

Abstract
The complexity of Alzheimer’s disease (AD) complicates the search for effective treatments. While the key roles of pathologically modified proteins has occupied a central role in hypotheses of the pathophysiology, less attention has been paid to the potential role for transition metals overload, subsequent oxidative stress, and tissue injury. The association of transition metals, the major focus heretofore iron and amyloid, the same can now be said for the likely pathogenic microtubular associated tau (MAPT). This review discusses the interplay between iron, pathologically modified tau and oxidative stress, and connects many related discoveries. Basic principles of the transition to pathological MAPT are discussed. Iron, its homeostatic mechanisms, the recently described phenomenon of ferroptosis and purported, although still controversial roles in AD are reviewed as well as considerations to overcome existing hurdles of iron-targeted therapeutic avenues that have been attempted in AD. We summarize the involvement of multiple pathological pathways at different disease stages of disease progression that supports the potential for a combinatorial treatment strategy targeting multiple factors.
Keywords: Alzheimer’s disease, tau, iron, ferroptosis, senescence, reactive oxygen species
1.1. Introduction to AD
AD is a progressive neurodegenerative disorder that currently afflicts 4.7 million people over the age of 65 in the United States and is expected to grow to 13.8 million people by 2050 (Hebert et al., 2013). Hallmark symptoms include progressive memory loss, altered behavior, delusions and hallucinations, and fine motor skill degradation (McKhann et al., 1984). The disease course leads to loss of personal independence, extended care given usually by family members that, as of 2011 amounted to $210 billion in unpaid services along with the $200 billion in medical expenses associated with that care (Alzheimer’s, 2012). AD is typically diagnosed over an age range > 65 yr and the life expectancy of patients depends largely on their minimental status exam (MMSE) score, a method to judge the progress of AD, with lower scores being associated with shortest life expectancies (Larson et al., 2004). AD is histologically characterized by the presence of senile plaques and neurofibrillary tangles (NFTs) found in and around pyramidal neurons in cortical tissue (Braak and Braak, 1991; George et al., 2011). Senile plaques are composed of amyloid-β (Aβ) and trace amounts of metals such as copper, zinc, iron, and aluminum (Bolognin et al., 2011; Faller, 2009; Zatta et al., 2009), although the amounts vary (Schrag et al, 2011). NFTs are composed primarily of pathologically phosphorylated microtubule-associated protein tau (MAPT). Pathologically modified tau and NFTs are found in a range of diseases collectively referred to as tauopathies (Williams, 2006), and include progressive supranuclear palsy (Gerson et al., 2014), chronic traumatic encephalopathy (McKee et al., 2009), corticobasal degeneration (Armstrong et al., 2013), and accompanies the amyloidosis observed in Down’s syndrome (Ballard et al., 2016). AD is classified as an amyloidosis with secondary tauopathy (Dickson, 2010). Similar to amyloid plaques, iron and aluminum have both been identified to be found resident in tau tangles (Good et al., 1992; Smith et al., 1997).
Given the lack of dramatic success in clinical trials of a focus on pathological amyloid and tau, additional targets are of great interest. Long standing interest in transition metal dysregulation (Kepp, 2012), and oxidative stress (Markesbery, 1997), and amyloid cascade (Selkoe and Hardy, 2016) have been proposed as etiologies of AD and may be interlinked. For instance, the transition metals implicated in AD: iron, copper, aluminum, and zinc have at least some indirect if not direct effect on the generation of reactive oxygen species (ROS) and all four elements have been found in Aβ plaques (Bolognin et al., 2011; Smith et al., 1997; Wang et al., 2012). Another repository of transition metals that may impart latent toxicity is neuromelanin, a dopamine-derived polymer produced extensively in the substantia nigra and locus coeruleus by catecholaminergic neurons (Zucca et al., 2017). Recently, both the oxidative stress and transition metal dysregulation hypotheses have gained some additional support with the discovery of a new mode of iron-mediated cell death, ferroptosis, reviewed below (Dixon et al., 2012).
To date, several disease-modifying treatments with different pathological targets have been proposed and tested; however, most have not shown to be effective at preventing the progression of the disease (Galimberti and Scarpini, 2011; Yiannopoulou and Papageorgiou, 2013). Current treatment is primarily supportive with small-molecule drugs such as memantine (Reisberg et al., 2003; Tariot et al., 2004) and donepezil but no permanent changes to the disease pathology are achieved (Reisberg et al., 2003; Yiannopoulou and Papageorgiou, 2013). The cost of developing effective drugs are staggering and the results are often not reassuring. For instance, may recent attempts at active and passive immune therapy for amyloid have failed to show improved outcomes (Jicha, 2009), the potential for more benefit in specific subgroups should be considered (Iqbal and Grundke-Iqbal, 2010). Recent failure of antibodies targeting amyloid precursor protein cleavage enzymes are consistent with the pattern that targeting amyloid may not be sufficient in treating AD (Egan et al., 2019; Henley et al., 2019).
The discovery of oligomeric tau (oTau), a soluble aggregation of tau molecules that can seed the growth of paired-helical filaments (PHFs), spurred new interest in the possible role of tau in AD. These findings are part of a growing body of work surrounding peptides with poorly-defined tertiary structures such as tau, (Lasagna-Reeves et al., 2010; Lasagna-Reeves et al., 2011; Ward et al., 2012) Aβ and α-synuclein that can easily form neurotoxic oligomers (Kayed et al., 2003; Lashuel et al., 2013). Additionally, tau is appears to be related to the regulation of intracellular iron by trafficking amyloid precursor protein (APP) to the cell membrane and maintenance of axonal function (Goldsbury et al., 2006; Lei et al., 2012). Because tau sits at the center of these relationships, the modification of tau function or structure may have an important role in AD. This review discusses some of the roles of tau in the brain under both physiological conditions and the pathological setting of AD; the interactions among tau and iron, and the diagnostic and therapeutic options that target tau and its etiologies in order to address the damage caused by AD.
2.1. Tau Structure and Function
Tau, or microtubule associated protein tau (MAPT) is a predominantly random-coil peptide translated from the 16 exons of MAPT found on the long arm of chromosome 17q21 (Buée et al., 2000). Because of splicing variations, the six isoforms of tau are between 352 and 441 (45-65 kDa) amino acids in length (Buée et al., 2000). Structurally, tau can be divided into two domains: projection and tubulin-binding (Figure 1). The projection domain can be further divided into the acidic N-terminal region and the proline-rich region whereas the microtubule-binding domain is divided into a tubulin binding repeat region (R1-R4) with three or four repeat units and finally a basic region nearest to the C-terminal (Buée et al., 2000). Two hexapeptide sequences in the microtubule binding region, 275VQIINK280 and 306VQIVYK311 have been shown to be necessary for the aggregation of tau as these form β-sheets (Mukrasch et al., 2009). Cryogenic transmission electron microscopy of tau paired helical fibrils (PHFs) and straight fibers (SFs) from AD patients has revealed that the monomers have very similar folding behavior but that the ultrastructure is very different (Fitzpatrick et al., 2017). In both cases the 306VQIVYK311 domain is essential for the formation of the filament, however the supramolecular interface between adjacent monomers is different. In PHFs, the 332PGGGQ336 residues form the interface whereas in SFs, packing is observed in two regions between 321KCGS324 on the first monomer and 313VDLSK317 on the second monomer (Fitzpatrick et al., 2017).
Figure 1.
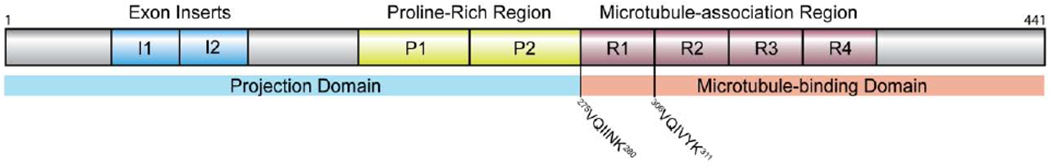
Schematic structure of human tau (htau40).
While the primary role of tau in neurons is to directly modulate the stability of axonal microtubules by binding adjacent α-β-tubulin heterodimers (Kadavath et al., 2015), secondary roles have also been observed including the trafficking of APP to the cell membrane to stabilize ferroportin (FPN1), the sole iron export channel found in mammalian cells (Lei et al., 2012; Stankowski et al., 2012). Another role for tau may be neuroprotective: work by Wallin et al. showed that full-length, 441-amino acid human tau without pathological modification prevents the aggregation of Aβ40, a 40 amino acid fragment of APP cleaved by β-secretase, in a substoichiometric, dose-dependent manner (Wallin et al., 2018). Wallin et al. hypothesize that a drop in native tau caused by an increase in phosphorylation of tau could lead to an increase in Aβ40 aggregation and fibrillization (Wallin et al., 2018).
2.2. Pathological Posttranslational Modification of Tau
Tau undergoes post-translational modification (PTM) at many different residues by phosphorylation (Stoothoff and Johnson, 2005), acetylation (Min et al., 2015), methylation (Thomas et al., 2012), O-GlcNAcylation (Yuzwa et al., 2012), and ubiquitylation (Wang et al., 2017b). PTM dysregulation appears to be largely responsible for the toxic gain of function that tau expresses in neurodegenerative disease (Medina et al., 2016). The most common PTM associated with tau is phosphorylation, where 48 of 85 distinct serine, threonine, or tyrosine residues have been observed in normal physiologically modified tau. (Luna-Muñoz et al., 2013; Wang and Mandelkow, 2016) The phosphorylation epitope of tau is regulated by kinases such as microtubule affinity-regulating kinase (MARK/Par-1) and glycogen synthase kinase 3A/3B (GSK3A/β), and the protein phosphatases (PP)1, PP2A, and PP2B (Billingsley and Kincaid, 1997). Additional phosphorylation is performed by a diverse set of other kinases (Ferrer et al., 2005).
Specified phosphorylation by microtubule-associated protein regulatory kinase 2 (MARK2) in the Lys-Xaa-Gly-Ser motifs of the microtubule binding repeat domain largely modulate tubulin affinity (Matenia and Mandelkow, 2009; Schwalbe et al., 2013). Additional phosphorylation by GSK3β has been shown to be stimulated by upstream activations of PI3K and later Akt (Uranga et al, 2009). Akt and GSK3β have both been shown to be overactive in homogenized AD brain fractions, with GSK3β activity approximately 200% greater than that in control fractions (p < 0.05), and Akt activity correlates moderately with Braak staging (pathology classification) (r2 = 0.564 p = 0.010) in the frontal cortex (Rickle et al., 2004).
Dephosphorylation of tau is largely performed by the protein phosphatase family of enzymes with PP2A contributing to nearly 70% of tau dephosphorylation activity (Wang and Mandelkow, 2016). Active PP2A is a heterotrimeric complex composed of a structural (PP2Aa), regulatory (PP2Ab), and one catalytic subunit (PP2Ac) (Martin et al, 2010). I1PP2A and I2PP2A are two endogenous inhibitors of the catalytic PP2Ac subunit and may become dysregulated in AD and contribute to hyperphosphorylation even if overall expression of MARK/Par-1 or GSK3β remain unchanged by reducing the rate that phosphorylated amino acid residues are dephosphorylated (Arif et al, 2014; Chen et al, 2008; Chohan et al, 2006). The endogenous PP2A inhibitor I2PP2A has been observed to translocate from the nucleus into the cytoplasm in tau expressing PC-12 cells (Arif et al., 2014). Dysregulation of the kinase/phosphatase system leads to the formation of hyperphosphorylated tau, a pathological form of tau that carries 3-4 times the normal number of phosphate groups (Iqbal et al., 2013).
Inhibition of PP2A by okadaic acid and calyculin also results in the hyperphosphorylation of tau both in vitro by okadaic acid in metabolically active brain sections and SH-SY5Y cells and in vivo by calyculin (caSun et al, 2003; Gong et al, 2000; Zhang and Simpkins, 2010). Rats exposed to calyculin show a loss of spatial memory as measured by the Morris water maze test, manifested by performing a random search for the platform as opposed to a straight path to a prelearned position (caSun et al., 2003). Additionally, qualitatively more-intense Western blot bands for anti-PHF-tau in rat brain homogenates were observed, whereas the intensity of the total tau bands remained the same, suggesting an increase in S396-phosphorylated tau (caSun et al., 2003). Iron can also modulate tau-phosphorylating kinases such as Akt, which may lead to aberrant phosphorylation of tau (Bautista et al., 2016; Egaña et al., 2003).
2.3. Oligomeric Tau
Beginning in 1999, epidemiological studies of AD patients at varying stages of the disease found that a large percentage of neuronal death could not be directly accounted for by the presence of NFTs (Kril et al., 2002; Morsch et al., 1999). As far back as 1993, Köpke et al. had reported small aggregates of tau accounted for a large percentage of precipitable tau in AD neuron isolates (Köpke et al., 1993a). By the mid-2000s, these effects had been identified: oligomeric tau serves as a seed for NFTs (Harris et al., 2012; Maeda et al., 2007).
Oligomeric tau (oTau) on average consists of 40 molecules of tau and are approximately 20 nm in diameter (Cowan et al., 2012) with a β-sheet conformation (Maeda et al., 2007) and are soluble (Cowan et al., 2012). Tau oligomers are enriched with acetylated K174 and may be necessary for accumulation to take place (Min et al., 2015; Rane et al., 2019). Several different mechanisms are known to generate oligomeric tau capable of forming PHFs (Figure 2) thus, the composition of oTau is diverse and each oligomer is not necessarily identical. One important species, hyperphosphorylated oTau, may make up a substantial portion of the total hyperphosphorylated tau in AD; Köpke et al. reported that only 26% of total hyperphosphorylated tau is derived from PHFs, the remaining 74% is derived from the non-PHF pool (Köpke et al., 1993b) a fraction of which is precipitable at 200000 × g and may have possibly represented oligomers (Köpke et al., 1993a). Hyperphosphorylation is not the only means to form oligomeric tau, however; oligomeric tau has been generated by disulfide crosslinking via oxidation, (Kim et al., 2015) polyanions such as heparin, (Barghom and Mandelkow, 2002) amphiphiles such as arachidonic acid, (Barghorn and Mandelkow, 2002) the transition metals aluminum(III) and iron(III) (Bader et al., 2011), and cross-templating by α-synuclein or Aβ42, a 42 amino acid APP fragment, oligomers (Götz et al., 2001; Lasagna-Reeves et al., 2010; Waxman and Giasson, 2011).
Figure 2.
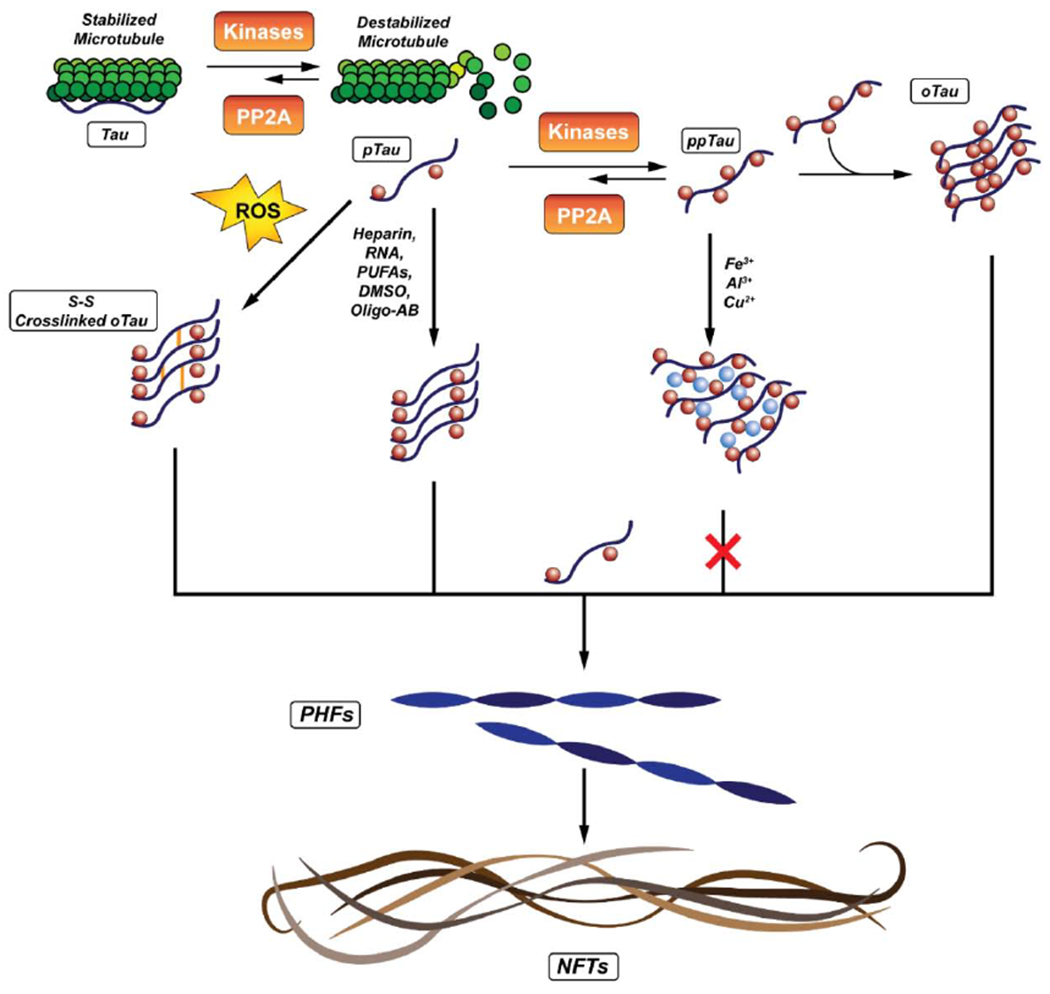
Possible pathways for generating NFTs from Oligomeric Tau.
Recently, work by DeVos et al. showed that in htau-expressing PS19 mice the antisense mRNA oligonucleotide ASO-12 could reduce transcription of htau by approximately 50% (p < 0.05) compared to untreated PS19 mice. The result of reducing htau mRNA transcription was a nearly 50% reduction (p < 0.05) of htau translation. Compared to scrambled mRNA, ASO-12 was able to reduce low molecular weight htau by nearly 25% (ns), high molecular weight aggregates (including oligomers) by 50% (p = 0.004), and phosphorylated tau by 60% (p = 0.0104). Reducing tau levels prolonged life expectancy (ASO-12: 348 vs. control: 312 days; p = 0.0052) oligomeric tau formation could be shunted in PS19 mice expressing human P301S tau by antisense mRNA oligonucleotides (ASOs) (DeVos et al., 2017). FRET studies on HEK-293 cells expressing CFP and YFP-labeled tau showed that lysate from 6-9 month old and 9-12 month old PS19 mice treated with ASO-120 added to the culture media formed fewer aggregates, as measured by the control-normalized integrated FRET density (6-9 m/o PS19 scrambled vs. 6-9 m/o PS19 ASO-120 compared to adding mouse brain lysate treated with scrambled mRNA) (DeVos et al., 2017). These findings show the potential of reducing tauopathy by reducing the overall cytosolic concentration of tau. By limiting fibril growth, the disease pathology may be slowed.
2.4. Tau Oligomers, Fast Axonal Transport and Oxidative Stress
Microtubules provide a means for kinesin and dynein-mediated transport of vesicles in the antero- and retrograde directions from soma respectively via fast axonal transport. The precise role of tau in mediating this process and the effect of dysfunctional tau on fast axonal transport is still somewhat unclear. Tau stabilizes microtubules by binding α,β-tubulin heterodimers, (Kadavath et al., 2015) and prevents their degradation. Tau also moderates vesicle transport: Dixit et al. and Mandelkow et al. independently showed that microtubule-associated tau causes the detachment of kinesin from microtubules, (Dixit et al., 2008; Mandelkow et al., 2004) thus attenuating the migration of vesicles and mitochondria toward the plus end of the axon. Mandelkow et al. presented the hypothesis that phosphorylation of tau by MARK/Par-1, which would lead to detachment of tau from the microtubules and was a cellular defense mechanism to accelerate the anterograde migration of mitochondria and cargo vesicles to the synapse to combat synaptotoxicity (Mandelkow et al., 2004). Later, Kanaan et al. showed that tau aggregates are capable of activating the PP1/GSK3β pathway leading to phosphorylation of the kinesin light chain and spontaneous detachment of cargo vesicles this leads to a retrograde accumulation of vesicles and an attenuation of their anterograde movement (Kanaan et al., 2011). This effect is more pronounced with aggregated tau due to better exposure of the phosphatase activating domain (PAD), which stimulates phosphorylation by PP1 and GSK3β (Kanaan et al., 2011). Stamer et al. reported that the transfection of T2a cells with hTau40, a gene coding for the longest human tau variant, were highly sensitive to oxidative stress (Stamer et al., 2002). 12.3% (triplicate measures of 150 cells) of normal N2a cells have neurites longer than 30 microns, treatment with 30 μM H2O2 for 40 minutes reduces this fraction to 5.9%. Cotreatment with 0.02 U/μL catalase reduces the loss to 8.7%. hTau40-N2a cells are much more sensitive to H2O2, where under the same conditions, only 0.3% and 5.6% of cells have long neurites after treatment with 30 μM H2O2 or catalase and 30 μM H2O2 respectively (Stamer et al., 2002).
The possible cause of the increased sensitivity to oxidative stress observed in chick neuroganglion cells (RGCs) expressing hTau40 is a loss of anterograde cargo and APP vesicle and mitochondria migration to the neurite terminus that reduces the local energy and materials supply for maintaining the synaptic terminus. These authors discuss that the reduction of vesicular transport to the synapse also limits the availability of peroxisomes which would otherwise detoxify hydrogen peroxide at the synaptic terminus leading to impaired antioxidant capacity (Stamer et al., 2002). Stamer et al. hypothesize that because neurons have elevated tau concentrations in the AD brain (Khatoon et al., 1994), it would impair axonal transport. Hyperphosphorylation may be one way to counteract this since it would reduce the amount of microtubule-bound tau and reduce the inhibition of vesicular transport (Stamer et al., 2002) Despite inhibition of anterograde APP-trafficking, Goldsbury et al. demonstrated that inhibition of APP-trafficking did not increase amyloid-β production in isolated axons (Goldsbury et al., 2006).
In vivo studies of axonal transport were performed by Bertrand et al. using manganese-enhanced magnetic resonance imaging (MEMRI) of the olfactory tract in wild-type and transgenic mice expressing P301L tau. The mice were intranasally injected with MnCl2 and the glomerular layer was imaged by MRI at different time points up to 2 days later. Bertrand et al. monitored the MRI signal intensity of the olfactory tract and found that the signal maximum was correlated to the tau status and age (WT vs. P301L) and age (3 mo vs. 6 mo). The normalized MRI signal from the glomerular layer of 6 month-old transgenic mice had lower peak MRI signal intensities than the wild-type (p < 0.01) of the same age indicating that less of the MnCl2 was transported to the glomerular cells, the maximum slope was lower (p < 0.001), indicating that less MnCl2 was being transported over time thus maintaining the tissue store of MnCl2 before the glomerular layer, and the time to peak intensity was later (p < 0.05), also another indicator of slow transport. Bertrand et al. were able to correlate rates with tauopathy degree in both dendritic cells and neuron cell bodies in the mitral layer, showing that excess tau lead to reduced maximum signal intensities, indicating that intercellular transport was impeded by excess tau.
Vossel et al. reported that in neuronal cultures from tau knockout (+/− and −/−) mice that the negative effect of treatment with hAPP monomer on anterograde axonal transport of mitochondria was reduced as tau expression (+/−, −/−) decreased (Vossel et al., 2010). Retrograde axonal transport was only reduced in (+/+) tau mice (p < 0.01) when treated with Aβ oligomers compared to (−/−) or (+/−) mice, indicating that the inhibitory action of Aβ oligomers requires tau in order to be active. Vossel et al. also reported that wildtype neurons treated with Aβ oligomers could be protected by cotreatment with a GSK3β inhibitor, suggesting that activation of GSK3β is involved in the pathology of Aβ and its interactions with tau also suggests an interaction between Aβ oligomers and tau in the axon, and that tau in the presence of Aβ seeds its own hyperphosphorylation (Vossel et al., 2015)
2.5. Senescence as a Tau-related Pathological Mechanism
Cellular senescence is a phenomenon first observed in cultured primary human cells by Hayflick and Moorehead, and is associated with aging and other causes, including pharmacological treatment where cells eventually cease to divide (Childs et al., 2015). Senescent cells are characterized by an active secretory phenotype including pro-inflammatory features that can influence its neighbors and cause a spread of the senescent phenotype (Childs et al., 2015). Some characteristics of senescent cells include expression of the cell cycle inhibitory protein p16INK4a (Baker et al., 2011), multiple genes related to inflammatory cascades as well as senescence-associated β-galactosidase activity, although these may be non-specific (Childs et al., 2015). While there are several reported stimuli for cellular senescence, persistent activation of the DNA damage response (DDR) appears to be especially active in neurons (Di Micco et al., 2006). Cellular senescence has been associated with aging (van Deursen, 2014), and one underlying mechanism may involve a persistent DDR, that tends to be less efficient in post-mitotic neurons. Importantly, senescence confers some resistance to certain forms of cell death, the most studied being the apoptotic pathway (Childs et al., 2014; Hampel et al., 2005), and the ferroptosis pathway (Masaldan et al., 2018)
In the brain, recent work suggests that the pathological deposition of tau is associated with cellular senescence and may contribute to many features of neurodegeneration including cognitive decline and brain atrophy (Kritsilis et al., 2018; Musi et al., 2018). Using mouse models of aberrant tau expression, senescence markers were seen in astrocytes, microglia and neurofibrillary tangles, but not in Aβ amyloid plaques (Musi et al., 2018). Musi et al. observed that quercetin, employed as a senolytic, an agent that selectively depletes senescent cells, reduced the density of NFTs in 23-month old rTg(tauP301L)4510 mice by 35% (p < 0.0001) and reduced ventricle size compared to the vehicle-treated mice by 28% (p = 0.05) (Musi et al., 2018).
Tau accumulation initiates senescence, as measured by the expression of tumor suppressing protein P16Ink4a, in the glial cells of MAPTP301sPS19 (PS19) rats (Bussian et al., 2018). Bussian et al. found that P16Ink4a expression in glial cells increased more in the hippocampus than in the cortex as the animals aged suggesting that hippocampal glial cells were more sensitive to tau accumulation than glial cells in the cortex, which may correlate with volume changes in the hippocampus in AD (Schuff et al., 2009). Senescence markers appeared before the appearance of NFTs, indicating that the cells were affected by smaller accumulations of tau (Bussian et al., 2018). Crossing PS19 rats with transgenic ATTAC (Apoptosis Throught Targeted Activation of Caspase) rats. ATTAC is a caspase signal which when dimerized triggers apoptosis to occur. In this case, apoptosis was triggered by the presence of the ligand AP20187 (AP). Treatment with AP in ATTAC-expressing PS19 rats lead to a reduction in senescent cells (p < 0.05; PS19 (−)AP vs PS19 (+)AP), phosphorylated tau (p < 0.01; PS19 (−)AP vs PS19 (+)AP), and cognitive loss (p < 0.001; PS19 (−)AP vs PS19 (+)AP) in the cross-bred rats (Baker et al., 2011; Bussian et al., 2018). The finding that senescence can be induced by exposure to tau aggregates suggests a new target to treating Alzheimer’s disease may be to target senescence pathways in the brain, especially since phosphorylation was shown to be enhanced in senescent cells.
As Musi et al. showed (Musi et al., 2018), senolytics, may possibly serve in the role of eliminating senescent cells from the brain. The anti-tumor compound ABT-263 (Navitoclax) has been trialed in two Phase I trials with its use in combinational therapy with Vistusertib (Medicine, 2019) or gemcitabine (Cleary et al., 2014) in lung cancer and solid tumors, respectively, and has been shown by Chang et al. to clear senescent hematopoetic cells (Chang et al., 2016). The use of CAR T treatment has also been proposed (Childs et al., 2015) as a means to clear senescent cells by targeting specific surface markers, potentially CD44, a surface receptor associated with senescent cells (Mun and Boo, 2010).
Overall, these results suggest that senescence may contribute to several of the pathological features associated with tau aggregates, and that non-neuronal cells, specifically astrocytes and microglia, contribute to neuronal degeneration, perhaps through the secretory properties of these cells. An interaction with ferroptosis is very likely, suggesting a cell’s ultimate fate may depend on a balance of competing survival factors.
2.6. Potential Therapeutic Methods to Target Tau
These findings suggest several potential targets involving tau may influence the progression of AD. Preventing the spread of pathologically-modified tau may be possible by downregulating the translation of normal tau (DeVos et al., 2017). As described earlier, work by DeVos et al. showed that antisense P301S mutant 1N4R mRNA oligonucleotides could be utilized to downregulate the translation of tau (DeVos et al., 2017). The principal advantage to this method is a direct and immediate reduction in available modifiable tau. However, Betrie et al. showed that tau plays a role in the cardiovascular system and that tau knockout mice develop cardiovascular problems during their life so that approach would like need to be specifically targeted (Betrie et al., 2017).
Preventing tau hyperphosphorylation may block the development of oligomeric tau. Tau phosphorylation occurs predominately on serine and threonine residues and largely by the kinase GSK3β, which requires initial phosphorylation substrate priming in order to catalytically modify adjacent serine residues (Cohen and Goedert, 2004). Because tau can become phosphorylated and detach from microtubules in the physiological context, designing drugs to target the initial phosphorylation event may have unintended adverse effects. However, inhibition of serine/threonine kinases such as GSK3β may provide an early point to reduce the rate of hyperphosphorylation and subsequent oligomerization. Additionally, GSK3β inhibition has also been shown to reduce APP cleavage by BACE1 making it possible that GSK3β modulation can have multiple therapeutic targets (Ly et al., 2013). Tideglusib, an irreversible GSK3β inhibitor ATP-mimetic that showed promise in vitro and in vivo, unfortunately failed to produce significant reductions in cognitive decline rate in humans (Del Ser et al., 2013; Lovestone et al., 2015). A follow up trial of tideglusib was also performed for supranuclear palsy with similar negative results. These negative results suggest that because phosphorylation may be a sufficient, but not necessary event for aggregation to occur (Tolosa et al., 2014). In addition to tideglusib, several other candidate compounds for GSK3β inhibition have been discovered using pharmacophore modeling (Fu et al., 2014).
Preventing tau oligomerization is another link in the pathology chain and two important trials have been conducted to study this. Methylene blue, a phenothiazine mainly used to treat methemoglobinemia, with the known contraindication of hemolytic anemia in individuals with glucose-6-phosphate dehydrogenase deficiency, (Wischik et al., 1996; Wischik et al., 2014) was observed by Wischik et al. (Wischik et al., 1996; Wischik et al., 2014) to prevent the aggregation of tau while not inhibiting the tau-tubulin interaction. A derivative, LMTM, (Wischik et al., 2014) was carried through a Phase III trial but failed to meet its endpoints of reducing the rate of cognitive decline (Gauthier et al., 2016). In 2018, Cascio et al. (Cascio and Kayed, 2018) reported the use of Azure C, a structural analog of methylene blue and its ability to modulate the formation of oligomeric tau and toxic tau aggregates. Aggregation of tau was not eliminated with Azure C but the resulting aggregates that were produced were not targetable in a dose-dependent fashion by existing antibodies that react with the known toxic tau aggregate epitope. The authors found Azure C partially protects SHSY-5Y cells from the toxic effects of 5 μM oligomeric tau. (Cascio and Kayed, 2018). Computationally aided drug discovery methods are being explored for the development of drugs that specifically bind to phosphorylated tau to inhibit its aggregation (Pradeepkiran and Reddy, 2019).
Preventing the spread of tau aggregates may also be another target: a joint venture by AbbVie and C2N Diagnostics completed Phase I tolerance trials in October 2016 for C2N 8E12, relabeled ABB-8E12, a humanized antibody targeting extracellular tau in patients with progressive supranuclear palsy which may also be applicable towards AD (Diagnostics, 2016). In a Phase I trial for tolerance, and safety, 32 patients with PSP were treated with the drug over a concentration range from 2.5 mg/kg to 50 mg/kg, few adverse effects were reported and were not dose-dependent (West et al., 2017).As of the time of this writing, no results have emerged from the Phase II trial. Active immunization is another possible route to reduce pathologically phosphorylated tau and oligomeric tau loading. Kontsekova et al. (Kontsekova et al., 2014) demonstrated that a peptide with the sequence KDNIKHVPGGGS coupled to keyhole limpet hemocyanin, otherwise known as Axon peptide 108, or AADvac1, induced the generation of pathology-selective antibodies that significantly reduced hyperphosphorylated tau and oligomeric tau loading in transgenic rats (Kontsekova et al., 2014). In a 12-week randomized, double-blind placebo-controlled study with 30 participants (24 in the AADvac1 group and 6 placebo members), AADvac1 was shown to generate a 1:31415 IgG antibody titer (Novak et al., 2017). Two clinical trials, ADAMANT and FUNDAMANT were started following the initial 30-member trial in humans (Health, 2016a, b; Novak et al., 2018). ADAMANT consists of 208 participants and is a 24-month two-arm (treatment and placebo) phase 2 safety and immunogenicity study that has not completed as of the time of this writing. FUNDAMANT was a 72-week safety study of 26 participants from the original 30-member trial. FUNDAMANT showed that AADvac1 was capable of raising IgG titers over the course of the treatment period and reduced hippocampal atrophy was associated with higher IgG titers (corrected r = 0.476, p = 0.0460) (Novak et al, 2018).
The inter-cellular transmission of oligomeric tau is another possible interdiction. The tau oligomer-specific monoclonal antibody (TOMA) has been raised by Castillo-Carranza et al. (Castillo-Carranza et al., 2014) to selectively bind tau oligomers as opposed to NFTs and promote clearance of oligomeric tau in 8-month-old P301L tau mutant transgenic mice (Castillo-Carranza et al., 2014). When treated with a single 30 μg/animal intravenous injection of TOMA, the motor coordination and memory deficits of P301L transgenic mice were restored to the levels of their wild-type counterparts for 60 days. Passive immunization with TOMA against NFTs was not seen and TOMA was not observed to enter the cellular compartment suggesting a strictly extracellular role of the antibody (Castillo-Carranza et al., 2014).
Monoclonal antibodies have some advantages and possible disadvantages compared to small molecules. Monoclonal antibodies can be made highly specific, for example antibodies for tau can target specific phosphorylation epitopes (Goedert et al., 1995; Petry et al., 2014), paired-helical fibrils (Otvos et al., 1994), or oligomeric tau (Castillo-Carranza et al., 2014). Second, experimental antibody therapies such as Aducanumab have shown that amyloid plaques can be cleared (Sevigny et al., 2016), though in the case of Aducanumab no therapeutic benefit emerged. On the other hand, monoclonal antibodies are difficult to pass through the blood brain barrier (Mitragotri et al., 2014) and require high doses in order to be effective in the brain. Efforts to improve blood brain barrier translocation, however, are ongoing (Mitragotri et al., 2014; Yu et al., 2011). At the same time, a small molecule with broad affinity for tau may be able to target several epitopes without requiring the administration of a second compound. The use of pharmacophore-based drug discover may aid in identifying existing compounds that can target aspects of tau aggregation (Mohamed et al., 2013; Pradeepkiran and Reddy, 2019) and the hyperphosphorylation pathway (Fu et al., 2014; Mazanetz and Fischer, 2007) to arrest or at least delay the progression of the disease.
Some upstream therapeutic targets may not be suitable because of the ubiquitous nature of each component in the cascade. For instance, broad-spectrum tyrosine kinase inhibitors have been shown to produce endocrine side effects such as hypo/hyperglycemia (Lodish and Stratakis, 2010) which theoretically may exacerbate neuronal oxidative stress (Martini and Kent, 2007). Extracellular antibodies that target Aβ may also potentially interfere with the function of amyloid precursor protein as a regulator of ferroportin (Wong et al., 2014) due to its exposure to the extracellular space though no reports of this were found. Regulation of APP translation and transcription from mRNA may also have adverse effects on iron regulation and must also be considered when targeting APP because APP is natively involved in the regulation of iron export from the cell (Dlouhy et al., 2019; Lei et al., 2012). Loss of APP may cause an accumulation of intracellular iron (Raha et al., 2013). Even reducing the translation of tau may have negative effects such as the appearance of a tau knockout-like phenotype which involves the appearance of a Parkinson-like phenotype (Lei et al., 2012; Lei et al., 2014).
3.1. Iron and Tau Pathology
There are at least two known interactions between tau and iron in the formation of paired helical fibrils and their final NFTs. First, iron overload can generate ROS (Smith et al., 1997) which may result in the formation of oligomeric tau via Cys-Cys binding or via kinase pathways and iron overload that hyperphosphorylate tau (Soeda et al., 2015; Uranga et al., 2009; Wan et al., 2019). Second, iron can generate oligomeric tau by the formation of intermolecular coordination complexes mediated via phosphorylated amino acid residues (Bader et al., 2011; Nubling et al., 2012). Two more possible interactions exist: tau has been shown to be necessary for ferroportin activity, loss of soluble tau may inhibit iron export in cells(Lei et al., 2012) and the aggregation of hyperphosphorylated tau and possibly the decoration of existing PHFs by iron over time (Yamamoto et al., 2002). The latter interaction only has circumstantial evidence, namely, the detection of iron in NFTs and the inability for PHF-phosphorylated tau to reform PHFs in the presence of iron (Yamamoto et al., 2002).
Good et al. observed the presence of both iron and aluminum using laser microprobe mass analysis in paired helical fibrils isolated from AD brains (Good et al., 1992). Early work by Shin et al. demonstrated that in vivo Al(III)-induced aggregation requires tau hyperphosphorylation (Shin et al., 1994). Later, work by Yamamoto et al. showed that Fe(III) is also capable of aggregating isolated PHF-tau in vitro and detailed many of the conditions necessary for transition metals-associated aggregation including oxidation state and tau phosphorylation (Yamamoto et al., 2002). In their work, Yamamoto et al. demonstrated that the aggregation of PHF-tau by iron and the subsequent reduction of Fe(III) to Fe(II) via dithiothreitol results in the disassembly of the tau aggregates, and, likewise, tau dephosphorylated by alkaline phosphatase is unable to aggregate in the presence of Fe(III). Yamamoto et al. indicated that iron-mediated tau aggregation was driven by the formation of Fe(III)-PO4 complexes but were not able to determine the structure or location of those complexes. Previous work by Shin et al. (Shin et al., 1994) on Al(III) complexation with tau and early work by Webb on Fe(III) (Webb et al., 1973) complexation with phosphorylated serines supports the hypothesis that the metal-phosphate complexes are responsible for triggering aggregation.
In the context of ROS-mediated damage, an important finding by Sayre et al. was the demonstration that tau, in the presence of Fe(III), can perform the catalytic oxidation of diaminobenzidine in the presence of H2O2 (Sayre et al., 2000). The mechanism of action may be similar to that of a typical peroxidase where ferric iron (Fe(III)) is oxidized by H2O2 and a reduced substrate to produce a ferryl (Fe(IV)) and substrate radical. The substrate is then oxidized by the ferryl radical and releases water and the oxidized substrate (Wang et al., 2010). Artificial peroxidases have also been developed using metal oxide nanocrystalline materials (Fe3O4, CeO2, FeHPO) (Celardo et al., 2011; Gao et al., 2007; Zhang et al., 2014) that exhibit similar oxidative chemistry in the presence of hydrogen peroxide (Zhang et al., 2014).
Work by Wan et al. (Wan et al., 2019) implicates the opposite pathway: that iron may stimulate phosphorylated tau. They showed that neurons treated with ferrous iron expressed 110% more S396-phosphorylated tau than untreated controls. In addition, iron reduced the phosphorylation of insulin receptor β (IRβ- pY1150/1151), the cytosolic subunit of insulin receptor that activates the insulin pathway (Wan et al., 2019) (−55%, p < 0.01), insulin signaling substrate (−70%, p < 0.01), and phosphoinositide (−28%, p < 0.01). Phosphorylation of insulin pathway targets (IRβ, IRS-1) and could be returned to normal with the introduction of additional insulin. Altered phosphorylation epitopes have also been reported in AD patients by Talbot et al. IRβ and multiple IRS-1 phosphorylation epitopes were reduced (IRβ: p = 0.0122). IRβ expression remains unchanged (p = 0.4691) in humans, but IRS-1 expression does increase in AD (p = 0.0009), perhaps as a compensatory mechanism to an in inactive IRβ receptor (Talbot et al., 2012). When tested in mice given high-iron diets, mice expressed lower IRβ-pY1150/1151 (~50%) and higher expression of tau phosphorylation epitopes (~175%) suggesting that an accumulation of iron, as has been reported in ageing in some regions of the human brain in AD (Acosta-Cabronero et al., 2016) may be sufficient to start a cascade of tau impairment. Additionally, Wan et al. demonstrate that insulin receptor B is impaired by a reduction in phosphorylation in cultured neurons but treatment with insulin enhanced the phosphorylation of IRβ, IRS, and PI3K to return them to baseline. Wan et al. showed that neurons cultured with excess iron, treatment with insulin had the effect of also reducing tau hyperphosphorylation, suggesting an insulin dependent pathway (Wan et al., 2019). Insulin resistance in the context of AD is an ongoing subject of discussion (Dineley et al., 2014). The case made by Wan et al. rests on the accumulation of iron in the brain, a case that has not yet been completely resolved (Schrag et al., 2011). In our view, it is possible that the most likely event where iron would play a significant role in tau phosphorylation would be in those regions already enriched in brain iron and thus vulnerable to changes in free iron levels.
Hare et al. had observed that while clinical assays of transferrin saturation indicated no change between control and AD patients, they were able to identify a significant difference in transferrin saturation using size-exclusion chromatography inductively coupled plasma mass spectrometry (SEC-ICP-MS) and revealed that while transferrin saturation in AD patients was lower (p < 0.05) than in healthy controls the total amount of transferrin remained the same (Hare et al., 2015). Ceruloplasmin is a copper-containing enzyme that facilitates the binding of iron to transferrin, they hypothesized this was due to inadequate loading of ferric iron from ceruloplasmin, the ferroxidase on the outward-facing side of ferroportin, however they were unclear of the precise mechanism in which ceruloplasmin was faulty. This subject has also been broached by Kristinsson et al. (Kristinsson et al., 2012) who found that ceruloplasmin concentration was unchanged but ceruloplasmin activity was reduced (patients: 89 U/mL vs. control 136 U/mL; p = 0.0005) in AD patients (N = 26 pairs). This could in theory lead to a dysregulation of intracellular iron concentration where the export of iron is shunted and the new equilibrium iron concentration is higher than normal, potentially leading to tau phosphorylation (Wan et al., 2019).
Tau is not alone in AD when it comes to iron-centered catalysis; work by Plascencia-Villa et al. found that Aβ plaques contain Fe3O4 nanocrystals, which may account for their apparent catalytic oxidation properties (Plascencia-Villa et al., 2016). In 2014, Everett et al. showed that amyloid plaques were capable of reducing Fe(III) to Fe(II) (Everett et al., 2014b). Everett et al. also showed that Aβ plaques were capable of converting ferrihydrite, an iron mineral found in holo-ferritin to Fe(II)-rich redox-active biominerals (Everett et al., 2014a). In 2017, Telling et al. found that the iron in Aβ plaques from AbPPswe/PS1dE9 transgenic mice varied in composition despite being morphologically similar (Telling et al., 2017). The findings with amyloid plaques are exciting and recommend that parallel experiments with tau in its various forms could be a worthwhile study.
Electrochemical studies by Ahmadi et al. detailed possible associations between normal and phosphorylated tau covalently attached to gold electrodes (Ahmadi et al., 2017). They showed that Fe(II) reduced the resistance of tau-functionalized electrodes more than Fe(III) indicating that Fe(II) was binding to the electrode-bound tau. This effect was confirmed with scanning tunneling microscopy which showed an increase in electrode surface roughness when treated the electrodes were treated with Fe(II) but not Fe(III) indicating that Fe(III) did not interact with the electrode-bound tau. Phosphorylation of tau by Src and GSK-3β showed that Fe(III) binding was inhibited by GSK-3β and no change occurred with Src, whereas GSK-3β caused no change to Fe(II) binding but Src eliminated binding of Fe(II). These results suggest that Fe(II) is responsible for forming iron-containing aggregates of tau as opposed to Fe(III).
Quantitative susceptibility mapping MRI (QSM-MRI) has been utilized to show a spatial colocalization of iron deposits and amyloid deposits in AD patients. Practically, this method first requires the collection of PET imaging for amyloid deposits using labels such as PiB followed by QSM-MRI to map the locations of iron deposits in the brain (van Bergen et al, 2016). Scanning electron microscopy of ultra-thin brain tissue sections containing amyloid plaques was found to contain spatially colocalized regions of iron and amyloid (Plascencia-Villa et al., 2016). The appearance of iron deposits was found by Ayton et al. to correlate with the progression of AD (Ayton et al., 2017). The mechanism of iron deposition is not currently understood, nor is the result of iron deposition in plaques. Iron may contribute to the disease course, perhaps through a ferroptotic pathway discussed below (Derry and Kent, 2017). The potential to use QSM-MRI on tau has also been studied by O’Callaghan et al. (O’Callaghan et al., 2017) in rTg4510 transgenic mice carrying an S301L mutation on MAPT. The authors noted that areas containing neurofibrillary tangles had higher magnetic susceptibilities than in their wild type controls (O’Callaghan et al., 2017).
3.2. Iron Metabolism and Tau
Iron metabolism is a tightly regulated process in neurons because of their high metabolic activity (Belaidi and Bush, 2016). Briefly, (Figure 3) iron import is predominantly handled by the endocytosis of Fe(III)-trafficking holo-transferrin by docking to transferrin receptor (TfR1) on luminal capillary endothelial cells (Moos et al, 2007). A second mechanism for iron import is through divalent cation transport (DMT1), a promiscuous divalent metal cation channel known for transporting ions such as zinc, cadmium, iron, nickel, manganese, copper, cobalt and lead across lipid membranes (Garrick et al., 2003). Fe(III) is released from transferrin by its reduction to Fe(II) by the endosomal ferrireductase six-transmembrane epithelial antigen of the prostate 3 (Steap3) (Ohgami et al., 2005) and transported into the cytosol via DMT1 as Fe(II) to join the labile iron pool (LIP) (Kahklon and Cabantchik, 2002). Other members of the Steap family such as Steap1 may play a role in this process but the relationship is still unclear (Kim et al., 2016). The LIP controls the activation of two iron regulatory proteins IRP1 and IRP2 which in turn bind to mRNA iron regulatory sequences known as Iron Response Elements (IREs) (Haile, 1999). Iron transport and storage genes contain IREs that regulate the import, storage, and export of intracellular iron (Haile, 1999). Labile iron is stored in ferritin complexes as an Fe(III) complex and is released via proteasomal or autophagosomal degradation (De Domenico et al., 2009). The export of iron is performed by the transmembrane channel, ferroportin (Moos et al., 2007). In neurons, ferroportin and APP have been thought to interact, and APP was suspected of acting as a ferrioxidase to convert Fe(II) to Fe(III) for transport out of the cell (Duce et al., 2010). However, it was recently discovered that hephaestin, a transmembrane ferroxidase is the active agent in that complex (Dlouhy et al., 2019). However, the supposed enzymatic properties of APP have since been resolved by the finding that ceruloplasmin also interacts closely with ferroportin and performs the ferrioxidase chemistry instead (Honarmand Ebrahimi et al., 2013; Wong et al., 2014). Finally, an intersection between ferroportin, tau, and APP is reached; tau transports APP to the cell membrane and stabilizes the FPN1-APP complex (Lei et al., 2012). Lei et al. demonstrated in tau knock-out mice that tau is essential to maintaining iron efflux and that cortical iron loads are higher (+73%, p = 0.007) than in wildtype mice. Most importantly, tau knock-out mice developed Parkinsonism during the course of their lives with cortical atrophy and a loss of motor abilities when compared with control animals. Lei et al. found that compared to sham-treated tau knockout mice, clioquinol-treated tau knockout mice performed better on the pole test. While over time, the sham-treated mice progressively became worse, the clioquinol-treated mice remained relatively the same with significant differences appearing at 11 mo (p < 0.005) (Lei et al., 2012). This latter experiment also showed that clioquinol, a metal chelator when given starting at 6.5 months of age, just prior to the expected seven months of age before symptoms appeared, for five months could prevent some of the symptoms of Parkinsonism and reduce the hippocampal and substantia nigra iron load by 11% (p = 0.025) and 25% (p = 0.029), respectively.(Lei et al., 2012). A follow-up study by Lei et al. in 2015 showed that ventricle enlargement (measured as volume) as a result of neurodegeneration was reduced by 43% (p = 0.032) and that cortical thickness in clioquinol-treated mice was 6.5% thicker (p = 0.043) than in the untreated tau knockout mice (Lei et al., 2015). The hypothesized mechanism of action was the chelation of iron by clioquinol since that would lower the cytosolic concentration of iron, effectively serving the same mechanism as the impaired ferroportin (Lei et al., 2012).
Figure 3.
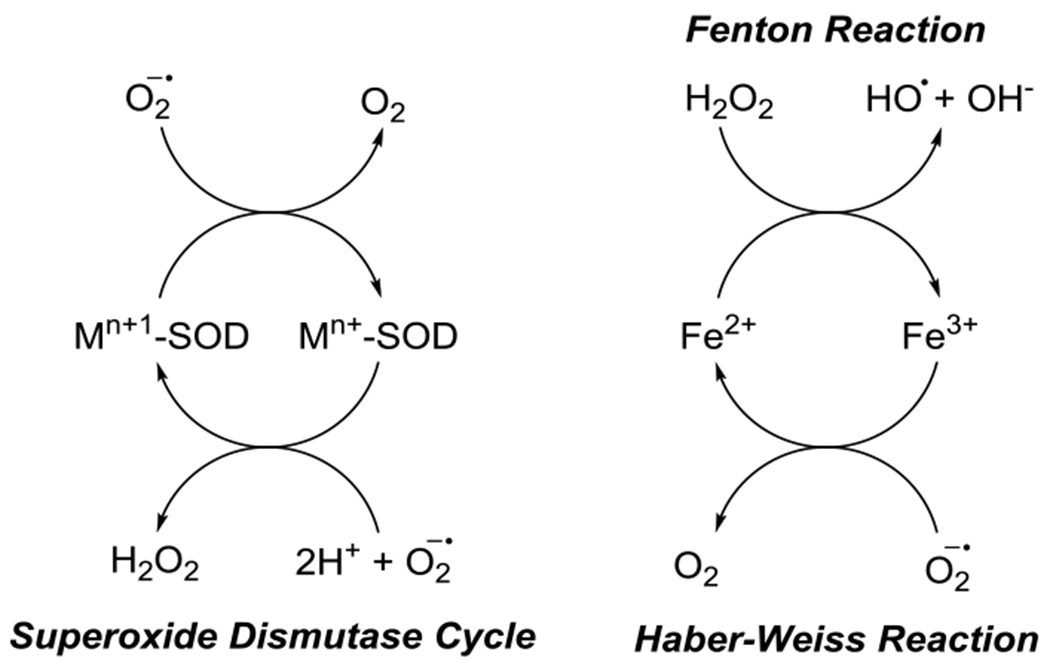
SOD Cycle and ROS-generating cycles of Fe(II)/Fe(III)
Considering that tau knockout mice suffer from a Parkinson-like phenotype and exhibit elevated iron loads, it would appear that tau interacts with ferroportin, somehow, with the new knowledge that hephaestin is responsible for iron oxidation at ferroportin, perhaps more clarity can be had for understanding that process in the context of neurodegenerative disease.
3.3. Iron and the ROS Hypothesis
Iron is a trace element in the body found as Fe(II) or Fe(III) in a variety of states, including as a cofactor for enzymes, sequestered by ferritin, and labile, unchelated, to name a few . The general mechanism of iron toxicity is primarily driven by the Fenton and Haber-Weiss reactions as shown in Figure 4 (Winterbourn, 1995). Fe(II) is oxidized to Fe(III) by hydrogen peroxide (H2O2) to form highly reactive hydroxyl radical through the Fenton reaction (Figure 4). A second reverse reaction, the Haber-Weiss reaction can convert Fe(III) into Fe(II) via superoxide dismutation to produce dioxygen similarly to superoxide dismutase (SOD). Both hydroxyl and superoxide radicals can induce oxidative stress in cells by lipid, nucleic acid, or protein oxidation (Berlett and Stadtman, 1997; Cooke et al., 2003; Doll and Conrad, 2017). To reduce the effect of reactive oxygen species production, superoxide dismutase and catalase convert O2.− radicals and hydrogen peroxide into dioxygen and water and dioxygen, respectively (Samuel et al., 2014). Increasing the rate of peroxide synthesis via superoxide reduction depletes intracellular stores of glutathione and NADPH increasing vulnerability to additional ROS.
Figure 4.
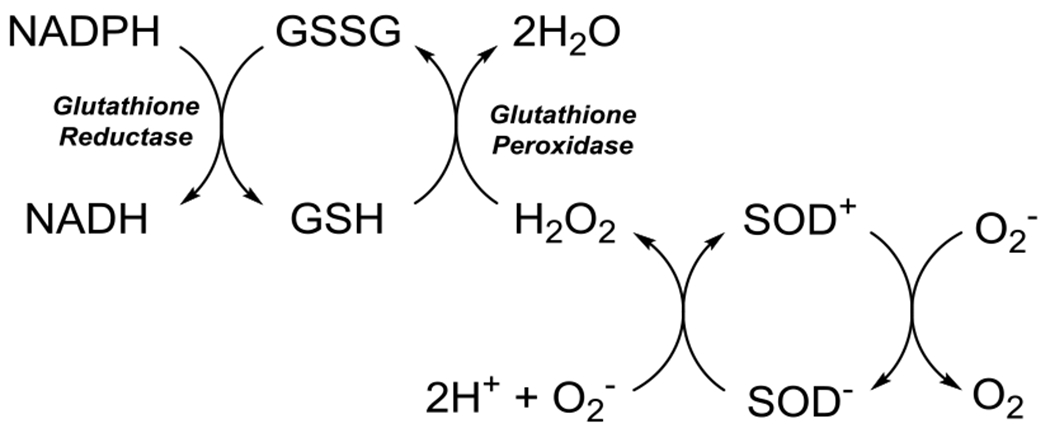
Quenching of ROS by Glutathione.
3.4. ARIAs and Hemorrhage as a Potential Source of Iron
In recent years, the strategy of immunotherapy for AD has been trialed with monoclonal anti-amyloid antibodies such as bapineuzumab. During the phase 2 trial for bapineuzumab magnetic resonance imaging of treated patients revealed the appearance of hyperintense regions by fluid-attenuated inversion recovery (FLAIR), and other hypointense regions by T2*-weighted MRI (Salloway et al., 2009; Sperling et al., 2012). The hyperintense FLAIR signals, referred to as amyloid-related imaging abnormalities with edema (ARIA-Es) were usually asymptomatic, however a few patients did develop symptoms such as an acute reduction in MMSE score, confusion, and gait difficulties; these symptoms resolved following steroid therapy or discontinuation of the immunotherapy (Salloway et al., 2009; Sperling et al., 2011). In the case of the hypointense regions seen with T2*-weighted MRI on the other hand did not resolve (Sperling et al., 2012; Sperling et al., 2011). These regions were believed to be hemosiderin deposits (Black et al., 2010), and were later termed amyloid-related imaging abnormalities with microhemorrhage or hemosiderin (ARIA-Hs) (Sperling et al., 2011). In a retrospective analysis of the phase 2 bapineuzumab trials, Sperling et al. found that of 210 patients treated with bapineuzumab at all doses, 36 developed ARIA-Es, 17 presented with concurrent ARIA-H (Sperling et al., 2012). The mechanism of ARIA-H formation proposed by Sperling et al. suggests that during treatment with bapineuzumab the arteriolar wall is weakened by depletion of trapped amyloid plaques leading to the extravasation of red blood cells into the vessel wall and surrounding tissue (Sperling et al., 2012). The extravasated red blood cells eventually lyse and release hemoglobin into the surrounding tissue.
Blood contains on average 2.5 mM hemoglobin potentially resulting in the generation of a solution with a concentration of 10 mM heme, and through oxidation, 10 mM hemin (Robinson et al., 2009). A concentration of 3-30 μM hemin is reported to be lethal to 60-70% of cultured neurons (Robinson et al., 2009). Due to the toxicity of hemin, and the abundance of hemoglobin in blood it is reasonable that hemin-induced cell death may accompany the appearance of microhemorrhages characterized as ARIA-Hs through either cytosolic ROS generation or lipid peroxidation (Robinson et al., 2009). Iron released from the degradation of hemin joins the iron storage pathway to form ferritin and eventually hemosiderin (Robinson et al., 2009). The long term consequences of ARIA’s are not known, but subject of investigation.
3.5. The Locus Coeruleus, Neuromelanin, and Iron
A small, but growing body of work is actively investigating the role of the locus coeruleus (LC) in Alzheimer’s disease (Weinshenker, 2018). The LC contains a cluster of catechol ami nergic neurons with projections throughout the cortex and midbrain. In AD, volumetric atrophy of the LC correlates to Braak stage as shown by Theofilas et al. (Theofilas et al., 2017). Destruction of LC neurons by N-(2-chloroethyl)-N-ethyl-bromo-benzylamine has been shown in APP transgenic mice to lead to the formation of amyloid plaques and neurodegeneration faster than in wild type mice with the same treatment, suggesting that the LC has some undetermined role in moderating memory loss, cell death, and cortical morphological changes (Heneka et al., 2006). Neuromelanin captures heavy metals with a strong affinity for iron, generating both an intercalated iron oxide compound as well as peripheral ferric catecholamine complexes (Zucca et al., 2017). The relationship of neuromelanin and neurodegeneration is complicated as it serves both as a buffer for excess iron in LC and SN neurons but can also generate reactive oxygen species during synthesis and degradation (Aguirre et al., 2012; Hare and Double, 2016; Urrutia et al., 2013; Zecca et al., 2001; Zucca et al., 2017). In AD, the number of LC neurons decreases progressively in the course of the disease as shown by Kelly et al. (Kelly et al, 2017). Using fresh brain slices and a tyrosine hydroxylase treatment, Kelly et al. compared the number of LC neurons in individuals that were cognitively normal, expressed mild cognitive impairment, and expressed full-blown Alzheimer’s disease post mortem and observed that a decline in catechol ami nergic neurons followed the progression of the disease (Kelly et al., 2017). In addition, Kelly et al. measured the mRNA expression of several genes responsible for redox homeostasis, mitochondrial function, and neuroplasticity and observed that in MCI and AD cases, there was a substantial reduction in gene expression compared to the cognitively normal cases. Cytochrome C (cytc1) for instance, was reduced by approximately 50% (p < 0.01) in MCI and AD cases as well as superoxide dismutase (SOD2) (50%, p < 0.01) (Kelly et al., 2017). Glutathione peroxidase (GPX1) was reduced by 55% (p < 0.001) in MCI and AD cases (Kelly et al., 2017). These findings indicate that not only is mitochondrial activity reduced in MCI and AD, the redox homeostasis of the affected cells is also altered (Kelly et al., 2017).
During the synthesis of neuromelanin, dopamine is oxidized to aminochrome which then tautomerizes to form 5,6-dihydroxyindole. Dioxygen finally oxidizes the 5,6-dihydroxyindole to 5,6-indolequinone which polymerizes to form neuromelanin (Munoz et al., 2012). The oxygen consumed in this reaction becomes superoxide and then leaves the reaction (Munoz et al., 2012). Theoretically, the resulting superoxide can reduce ferric iron to ferrous iron, which then subsequently can react with hydrogen peroxide, possibly generated by superoxide dismutase, to generate hydroxyl radical in situ. Work by Salgado et al. showed that several comparable 1,2-dihydroxybenzenes, when chelated to iron generate reactive oxygen species such as hydroxyl radical freely in the presence of hydrogen peroxide through a Fenton-like reaction (Salgado et al., 2017). Consequently, dopaminergic neurons may seed their own doom by generating and storing iron-laden neuromelanin.
3.6. Is AD a Ferroptopathy?
A recent addition to the transition metal dysregulation hypothesis is the observation of a nonapoptotic mode of cell death linked to iron known as ferroptosis. First defined by Dixon et al. in 2012, ferroptosis was initially discovered as a consequence of treating RAS mutant HT-1080 cell lines with erastin, a small molecule that inhibited voltage dependent anion channels 2 and 3 (VDAC2/3) and activating specific iron-related pathways thereafter (Dixon et al., 2012). Dixon et al. reported that treating HT-1080 cells with 10 μM erastin lead to a time-dependent loss of cell viability that could be prevented with the cotreatment of 100 μM desferrioxamine, but this treatment did not protect cells from rotenone, an inhibitor of Complex I (Dixon et al., 2012). Inhibition of ferroptosis was also achieved by the use of the small molecule ferrostatin-1, a highly lipophilic compound presumed to accumulate in the membranes of the cell (Dixon et al., 2012; Gaschler et al., 2018; Zilka et al., 2017). Notably, like deferoxamine, ferrostatin-1 does not protect cells from rotenone (Dixon et al., 2012). The authors determined the upstream causation of the downstream iron-mediated toxicity as inhibition of the Xc− system which lead to a depletion of mitochondrial cystine, a crucial substrate for the synthesis of GSH and cysteine (Dixon et al., 2012). Dixon et al. also showed that HT-1080 cells can be potentiated for ferroptosis specifically by treatment with Fe(III) salts (Dixon et al., 2012).
Ferroptosis is largely wrapped around the glutathione synthesis and utilization pathways. Figure 6 summarizes the pathway and the anomalous variations in substituent levels and activities. The synthetic pathway starts with the import of the cysteine dimer cystine by the Xc− cystine-glutamate antiporter. Next, cystine is reduced by thioredoxin to two cysteine monomers after which it undergoes ligation with glutamate by glutamyl cysteine ligase (Doll and Conrad, 2017). Finally, GluCys is cleaved by glutathione synthase to produce glutathione (Doll and Conrad, 2017). Glutathione serves as a substrate for several enzymes involved in the antioxidant systems of the cell (Dringen et al., 2000). Glutathione, when bound to glutathione peroxidases reacts with lipid peroxides to produce a hydroxyl-functionalized lipid (Hambright et al., 2017). The resulting glutathione disulfide is then subsequently reduced back to glutathione by glutathione reductase which uses NADPH as its reducing substrate (Doll and Conrad, 2017). In the classical model of ferroptosis, the Xc− system antiporter is inhibited which leads to an overall reduction in glutathione available for the cell and subsequent accumulation of lipid peroxides, the hallmark indicator of ferroptosis, and eventual cell death (Dixon et al., 2012).
Figure 6.
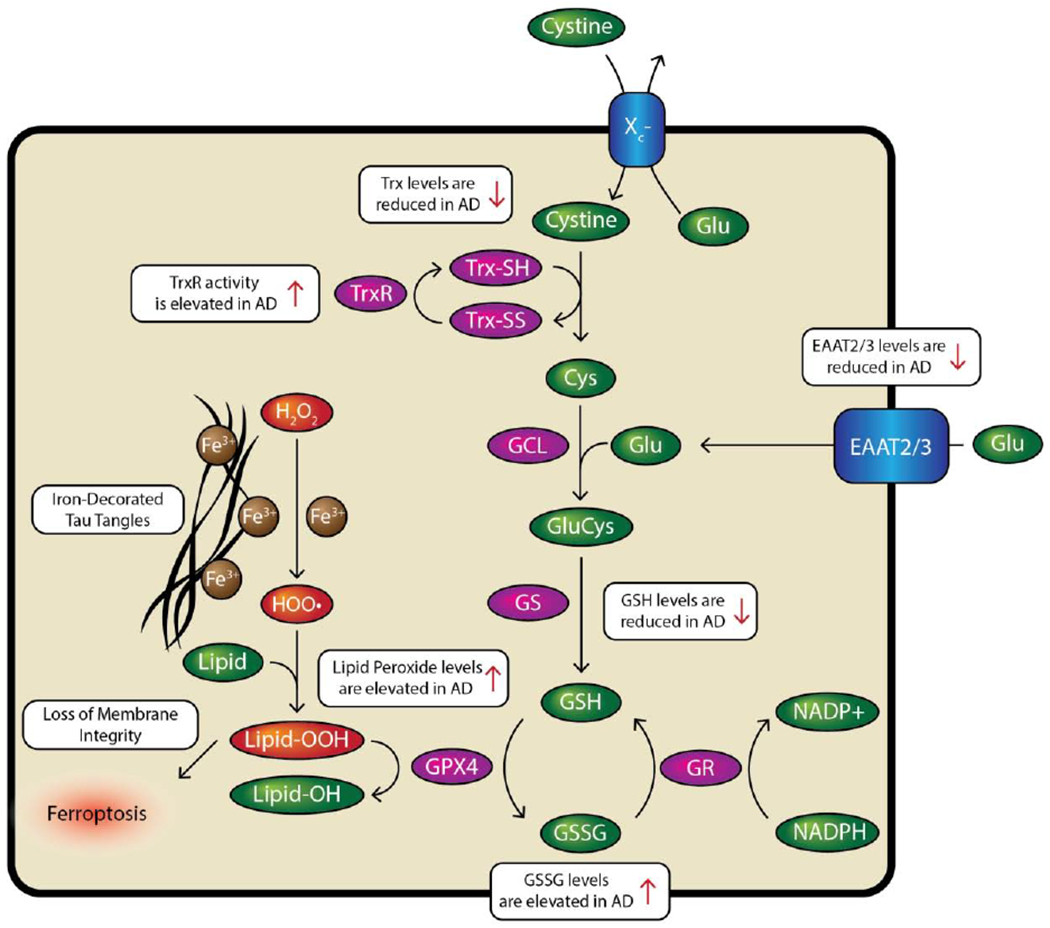
Pathways of Ferroptosis involving iron and the mitigation and fate of lipid peroxides. Adapted from Derry and Kent, 2017 and Doll and Conrad, 2017.
In Alzheimer’s disease some aspects of the glutathione synthesis and utilization pathways are perturbed (Figure 6.) Lovell et al. reported that in AD, TrxR activity was significantly increased when compared to normal age-controlled healthy controls in the amygdala (~35%, p ≤ 0.01) and cerebellum (~40%, p ≤ 0.01), and Trx concentrations were significantly reduced in AD brains in the amygdala (58.9±10.8%%, p = 0.01), hippocampus (67.9±8.0%, p = 0.01), and superior and middle temporal gyri (80.4±7.9%, p = 0.08) (Lovell et al., 2000). Reduced thioredoxin levels could lead to a reduction in cysteine synthesis. The increase in TrxR activity may be a compensatory factor to increase the rate of turnover of the depleted Trx (Lovell et al., 2000). Lovell et al. observed that cultured neurons treated with Aβ could be rescued by the inclusion of thioredoxin or thioredoxin reductase suggesting that Aβ has an influence on some upstream process that leads to an oxidative stress condition (Lovell et al., 2000).
Downstream of cystine, Eto et al. did not show that cysteine levels were lower in AD patients (7.4 nmol/mg vs. 7.4 nmol/mg) (Eto et al., 2002). However, Eto et al. found that S-adenosyl-methionine levels were significantly lower in AD brains versus normal controls (0.16 ± 0.03 nmol/mg vs. 0.53 ± 0.04 nmol/mg, p < 0.001) (Eto et al., 2002). This finding suggests that the SAM pathway is somehow inhibited while the Xc− system remains functional. Glutathione levels in AD patients have been shown by several groups to be perturbed in the brain and are inversely correlated to cognitive function. Chiang et al. showed by magnetic resonance imaging that glutathione levels were inversely correlated to brain Pittsburgh Compound B uptake indicating that in areas with high amyloid plaque density there was a reduction in total glutathione with significance achieved in the temporal (p = 0.03) and pariet al lobes (p = 0.05) but not the frontal (p = 0.67) or cingulate lobes (p = 0.88) (Chiang et al., 2017). Similarly, Mandal et al. showed that glutathione levels were suppressed even in individuals with mild cognitive impairment (0.69 mM vs. 1.02 in healthy control, p < 0.01 in MCI; 0.63 mM vs. 1.02 in healthy control; p < 0.001 in AD) (Mandal et al., 2015). Outside the brain, McCaddon et al. showed that plasma concentrations of glutathione were inversely correlated with ADAS-COG scores (p = 0.002) but that on average glutathione levels were not perturbed from healthy controls (2.5 μmol/L vs. 2.7 μmol/L) (McCaddon et al, 2003). Conversely, Cecchi et al. showed that familial AD have lower on average plasma glutathione levels than healthy control patients (55% of healthy controls; p < 0.01) but also showed that there was no difference in GSH levels between individuals with sporadic AD and healthy controls (Cecchi et al., 1999). Ansari et al. found that reduced glutathione (GSH) levels in isolated frontal cortex mitochondria and synaptosomes from the brains of recently deceased AD patients are on average lower than in healthy controls (mitochondria: ~70% vs. healthy control, p < 0.01; synaptosomes: ~50% vs. healthy control, p < 0.01) and that glutathione disulfide (GSSG) levels are elevated (mitochondria: ~300% vs. healthy control, p < 0.01; synaptosomes: 350% vs healthy control, p < 0.01) leading to negative shifts in the GSH/GSSG ratio indicating a more oxidizing environment than in normal individuals (mitochondria: ~25% vs. healthy controls, p < 0.01; synaptosomes: ~25% vs. healthy controls, p < 0.01) (Ansari and Scheff, 2010). Finally, Ghosh et al. showed that glutathione concentration reduction caused by treatment with buthionine sulfoximine in ageing neurons obtained from LaFerla’s triple transgenic mouse model (3xTg-AD) increased neuronal death overall compared to non-transgenic neurons (Ghosh et al., 2014).
Another aspect of glutathione synthesis is glutamate import (Figure 6.) Glutamate is transported into neurons via the excitatory amino acid transporters (EAATs). Glutamate is required to synthesize glutathione and the expression of glutamate transporters in AD brains is heterogeneous by region and receptor type. For instance, work by Jacob et al. demonstrated that EAAT2 and EAAT3 expression levels in the hippocampus were significantly lower (p < 0.01) in AD patients at all Braak stages than healthy controls (Jacob et al., 2007). Because these transporters are not expressed as readily, it may be possible that glutathione synthesis suffers even though thiol-containing cysteine levels are unchanged.
The utilization and recycling pathways of glutathione have also been studied in human AD patients (Figure 6.) Ansari et al. found that glutathione peroxidase (GPx) levels were lower in the isolated mitochondria (~15 nmol NADPH oxidized/min/mg protein vs. ~22 in healthy controls) and synaptosomes (~20 nmol NADPH oxidized/min/mg protein vs. ~35 in healthy controls) of AD patients (Ansari and Scheff, 2010). However this effect is likely region-specific because work from Lovell et al. indicated substantial regional variations (Lovell et al., 1995). On the recycling side, glutathione reductase levels were shown by Lovell and Ansari et al. to be suppressed in AD patients (Ansari and Scheff, 2010; Lovell et al., 1995). Additionally, GPX4 translation was found to be downregulated in a doxycycline-inducible mouse model of AD and controlled by guanine-rich sequence binding protein (GRSF1) by Yoo et al. who found that lipid peroxidation is consequently promoted (Yoo et al., 2010). The neurodegeneration found in Alzheimer’s can be mimicked by the ablation of GPX4 in mice (Hambright et al., 2017). Hambright et al. reported that GPX4 knockout mice brain lysates contained approximately 50% more HNE adducts than their normal counterparts, demonstrated a 100% increase in IL-6 activation, and a 200% increase in TNF-α activation showing significant neuroinflammation and oxidative damage from the removal of GPX4 (Hambright et al., 2017). Zhang et al. showed in P301S mice that GPX4 expression could be upregulated using lipoic acid (~280% of vehicle-treated control in lipoic acid-treated mice; p < 0.01). This also had the knock-on effect of increasing system Xc− activity (175% of vehicle-treated control in lipoic-acid treated mice; p < 0.01) (Zhang et al., 2018).
The late stage pathway component of ferroptosis, lipid peroxidation has been shown to be perturbed in AD. Lovell et al. demonstrated that lipid peroxidation is region-specific and is elevated in AD (Lovell et al, 1995). Ansari et al. similarly showed that lipid peroxidation was elevated in isolated mitochondria and synaptosomes (Ansari and Scheff, 2010). Furthermore, they showed that MMSE scores were negatively correlated to the lipid peroxidation markers protein carbonyls (r = 0.536), 3-nitrotyrosine (r = 0.560), 4-hydroxynonenal (4-HNE; r = 0.675) and acrolein (r = 0.786) (Ansari and Scheff, 2010).
The state of iron the Alzheimer’s disease brain is debatable. In a meta-analysis performed by Schrag et al. of 20 publications (Schrag et al., 2011) on the concentrations of iron, zinc, and copper in the neocortex the authors found that most publications found little overall increase in iron concentration in frontal cortex in AD, and that reports of higher levels were restricted to a single institution. Furthermore, Schrag et al. found that papers which reported a large difference between healthy controls and AD brains were most likely to be cited, suggesting a citation bias that could skew the interpretation of the literature, especially from review articles (Schrag et al., 2011). Bearing this in mind, the overall concentration of iron in AD is not fully clarified, however it seems likely that its regulation becomes dysfunctional as other studies have shown an increase in CSF ferritin (Ayton et al., 2018), and an increase in signal shift by QSM as well as in the pathological lesions plaques and NFTs (Ayton et al., 2017; Mander et al., 2017; van Bergen et al., 2016). Furthermore, the findings by Schrag et al. do not rule out the findings that transition metals are colocalized with amyloid plaques (van Bergen et al., 2016) and tau tangles (Smith et al., 1997) or that iron handling systems such as ferroportin are dysfunctional (Raha et al., 2013). Work on improving the quantitation of metals in brain tissue is ongoing (House et al., 2012) as the instrumental method, and handling procedures can influence the quality of the resulting data.
Raha et al. reported via histology that ferroportin is aberrantly downregulated in AD (Raha et al., 2013). Downregulation would have the effect of reducing iron export which may lead to elevated iron levels in the affected cells. Additionally, Lei et al. found that tau is necessary for ferroportin activity as shown in tau knockout mice. They hypothesized that that the elevated iron loads in AD are in part due to loss of soluble tau (Lei et al., 2012). The loss of soluble tau is supported by Khatoon et al. who found that brain tissue supernates containing cytosolic constituents contained on average 40% (p < 0.05) less soluble tau than normal healthy controls (Khatoon et al., 1994). However, they did also find that tissue homogenates from AD patients contained more tau than healthy controls, possibly due to tau tangles (Khatoon et al., 1994).
Tau may play a role when coupled to iron in ferroptosis as well. As reported previously by Yamamoto et al. and Smith et al., tau tangles can contain ferric iron (Smith et al., 1997; Yamamoto et al., 2002). Good et al. used laser microprobe mass analysis (LAMMA) to determine the concentration or iron in the cytoplasm of NFT-bearing neurons in cadaver brains. The NFT-bearing neurons from 10 AD patients on average contained more iron than in NFT-free neurons in 4 healthy cadaver control brains (mean = 20 vs. 4.3, arbitrary units;). Additionally, NFTs contained substantially more iron than the cytosol of NFT-containing neurons (mean = 62 vs. 20, arbitrary units;).
Because iron accumulates in ageing (Acosta-Cabronero et al., 2016; Connor et al., 1990; Hallgren and Sourander, 1958), and in recent work, has been shown to be stored in large quantities by senescent mouse embryonic fibroblasts (~20-fold greater than non-senescent cells) (Masaldan et al., 2018). Iron is a key figure in the potentiation of ferroptosis, (Ramos et al., 2014; Ward et al., 2014) it may be possible that both physiological and pathological sources of reactive oxygen species contribute for ferroptosis in the brain (Figure 5). In this case, hydrogen peroxide generated through oxidative stress may react with iron contained in the labile iron pool in its either Fe(II) or Fe(III) oxidation states to generate hydroxyl or peroxyl radical via Fenton and Haber-Weiss cycles, respectively (Figure 4). Peroxyl radicals are known to react with lipids to generate lipid peroxides (Ayala et al., 2014; Doll and Conrad, 2017; Latunde-Dada, 2017). Lipid peroxides are either reduced to lipid alcohols by glutathione peroxidase or accumulate leading to membrane permeability and eventual cell death.(Doll and Conrad, 2017)
Figure 5.
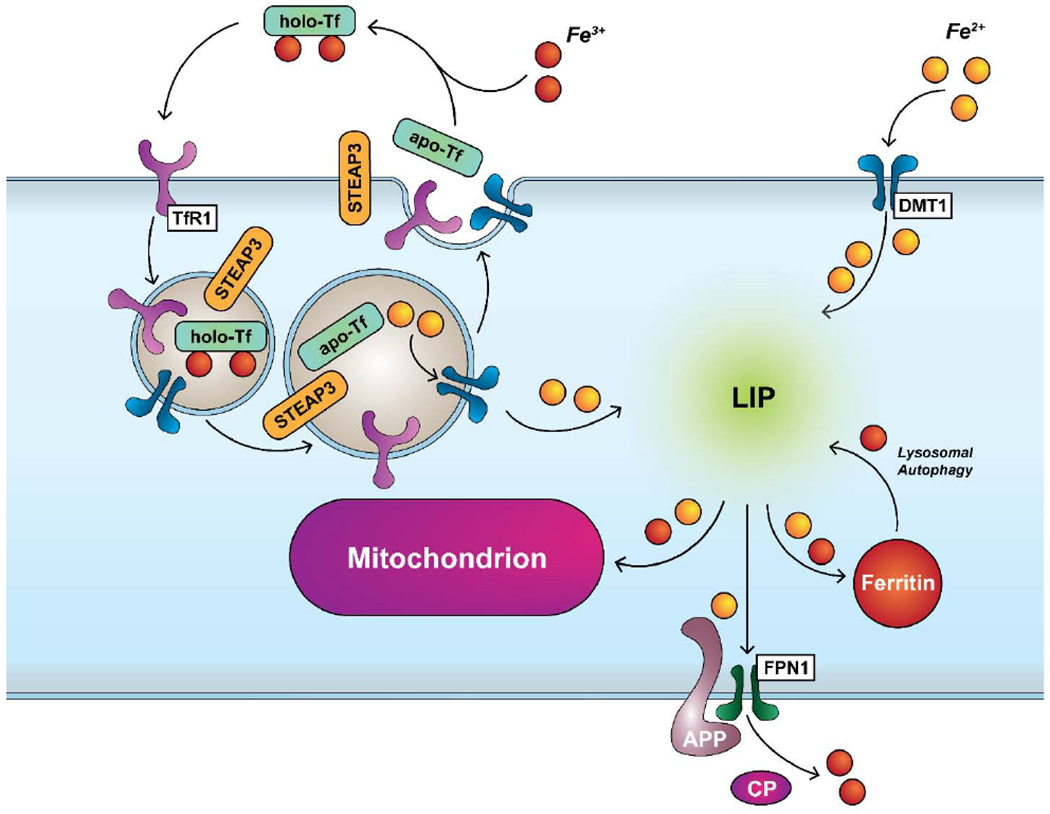
Schematic of import, storage and export pathways in iron metabolism.
Inhibitors of ferroptosis may be a treatment approach in AD. These compounds are typically antioxidants and highly lipophilic. The prototypical example provided by Dixon et al. is ferrostatin-1 (Dixon et al., 2012). This compound has been studied for its unique effect, and work by Zilka et al. showed that ferrostatin-1 is a radical trapping antioxidant that inhibits the peroxidation of membrane lipids. Gaschler et al. showed that a diyne-functionalized ferrostatin-1 preferentially accumulated in the membranes of lysosomes, mitochondria and the endoplasmic reticulum, with the latter perhaps the most important target (Gaschler et al., 2018). The endoplasmic reticulum is the site of oxidative protein folding by protein disulfide isomerase (PDI) (Wilkinson and Gilbert, 2004). The redox state of PDI is controlled by the availability of glutathione and oxygen within the membrane of the ER (Wilkinson and Gilbert, 2004), and impaired redox buffering by the depletion of glutathione as in the case of erastin treatment can lead to improper folding of proteins (Bhandary et al., 2012)
Vitamin E is a ferroptosis inhibitor, possibly due to its lipophilicity, similar to ferrostatin and liproxstatin but mechanistically different (Dixon et al, 2012; Zilka et al, 2017). It has been shown in vitro by Seiler et al. that lipid peroxidation in GPX4 knockout mice could be inhibited by administering α-tocopherol (vitamin E), suggesting that it could be used as a drug to protect against ferroptosis (Seiler et al., 2008). Other work by Veinbergs et al. showed that in APOE4-deficient mice lipid peroxidation was 2.5-fold higher than wildtype controls (Veinbergs, 2000). When APOE4-deficient mice were treated with vitamin E, lipid peroxidation returned to the level of the wildtype controls (Veinbergs, 2000). Despite the promising preclinical results showing that vitamin E was capable of reducing lipid peroxidation, placebo-controlled trials of vitamin E in humans with AD or MCI have not shown any benefit (Farina et al., 2017). In one trial, treatment with vitamin E showed a treated subgroup had accelerated cognitive decline when compared to the placebo control (Lloret et al., 2009). Similarly, in the SELECT trial, both vitamin E and selenium increased the risk of developing prostate cancer (vitamin E HR: 1.17, selenium HR: 1.09) (Klein et al., 2011), a finding not dissimilar to other studies with antioxidants in healthy and afflicted populations (Bjelakovic et al., 2012).
Selenium-containing compounds have also been explored as means to reduce ferroptosis because selenocysteine is in the active site of GPX4, without which cells become sensitized to ferroptosis (Cardoso et al., 2017). Zhang et al. demonstrated that selenomethionine (Se-Met) reduced tau hyperphosphorylation and autophagic clearance in a 3xTg tau mutant mouse model (Zhang et al., 2017). Zhang et al. showed that spatial memory loss was reduced in Se-Met-treated 3xTg mice and that both total hyperphosphorylated and otherwise soluble and insoluble tau (p < 0.05) were also reduced. In a study of intracranial hemorrhage and ferroptosis, Alim et al. showed that sodium selenite (Na2SeO3) could protect mouse primary cortical neurons against 5 uM erastin, an inducer of ferroptosis, and partially protect primary cortical neurons isolated from embryonic CD1 mice from 80 μM hemin in a dose-dependent fashion with a maximum of 1 μM before becoming toxic at 10 μM (Alim et al., 2019). GPX4 expression was elevated in cells treated with selenium and hemin (selenium + hemin vs. untreated control p < 0.01) meaning that the cells were more able to respond to reactive oxygen species produced by the hemin in situ. Alim et al. also showed that selenium raised GPX4 levels when cells were exposed to erastin, a classic activator of ferroptosis that operates by inhibition of VDAC2/3 (p < 0.01). The effects of selenium and vitamin E on dementia advancement were studied in the PREADViSE trial, the researchers found that there was a detrimental effect of both vitamin E (HR: 0.88) and selenium (HR: 0.83) compared to the untreated placebo control (HR: 1.00) (Kryscio et al, 2017). Based on the clinical trials that have been performed on either vitamin E or selenium there appears to be no benefit in their administration in treating Alzheimer’s disease and may in fact be harmful in the long run. This is consistent with previous studies with antioxidants in the treatment of Parkinson’s disease where no benefit was observed with vitamin E, CoQ10, MitoQ, or creatine (Filograna et al, 2016).
3.7. Pharmacological Methods to Target Iron-induced Neurotoxicity
The general strategy to address iron toxicity in vivo is chelation therapy. For example, iron overload associated with hemochromatosis can be addressed by the administration of large doses of deferoxamine (DFO) (Figure 7) on the order of 2 g/day due to its poor cellular uptake, low lipophilicity and relatively high molecular weight (Nielsen et al., 2003; Porter et al., 2005). However, chelation therapy in general is not without its detractors (Hegde et al., 2009). Depletion of iron can induce multisystem toxicity (Wong et al., 1997), and as in the case of EDTA, which can form a basket complex and become a more active catalyst (Flora and Pachauri, 2010). Two additional iron chelators, deferiprone and deferasirox (Figure 7) have been demonstrated to be clinically effective in reducing serum iron concentrations (Kontoghiorghes et al., 2000; Yang et al., 2007). Because ferroptosis may a contributing mechanism in the neurotoxicity of AD, removing iron from the brain may be a benefit by reducing the catalytic properties of NFTs (Sayre et al, 2000).
Figure 7.
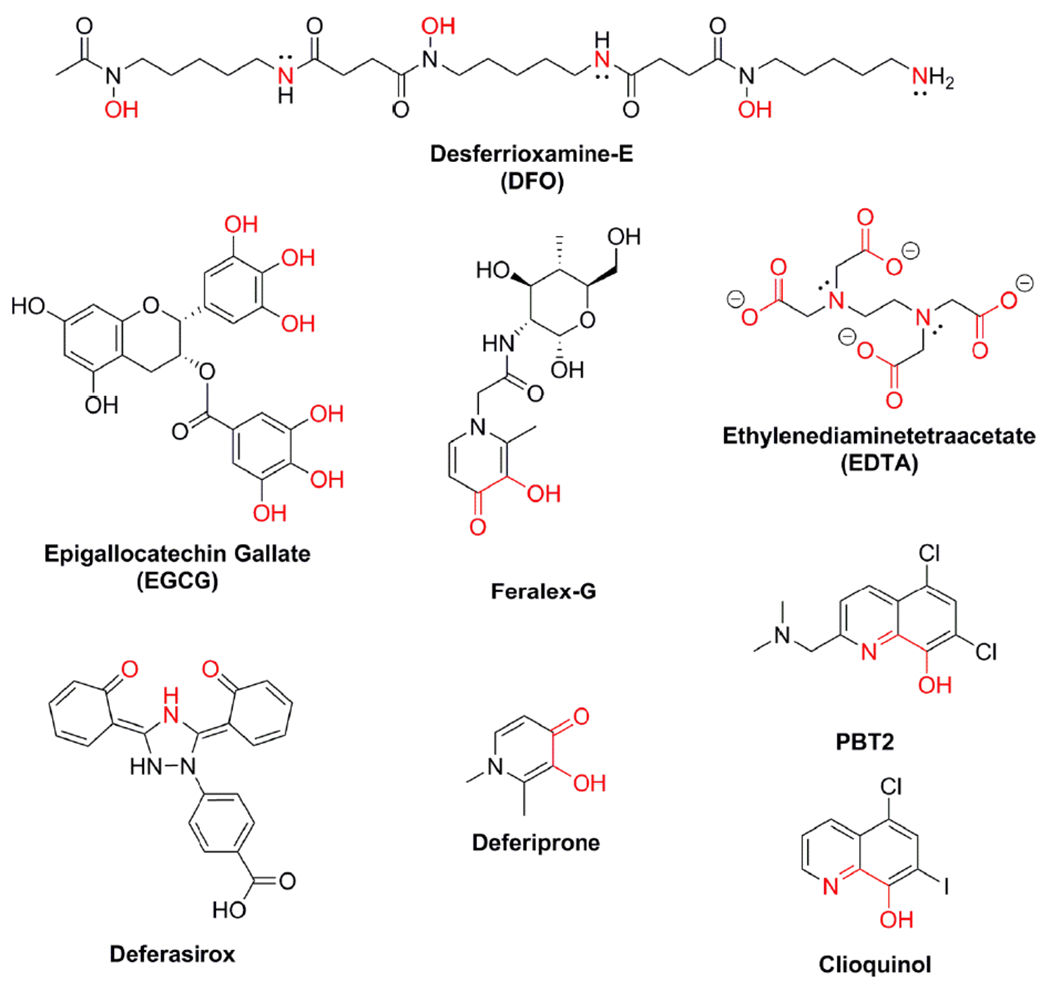
Structures of Iron Chelators Tested in AD models.
Because of the potential role of iron in AD, McLachlan et al. demonstrated that intramuscular administration of 125 mg/kg/day of DFO mesylate reduced the rate of AD dementia progression by a factor of two by several cognitive and behavioral measures performed at the start of the study and every 12 months thereafter to judge cognitive function among 48 adults with an average age of 63 years (McLachlan et al., 1991). Five members of the DFO-treated group reported side effects including appetite loss (n = 4) and weight loss (n = 1) (McLachlan et al., 1991). We are not aware of a follow up confirmation study.
Later, in vitro studies of DFO on Fe(III) dissociation from NFTs suggest that deferoxamine alone may have limited effectiveness by finding that labile iron is targeted by DFO opposed to NFT or Aβ plaque-bound iron because DFO does not function in this capacity at physiological temperatures but instead requires substantially elevated, autoclave temperatures of 121 °C (Murayama et al., 1999; Yamamoto et al., 2002), at least in-vitro. To address the temperature limitation of DFO vis a vis plaque and NFT bound iron, the bidentate chelator Feralex-G was developed to dissociate Fe(III) from NFTs at 37 °C (Shin et al., 2003; Shin et al., 1994). In addition to functioning as an iron chelator, Feralex-G was demonstrated to effectively bind Al(III) another constituent associated with PHFs at 37 °C (Shin et al., 2003).
Of the bidentate chelators, the most extensively studied in neurodegeneration is clioquinol, which was demonstrated by Cherny et al. (Cherny et al., 2001) to reduce the loading of Aβ plaques in 12-month-old APP2576 transgenic mice by 49% (p = 0.0001) after 12 weeks of treatment as a function of zinc and copper chelation. Clioquinol has two major advantages over other zinc or copper chelators such as penicillamine. Because clioquinol is less hydrophilic it penetrates cell membranes more effectively (Cherny et al., 2001). Also, clioquinol has already been utilized as an antimicrobial and has known tolerability in mammals (Bareggi and Cornelli, 2012). While often thought of as a Cu(II) or Zn(II) chelator, also has an affinity for iron. Kaur et al. showed that clioquinol decreases substantia nigra iron load (~30%, p < 0.01), and in cotreatment with 1-methyl-4-phenyl-1,2,3,6-tetrapyridine (MPTP), attenuated loss of endogenous GSH (ns) and 4-HNE (ns) (Kaur et al., 2003).
As mentioned previously, Lei et al. demonstrated that clioquinol prevented the development of Parkinsonism in 6.5-month-old Tau knockout mice (Lei et al., 2012) by binding iron in tau deficient cells. According to the theory of ferroptosis, removal of excess iron should decrease the amount of ROS damage in the brain (Doll and Conrad, 2017). In humans, clioquinol has been historically used as an antifungal and antiprotozoal drug (Bareggi and Cornelli, 2012). Its efficacy against AD progression has been tested in a 36-week study by Ritchie et al. that showed that in individuals with ADAS-cog scores less than 25 at baseline, serum concentrations of Aβ42 were significantly lower (approximately −1.2 ng/mL, p < 0.05 after week 20, and approximately −1 ng/mL, p < 0.001 after week 28) in 16 patients (Ritchie et al., 2003). As the safety of clioquinol could not be verified because of a synthetic contaminant (Adlard et al., 2008), an iodine-free variant, PBT2, was synthesized and tested as part of a Phase Ha trial (Lannfelt et al., 2008) with significant effect on cerebrospinal fluid (CSF) Aβ42 biomarkers in patients given 250 mg/day (−56.0 μg/mL, p = 0.006), but no significant effect was observed on CSF total tau, phosphorylated tau, or Aβ40 concentrations (Lannfelt et al, 2008). Improvement in ADAS-cog scores was observed at week 12 (placebo: +1.3; 250 mg/day: −1.1; p = 0.041) (Lannfelt et al, 2008). The IMAGINE Phase II trial, which was completed in 2014 did not show a statistically significant improvement in hippocampal volume or MMSE scores. In addition to studying the effect of PBT2 on Aβ, A structurally similar derivative of PBT2, PBT434 a quinazoline derivative (Finkelstein et al, 2017), was announced by Prana (now Alterity) in 2017 to test as an iron chelator for use in Parkinson’s disease to reduce reactive oxygen species load (Finkelstein et al., 2017). To further explore the possible uses of PBT434, a trial has also recently started to study the tolerability of PBT434 in patients with Huntington’s disease, no results have been reported as of the time of this writing
Deferiprone has been shown to reduce tau phosphorylation (p < 0 · 001) in 1.5–2-year-old white male rabbits fed a high cholesterol diet at a concentration of 50 mg/kg/day for 12 weeks by reducing the ratio of pGSK3β:GSK3β and casein kinase 1δ activity (Prasanthi et al., 2012). In this study, Prasanthi et al. demonstrated that whereas deferiprone by itself did not reduce cortical ROS or iron concentration, it did reduce the plasma concentration of iron while reducing the total Aβ40, Aβ42, and BACE1 load (Prasanthi et al., 2012). Prasanthi et al. concluded that the effect on Aβ accumulation was a result of altering iron metabolism by chelating internal iron and reducing the rate that APP was translated, while not changing the intracellular concentration of iron.
A major limitation of chelation therapy as the sole approach in AD comes from the complex and dynamic nature of iron toxicity in distinct regions of the central nervous system (Acosta-Cabronero et al., 2016; Mitra et al., 2014). Except in a small number of AD patients with a known occupational/environmental or dietary iron exposure, most AD patients show iron accumulation in affected regions, redistributed from neighboring brain regions, or CSF, where iron level is either reduced or not changed. Thus, targeting a chelator to the brain regions with higher free iron load may be critical. In addition, the dynamic perturbation in the metal homeostasis with disease progression, typically characterized by increased iron, copper or aluminum, adds a second level of complexity requiring chelation of excess metal ions (Hegde et al., 2009). Supplementation of a depleted essential metal may also be important to prevent long term adverse effects of chelation therapy. However, there is limited knowledge on trace metal homeostasis or inter-metal relationships in the AD brain (Mitra et al., 2014). Differential overload of iron in mitochondria vs. nucleus or other cellular organelles in neurons may further limit the efficacy of chelation approach. Thus, a multi-modal toxicity of iron towards biomolecules suggests the need for a deeper molecular understanding of the phenomenon to formulate an effective iron-targeted intervention.
Given these uncertain effects of chelation alone, another strategy beyond simple chelation is a dual-modality treatment that combines both iron chelation and oxidative stress quenching (Mitra et al., 2014). An example of this approach is the naturally occurring (−)-epigallocatechin gallate (EGCG), an iron chelator and antioxidant found in green tea (Weinreb et al., 2009). By treating with a broad-spectrum antioxidant simultaneously with an iron chelator infusion, a dual-modality treatment may be useful in treating neurodegenerative disorders by removing iron from the LIP and quenching oxidative stress.
One final consideration that may limit the utility of only targeting oxidative stress with an antioxidant is the potential for antioxidants to behave in a pro-oxidant fashion. For instance, ascorbic acid is known to reduce Fe(III) to Fe(II) through a single electron reduction followed by oxidation by dioxygen or peroxide to Fe(II) giving either superoxide or hydroxyl radical, respectively (Rietjens et al., 2002). In addition to ascorbic acid, EGCG has known pro-oxidant pathways with iron (Lambert and Elias, 2010). The close interaction of multiple sources of pathology and pathways for disease propagation suggests that a single target may be insufficient in a broad population of AD patients with different stages of disease or pathological typology. Table 1 shows some of the known and possible side-effects of different pathway targeting therapeutics, judicious selection of therapeutic agents may limit these. By using a multi-pronged strategy, the damage from a pathological condition may be attenuated more effectively than only addressing either the reactive oxygen species stress or the transition metal dysregulation alone (Wang et al., 2017a). Furthermore, coupling treatment with a bifunctional drug and immunotherapy may help shunt the oligomeric tau propagation pathway and slow the spread of the disease more effectively than a single agent by itself (Sharma et al., 2018).
Table 1.
Targets Associated with Tauopathy in Alzheimer’s Disease.
| Targets | Intended Effects | Side-Effects |
|---|---|---|
| Serine-threonine kinases | Reduce GSK-3B and other Ser/Thr kinase activity to reduce phosphorylation of tau. | Ser/Thr inhibitors may negatively affect endocrine and other systems.109 |
| Tau oligomerization inhibition | Prevent the formation of tau oligomers. | Covalent modification may be nonspecific and impair protein-protein interactions leading to signaling pathway dysfunction. |
| Oligomeric tau | Reduces the propagation of pathologically modified Tau. | Unknown |
| Iron | Reduces ROS generation and other excess free Fe mediated toxicity. | Hepatotoxicity,183 Depletion of essential metals.184 |
| Reactive Oxygen Species | Reduces rate of kinase activation; prevents oxidative inactivation of key biomolecules including genome repair enzymes. | Antioxidants have prooxidant properties under certain conditions.215 May interfere with electron transport chain. Can generate reactive intermediates. |
| Tau mRNA | Reduces synthesis rate of tau, proliferation and spread of oligomeric tau. | Halting tau translation may impart a tau-knockout-like phenotype. |
| Senolytics | Reduce contribution of senescent cells to neurodegeneration | Still under investigation, include low dose cancer chemotherapy |
4.1. Conclusions
The ubiquitous appearance of pathological tau in AD and other neurodegenerative diseases makes it a prime candidate for therapeutic targeting. Regardless, because Alzheimer’s is an insidious, multi-etiological disease, it is still difficult to ascertain precisely what dysfunction is necessary to initiate the disease course. Thus developing a truly disease-modifying, clinically useful pharmaceutical agent may remain elusive and may require multi-modal targeting. With the discovery of oligomeric tau, the numerous mechanisms that lead to its formation including iron, which itself has a well-developed therapeutic arsenal, and the observation of tau transmission between connected neurons, a new set of pharmaceutical targets have emerged that potentially can be exploited. Because maximal interference with an individual target may be associated with toxicity due to multiple roles, there may be a benefit in a more holistic targeting of multiple specific chokepoints to address iron and pathological tau propagation.
Highlights.
Iron can facilitate oxidative stress and the cell death pathway ferroptosis.
The Alzheimer disease related pathological proteins, amyloid-β and tau, bind iron in various forms.
In-vitro work suggests these combination entities become catalytic oxidants
Senescence, a pathological state associated with aging and AD, increases intracellular iron, and may contribute to sensitivity to ferroptosis
Therapies that address multiple pathways, particularly iron-mediated, are proposed
Acknowledgments
Funding Sources
PJD, TAK: Padfield Foundation
PJD, TAK, AT, JMT: National Institutes of Health, R01 NS094535
TAK: Welch Foundation grant No. BE-0048
MH: National Institute of Neurological Disorders and Stroke, R01 NS088645
GRJ, RK: No funding to report.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts
TAK and JMT are founders and own shares of Accelerox, LLC
Contributor Information
Paul J. Derry, Assistant Professor, College of Medicine Institute of Biosciences and Technology Center for Genomics and Precision Medicine Texas A&M Health Science Center Houston, TX.
Muralidhar L. Hegde, Associate Professor, Institute for Academic Medicine Houston Methodist Weill Cornell Medical College Houston, Texas.
George R. Jackson, Professor, Department of Neurology Baylor College of Medicine and Associate Director of Research, Parkinson’s Disease Research, Education and Clinical Center (PADRECC) Michael E. DeBakey VA Medical Center Houston, Texas.
Rakez Kayed, Associate Professor, Mitchell Center for Neurodegenerative Disorders Department of Neurology University of Texas Medical Branch Galveston, Texas.
James M. Tour, T.T. and W.F. Chao Professor of Chemistry, Professor of Computer Science, Professor of Materials Science and NanoEngineering Smalley Institute for Nanoscale Science and Technology Rice University Houston, Texas.
Thomas A. Kent, Robert A. Welch Chair Professor, Texas A&M College of Medicine Adjunct Professor of Chemistry Rice University Adjunct Professor of Neurology Houston Methodist Hospital Houston, Texas.
Ah-Lim Tsai, Professor of Biochemistry and Hematology, McGovern School of Medicine UT Health Science Center Houston, Texas.
References
- Acosta-Cabronero J, Betts MJ, Cardenas-Blanco A, Yang S, Nestor PJ, 2016. In Vivo MRI Mapping of Brain Iron Deposition across the Adult Lifespan. J Neurosci 36, 364–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlard PA, Cherny RA, Finkelstein DI, Gautier E, Robb E, Cortes M, Volitakis I, Liu X, Smith JP, Perez K, Laughton K, Li QX, Charman SA, Nicolazzo JA, Wilkins S, Deleva K, Lynch T, Kok G, Ritchie CW, Tanzi RE, Cappai R, Masters CL, Barnham KJ, Bush AI, 2008. Rapid restoration of cognition in Alzheimer’s transgenic mice with 8-hydroxy quinoline analogs is associated with decreased interstitial Abeta. Neuron 59, 43–55. [DOI] [PubMed] [Google Scholar]
- Aguirre P, Urrutia P, Tapia V, Villa M, Paris I, Segura-Aguilar J, Nunez MT, 2012. The dopamine metabolite aminochrome inhibits mitochondrial complex I and modifies the expression of iron transporters DMT1 and FPN1. Biometals 25, 795–803. [DOI] [PubMed] [Google Scholar]
- Ahmadi S, Ebralidze II, She Z, Kraatz H-B, 2017. Electrochemical studies of tau protein-iron interactions—Potential implications for Alzheimer’s Disease. Electrochimica Acta 236, 384–393. [Google Scholar]
- Alim I, Caulfield JT, Chen Y, Swarup V, Geschwind DH, Ivanova E, Seravalli J, Ai Y, Sansing LH, Ste Marie EJ, Hondal RJ, Mukherjee S, Cave JW, Sagdullaev BT, Karuppagounder SS, Ratan RR, 2019. Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 177, 1262–1279 e1225. [DOI] [PubMed] [Google Scholar]
- Alzheimer’s A, 2012. 2012 Alzheimer’s disease facts and figures. Alzheimers Dement 8, 131–168. [DOI] [PubMed] [Google Scholar]
- Ansari MA, Scheff SW, 2010. Oxidative stress in the progression of Alzheimer disease in the frontal cortex. J Neuropathol Exp Neurol 69, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arif M, Wei J, Zhang Q, Liu F, Basurto-Islas G, Grundke-Iqbal I, Iqbal K, 2014. Cytoplasmic retention of protein phosphatase 2A inhibitor 2 (I2PP2A) induces Alzheimer-like abnormal hyperphosphorylation of Tau. J Biol Chem 289, 27677–27691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong MJ, Litvan I, Lang AE, Bak TH, Bhatia KP, Borroni B, Boxer AL, Dickson DW, Grossman M, Hallett M, Josephs KA, Kertesz A, Lee SE, Miller BL, Reich SG, Riley DE, Tolosa E, Troster AI, Vidailhet M, Weiner WJ, 2013. Criteria for the diagnosis of corticobasal degeneration. Neurology 80, 496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala A, Munoz MF, Arguelles S, 2014. Lipid peroxidation: production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014, 360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayton S, Diouf I, Bush AI, Alzheimer’s disease Neuroimaging I, 2018. Evidence that iron accelerates Alzheimer’s pathology: a CSF biomarker study. J Neurol Neurosurg Psychiatry 89, 456–460. [DOI] [PubMed] [Google Scholar]
- Ayton S, Fazlollahi A, Bourgeat P, Raniga P, Ng A, Lim YY, Diouf I, Farquharson S, Fripp J, Ames D, Doecke J, Desmond P, Ordidge R, Masters CL, Rowe CC, Maruff P, Villemagne VL, Australian Imaging B, Lifestyle Research G, Salvado O, Bush AI, 2017. Cerebral quantitative susceptibility mapping predicts amyloid-beta-related cognitive decline. Brain 140, 2112–2119. [DOI] [PubMed] [Google Scholar]
- Bader B, Nübling G, Mehle A, Nobile S, Kretzschmar H, Giese A, 2011. Single particle analysis of tau oligomer formation induced by metal ions and organic solvents. Biochem Biophys Res Commun 411, 190–196. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM, 2011. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard C, Mobley W, Hardy J, Williams G, Corbett A, 2016. Dementia in Down’s syndrome. The Lancet Neurology 15, 622–636. [DOI] [PubMed] [Google Scholar]
- Bareggi SR, Cornelli U, 2012. Clioquinol: review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci Ther 18, 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barghorn S, Mandelkow E, 2002. Toward a Unified Scheme for the Aggregation of Tau into Alzheimer Paired Helical Filaments†. Biochemistry 41, 14885–14896. [DOI] [PubMed] [Google Scholar]
- Bautista E, Vergara P, Segovia J, 2016. Iron-induced oxidative stress activates AKT and ERK1/2 and decreases Dyrk1B and PRMT1 in neuroblastoma SH-SY5Y cells. J Trace Elem Med Biol 34, 62–69. [DOI] [PubMed] [Google Scholar]
- Belaidi AA, Bush AI, 2016. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J Neurochem 139 Suppl 1, 179–197. [DOI] [PubMed] [Google Scholar]
- Berlett BS, Stadtman ER, 1997. Protein oxidation in aging, disease, and oxidative stress. J Biol Chem 272, 20313–20316. [DOI] [PubMed] [Google Scholar]
- Betrie AH, Ayton S, Bush AI, Angus JA, Lei P, Wright CE, 2017. Evidences of a Cardiovascular Function for Microtubule-Associated Protein Tau. J Alzheimer’s Disease 56, 849–860. [DOI] [PubMed] [Google Scholar]
- Bhandary B, Marahatta A, Kim HR, Chae HJ, 2012. An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J Mol Sci 14, 434–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley ML, Kincaid RL, 1997. Regulated phosphorylation and dephosphorylation of tau protein: effects on microtubule interaction, intracellular trafficking and neurodegeneration. Biochem J 323 ( Pt 3), 577–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C, 2012. Antioxidant supplements for prevention of mortality in healthy participants and patients with various diseases. Cochrane Database Syst Rev, CD007176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black RS, Sperling RA, Safirstein B, Motter RN, Pallay A, Nichols A, Grundman M, 2010. A single ascending dose study of bapineuzumab in patients with Alzheimer disease. Alzheimer Dis Assoc Disord 24, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognin S, Messori L, Drago D, Gabbiani C, Cendron L, Zatta P, 2011. Aluminum, copper, iron and zinc differentially alter amyloid-Abeta(1-42) aggregation and toxicity. Int J Biochem Cell Biol 43, 877–885. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Buée L, Bussière T, Buée-Scherrer V, Delacourte A, Hof PR, 2000. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Brain Res Rev. 33, 95–130. [DOI] [PubMed] [Google Scholar]
- Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ, 2018. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562, 578–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso BR, Hare DJ, Bush AI, Roberts BR, 2017. Glutathione peroxidase 4: a new player in neurodegeneration? Mol Psychiatry 22, 328–335. [DOI] [PubMed] [Google Scholar]
- Cascio FL, Kayed R, 2018. Azure C Targets and Modulates Toxic Tau Oligomers. ACS Chem Neurosci B, 1317–1326. [DOI] [PubMed] [Google Scholar]
- Castillo-Carranza DL, Sengupta U, Guerrero-Munoz MJ, Lasagna-Reeves CA, Gerson JE, Singh G, Estes DM, Barrett AD, Dineley KT, Jackson GR, Kayed R, 2014. Passive immunization with Tau oligomer monoclonal antibody reverses tauopathy phenotypes without affecting hyperphosphorylated neurofibrillary tangles. J Neurosci 34, 4260–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- caSun L, Liu SY, Zhou XW, Wang XC, Liu R, Wang Q, Wang JZ, 2003. Inhibition of protein phosphatase 2A- and protein phosphatase 1-induced tau hyperphosphorylation and impairment of spatial memory retention in rats. Neuroscience 118, 1175–1182. [DOI] [PubMed] [Google Scholar]
- Cecchi C, Latorraca S, Sorbi S, lantomasi T, Favilli F, Vencenzini MT, Liguri G, 1999. Gluthatione level is altered in lymphoblasts from patients with familial Alzheimer’s disease. Neuroscience Letters 275, 152–154. [DOI] [PubMed] [Google Scholar]
- Celardo I, Pedersen JZ, Traversa E, Ghibelli L, 2011. Pharmacological potential of cerium oxide nanoparticles. Nanoscale 3, 1411–1420. [DOI] [PubMed] [Google Scholar]
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, Krager K, Ponnappan U, Hauer-Jensen M, Meng A, Zhou D, 2016. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Li B, Grundke-Iqbal I, Iqbal K, 2008. I1PP2A affects tau phosphorylation via association with the catalytic subunit of protein phosphatase 2A. J Biol Chem 283, 10513–10521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y-S, Huang X, Goldstein LE, Moir RD, Lim JT, Beyreuther K, Zheng H, Tanzi RE, Masters CL, Bush AI, 2001. Treatment with a Copper-Zinc Chelator Markedly and Rapidly Inhibits β-Amyloid Accumulation in Alzheimer’s Disease. Neuron 30, 665–676. [DOI] [PubMed] [Google Scholar]
- Chiang GC, Mao X, Kang G, Chang E, Pandya S, Vallabhajosula S, Isaacson R, Ravdin LD, Alzheimer’s Disease Neuroimaging I, Shungu DC, 2017. Relationships among Cortical Glutathione Levels, Brain Amyloidosis, and Memory in Healthy Older Adults Investigated In Vivo with (1)H-MRS and Pittsburgh Compound-B PET. AJNR Am J Neuroradiol 38, 1130–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Kirkland JL, Campisi J, van Deursen JM, 2014. Senescence and apoptosis: dueling or complementary cell fates? EMBO Rep 15, 1139–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Durik M, Baker DJ, van Deursen JM, 2015. Cellular senescence in aging and age-related disease: from mechanisms to therapy. Nat Med 21, 1424–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chohan MO, Khatoon S, Iqbal IG, Iqbal K, 2006. Involvement of I2PP2A in the abnormal hyperphosphorylation of tau and its reversal by Memantine. FEBS Lett 580, 3973–3979. [DOI] [PubMed] [Google Scholar]
- Cleary JM, Lima CM, Hurwitz HI, Montero AJ, Franklin C, Yang J, Graham A, Busman T, Mabry M, Holen K, Shapiro GI, Uronis H, 2014. A phase I clinical trial of navitoclax, a targeted high-affinity Bcl-2 family inhibitor, in combination with gemcitabine in patients with solid tumors. Invest New Drugs 32, 937–945. [DOI] [PubMed] [Google Scholar]
- Cohen P, Goedert M, 2004. GSK3 inhibitors: development and therapeutic potential. Nat Rev Drug Discov 3, 479–487. [DOI] [PubMed] [Google Scholar]
- Connor JR, Menzies SL, St. Martin SM, Mufson EJ, 1990. Cellular Distribution of Transferrin, Ferritin, and Iron in Normal and Aged Human Brains. J Neuroscience Research 27. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J, 2003. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17, 1195–1214. [DOI] [PubMed] [Google Scholar]
- Cowan CM, Quraishe S, Mudher A, 2012. What is the pathological significance of tau oligomers? Biochem Soc Trans 40, 693–697. [DOI] [PubMed] [Google Scholar]
- De Domenico I, Ward DM, Kaplan J, 2009. Specific iron chelators determine the route of ferritin degradation. Blood 114, 4546–4551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Ser T, Steinwachs KC, Gertz HJ, Andres MV, Gomez-Carrillo B, Medina M, Vericat JA, Redondo P, Fleet D, Leon T, 2013. Treatment of Alzheimer’s Disease with the GSK-3 Inhibitor Tideglusib: A Pilot Study. Journal of Alzheimer’s Disease 33, 205–215. [DOI] [PubMed] [Google Scholar]
- Derry PJ, Kent TA, 2017. Correlating quantitative susceptibility mapping with cognitive decline in Alzheimer’s disease. Brain 140, 2066–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, Miller RL, Schoch KM, Holmes BB, Kebodeaux CS, Wegener AJ, Chen G, Shen T, Tran H, Nichols B, Zanardi TA, Kordasiewicz HB, Swayze EE, Bennett CF, Diamond MI, Miller TM, 2017. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d’Adda di Fagagna F, 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444, 638–642. [DOI] [PubMed] [Google Scholar]
- Diagnostics, C.N. (2016) C2N Diagnostics Completes Phase 1 Clinical Study of C2N-8E12 (ABBV-8E12) Among Individuals with Progressive Supranuclear Palsy.
- Dickson DW, 2010. Neuropathology of non-Alzheimer degenerative disorders. Int J Clin Exp Pathol 3, 1–23. [PMC free article] [PubMed] [Google Scholar]
- Dineley KT, Jahrling JB, Denner L, 2014. Insulin resistance in Alzheimer’s disease. Neurobiol Dis 72 Pt A, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Ross JL, Goldman YE, Holzbaur EL, 2008. Differential regulation of dynein and kinesin motor proteins by tau. Science 319, 1086–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison BI, Stockwell BR, 2012. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dlouhy AC, Bailey DK, Steimle BL, Parker HV, Kosman DJ, 2019. Fluorescence resonance energy transfer links membrane ferroportin, hephaestin but not ferroportin, amyloid precursor protein complex with iron efflux. J Biol Chem 294, 4202–4214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Conrad M, 2017. Iron and ferroptosis: A still ill-defined liaison. IUBMB Life 69, 423–434. [DOI] [PubMed] [Google Scholar]
- Dringen R, Gutterer JM, Hirrlinger J, 2000. Glutathione metabolism in brain: Metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem 267, 4912–4916. [DOI] [PubMed] [Google Scholar]
- Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, Leong SL, Perez K, Johanssen T, Greenough MA, Cho HH, Galatis D, Moir RD, Masters CL, McLean C, Tanzi RE, Cappai R, Barnham KJ, Ciccotosto GD, Rogers JT, Bush AI, 2010. Iron-export ferroxidase activity of beta-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142, 857–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, Tariot PN, Vellas B, van Dyck CH, Boada M, Zhang Y, Li W, Furtek C, Mahoney E, Harper Mozley L, Mo Y, Sur C, Michelson D, 2019. Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N Engl J Med 380, 1408–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egaña JT, Zambrano C, Nuñez MT, Gonzales-Billault C, Maccioni RB, 2003. Iron-induced oxidative stress modify tau phosphorylation patterns in hippocampal cell cultures. BioMetals 16, 215–223. [DOI] [PubMed] [Google Scholar]
- Eto K, Asada T, Arima K, Makifuchi T, Kimura H, 2002. Brain hydrogen sulfide is severely decreased in Alzheimer’s disease. Biochemical and Biophysical Research Communications 293, 1485–1488. [DOI] [PubMed] [Google Scholar]
- Everett J, Cespedes E, Shelford LR, Exley C, Collingwood JF, Dobson J, van der Laan G, Jenkins CA, Arenholz E, Telling ND, 2014a. Evidence of redox-active iron formation following aggregation of ferrihydrite and the Alzheimer’s disease peptide beta-amyloid. Inorg Chem 53, 2803–2809. [DOI] [PubMed] [Google Scholar]
- Everett J, Cespedes E, Shelford LR, Exley C, Collingwood JF, Dobson J, van der Laan G, Jenkins CA, Arenholz E, Telling ND, 2014b. Ferrous iron formation following the co-aggregation of ferric iron and the Alzheimer’s disease peptide beta-amyloid (1-42). J R Soc Interface 11, 20140165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faller P, 2009. Copper and zinc binding to amyloid-beta: coordination, dynamics, aggregation, reactivity and metal-ion transfer. Chembiochem 10, 2837–2845. [DOI] [PubMed] [Google Scholar]
- Farina N, Llewellyn D, Isaac M, Tabet N, 2017. Vitamin E for Alzheimer’s dementia and mild cognitive impairment. Cochrane Database Syst Rev 4, CD002854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribé E, Dalfó E, Avila J, 2005. Current Advances on Different Kinases Involved in Tau Phosphorylation, and Implications in Alzheimer’s Disease and Tauopathies. Current Alzheimer Research 2, 3–18. [DOI] [PubMed] [Google Scholar]
- Filograna R, Beltramini M, Bubacco L, Bisaglia M, 2016. Anti-Oxidants in Parkinson’s Disease Therapy: A Critical Point of View. Current Neuropharmacology 14, 260–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelstein DI, Billings JL, Adlard PA, Ayton S, Sedjahtera A, Masters CL, Wilkins S, Shackleford DM, Charman SA, Bal W, Zawisza IA, Kurowska E, Gundlach AL, Ma S, Bush AI, Hare DJ, Doble PA, Crawford S, Gautier EC, Parsons J, Huggins P, Barnham KJ, Cherny RA, 2017. The novel compound PBT434 prevents iron mediated neurodegeneration and alpha-synuclein toxicity in multiple models of Parkinson’s disease. Acta Neuropathol Commun 5, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick AWP, Falcon B, He S, Murzin AG, Murshudov G, Garringer HJ, Crowther RA, Ghetti B, Goedert M, Scheres SHW, 2017. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flora SJ, Pachauri V, 2010. Chelation in metal intoxication. Int J Environ Res Public Health 7, 2745–2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu G, Sivaprakasam P, Dale OR, Manly SP, Cutler SJ, Doerksen RJ, 2014. Pharmacophore Modeling, Ensemble Docking, Virtual Screening, and Biological Evaluation on Glycogen Synthase Kinase-3beta. Mol Inform 33, 610–626. [DOI] [PubMed] [Google Scholar]
- Galimberti D, Scarpini E, 2011. Disease-modifying treatments for Alzheimer’s disease. Ther Adv Neurol Disord 4, 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Zhuang J, Nie L, Zhang J, Zhang Y, Gu N, Wang T, Feng J, Yang D, Perrett S, Yan X, 2007. Intrinsic peroxidase-like activity of ferromagnetic nanoparticles. Nat Nanotechnol 2, 577–583. [DOI] [PubMed] [Google Scholar]
- Garrick MD, Dolan KG, Horbinski C, Ghio AJ, Higgins D, Porubcin M, Moore EG, Hainsworth LN, Umbreit JN, Conrad ME, Feng L, Lis A, Roth JA, Singleton S, Garrick LM, 2003. DMT1: A mammalian transporter for multiple metals. Biometals 16, 41–54. [DOI] [PubMed] [Google Scholar]
- Gaschler MM, Hu F, Feng H, Linkermann A, Min W, Stockwell BR, 2018. Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem Biol 13, 1013–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier S, Feldman HH, Schneider LS, Wilcock GK, Frisoni GB, Hardlund JH, Moebius HJ, Bentham P, Kook KA, Wischik DJ, Schelter BO, Davis CS, Staff RT, Bracoud L, Shamsi K, Storey JMD, Harrington CR, Wischik CM, 2016. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. The Lancet 388, 2873–2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George DR, Whitehouse PJ, Ballenger J, 2011. The evolving classification of dementia: placing the DSM-V in a meaningful historical and cultural context and pondering the future of “Alzheimer’s”. Cult Med Psychiatry 35, 417–435. [DOI] [PubMed] [Google Scholar]
- Gerson JE, Sengupta U, Lasagna-Reeves CA, Guerrero-Munoz MJ, Troncoso J, Kayed R, 2014. Characterization of tau oligomeric seeds in progressive supranuclear palsy. Acta Neuropathol Commun 2, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh D, LeVault KR, Brewer GJ, 2014. Dual-energy precursor and nuclear erythroid-related factor 2 activator treatment additively improve redox glutathione levels and neuron survival in aging and Alzheimer mouse neurons upstream of reactive oxygen species. Neurobiol Aging 35, 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Jakes R, Vanmechelen E, 1995. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett 189, 167–170. [DOI] [PubMed] [Google Scholar]
- Goldsbury C, Mocanu MM, Thies E, Kaether C, Haass C, Keller P, Biernat J, Mandelkow E, Mandelkow EM, 2006. Inhibition of APP trafficking by tau protein does not increase the generation of amyloid-beta peptides. Traffic 7, 873–888. [DOI] [PubMed] [Google Scholar]
- Gong C-X, Lidsky T, Wegiel J, Zuck L, Grundke-Iqbal I, Iqbal K, 2000. Phosphorylation of Microtubule-associated Protein Tau Is Regulated by Protein Phosphatase 2A in Mammalian Brain. Journal of Biological Chemistry 275, 5535–5544. [DOI] [PubMed] [Google Scholar]
- Good PF, Perl DP, Bierer LM, Schmeidler J, 1992. Selective Accumulation of Aluminum and Iron in the Neurofibrillary Tangles of Alzheimer’s Disease: A Laser Microprobe (LAMMA) Study. Ann Neurol 31, 286–292. [DOI] [PubMed] [Google Scholar]
- Götz J, Chen F, van Dorpe J, Nitsch RM, 2001. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science 293, 1491–1495. [DOI] [PubMed] [Google Scholar]
- Haile DJ, 1999. Regulation of Genes of Iron Metabolism by the Iron-Response Proteins. The American Journal of the Medical Sciences 318, 230–240. [DOI] [PubMed] [Google Scholar]
- Hallgren B, Sourander P, 1958. The effect of non-haemin iron in the human brain. J. Neurochemistry 3, 41–51. [DOI] [PubMed] [Google Scholar]
- Hambright WS, Fonseca RS, Chen L, Na R, Ran Q, 2017. Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol 12, 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampel B, Wagner M, Teis D, Zwerschke W, Huber LA, Jansen-Durr P, 2005. Apoptosis resistance of senescent human fibroblasts is correlated with the absence of nuclear IGFBP-3. Aging Cell 4, 325–330. [DOI] [PubMed] [Google Scholar]
- Hare DJ, Doecke JD, Faux NG, Rembach A, Volitakis I, Fowler CJ, Grimm R, Doble PA, Cherny RA, Masters CL, Bush AI, Roberts BR, 2015. Decreased plasma iron in Alzheimer’s disease is due to transferrin desaturation. ACS Chem Neurosci 6, 398–402. [DOI] [PubMed] [Google Scholar]
- Hare DJ, Double KL, 2016. Iron and dopamine: a toxic couple. Brain 139, 1026–1035. [DOI] [PubMed] [Google Scholar]
- Harris JA, Koyama A, Maeda S, Ho K, Devidze N, Dubal DB, Yu GQ, Masliah E, Mucke L, 2012. Human P301L-mutant tau expression in mouse entorhinal-hippocampal network causes tau aggregation and presynaptic pathology but no cognitive deficits. PLoS One 7, e45881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Health, U.S.N.I.o. (2016a) 18-months Safety Follow-up Study of AADvac1, an Active Tau Vaccine for Alzheimer’s Disease (FUNDAMANT).
- Health, U.S.N.I.o. (2016b) 24 Months Safety and Efficacy Study of AADvac1 in Patients with Mild Alzheimer’s Disease (ADAMANT).
- Hebert L, Weuve J, Scherr P, Evans D, 2013. Alzheimer disease in the United States (2010-2050) estimated using the 2010 census. Neurology 80, 1778–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde ML, Bharathi P, Suram A, Venugopal C, Jagannathan R, Poddar P, Srinivas P, Sambamurti K, Rao KJ, Scancar J, Messori L, Zecca L, Zatta P, 2009. Challenges associated with metal chelation therapy in Alzheimer’s disease. J Alzheimers Dis 17, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Ramanathan M, Jacobs AH, Dumitrescu-Ozimek L, Bilkei-Gorzo A, Debeir T, Sastre M, Galldiks N, Zimmer A, Hoehn M, Heiss WD, Klockgether T, Staufenbiel M, 2006. Locus ceruleus degeneration promotes Alzheimer pathogenesis in amyloid precursor protein 23 transgenic mice. J Neurosci 26, 1343–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henley D, Raghavan N, Sperling R, Aisen PS, Raman R, Romano G, 2019. Preliminary Results of a Trial of Atabecestat in Preclinical Alzheimer’s Disease. N Engl J Med 380, 1483–1485. [DOI] [PubMed] [Google Scholar]
- Honarmand Ebrahimi K, Dienemann C, Hoefgen S, Than ME, Hagedoorn PL, Hagen WR, 2013. The amyloid precursor protein (APP) does not have a ferroxidase site in its E2 domain. PLoS One 8, e72177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- House E, Esiri M, Forster G, Ince PG, Exley C, 2012. Aluminium, iron and copper in human brain tissues donated to the Medical Research Council’s Cognitive Function and Ageing Study. Metallomics 4, 56–65. [DOI] [PubMed] [Google Scholar]
- Iqbal K, Gong CX, Liu F, 2013. Hyperphosphorylation-induced tau oligomers. Front Neurol 4, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Grundke-Iqbal I, 2010. Alzheimer’s disease, a multifactorial disorder seeking multitherapies. Alzheimers Dement 6, 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob CP, Koutsilieri E, Bartl J, Neuen-Jacob E, Arzberger T, Zander N, Ravid R, Roggendorf W, Riederer P, Grunblatt E, 2007. Alterations in Expression of Glutamatergic Transporters and Receptors in Sporadic Alzheimer’s Disease. Journal of Alzheimer’s Disease 11, 97–116. [DOI] [PubMed] [Google Scholar]
- Jicha GA, 2009. Is passive immunization for Alzheimer’s disease ‘alive and well’ or ‘dead and buried’? Expert Opin Biol Ther 9, 481–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadavath H, Hofele RV, Biernat J, Kumar S, Tepper K, Urlaub H, Mandelkow E, Zweckstetter M, 2015. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc Natl Acad Sci U S A 112, 7501–7506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahklon O, Cabantchik ZI, 2002. The labile iron pool: characterization, measurement, and participation in cellular processes. Free Radic Biol Med 33, 1037–1046. [DOI] [PubMed] [Google Scholar]
- Kanaan NM, Morfini GA, LaPointe NE, Pigino GF, Patterson KR, Song Y, Andreadis A, Fu Y, Brady ST, Binder LI, 2011. Pathogenic forms of tau inhibit kinesin-dependent axonal transport through a mechanism involving activation of axonal phosphotransferases. J Neurosci 31, 9858–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, Viswanath V, Jacobs R, Yang L, Beal MF, DiMonte D, Volitaskis I, Ellerby L, Cherny RA, Bush AI, Andersen JK, 2003. Genetic or Pharmacological Iron Chelation Prevents MPTP-Induced Neurotoxicity In Vivo. Neuron 37, 899–909. [DOI] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG, 2003. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 300, 486–489. [DOI] [PubMed] [Google Scholar]
- Kelly SC, He B, Perez SE, Ginsberg SD, Mufson EJ, Counts SE, 2017. Locus coeruleus cellular and molecular pathology during the progression of Alzheimer’s disease. Acta Neuropathol Commun 5, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepp KP, 2012. Bioinorganic chemistry of Alzheimer’s disease. Chem Rev 112, 5193–5239. [DOI] [PubMed] [Google Scholar]
- Khatoon S, Grundke-Iqbal I, Iqbal K, 1994. Levels of normal and abnormally phosphorylated tau in different cellular and regional compartments of Alzheimer disease and control brains. FEBS Lett 351, 80–84. [DOI] [PubMed] [Google Scholar]
- Kim D, Lim S, Haque MM, Ryoo N, Hong HS, Rhim H, Lee DE, Chang YT, Lee JS, Cheong E, Kim DJ, Kim YK, 2015. Identification of disulfide cross-linked tau dimer responsible for tau propagation. Sci Rep 5, 15231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K, Mitra S, Wu G, Berka V, Song J, Yu Y, Poget S, Wang DN, Tsai AL, Zhou M, 2016. Six-Transmembrane Epithelial Antigen of Prostate 1 (STEAP1) Has a Single b Heme and Is Capable of Reducing metal Ion Complexes and Oxygen. Biochemistry 55, 6673–6684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein EA, Thompson IM Jr., Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL, Gaziano JM, Karp DD, Lieber MM, Walther PJ, Klotz L, Parsons JK, Chin JL, Darke AK, Lippman SM, Goodman GE, Meyskens FL Jr., Baker LH, 2011. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 306, 1549–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontoghiorghes GJ, Pattichi K, Hadjigavriel M, Kolnagou A, 2000. Transfusional iron overload and chelation therapy with deferoxamine and deferiprone (L1). Transfusional Science 23, 211–223. [DOI] [PubMed] [Google Scholar]
- Kontsekova E, Zilka N, Kovacech B, Novak P, Novak M, 2014. First-in-man tau vaccine targeting structural determinants essential for pathological tau-tau interaction reduces tau oligomerisation and neurofibrillary degeneration in an Alzheimer’s disease model. Alzheimers Res Ther 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köpke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I, 1993a. Microtubule-associated Protein Tau: Abnormal Phosphorylation of a Non-paired Helical Filament Pool in Alzheimer Disease. J Biol Chem 268, 24374–24384. [PubMed] [Google Scholar]
- Köpke E, Tung YC, Shaikh S, Alonso AC, Iqbal K, Grundke-Iqbal I, 1993b. Microtubule-associated Protein Tau: Abnormal Phosphorylatoin of a Non-paired Helical Filament Pool in Alzheimer Disease. J Biol Chem 268, 24374–24384. [PubMed] [Google Scholar]
- Kril JJ, Patel S, Harding AJ, Halliday GM, 2002. Neuron loss from the hippocampus of Alzheimer’s disease exceeds extracellular neurofibrillary tangle formation. Acta Neuropathol 103, 370–376. [DOI] [PubMed] [Google Scholar]
- Kristinsson J, Snaedal J, Torsdottir G, Johannesson T, 2012. Ceruloplasmin and iron in Alzheimer’s disease and Parkinson’s disease: a synopsis of recent studies. Neuropsychiatr Dis Treat 8, 515–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritsilis M, S VR, Koutsoudaki PN, Evangelou K, Gorgoulis VG, Papadopoulos D, 2018. Ageing, Cellular Senescence and Neurodegenerative Disease. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryscio RJ, Abner EL, Caban-Holt A, Lovell M, Goodman P, Darke AK, Yee M, Crowley J, Schmitt FA, 2017. Association of Antioxidant Supplement Use and Dementia in the Prevention of Alzheimer’s Disease by Vitamin E and Selenium Trial (PREADViSE). JAMA Neurol 74, 567–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JD, Elias RJ, 2010. The antioxidant and pro-oxidant activities of green tea polyphenols: a role in cancer prevention. Arch Biochem Biophys 501, 65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, Masters CL, Targum S, Bush AI, Murdoch R, Wilson J, Ritchie CW, 2008. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: a phase IIa, double-blind, randomised, placebo-controlled trial. The Lancet Neurology 7, 779–786. [DOI] [PubMed] [Google Scholar]
- Larson EB, Shadlen MF, Wang L, McCormick WC, Bowen JD, Teri L, Kukull WA, 2004. Survival after initial diagnosis of Alzheimer disease. Ann Intern Med 140, 501–509. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Guerrero-Muoz MJ, Jackson GR, Kayed R, 2010. Preparation and characterization of neurotoxic tau oligomers. Biochemistry 49, 10039–10041. [DOI] [PubMed] [Google Scholar]
- Lasagna-Reeves CA, Castillo-Carranza DL, Sengupta U, Clos AL, Jackson GR, Kayed R, 2011. Tau oligomers impair memory and induce synaptic and mitochondrial dysfunction in wild-type mice. Molecular Neurodegeneration 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E, 2013. The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latunde-Dada GO, 2017. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim Biophys Acta 1861, 1893–1900. [DOI] [PubMed] [Google Scholar]
- Lei P, Ayton S, Appukuttan AT, Volitakis I, Adlard PA, Finkelstein DI, Bush AI, 2015. Clioquinol rescues Parkinsonism and dementia phenotypes of the tau knockout mouse. Neurobiol Dis 81, 168–175. [DOI] [PubMed] [Google Scholar]
- Lei P, Ayton S, Finkelstein DI, Spoerri L, Ciccotosto GD, Wright DK, Wong BX, Adlard PA, Cherny RA, Lam LQ, Roberts BR, Volitakis I, Egan GF, McLean CA, Cappai R, Duce JA, Bush AI, 2012. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 18, 291–295. [DOI] [PubMed] [Google Scholar]
- Lei P, Ayton S, Moon S, Zhang Q, Volitakis I, Finkelstein DI, Bush AI, 2014. Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol Neurodegener 9, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloret A, Badia MC, Mora NJ, Pallardo FV, Alonso MD, Vina J, 2009. Vitamin E paradox in Alzheimer’s disease: it does not prevent loss of cognition and may even be detrimental. J Alzheimers Dis 17, 143–149. [DOI] [PubMed] [Google Scholar]
- Lodish MB, Stratakis CA, 2010. Endocrine side effects of broad-acting kinase inhibitors. Endocr Relat Cancer 17, R233–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell MA, Ehmann WD, Butler SM, Markesbery WR, 1995. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology 45, 1594–1601. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Xie C, Gabbita P, Markesbery WR, 2000. Decreased thioredoxin and increased thioredoxin reductase levels in Alzheimer’s disease brain. Free Radic Biol Med 28, 418–427. [DOI] [PubMed] [Google Scholar]
- Lovestone S, Boada M, Dubois B, Hüll M, Rinne JO, Huppertz H-J, Calero M, Andres MV, Gomez-Carrillo B, Leon T, del ser T, Investigators, A., 2015. A Phase II Trial of Tideglusib in Alzheimer’s Disease. Journal of Alzheimer’s Disease 45, 75–88. [DOI] [PubMed] [Google Scholar]
- Luna-Muñoz J, R C, M C, Flores-Rodriguez P, Avila J, R S, la Cruz FD, Mena R, A M, Floran-Garduno B 2013. Phosphorylation of Tau Protein Associated as a Protective Mechanism in the Presence of Toxic, C-Terminally Truncated Tau in Alzheimer’s Disease. In: Understanding Alzheimer’s Disease. [Google Scholar]
- Ly PTT, Wu Y, Zou H, Wang R, Zhou W, Kinoshita A, Zhang M, Yang Y, Cai F, Woodgett J, Song W, 2013. Inhibition of GSK3beta-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest 123, 224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama M, Yoshiike Y, Kim H, Miyasaka T, Murayama S, Ikai A, Takashima A, 2007. Granular tau oligomers as intermediates of tau filaments. Biochemistry 46, 3856–3861. [DOI] [PubMed] [Google Scholar]
- Mandal PK, Saharan S, Tripathi M, Murari G, 2015. Brain glutathione levels--a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol Psychiatry 78, 702–710. [DOI] [PubMed] [Google Scholar]
- Mandelkow EM, Thies E, Trinczek B, Biernat J, Mandelkow E, 2004. MARK/PAR1 kinase is a regulator of microtubule-dependent transport in axons. J Cell Biol 167, 99–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander BA, Winer JR, Walker MP, 2017. Correlating quantitative susceptibility mapping with cognitive decline in Alzheimer’s disease. Brain 140, 2066–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markesbery WR, 1997. Oxidative stress hypothesis in Alzheimer’s disease. Free Radic Biol Med 23, 134–147. [DOI] [PubMed] [Google Scholar]
- Martin M, Kettmann R, Dequiedt F, 2010. Recent insights into Protein Phosphatase 2A structure and regulation: the reasons why PP2A is no longer considered as a lazy passive housekeeping enzyme. Biotechnol Agron Soc Environ 14, 243–252. [Google Scholar]
- Martini SR, Kent TA, 2007. Hyperglycemia in acute ischemic stroke: a vascular perspective. J Cereb Blood Flow Metab 27, 435–451. [DOI] [PubMed] [Google Scholar]
- Masaldan S, Clatworthy SAS, Gamell C, Meggyesy PM, Rigopoulos AT, Haupt S, Haupt Y, Denoyer D, Adlard PA, Bush AI, Cater MA, 2018. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol 14, 100–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matenia D, Mandelkow EM, 2009. The tau of MARK: a polarized view of the cytoskeleton. Trends Biochem Sci 34, 332–342. [DOI] [PubMed] [Google Scholar]
- Mazanetz MP, Fischer PM, 2007. Untangling tau hyperphosphorylation in drug design for neurodegenerative diseases. Nat Rev Drug Discov 6, 464–479. [DOI] [PubMed] [Google Scholar]
- McCaddon A, Hudson P, Hill D, Barber J, Lloyd A, Davies G, Regland B.j., 2003. Alzheimer’s disease and total plasma aminothiols. Biological Psychiatry 53, 254–260. [DOI] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA, 2009. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68, 709–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM, 1984. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 2, 939–944. [DOI] [PubMed] [Google Scholar]
- McLachlan DRC, Dalton AJ, Kruck TPA, Bell MY, Smith WL, Kalow W, Andrews DF, 1991. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet 337, 1304–1308. [DOI] [PubMed] [Google Scholar]
- Medicine U.S.N.L.o. (2019) Navitoclax and Vistusertib in Treating Patients With Relapsed Small Cell Lung Cancer and Other Solid Tumors.
- Medina M, Hernandez F, Avila J, 2016. New Features about Tau Function and Dysfunction. Biomolecules 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min SW, Chen X, Tracy TE, Li Y, Zhou Y, Wang C, Shirakawa K, Minami SS, Defensor E, Mok SA, Sohn PD, Schilling B, Cong X, Ellerby L, Gibson BW, Johnson J, Krogan N, Shamloo M, Gestwicki J, Masliah E, Verdin E, Gan L, 2015. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med 21, 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra J, Vasquez V, Hegde PM, Boldogh I, Mitra S, Kent TA, Rao KS, Hegde ML, 2014. Revisiting metal Toxicity in Neurodegenerative Diseases and Stroke: Therapeutic Potential. Neurol Res Ther 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitragotri S, Burke PA, Langer R, 2014. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov 13, 655–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamed T, Hoang T, Jelokhani-Niaraki M, Rao PP, 2013. Tau-derived-hexapeptide 306VQIVYK311 aggregation inhibitors: nitrocatechol moiety as a pharmacophore in drug design. ACS Chem Neurosci 4, 1559–1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moos T, Rosengren Nielsen T, Skjorringe T, Morgan EH, 2007. Iron trafficking inside the brain. J Neurochem 103, 1730–1740. [DOI] [PubMed] [Google Scholar]
- Morsch R, Simon W, Coleman PD, 1999. Neurons May Live for Decades with Neurofibrillary Tangles. J Neuropathol Exp Neurol 58, 188–197. [DOI] [PubMed] [Google Scholar]
- Mukrasch MD, Bibow S, Korukottu J, Jeganathan S, Biernat J, Griesinger C, Mandelkow E, Zweckstetter M, 2009. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol 7, e34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mun GI, Boo YC, 2010. Identification of CD44 as a senescence-induced cell adhesion gene responsible for the enhanced monocyte recruitment to senescent endothelial cells. Am J Physiol Heart Circ Physiol 298, H2102–2111. [DOI] [PubMed] [Google Scholar]
- Munoz P, Huenchuguala S, Paris I, Segura-Aguilar J, 2012. Dopamine oxidation and autophagy. Parkinsons Dis 2012, 920953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murayama H, Shin R-W, Higuchi J, Shibuya S, Muramoto T, Kitamoto T, 1999. Interaction of Aluminum with PHFτ in Alzheimer’s Disease Neurofibrillary Degeneration Evidenced by Desferrioxamine-Assisted Chelating Autoclave Method. The American Journal of Pathology 155, 877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, Shen Q, Orr ME, 2018. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17, e12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen P, Fischer R, Buggisch P, Janka-Schaub G, 2003. Effective treatment of hereditary haemochromatosis with desferrioxamine in selected cases. British Journal of Haematology 123, 952–953. [DOI] [PubMed] [Google Scholar]
- Novak P, Schmidt R, Kontsekova E, Kovacech B, Smolek T, Katina S, Fialova L, Prcina M, Parrak V, Dal-Bianco P, Brunner M, Staffen W, Rainer M, Ondrus M, Ropele S, Smisek M, Sivak R, Zilka N, Winblad B, Novak M, 2018. FUNDAMANT: an interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res Ther 10, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak P, Schmidt R, Kontsekova E, Zilka N, Kovacech B, Skrabana R, Vince-Kazmerova Z, Katina S, Fialova L, Prcina M, Parrak V, Dal-Bianco P, Brunner M, Staffen W, Rainer M, Ondrus M, Ropele S, Smisek M, Sivak R, Winblad B, Novak M, 2017. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase 1 trial. The Lancet Neurology 16, 123–134. [DOI] [PubMed] [Google Scholar]
- Nubling G, Bader B, Levin J, Hildebrandt J, Kretzschmar H, Giese A, 2012. Synergistic influence of phosphorylation and metal ions on tau oligomer formation and coaggregation with alpha-synuclein at the single molecule level. Mol Neurodegener 7, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan J, Holmes H, Powell N, Wells JA, Ismail O, Harrison IF, Siow B, Johnson R, Ahmed Z, Fisher A, Meftah S, O’Neill MJ, Murray TK, Collins EC, Shmueli K, Lythgoe MF, 2017. Tissue magnetic susceptibility mapping as a marker of tau pathology in Alzheimer’s disease. Neuroimage 159, 334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohgami RS, Campagna DR, Greer EL, Antiochos B, McDonald A, Chen J, Sharp JJ, Fujiwara Y, Barker JE, Fleming MD, 2005. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat Genet 37, 1264–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otvos LJ, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VM-Y, 1994. Monoclonal Antibody PHF4 Recognizes Tau Protein Phosphorylated at Serine Residues 396 and 404. J Neurosci Res 39, 669–673. [DOI] [PubMed] [Google Scholar]
- Petry FR, Pelletier J, Bretteville A, Morin F, Calon F, Hebert SS, Whittington RA, Planel E, 2014. Specificity of anti-tau antibodies when analyzing mice models of Alzheimer’s disease: problems and solutions. PLoS One 9, e94251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plascencia-Villa G, Ponce A, Collingwood JF, Arellano-Jimenez MJ, Zhu X, Rogers JT, Betancourt I, Jose-Yacaman M, Perry G, 2016. High-resolution analytical imaging and electron holography of magnetite particles in amyloid cores of Alzheimer’s disease. Sci Rep 6, 24873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter JB, Rafique R, Srichairatanakool S, Davis BA, Shah FT, Hair T, Evans P, 2005. Recent insights into interactions of deferoxamine with cellular and plasma iron pools: Implications for clinical use. Ann N Y Acad Sci 1054, 155–168. [DOI] [PubMed] [Google Scholar]
- Pradeepkiran JA, Reddy PH, 2019. Structure Based Design and Molecular Docking Studies for Phosphorylated Tau Inhibitors in Alzheimer’s Disease. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanthi JR, Schrag M, Dasari B, Marwarha G, Dickson A, Kirsch WM, Ghribi O, 2012. Deferiprone reduces amyloid-beta and tau phosphorylation levels but not reactive oxygen species generation in hippocampus of rabbits fed a cholesterol-enriched diet. J Alzheimers Dis 30, 167–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha AA, Vaishnav RA, Friedland RP, Bomford A, Raha-Chowdhury R, 2013. The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathologica Communications 1, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos P, Santos A, Pinto NR, Mendes R, Magalhaes T, Almeida A, 2014. Iron levels in the human brain: a post-mortem study of anatomical region differences and age-related changes. J Trace Elem Med Biol 28, 13–17. [DOI] [PubMed] [Google Scholar]
- Rane JS, Kumari A, Panda D, 2019. An acetylation mimicking mutation, K274Q, in tau imparts neurotoxicity by enhancing tau aggregation and inhibiting tubulin polymerization. Biochem J 476, 1401–1417. [DOI] [PubMed] [Google Scholar]
- Reisberg B, Doody R, Stöffler A, Schmitt F, Ferris S, Möbius HJ, Group MS, 2003. Memantine in Moderate-to-Severe Alzheimer’s Disease. N Engl J Med 348, 1333–1341. [DOI] [PubMed] [Google Scholar]
- Rickie A, Bogdanovic N, Volkman I, Winblad I, Ravid R, Cowburn RF, 2004. Akt activity in Alzheimer’s disease and other neurodegenerative disorders. Neurochemistry 15, 955–959. [DOI] [PubMed] [Google Scholar]
- Rietjens IMCM, Boersma MG, de Haan L, Spenkelink B, Awad HM, Cnubben NHP, van Zenden JJ, van der Woude H, Alink GM, Koeman JH, 2002. The pro-oxidant chemistry of the natural antioxidants vitamin C, vitamin E, carotenoids and flavonoids. Environmental Toxicology and Pharmacology 11, 321–333. [DOI] [PubMed] [Google Scholar]
- Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, Carrington D, Mavros C, Volitakis I, Xilinas M, Ames D, Davis S, Beyreuther K, Tanzi RE, Masters CL, 2003. metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol 60, 1685–1691. [DOI] [PubMed] [Google Scholar]
- Robinson SR, Dang TN, Dringen R, Bishop GM, 2009. Hemin toxicity: a preventable source of brain damage following hemorrhagic stroke. Redox Rep 14, 228–235. [DOI] [PubMed] [Google Scholar]
- Salgado P, Melin V, Duran Y, Mansilla H, Contreras D, 2017. The Reactivity and Reaction Pathway of Fenton Reactions Driven by Substituted 1,2-Dihydroxybenzenes. Environ Sci Technol 51, 3687–3693. [DOI] [PubMed] [Google Scholar]
- Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, Sabbagh M, Honig LS, Doody R, van Dyck CH, Mulnard R, Barakos J, Gregg KM, Liu E, Lieberburg I, Schenk D, Black R, Grundman M, Bapineuzumab 201 Clinical Trial, I., 2009. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology 73, 2061–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel EL, Duong MT, Bitner BR, Marcano DC, Tour JM, Kent TA, 2014. Hydrophilic carbon clusters as therapeutic, high-capacity antioxidants. Trends Biotechnol 32, 501–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Harris PLR, Liu Y, Schubert KA, Smith MA, 2000. In Situ Oxidative Catalysis by Neurofibrillary Tangles and Senile Plaques in Alzheimer’s Disease: A Central Role for Bound Transition metals. Journal of Neurochemistry 74, 270–279. [DOI] [PubMed] [Google Scholar]
- Schrag M, Mueller C, Oyoyo U, Smith MA, Kirsch WM, 2011. Iron, zinc and copper in the Alzheimer’s disease brain: a quantitative meta-analysis. Some insight on the influence of citation bias on scientific opinion. Prog Neurobiol 94, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuff N, Woerner N, Boreta L, Kornfield T, Shaw LM, Trojanowski JQ, Thompson PM, Jack CR Jr., Weiner MW, Alzheimer’s Disease Neuroimaging I, 2009. MRI of hippocampal volume loss in early Alzheimer’s disease in relation to ApoE genotype and biomarkers. Brain 132, 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwalbe M, Biernat J, Bibow S, Ozenne V, Jensen MR, Kadavath H, Blackledge M, Mandelkow E, Zweckstetter M, 2013. Phosphorylation of human Tau protein by microtubule affinity-regulating kinase 2. Biochemistry 52, 9068–9079. [DOI] [PubMed] [Google Scholar]
- Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, Bornkamm GW, Schweizer U, Conrad M, 2008. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8, 237–248. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Hardy J, 2016. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol Med 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sevigny J, Chiao P, Bussiere T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, O’Gorman J, Qian F, Arastu M, Li M, Chollate S, Brennan MS, Quintero-Monzon O, Scannevin RH, Arnold HM, Engber T, Rhodes K, Ferrero J, Hang Y, Mikulskis A, Grimm J, Hock C, Nitsch RM, Sandrock A, 2016. The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537, 50–56. [DOI] [PubMed] [Google Scholar]
- Sharma A, Pachauri V, Flora SJS, 2018. Advances in Multi-Functional Ligands and the Need for metal-Related Pharmacology for the Management of Alzheimer Disease. Front Pharmacol 9, 1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin R-W, Kruck TPA, Murayama H, Kitamoto T, 2003. A novel trivalent cation chelator Feralex dissociates binding of aluminum and iron associated with hyperphosphorylated τ of Alzheimer’s disease. Brain Research 961, 139–146. [DOI] [PubMed] [Google Scholar]
- Shin R-W, Lee VM-Y, Trojanowski JQ, 1994. Aluminum Modifies the Properties of Alzheimer’s Disease PHFT Proteins in vivo and in vitro. Journal of Neuroscience 14, 7221–7233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Harris PL, Sayre LM, Perry G, 1997. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A 94, 9866–9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeda Y, Yoshikawa M, Almeida OF, Sumioka A, Maeda S, Osada H, Kondoh Y, Saito A, Miyasaka T, Kimura T, Suzuki M, Koyama H, Yoshiike Y, Sugimoto H, Ihara Y, Takashima A, 2015. Toxic tau oligomer formation blocked by capping of cysteine residues with 1,2-dihydroxybenzene groups. Nat Commun 6, 10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling R, Salloway S, Brooks DJ, Tampieri D, Barakos J, Fox NC, Raskind M, Sabbagh M, Honig LS, Porsteinsson AP, Lieberburg I, Arrighi HM, Morris KA, Lu Y, Liu E, Gregg KM, Brashear HR, Kinney GG, Black R, Grundman M, 2012. Amyloid-related imaging abnormalities in patients with Alzheimer’s disease treated with bapineuzumab: a retrospective analysis. The Lancet Neurology 11, 241–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, Jack CR Jr., Black SE, Frosch MP, Greenberg SM, Hyman BT, Scheltens P, Carrillo MC, Thies W, Bednar MM, Black RS, Brashear HR, Grundman M, Siemers ER, Feldman HH, Schindler RJ, 2011. Amyloid-related imaging abnormalities in amyloidmodifying therapeutic trials: recommendations from the Alzheimer’s Association Research Roundtable Workgroup. Alzheimers Dement 7, 367–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM, 2002. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol 156, 1051–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stankowski JN, Dawson VL, Dawson TM, 2012. Ironing out tau’s role in parkinsonism. Nat Med 18, 197–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoothoff WH, Johnson GV, 2005. Tau phosphorylation: physiological and pathological consequences. Biochim Biophys Acta 1739, 280–297. [DOI] [PubMed] [Google Scholar]
- Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE, 2012. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122, 1316–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tariot PN, Farlow MR, Grossberg GT, Graham SM, McDonald S, Gergel I, Group MS, 2004. Memantine Treatment in Patients With Moderate to Severe Alzheimer Disease Already Receiving Donepezil. JAMA 291, 317–324. [DOI] [PubMed] [Google Scholar]
- Telling ND, Everett J, Collingwood JF, Dobson J, van der Laan G, Gallagher JJ, Wang J, Hitchcock AP, 2017. Iron Biochemistry is Correlated with Amyloid Plaque Morphology in an Established Mouse Model of Alzheimer’s Disease. Cell Chem Biol 24, 1205–1215 e1203. [DOI] [PubMed] [Google Scholar]
- Theofilas P, Ehrenberg AJ, Dunlop S, Di Lorenzo Alho AT, Nguy A, Leite REP, Rodriguez RD, Mejia MB, Suemoto CK, Ferretti-Rebustini REL, Polichiso L, Nascimento CF, Seeley WW, Nitrini R, Pasqualucci CA, Jacob Filho W, Rueb U, Neuhaus J, Heinsen H, Grinberg LT, 2017. Locus coeruleus volume and cell population changes during Alzheimer’s disease progression: A stereological study in human postmortem brains with potential implication for early-stage biomarker discovery. Alzheimers Dement 13, 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas SN, Funk KE, Wan Y, Liao Z, Davies P, Kuret J, Yang AJ, 2012. Dual modification of Alzheimer’s disease PHF-tau protein by lysine methylation and ubiquitylation: a mass spectrometry approach. Acta Neuropathol 123, 105–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolosa E, Litvan I, Hoglinger GU, Burn D, Lees A, Andres MV, Gomez-Carrillo B, Leon T, Del Ser T, Investigators T, 2014. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord 29, 470–478. [DOI] [PubMed] [Google Scholar]
- Uranga RM, Giusto NM, Salvador GA, 2009. Iron-induced oxidative injury differentially regulates PI3K/Akt/GSK3beta pathway in synaptic endings from adult and aged rats. Toxicol Sci 111, 331–344. [DOI] [PubMed] [Google Scholar]
- Urrutia P, Aguirre P, Esparza A, Tapia V, Mena NP, Arredondo M, Gonzalez-Billault C, Nunez MT, 2013. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J Neurochem 126, 541–549. [DOI] [PubMed] [Google Scholar]
- van Bergen JM, Li X, Hua J, Schreiner SJ, Steininger SC, Quevenco FC, Wyss M, Gietl AF, Treyer V, Leh SE, Buck F, Nitsch RM, Pruessmann KP, van Zijl PC, Hock C, Unschuld PG, 2016. Colocalization of cerebral iron with Amyloid beta in Mild Cognitive Impairment. Sci Rep 6, 35514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM, 2014. The role of senescent cells in ageing. Nature 509, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veinbergs I, Mallory M, Sagara Y, Masliah E, 2000. Vitamin E supplementation prevents spatial learning deficits and dendritic alterations in aged apolipoprotein E-deficient mice. Eur J Neurosci 12, 4541–4546. [PubMed] [Google Scholar]
- Vossel KA, Xu JC, Fomenko V, Miyamoto T, Suberbielle E, Knox JA, Ho K, Kim DH, Yu GQ, Mucke L, 2015. Tau reduction prevents Abeta-induced axonal transport deficits by blocking activation of GSK3beta. J Cell Biol 209, 419–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, Zhang K, Brodbeck J, Daub AC, Sharma P, Finkbeiner S, Cui B, Mucke L, 2010. Tau Reduction Prevents Ab-Induced Defects in Axonal Transport. Science 330, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallin C, Hiruma Y, Warmlander S, Huvent I, Jarvet J, Abrahams JP, Graslund A, Lippens G, Luo J, 2018. The Neuronal Tau Protein Blocks in Vitro Fibrillation of the Amyloid-beta (Abeta) Peptide at the Oligomeric Stage. J Am Chem Soc 140, 8138–8146. [DOI] [PubMed] [Google Scholar]
- Wan W, Cao L, Kalionis B, Murthi P, Xia S, Guan Y, 2019. Iron Deposition Leads to Hyperphosphorylation of Tau and Disruption of Insulin Signaling. Front Neurol 10, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Dharmalingam P, Vasquez V, Mitra J, Boldogh I, Rao KS, Kent TA, Mitra S, Hegde ML, 2017a. Chronic oxidative damage together with genome repair deficiency in the neurons is a double whammy for neurodegeneration: Is damage response signaling a potential therapeutic target? Mech Ageing Dev 161, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Xi G, Keep RF, Hua Y, 2012. Iron enhances the neurotoxicity of amyloid beta. Transl Stroke Res 3, 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Bai HW, Zhu BT, 2010. Structural basis for certain naturally occurring bioflavonoids to function as reducing co-substrates of cyclooxygenase I and II. PLoS One 5, e12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Joberty G, Buist A, Vanoosthuyse A, Stancu IC, Vasconcelos B, Pierrot N, Faelth-Savitski M, Kienlen-Campard P, Octave JN, Bantscheff M, Drewes G, Moechars D, Dewachter I, 2017b. Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol 133, 731–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mandelkow E, 2016. Tau in physiology and pathology. Nat Rev Neurosci 17, 5–21. [DOI] [PubMed] [Google Scholar]
- Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L, 2014. The role of iron in brain ageing and neurodegenerative disorders. The Lancet Neurology 13, 1045–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward SM, Himmelstein DS, Lancia JK, Binder LI, 2012. Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans 40, 667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman EA, Giasson BI, 2011. Induction of intracellular tau aggregation is promoted by alpha-synuclein seeds and provides novel insights into the hyperphosphorylation of tau. J Neurosci 31, 7604–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb J, Multani JS, Saltman P, Beach NA, Gray HB, 1973. Spectroscopic and Magnetic Studies of Iron( 111) Phosvitins. Biochemistry 12, 1979–1801. [DOI] [PubMed] [Google Scholar]
- Weinreb O, Amit T, Mandel S, Youdim MB, 2009. Neuroprotective molecular mechanisms of (−)-epigallocatechin-3-gallate: a reflective outcome of its antioxidant, iron chelating and neuritogenic properties. Genes Nutr 4, 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinshenker D, 2018. Long Road to Ruin: Noradrenergic Dysfunction in Neurodegenerative Disease. Trends Neurosci 41, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West T, Hu Y, Verghese PB, Bateman RJ, Braunstein JB, Fogelman I, Budur K, Florian H, Mendonca N, Holtzman DM, 2017. Preclinical and Clinical Development of ABBV-8E12, a Humanized Anti-Tau Antibody, for Treatment of Alzheimer’s Disease and Other Tauopathies. J Prev Alzheimers Dis 4, 236–241. [DOI] [PubMed] [Google Scholar]
- Wilkinson B, Gilbert HF, 2004. Protein disulfide isomerase. Biochim Biophys Acta 1699, 35–44. [DOI] [PubMed] [Google Scholar]
- Williams DR, 2006. Tauopathies: classification and clinical update on neurodegenerative diseases associated with microtubule-associated protein tau. Intern Med J 36, 652–660. [DOI] [PubMed] [Google Scholar]
- Winterbourn CC, 1995. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicology Letters 82/83, 969–974. [DOI] [PubMed] [Google Scholar]
- Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR, 1996. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci U S A 93, 11213–11218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wischik CM, Harrington CR, Storey JM, 2014. Tau-aggregation inhibitor therapy for Alzheimer’s disease. Biochem Pharmacol 88, 529–539. [DOI] [PubMed] [Google Scholar]
- Wong A, Alder V, Robertson D, Papadimitriou J, Maserei J, Berdoukas V, Kontoghiorghes G, Taylor E, Baker E, 1997. Liver iron depletion and toxicity of the iron chelator deferiprone (L1, CP20) in the guinea pig. Biometals 10, 247–256. [DOI] [PubMed] [Google Scholar]
- Wong BX, Tsatsanis A, Lim LQ, Adlard PA, Bush AI, Duce JA, 2014. beta-Amyloid precursor protein does not possess ferroxidase activity but does stabilize the cell surface ferrous iron exporter ferroportin. PLoS One 9, e114174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, Shin R-W, Hasegawa K, Naiki H, Sato H, Yoshimasu F, Kitamoto T, 2002. Iron (III) induces aggregation of hyperphosphorylated s and its reduction to iron (II) reverses the aggregation: implications in the formation of neurofibrillary tangles of Alzheimer’s disease. Journal of Neurochemistry 82, 1137–1147. [DOI] [PubMed] [Google Scholar]
- Yang LPH, Keam SJ, Keating GM, 2007. Deferasirox: a review of its use in the management of transfusional chronic iron overload. Drugs 67, 2211–2230. [DOI] [PubMed] [Google Scholar]
- Yiannopoulou KG, Papageorgiou SG, 2013. Current and future treatments for Alzheimer’s disease. Ther Adv Neurol Disord 6, 19–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo M-H, Gu X, Xu X-M, Kim J-Y, Carlson BA, Patterson AD, Cai H, Gladyshev VN, Hatfield DL, 2010. Delineating the Role of Glutathione Peroxidase 4 in Protecting Cells Against Lipid Hydroperoxide Damage and in Alzheimer’s Disease. Antioxidants and Redox Signalling 12, 819–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YJ, Zhang Y, Kenrick M, Hoyte M, Luk W, Lu Y, Atwal J, Elliot JM, Prabhu S, Watts RJ, Dennis MS, 2011. Boosting Brain Uptake of a Therapeutic Antibody by Reducing Its Affinity for a Transcytosis Target. Sci Transl Med 3, 84ra44. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, Vocadlo DJ, 2012. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nat Chem Biol 8, 393–399. [DOI] [PubMed] [Google Scholar]
- Zatta P, Drago D, Bolognin S, Sensi SL, 2009. Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol Sci 30, 346–355. [DOI] [PubMed] [Google Scholar]
- Zecca L, Tampellini D, Gerlach M, Riederer P, Fariello RG, Sulzer D, 2001. Substantia nigra neuromelanin: structure, synthesis, and molecular behaviour. Mol Pathol 54, 414–418. [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Lu Y, Luo G, 2014. Synthesis of hierarchical iron hydrogen phosphate crystal as a robust peroxidase mimic for stable H(2)O(2) detection. ACSAppI Mater Interfaces 6, 14433–14438. [DOI] [PubMed] [Google Scholar]
- Zhang YH, Wang DW, Xu SF, Zhang S, Fan YG, Yang YY, Guo SQ, Wang S, Guo T, Wang ZY, Guo C, 2018. alpha-Lipoic acid improves abnormal behavior by mitigation of oxidative stress, inflammation, ferroptosis, and tauopathy in P301S Tau transgenic mice. Redox Biol 14, 535–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Simpkins JW, 2010. Okadaic acid induces tau phosphorylation in SH-SY5Y cells in an estrogen-preventable manner. Brain Res 1345, 176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZH, Wu QY, Zheng R, Chen C, Chen Y, Liu Q, Hoffmann PR, Ni JZ, Song GL, 2017. Selenomethionine Mitigates Cognitive Decline by Targeting Both Tau Hyperphosphorylation and Autophagic Clearance in an Alzheimer’s Disease Mouse Model. J Neurosci 37, 2449–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, Pratt DA, 2017. On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent Sci 3, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucca FA, Segura-Aguilar J, Ferrari E, Munoz P, Paris I, Sulzer D, Sarna T, Casella L, Zecca L, 2017. Interactions of iron, dopamine and neuromelanin pathways in brain aging and Parkinson’s disease. Prog Neurobiol 155, 96–119. [DOI] [PMC free article] [PubMed] [Google Scholar]