

Abstract
The abnormal neurites have long been regarded as the main player contributing to the poor outcome of patients with subarachnoid hemorrhage (SAH). (-)-Eigallocatechin-3-gallate (EGCG), the major biological component of tea catechin, exhibited strong neuroprotective effects against central nervous system diseases; however, the role of EGCG-mediated neurite outgrowth after SAH has not been delineated. Here, the effect of reactive oxygen species (ROS)/integrin β1/FAK/p38 pathway on neurite outgrowth was investigated. As expected, oxyhemoglobin- (OxyHb-) induced excessive ROS level was significantly reduced by EGCG as well as antioxidant N-acetyl-l-cysteine (NAC). Consequently, the expression of integrin β1 was significantly inhibited by EGCG and NAC. Meanwhile, EGCG significantly inhibited the overexpression of phosphorylated FAK and p38 to basal level after SAH. As a result, the abnormal neurites and cell injury were rescued by EGCG, which eventually increased energy generation and neurological score after SAH. These results suggested that EGCG promoted neurite outgrowth after SAH by inhibition of ROS/integrin β1/FAK/p38 signaling pathway. Therefore, EGCG might be a new pharmacological agent that targets neurite outgrowth in SAH therapy.
1. Introduction
A growing body of epidemiological and animal evidence demonstrated that green tea dramatically lowered the incidence of strokes and Alzheimer's disease risk. Moreover, people who drank more than two cups of green tea per day exhibited less cognitive impairment after neuronal injury [1, 2]. The main components of green tea include (-)-epigallocatechin (EGC), (-)-epicatechin-3-gallate (ECG), (-)-epicatechin (EC), and (-)-epigallocatechin-3-gallate (EGCG). Notably, EGCG played pivotal roles in the prevention and treatment of almost all diseases largely dependent on its well-known anti-oxidative activity [3, 4]. Besides, we previously reported that EGCG exhibited strong neuroprotective effects against subarachnoid hemorrhage (SAH) by inhibition of oxidative stress, Ca2+ overloading, mitochondrial dysfunction, and cell death [5–8].
Being a devastating subtype of strokes, the mortality rate of SAH can be as high as 67% [9]. More importantly, it has been estimated that 36–55% survivors suffered impairment, disability, handicap, and poor quality of life during the first year after SAH, which might predict functional outcome of neuronal injury [10]. Neuritis, composed of axon and dendrites, is the structural unit determining the function of the brain [11]. Contrary to the reversible axonotmesis in myelinated and unmyelinated white matter, neurite degeneration might be less likely to be rescued in gray matter, which is mainly made up of neuronal cell bodies and dendrites. Therefore, abnormal neurite morphology and length have been regarded as a common factor contributing to aging, neurodegenerative diseases, and central nervous system (CNS) injury [12–14]. Recently, multifocal axonal injury has also been observed in the early stage after SAH. Most importantly, axonal degeneration has been a major player determining primary injury and the outcome of SAH patients. Few molecules, such as ROS, Ca2+, neurofilament high chain (NfH), and S100, have been identified regulating axonal damage after SAH. However, the precise mechanism of axonal degeneration after SAH has not been fully understood.
EGCG has been demonstrated to regulate nerve growth factor- (NGF-) induced neurite outgrowth [15]. It has been reported that EGCG bound to 67 LR and inhibited ROS generation to promoting axonal regeneration. However, whether EGCG induces neurite outgrowth after SAH and the underlying mechanisms remain unclear. Many intracellular signaling pathways have been identified to regulate neurite outgrowth, of which the integrin family of adhesion molecules played pivotal roles and might be regarded as important therapeutic targets by activating “outside-in” signaling molecules, such as FAK, ERK, p38, and JNK [16–18]. In our previous study, EGCG (50 μM in vitro or 50 mg/kg in vivo) significantly inhibited ROS generation and the overexpression of p38 and eventually rescued neuronal cells injury after SAH. Based on these findings, we aimed to investigate the neuroprotective effects of EGCG on neurite outgrowth via ROS/integrin β1/FAK/p38 pathway by using SAH cell and mice models [5–8].
2. Materials and Methods
2.1. Materials
PC12 cells were bought from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). EGCG with a 95% purity (Huike, Shanxi, China) was diluted by water (pH 3.0) and stored at −20°C. NGF was gained from Sigma (St. Louis, MO, USA). OxyHb was obtained from Shanghai Sangon Biotech (China). FAK inhibitor Y15 and p38 inhibitor SB203580 were obtained from Selleck (Houston, TX, USA). Primary antibodies against integrin beta 1, pFAK, and p-p38 (Thr180/Tyr182) were purchased from Cell Signaling Technologies (Danvers, MA, USA). Penicillin and streptomycin, N-Acetyl-L-cysteine (NAC), and an enhanced chemiluminescence (ECL) kit were purchased from Beyotime (Shanghai, China). ATP and lactate dehydrogenase (LDH) assay kit were obtained from Nanjing Jiancheng Bioengineering Institute (Jiangshu, China). The First-Strand Complementary DNA (cDNA) Synthesis Kit (11483188001) was purchased from Roche Applied Sciences (Basel, Switzerland). Power SYBR® Green PCR Master Mix was obtained from Thermo Fisher (Waltham, Massachusetts, USA).
2.2. Cell Culture and Treatment
The cells were cultured at 37°h in 5% CO2 in RPMI 1640 medium supplemented with 1% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA, USA), 1% penicillin/streptomycin, and 50 ng/ml NGF for 24 h. Then, OxyHb was added to the medium to a final concentration of 10 μM. EGCG (50 μM) was added 30 min before the addition of OxyHb as EGCG pretreatment group [7]. NAC (2.5 μM) and p38 inhibitor (20 μM) were added 30 min after the addition of OxyHb as NAC and p38 inhibitor treatment groups [19].
2.3. Animals
The animal use and care protocols were approved by the Institutional Animal Care and Use Committee (IACUC) of Henan Normal University. Male Kunming mice (n = 150, weighing from 20 to 25 g) were bought from Zhengzhou University. The animals were kept in a constant temperature (22 ± 3°C) and relative humidity (60%) animal rooms in a 12 h light/dark cycle. All animals were allowed to undergo appropriate quarantine procedures.
2.4. Mouse SAH Model
SAH mice model was established based on what was previously reported by Huang et al. [20]. After being given 1% pentobarbital general anesthesia (50 mg/kg, intraperitoneally), the posterior cervical muscles of mice were dissected through midline suboccipital approach to penetrate the transparent atlanto-occipital membrane. Then, a hole was made in the cross position at 2 mm from the sagittal suture and 1 mm from the coronaria suture. Subsequently, 30 μL of OxyHb (150 μmol/L) was injected into the subarachnoid space, whereas the isotonic saline was administered as the sham group. As for EGCG treatment group, EGCG was intragastrically administered at a dose of 50 mg/kg/d for 14 days before OxyHb injection. As for NAC, FAK inhibitor, and p38 inhibitor treated groups, a total of NAC, Y15, and SB203580 (150 mg/kg, 30 mg/kg, and 200 μg/kg, respectively) was injected into subarachnoid space after 30 min of SAH established [20]. The mice were sacrificed and brain tissue was collected after 48h of SAH.
2.5. Experimental Design
2.5.1. Experimental Design for Exploring the Roles of Superoxide Anion and Neurite Outgrowth in In Vitro SAH Model
NGF-induced PC12 were divided into 4 groups to investigate superoxide anion concentration (control, SAH, SAH + EGCG, SAH + NAC) and the changes of neurite outgrowth (control, SAH, SAH + EGCG, SAH + p38 inhibitor).
2.5.2. Experimental Design for Exploring the Roles of Integrin Beta 1, FAK, and p38 in In Vivo SAH Model
In this experiment, mice (n = 150) were randomly divided into the following 6 groups: control, SAH, SAH + EGCG, SAH + NAC, SAH + FAK inhibitor, and SAH + p38 inhibitor:
The four groups (n = 10 per group) (control, SAH, SAH + EGCG, and SAH + NAC) were used to detect the expression of integrin.
The four groups (n = 10 per group) (control, SAH, SAH + EGCG, and SAH + p38 inhibitor) were used to detect the expression of pFAK.
The four groups (n = 10 per group) (control, SAH, SAH + EGCG, and SAH + FAK inhibitor) were used to detect the expression of p-p38.
The three groups (n = 10 per group) (control, SAH, and SAH + EGCG) were used to detect ATP, LDH, and neurological score.
2.6. Measurement of Superoxide Anion Levels
The cell permeable non-fluorescent DHE was used to detect the intracellular superoxide anion levels according to the manufacturer's instruction. After the experiment, serum-free culture medium was used to wash the cells. Then, DHE probe was added to a final concentration of 10 μM and incubated at 37°C for 15 min. The fluorescence was measured by a microplate reader using 300 nm excitation and 610 nm emission wavelength.
2.7. Fluorescence Microscopy
The cells were seeded on glass coverslips. After washing with PBS, the cells were incubated with DHE (10 μM) for 15 min as described previously [8]. Fluorescence signals were visualized with a Delta Vision Spectris fluorescence microscopy (AX10, Zeiss).
2.8. Measurement of Neurite Outgrowth
To study neurite outgrowth, the total length of neurites and neurite outgrowth cells were observed by microscope (Zeiss Oberkochen, Germany). The length of neurites was measured using Imaging software Presage (Brooksville, FL, USA). The cell was counted as a neurite outgrowth cell when the length was equal to or greater than the diameter of the cell body. At least, 50 cells were measured and all experiments were performed in triplicate three times.
2.9. Neurological Functions Assessment
Animal neurological behavior and function were evaluated using the Garcia scoring system [21]. Briefly, the neurobehavioral examination was performed at 48 h. An 18-point scoring system was used based on (1) spontaneous activity, (2) symmetry of limb movement, (3) climbing, (4) body proprioception, (5) the movement of forelimbs, and (6) response to vibrissae touch.
2.10. Brain Cortex ATP Content Assessment
Boiling double distilled water was used to homogenize cerebral cortex to denature endogenous ATPase. After being centrifugated at 10,000 g for 10 min, the supernatant fraction was collected. ATP levels were measured according to manufacturer's instructions.
2.11. Lactate Dehydrogenase (LDH) Assay from Brain Cortex
The supernatant fraction of all the samples was collected to detect LDH activity according to the manufacturer's instructions. LDH cytotoxicity was calculated using OD as LDH cytotoxicity (U/g protein) = (OD sample – OD blank)/(OD standard solution – OD blank standard solution) × standard solution concentration/sample protein concentration.
2.12. Western Blot Assay
Brain cortex was homogenized (1% proteinase K inhibitor, PMSF 1 mM, and 1% (v/v) protease inhibitor cocktail) and centrifugated at 10000 g for 10 min at 4°C. The collected supernatant was frozen at −80°C. Total protein (50 μg) from each group was loaded and separated by 12% SDS-PAGE and electro-transferred to nitrocellulose membrane. After being blocked with non-fat milk in PBS for 1 h, the membrane was incubated with monoclonal antibody against integrin beta 1 (1 : 1000), pFAK (1 : 500), and GAPDH (1 : 400) overnight, respectively. Being washed three times with PBS containing 0.05% Tween-20, the membrane was incubated with a secondary antibody for 1 h. The densities of the bands were visualized and analyzed by using enhanced chemiluminescence (ECL) kit and ImageQuant software. All the experiments were independently repeated three times.
2.13. Quantification of mRNA Expression of Integrin Genes Using RT-PCR
qRT-PCR was carried out based on our previous report [7]. Briefly, the total RNA was isolated and each RNA sample was reverse-transcribed. Then, qRT-PCR was performed to identify the mRNA expression of integrin beta 1 (NM_017022.2) and integrin beta 5 (NM_147139.2) by using the BIO-RAD CFX ConnectTM platform (Hercules, California, USA). Briefly, qRT-PCR reactions were performed in a total volume of 25 μl containing 2.5 μl of cDNA, 0.5 μl of primes (5 μM), 12.5 μl of PCR Master Mix, and 9.5 μl of deionized water. GAPDH was used to normalize each target gene by using the comparative 2−ΔΔCt method. The sequences of mRNA primers were listed as follows:
- Integrin beta 1: Fw: 5′-ACAGAAGAAGTAGAGGTGGTCC-3′,
- Rw: 5′-GCTACATTCACAGTGTCTCCC-3′;
- Integrin beta 5: Fw: 5′-TTGCGGAGGAGATGAGGAAG-3′,
- Rw: 5′-AAGGGACACAGTTGGGGAAT-3′;
- GAPDH: Fw: 5′-TGACTCTACCCACGGCAAG-3′,
- Rw: 5′-ACTCAGCACCAGCATCACC-3′.
2.14. Immunohistochemistry Assay
Deparaffinized sections were heated in sodium citrate buffer (0.01 M, pH 6.5) for 10 min at 121°C. Subsequently, sections were incubated with 0.3% hydrogen peroxide to block endogenous peroxidase activity. After being blocked with 10% normal goat serum, primary anti-p-p38 antibody was used at a 1 : 1000 dilution and incubated overnight. After further washing, slides were treated with Histostain™-Plus kit (SP-9001, Zymed, USA). Immunoperoxidase staining was developed using DAB (ZLI-9032, Zhongshan Golden Bridge Biotechnology Co., Ltd., China). At least 10 visual fields were captured and more than 500 cells were counted using the IDA-2000 software (Beijing Konghai Technology Company, China).
2.15. Statistical Analysis
Data were expressed as means ± SD. Significant differences were verified with a one-way ANOVA analysis, followed by Tukey's test for multiple comparisons by using SPSS 17.0 software. Kruskal–Wallis nonparametric test followed by Duncan's multiple-comparison procedures was used for the neurological scores. p < 0.05 was considered as statistically significant.
3. Results
3.1. EGCG Inhibited Superoxide Anion Generation after SAH
OxyHb-induced oxidative stress has long been regarded as an early event in the progression of SAH. DHE is oxidized by superoxide anion to form ethidium, which binds to DNA. Therefore, more red fluorescence indicated higher superoxide anion levels. Compared with the control group (2.2 ± 0.2), strong signals were captured in SAH group showing an increase in red fluorescence intensity (2.9 ± 0.1, p < 0.01 vs. control), whereas less signals were detected in the EGCG and NAC treatment group (p < 0.01 vs. SAH) (Figure 1).
Figure 1.
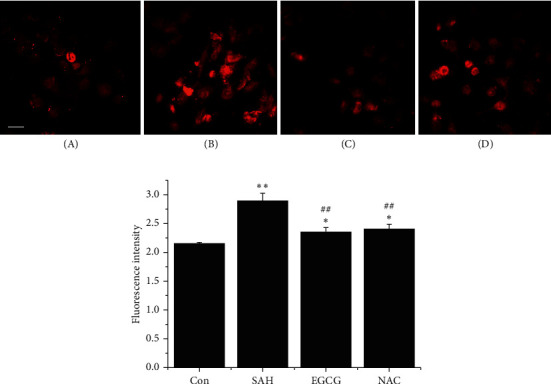
EGCG inhibited superoxide anion generation after SAH. (a) Superoxide anion fluorescence distribution in cells detected by fluorescence microscopy. Normal cells showed less fluorescence (A). OxyHb at a final concentration of 10 μM significantly increased fluorescence intensity (B). EGCG, added 30 min before the addition of OxyHb, significantly reduced superoxide anion level almost to basal level (C). Similar trend of superoxide anion level was also observed in NAC group, which was added 30 min after the addition of OxyHb (D). Bar, 200 μm. (b) Quantification of superoxide anion fluorescence intensity. The values represent three independent experiments. ∗p < 0.05 vs. control, ∗∗p < 0.01 vs. control, and ##p < 0.01 vs. SAH.
3.2. EGCG Downregulated Integrin β1 Expression through ROS
Given that ROS functioned as an inducer to activate integrins, the expression of integrins β1 and β5 was measured after SAH. Contrary to the high ROS concentrations in SAH group, the downregulated mRNA expression of integrin β5 was observed (p < 0.01 vs. control), whereas the expression of integrin β1 was significantly increased 1.25-fold after SAH (p < 0.01 vs. control) (Figure 2(a)). However, EGCG significantly restored the imbalance in the expression of integrin β5 (p < 0.05 vs. SAH) to basal level but not integrin β1 (p < 0.05 vs. control, p < 0.01 vs. SAH). NAC has the similar anti-oxidative capacity in regulation of the elevated expression of integrin β1 (p < 0.01 vs. SAH) and β5 (p > 0.05 vs. SAH). Notably, the protein expression level of integrin β1 is similar to the mRNA expression trend, showing that both EGCG and NAC significantly inhibited the overexpression of integrin β1 after OxyHb stimuli (p < 0.01 vs. SAH), demonstrating that ROS triggered the overexpression of integrin β1 after SAH (Figure 2(b)).
Figure 2.
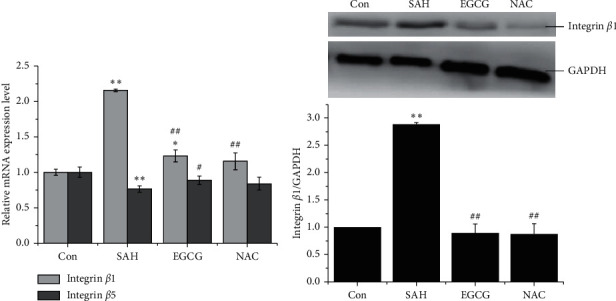
EGCG inhibited the overexpression of integrin β1 after SAH. (a) EGCG regulated integrin β1 and integrin β5 mRNA expression level in in vivo SAH model. (b) Identification of integrin β1 protein level in in vivo SAH model (upper panel) (n = 3), and quantification of the expression of integrin β1 (lower panel). SAH: 30 μL of OxyHb (150 μmol/L); EGCG: 50 mg/kg/d; NAC: 150 mg/kg. A representative trace of three repeats of each experiment is shown. ∗p < 0.055 vs. control, ∗∗p < 0.01 vs. control, #p < 0.01 vs. SAH, and ##p < 0.01 vs. SAH.
3.3. EGCG Inhibited FAK and p38 Signaling after SAH
To elucidate the mechanism through which EGCG modulated integrin signaling pathway, the downstream effectors, FAK and p38, were examined. In line with the finding of integrin β1, the expression of phosphorylated FAK and p38 significantly reached 4.2 ± 0.3 and 6.3 ± 0.5 in SAH group (p < 0.01 vs. control), whereas EGCG dramatically downregulated the expression of FAK and p38 phosphorylation (2.2 ± 0.2 and 2.4 ± 0.2, respectively, p < 0.01 vs. SAH). More importantly, FAK inhibitor significantly antagonized the expression of phosphorylated p38 after SAH, whereas the expression of pFAK showed no significant difference between p38 inhibitor and SAH group, indicating that p38 might be the downstream effector of FAK in response to integrin β1 overexpression after OxyHb stimuli (Figures 3(a) and 3(b)).
Figure 3.
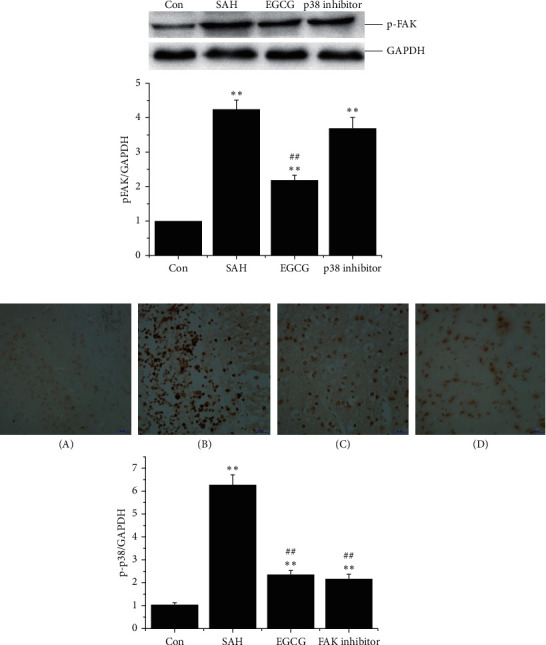
The expression of pFAK and p-p38 after SAH in vivo. (a) EGCG downregulated the expression of pFAK (upper panel) (n = 3). SAH: 30 μL of OxyHb (150 μmol/L); EGCG: 50 mg/kg/d; p38 inhibitor: 200 μg/kg. Quantification of the expression of pFAK (lower panel). (b) EGCG inhibited p-p38 overexpression after SAH (upper panel) (n = 3) (bar, 50 μm), and quantification of the expression of p-p38 (lower panel). (A) control; (B) SAH (30 μL (150 μmol/L) of OxyHb); (C) EGCG (50 mg/kg/d); (D) FAK inhibitor (30 mg/kg). A representative trace of three repeats of each experiment is shown. ∗∗p < 0.01 vs. control and ##p < 0.01 vs. SAH.
3.4. EGCG Promoted Neurite Outgrowth by Inhibition of Excessive Activation of Integrin β1/FAK/p38 Pathway
To examine the effect of EGCG on neurite outgrowth after SAH, the NGF-induced PC12 cells were used to observe the length of neurites and number of neurite cells. As shown in Figure 4(a), OxyHb impaired the neuritogenic action. The length of neurites and the number of neurite cells significantly decreased from 168.6 ± 21.1 and 9.3 ± 1.2 in control group to 78.1 ± 12.3 and 3.7 ± 0.6 in SAH group (p < 0.01). However, after EGCG treatment, the length of neurites was significantly increased and more neurite cells were observed (152.7 ± 18.9, p < 0.01 vs. SAH; 7.3 ± 1.5, p < 0.01 vs. SAH), suggesting that neurite outgrowth was induced by EGCG (Figures 4(b) and 4(c)). Given that integrin β1/FAK/p38 pathway was involved in NGF-induced neurite outgrowth, we next examined the effects of the specific p38 inhibitor on neuritogenesis. As expected, SB203580 significantly promoted neurite outgrowth after SAH as well as the number of neurite cells (124.9 ± 15.2, 6.7 ± 0.6) (Figure 4).
Figure 4.
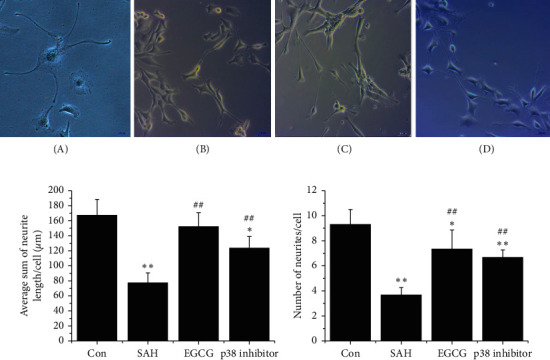
EGCG induced PC12 cells neurite outgrowth. (a) The morphology of NGF-induced PC12 cells after SAH in vitro. (A) Control; (B SAH (at a final concentration of 10 μM OxyHb; (C) EGCG (50 μM); (D) NAC (2.5 μM). Bar, 200 μm for (A) and 100 μm for (B–D). (b) The length of neurites after SAH. (c) The average number of neurites per cell after SAH. A representative trace of three repeats of each experiment is shown. ∗p < 0.05 vs. control, ∗∗p < 0.01 vs. control, and ##p < 0.01 vs. SAH.
3.5. EGCG Protects Neurological Function after SAH
LDH activity is the most widely used marker in cytotoxic studies. Using this assay, we detected the neuroprotective role of EGCG against SAH. LDH activity was lower in the cortical tissue of the control group (10909.0 ± 550.2) (Figure 5(a)). After SAH, LDH levels significantly increased to 17379.6 ± 602.8 (p < 0.01 vs. control). However, EGCG induced a dramatic decrease in LDH activity (9424.1 ± 595.4, p < 0.01 vs. SAH).
Figure 5.
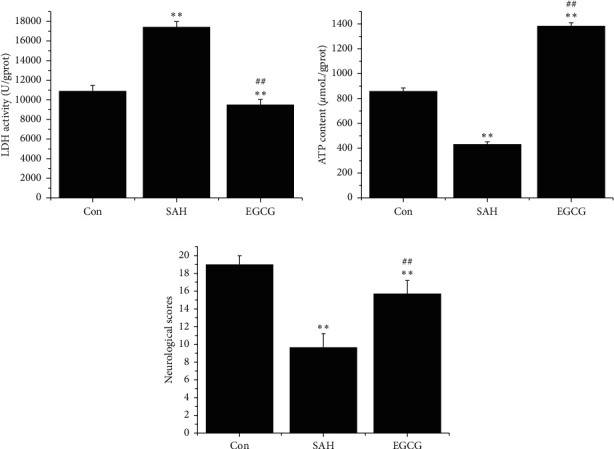
Neuroprotective effects of EGCG after SAH. (a) Effect of EGCG on LDH activity after SAH (n = 3). (b) EGCG increased ATP content after SAH (n = 3). (c) Neurological deficits after SAH (n = 6). SAH: 30 μL of OxyHb (150 μmol/L); EGCG: 50 mg/kg/d. A representative trace of three repeats of each experiment is shown. ∗∗p < 0.01 vs. control; ##p < 0.01 vs. SAH.
Similar to the finding of LDH, the ATP level was significantly decreased from 859.4 ± 23.5 in the control group to 432.9 ± 18.3 in the SAH group (p < 0.01 vs. control), whereas EGCG restored ATP level to basal level (1382.1 ± 26.2, p < 0.01 vs. control, p < 0.01 vs. SAH), suggesting that EGCG significantly increased ATP content after SAH (Figure 5(b)).
Following the higher energy generation and lower toxicity in the EGCG treatment, the preconditioned EGCG animals showed improved neurological scores (p < 0.01 vs. SAH) contrary to severe behavioral disabilities after SAH (p < 0.01 vs. control), indicating that impaired behavioral function caused by SAH could be restored by EGCG (Figure 5(c)).
4. Discussion
Neuronal cells injury, particularly for the detected axonal degeneration, has been demonstrated to be the major player contributing to the outcome of SAH patients by leading to irreversible neurological deficit [22–24]. Mounting evidence has demonstrated the positive neuroprotective role of EGCG in regulating neurite outgrowth [15, 25]. However, few studies have focused on the mechanisms of EGCG against neurite degeneration after SAH. In the present study, in vitro and in vivo SAH models were used to investigate the mechanism through which EGCG promoted neurite outgrowth and associated signaling pathway.
Neuronal cells are especially vulnerable to oxidative damage, partly because of the high level of oxygen consumption and the low level of ROS scavenging ability. After SAH, OxyHb oxidized to methaemoglobin (MetHb), which induced ROS generation. Given that oxidative stress was positively associated with the poor outcome of SAH patients, antioxidant therapy has long been regarded as a promising strategy promoting neurons survival under the harmful conditions [26, 27]. Even though EGCG playes a dual role in its physiologicol activities, i.e., the harmful pro-oxidative (high-dose) and the benificial antioxidative activities (low-dose), most studies have, recently, reported that the harmful effect of EGCG was overwhelmed by its benificial role under pathological conditions [28]. These findings were supported by our previous studies showing that EGCG exhibited neuroprotective effects by inhibition of mitochondrial dysfunction-induced oxygen stress after SAH.
Several research groups reported that ROS activated integrin-mediated “outside-in” signaling pathways, whereas others found that integrin, in turn, induced ROS generation and activated “inside-out” signaling [29]. Integrins are heterodimeric membrane adhesion molecules composed of noncovalently bound α- and β-subunits, of which integrin β1 is widely expressed in the brain and involved in morphogenesis, cell differentiation, and proliferation as well as cell survival. The overexpression of integrin β1 and FAK aggravated neurological deficit in Alzheimer's disease (AD) and cerebral ischemia-reperfusion [30, 31]. However, after intracerebral hemorrhage, increased expression of integrin β1 in the brain significantly improved neurobehavioural score with decrease of neutrophil infiltration through JAK2/STAT1 pathway [32]. Besides, a growing body of evidence showed that integrin β1 modulated neuronal migration to regulate neurovascular remodeling, brain edema, and functional outcomes following stroke [33–36]. Therefore, β1 integrin has been regarded as a potential target in strokes therapy. Unfortunately, even though there are a few studies about the role of integrin after SAH, inconsistent results have been reported. The expression of integrin β3 reduced oxidative stress to inhibit cell death after SAH, whereas the overexpression of GPIIB/IIa integrin has also been reported inducing cell death and neurobehavioral injury in early brain injury after SAH [37–39]. Given that EGCG has been demonstrated to regulate the expression of αIIb, α5, β1, and β3 integrins, in the present study, we focused on the role of integrin β1 after EGCG treatment of SAH and demonstrated that EGCG inhibited ROS-induced overexpression of integrin β1 after SAH [37, 40]. FAK is a well-known downstream effector of ROS-integrin signaling pathway under oxidative stress condition. Inactivation of FAK in mouse endothelium caused brain hemorrhage, and several researches reported that increased phosphorylated FAK resulted in the attenuation of neuronal cell death and improved neurological status after SAH [41, 42]. However, we observed a significant increase in the expression of FAK phosphorylation, whereas EGCG restored the expression of phosphorylated FAK to basal level, indicating the higher expression of phosphorylated FAK and the severe pathological state after SAH. Recently, several substrates of integrin β1/FAK have been identified in the SAH progression, such as p38 [43]. FAK inhibitor significantly inhibited the expression of p38, whereas the expression of FAK was maintained at the same level after p38 inhibitor treatment, indicating that p38 was the downstream signal molecule of FAK. In addition, the overexpression of p38 has been reported to induce mitochondrial dysfunction, oxidative stress, apoptosis, and autophagy after SAH. Notably, EGCG inhibited the overexpression of p-p38 against mitochondrial dysfunction and cell death after SAH [5, 20]. Therefore, the neuroprotective role of EGCG partly relied on integrin β1/FAK/p38 signaling pathway after SAH [44].
Under serum-free condition, NGF induced neurite outgrowth of PC12 cells. This well-defined neuronal model is often employed for studying neurotoxicity in some CNS diseases. Integrins affect neurite outgrowth in adult healthy brains and pathological conditions [45–47]. Farizatto et al. found that the overexpression of integrin β1 regulated synapse maintenance and plasticity [48]. However, some research showed that the lower level of integrin β1 reduced dendrite growth, suggesting that integrin β1/FAK signaling pathway was involved in neurite outgrowth, which contributed to the differentiation of neuronal cells and determined the outcome of patients [31, 49]. In the present study, EGCG dramatically restored the degenerative neurite outgrowth after SAH. Furthermore, inhibition of the expression of p38 also abolished neurites degeneration after SAH. These findings suggested that EGCG promoted neurite outgrowth after SAH via integrin β1/FAK/p38 pathway, and the excessive activation of integrin β1/FAK/p38 signaling pathway might indicate the severe injury after SAH.
As a result, EGCG increased energy generation, decreased cell injury, and eventually improved neurological function. These results revealed the mechanisms behind the neuroprotective effects of EGCG by inhibition of OxyHb-induced neurite degeneration via integrin β1/FAK/p38 signaling pathway and the following cell injury. Therefore, simultaneous inhibition and elimination of neurite degeneration could determine the therapeutic effect of EGCG. Further investigation will be necessary to explore the mechanisms of EGCG transmembrane transport and the ways to improve the bio-availability, which may promote the potential for application to SAH disease.
Acknowledgments
This work was supported by the Natural Science Foundation of Henan Province (No. 182300410299).
Data Availability
Data are available upon request to the corresponding author.
Ethical Approval
This work is in compliance with ethical standards.
Conflicts of Interest
The authors declare no conflicts of interest.
Authors' Contributions
Yuyuan Zhang and Mengguo Han are the co-senior authors and contributed equally to this work.
References
- 1.Kuriyama S., Hozawa A., Ohmori K., et al. Green tea consumption and cognitive function: a cross-sectional study from the Tsurugaya Project. The American Journal of Clinical Nutrition. 2006;83(2):355–361. doi: 10.1093/ajcn/83.2.355. [DOI] [PubMed] [Google Scholar]
- 2.Liang W., Lee A. H., Binns C. W., Huang R., Hu D., Zhou Q. Tea consumption and ischemic stroke risk. Stroke. 2009;40(7):2480–2485. doi: 10.1161/strokeaha.109.548586. [DOI] [PubMed] [Google Scholar]
- 3.Itoh T., Tabuchi M., Mizuguchi N., et al. Neuroprotective effect of (-)-epigallocatechin-3-gallate in rats when administered pre- or post-traumatic brain injury. Journal of Neural Transmission. 2013;120(5):767–783. doi: 10.1007/s00702-012-0918-4. [DOI] [PubMed] [Google Scholar]
- 4.Hyung S.-J., DeToma A. S., Brender J. R., et al. Insights into antiamyloidogenic properties of the green tea extract (-)-epigallocatechin-3-gallate toward metal-associated amyloid- species. in Proceedings of the National Academy of Sciences. 2013;110(10):3743–3748. doi: 10.1073/pnas.1220326110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y., Huang L., Zhang H., Sun H., Zhou W. EGCG protective mitochondrial dysfunction after subarachnoid haemorrhage via inhibition p38 α pathway. Journal of Functional Foods. 2016;23:115–123. doi: 10.1016/j.jff.2016.02.035. [DOI] [Google Scholar]
- 6.Chen Y., Huang L., Wang L., Chen L., Ren W., Zhou W. Differential expression of microRNAs contributed to the health efficacy of EGCG in vitrosubarachnoid hemorrhage model. Food & Function. 2017;8(12):4675–4683. doi: 10.1039/c7fo01064h. [DOI] [PubMed] [Google Scholar]
- 7.Chen Y., Huang L., Zhang H., Diao X., Zhao S., Zhou W. Reduction in autophagy by (-)-epigallocatechin-3-gallate (EGCG): a potential mechanism of prevention of mitochondrial dysfunction after subarachnoid hemorrhage. Molecular Neurobiology. 2017;54(1):392–405. doi: 10.1007/s12035-015-9629-9. [DOI] [PubMed] [Google Scholar]
- 8.Chen Y., Chen J., Sun X., et al. Evaluation of the neuroprotective effect of EGCG: a potential mechanism of mitochondrial dysfunction and mitochondrial dynamics after subarachnoid hemorrhage. Food & Function. 2018;9(12):6349–6359. doi: 10.1039/c8fo01497c. [DOI] [PubMed] [Google Scholar]
- 9.Sehba F. A., Hou J., Pluta R. M., Zhang J. H. The importance of early brain injury after subarachnoid hemorrhage. Progress in Neurobiology. 2012;97(1):14–37. doi: 10.1016/j.pneurobio.2012.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nieuwkamp D. J., Setz L. E., Algra A., Linn F. H., de Rooij N. K., Rinkel G. J. Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta-analysis. The Lancet Neurology. 2009;8(7):635–642. doi: 10.1016/s1474-4422(09)70126-7. [DOI] [PubMed] [Google Scholar]
- 11.Barnes A. P., Polleux F. Establishment of axon-dendrite polarity in developing neurons. Annual Review of Neuroscience. 2009;32(1):347–381. doi: 10.1146/annurev.neuro.31.060407.125536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arimura N., Kaibuchi K. Neuronal polarity: from extracellular signals to intracellular mechanisms. Nature Reviews Neuroscience. 2007;8(3):194–205. doi: 10.1038/nrn2056. [DOI] [PubMed] [Google Scholar]
- 13.Reddy P. H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Research. 2011;1415:136–148. doi: 10.1016/j.brainres.2011.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwab M. E., Bartholdi D. Degeneration and regeneration of axons in the lesioned spinal cord. Physiological Reviews. 1996;76(2):319–370. doi: 10.1152/physrev.1996.76.2.319. [DOI] [PubMed] [Google Scholar]
- 15.Gundimeda U., McNeill T. H., Schiffman J. E., Hinton D. R., Gopalakrishna R. Green tea polyphenols potentiate the action of nerve growth factor to induce neuritogenesis: possible role of reactive oxygen species. Journal of Neuroscience Research. 2010;88(16):3644–3655. doi: 10.1002/jnr.22519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andrews M. R., Czvitkovich S., Dassie E., et al. 9 integrin promotes neurite outgrowth on tenascin-C and enhances sensory axon regeneration. Journal of Neuroscience. 2009;29(17):5546–5557. doi: 10.1523/jneurosci.0759-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blackmore M., Letourneau P. C. L1, β1 integrin, and cadherins mediate axonal regeneration in the embryonic spinal cord. Journal of Neurobiology. 2006;66(14):1564–1583. doi: 10.1002/neu.20311. [DOI] [PubMed] [Google Scholar]
- 18.Lemons M. L., Condic M. L. Integrin signaling is integral to regeneration. Experimental Neurology. 2008;209(2):343–352. doi: 10.1016/j.expneurol.2007.05.027. [DOI] [PubMed] [Google Scholar]
- 19.Huang L., Hou Y., Wang L., et al. p38 inhibitor protects mitochondrial dysfunction by induction of DJ-1 mitochondrial translocation after subarachnoid hemorrhage. Journal of Molecular Neuroscience. 2018;66(2):163–171. doi: 10.1007/s12031-018-1131-1. [DOI] [PubMed] [Google Scholar]
- 20.Huang L., Wan J., Chen Y., et al. Inhibitory effects of p38 inhibitor against mitochondrial dysfunction in the early brain injury after subarachnoid hemorrhage in mice. Brain Research. 2013;1517:133–140. doi: 10.1016/j.brainres.2013.04.010. [DOI] [PubMed] [Google Scholar]
- 21.Garcia J. H., Wagner S., Liu K.-F., Hu X.-J. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Stroke. 1995;26(4):627–635. doi: 10.1161/01.str.26.4.627. [DOI] [PubMed] [Google Scholar]
- 22.Petzold A., Rejdak K., Belli A., et al. Axonal pathology in subarachnoid and intracerebral hemorrhage. Journal of Neurotrauma. 2005;22(3):407–414. doi: 10.1089/neu.2005.22.407. [DOI] [PubMed] [Google Scholar]
- 23.van Gijn J., Rinkel G. J. E. Subarachnoid haemorrhage: diagnosis, causes and management. Brain. 2001;124(2):249–278. doi: 10.1093/brain/124.2.249. [DOI] [PubMed] [Google Scholar]
- 24.Nzali J. J., Mayer S. A. Cerebral vasospasm after subarachnoid hemorrhage. Current Opinion in Critical Care. 2003;9:113–119. doi: 10.1097/00075198-200304000-00006. [DOI] [PubMed] [Google Scholar]
- 25.Unno K., Pervin M., Nakagawa A., et al. Blood-brain barrier permeability of green tea catechin metabolites and their neuritogenic activity in human neuroblastoma SH-SY5Y cells. Molecular Nutrition & Food Research. 2017;61(12):p. 1700294. doi: 10.1002/mnfr.201700294. [DOI] [PubMed] [Google Scholar]
- 26.Gaetani P., Pasqualin A., Rodriguez y Baena R., Borasio E., Marzatico F. Oxidative stress in the human brain after subarachnoid hemorrhage. Journal of Neurosurgery. 1998;89(5):748–754. doi: 10.3171/jns.1998.89.5.0748. [DOI] [PubMed] [Google Scholar]
- 27.Hsieh Y.-P., Lin C.-L., Shiue A.-L., et al. Correlation of F4-neuroprostanes levels in cerebrospinal fluid with outcome of aneurysmal subarachnoid hemorrhage in humans. Free Radical Biology and Medicine. 2009;47(6):814–824. doi: 10.1016/j.freeradbiomed.2009.06.026. [DOI] [PubMed] [Google Scholar]
- 28.Miao Y., Sun X., Gao G., et al. Evaluation of (-)-epigallocatechin-3-gallate (EGCG)-induced cytotoxicity on astrocytes: a potential mechanism of calcium overloading-induced mitochondrial dysfunction. Toxicology in Vitro. 2019;61 doi: 10.1016/j.tiv.2019.104592.104592 [DOI] [PubMed] [Google Scholar]
- 29.Dayan A., Fleminger G., Ashur-Fabian O. Targeting the Achilles’ heel of cancer cells via integrin-mediated delivery of ROS-generating dihydrolipoamide dehydrogenase. Oncogene. 2019;38(25):5050–5061. doi: 10.1038/s41388-019-0775-9. [DOI] [PubMed] [Google Scholar]
- 30.Liu T., Roh S. E., Woo J. A., Ryu H., Kang D. E. Cooperative role of RanBP9 and P73 in mitochondria-mediated apoptosis. Cell Death & Disease. 2013;4(1):p. e476. doi: 10.1038/cddis.2012.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li X. L., Guo Y., Zhang Y. S., Zhao Y., Zhang L. Effects of integrin β1 on behavior and neurovascular regeneration in rats with cerebral ischemia-reperfusion injury. Eur Rev Med Pharmaco. 2019;23(8):3487–3494. doi: 10.26355/eurrev_201904_17714. [DOI] [PubMed] [Google Scholar]
- 32.Gong L., Manaenko A., Fan R., et al. Osteopontin attenuates inflammation via JAK2/STAT1 pathway in hyperglycemic rats after intracerebral hemorrhage. Neuropharmacology. 2018;138:160–169. doi: 10.1016/j.neuropharm.2018.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lathia J. D., Chigurupati S., Thundyil J., et al. Pivotal role for beta-1 integrin in neurovascular remodelling after ischemic stroke. Experimental Neurology. 2010;221(1):107–114. doi: 10.1016/j.expneurol.2009.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Fujioka T., Kaneko N., Ajioka I., et al. β1 integrin signaling promotes neuronal migration along vascular scaffolds in the post-stroke brain. EBioMedicine. 2017;16:195–203. doi: 10.1016/j.ebiom.2017.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Izawa Y., Gu Y.-H., Osada T., et al. β1-integrin-matrix interactions modulate cerebral microvessel endothelial cell tight junction expression and permeability. Journal of Cerebral Blood Flow & Metabolism. 2018;38(4):641–658. doi: 10.1177/0271678x17722108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Milner R., Hung S., Wang X., Berg G. I., Spatz M., del Zoppo G. J. Responses of endothelial cell and astrocyte matrix-integrin receptors to ischemia mimic those observed in the neurovascular unit. Stroke. 2008;39(1):191–197. doi: 10.1161/strokeaha.107.486134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu F., Chen Y., Hu Q., et al. MFGE8/integrin β3 pathway alleviates apoptosis and inflammation in early brain injury after subarachnoid hemorrhage in rats. Experimental Neurology. 2015;272:120–127. doi: 10.1016/j.expneurol.2015.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu F., Hu Q., Li B., et al. Recombinant milk fat globule-EGF factor-8 reduces oxidative stress via integrin β3/nuclear factor erythroid 2-related factor 2/heme oxygenase pathway in subarachnoid hemorrhage rats. Stroke. 2014;45(12):3691–3697. doi: 10.1161/strokeaha.114.006635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu Z.-W., Gu H., Zhang B.-F., et al. Rapidly increased vasopressin promotes acute platelet aggregation and early brain injury after experimental subarachnoid hemorrhage in a rat model. Brain Research. 2016;1639:108–119. doi: 10.1016/j.brainres.2016.02.038. [DOI] [PubMed] [Google Scholar]
- 40.Sen T., Chatterjee A. Epigallocatechin-3-gallate (EGCG) downregulates EGF-induced MMP-9 in breast cancer cells: involvement of integrin receptor α5β1 in the process. European Journal of Nutrition. 2011;50(6):465–478. doi: 10.1007/s00394-010-0158-z. [DOI] [PubMed] [Google Scholar]
- 41.Shen T.-L., Park A. Y.-J., Alcaraz A., et al. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. Journal of Cell Biology. 2005;169(6):941–952. doi: 10.1083/jcb.200411155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Topkoru B. C., Altay O., Duris K., Krafft P. R., Yan J., Zhang J. H. Nasal administration of recombinant osteopontin attenuates early brain injury after subarachnoid hemorrhage. Stroke. 2013;44(11):3189–3194. doi: 10.1161/strokeaha.113.001574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kusaka G., Kimura H., Kusaka I., Perkins E., Nanda A., Zhang J. H. Contribution of Src tyrosine kinase to cerebral vasospasm after subarachnoid hemorrhage. Journal of Neurosurgery. 2003;99(2):383–390. doi: 10.3171/jns.2003.99.2.0383. [DOI] [PubMed] [Google Scholar]
- 44.Milner R., Campbell I. L. Increased expression of the β4 and α5 integrin subunits in cerebral blood vessels of transgenic mice chronically producing the pro-inflammatory cytokines IL-6 or IFN-α in the central nervous system. Molecular and Cellular Neuroscience. 2006;33(4):429–440. doi: 10.1016/j.mcn.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimamura N., Matchett G., Solaroglu I., Tsubokawa T., Ohkuma H., Zhang J. Inhibition of integrin αvβ3 reduces blood-brain barrier breakdown in focal ischemia in rats. Journal of Neuroscience Research. 2006;84(8):1837–1847. doi: 10.1002/jnr.21073. [DOI] [PubMed] [Google Scholar]
- 46.Ellison J. A., Barone F. C., Feuerstein G. Z. Matrix remodeling after stroke: de novo expression of matrix proteins and integrin receptors. Annals of the New York Academy of Sciences. 1999;890(1):204–222. doi: 10.1111/j.1749-6632.1999.tb07996.x. [DOI] [PubMed] [Google Scholar]
- 47.Ellison J. A., Velier J. J., Spera P., et al. Osteopontin and its integrin receptor αvβ3Are upregulated during formation of the glial scar after focal stroke. Stroke. 1998;29(8):1698–1707. doi: 10.1161/01.str.29.8.1698. [DOI] [PubMed] [Google Scholar]
- 48.Farizatto K. L. G., Almeida M. F., Long R. T., Bahr B. A. Early synaptic alterations and selective adhesion signaling in hippocampal dendritic zones following organophosphate exposure. Science Reports. 2019;9(1):p. 6532. doi: 10.1038/s41598-019-42934-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Santos A. R., Corredor R. G., Obeso B. A., Trakhtenberg E. F., Wang Y., Ponmattam J. β1 integrin-focal adhesion kinase (FAK) signaling modulates retinal ganglion cell (RGC) survival. PLoS One. 2012;7(10) doi: 10.1371/journal.pone.0048332.e48332 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data are available upon request to the corresponding author.