms Using Hplc-Ms/Ms and Cobalt(Iii) Protoporphyrin Ix Inhibition of Heme Acquisition in Escherichia coli. Anal. Bioanal. Chem 2017, 409, 6999–7010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Sak A; Stueben G; Groneberg M; Bocker W; Stuschke M, Targeting of Rad51-Dependent Homologous Recombination: Implications for the Radiation Sensitivity of Human Lung Cancer Cell Lines. Br. J. Cancer 2005, 92, 1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Sarker AH; Chatterjee A; Williams M; Lin S; Havel C; Jacob P 3rd; Boldogh I; Hazra TK; Talbot P; Hang B Neil2 Protects against Oxidative DNA Damage Induced by Sidestream Smoke in Human Cells. PLoS One 2014, 9, No. e90261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Malinouski M; Zhou Y; Belousov VV; Hatfield DL; Gladyshev VN Hydrogen Peroxide Probes Directed to Different Cellular Compartments. PLoS One 2011, 6, No. e14564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Nakamura T; Xi G; Hua Y; Schallert T; Hoff JT; Keep RF Intracerebral Hemorrhage in Mice: Model Characterization and Application for Genetically Modified Mice. J. Cereb. Blood Flow Metab 2004, 24, 487–494. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.