

Abstract
Neuroinflammation triggered by the expression of damaged-associated molecular patterns released from dying cells plays a critical role in the pathogenesis of ischemic stroke. However, the benefits from the control of neuroinflammation in the clinical outcome have not been established. In this study, the effectiveness of intranasal, a highly efficient route to reach the central nervous system, and intraperitoneal dexamethasone administration in the treatment of neuroinflammation was evaluated in a 60-min middle cerebral artery occlusion (MCAO) model in C57BL/6 male mice. We performed a side-by-side comparison using intranasal versus intraperitoneal dexamethasone, a timecourse including immediate (0 h) or 4 or 12 h poststroke intranasal administration, as well as 4 intranasal doses of dexamethasone beginning 12 h after the MCAO versus a single dose at 12 h to identify the most effective conditions to treat neuroinflammation in MCAO mice. The best results were obtained 12 h after MCAO and when mice received a single dose of dexamethasone (0.25 mg/kg) intranasally. This treatment significantly reduced mortality, neurological deficits, infarct volume size, blood–brain barrier permeability in the somatosensory cortex, inflammatory cell infiltration, and glial activation. Our results demonstrate that a single low dose of intranasal dexamethasone has neuroprotective therapeutic effects in the MCAO model, showing a better clinical outcome than the intraperitoneal administration. Based on these results, we propose a new therapeutic approach for the treatment of the damage process that accompanies ischemic stroke.
Electronic supplementary material
The online version of this article (10.1007/s13311-020-00884-9) contains supplementary material, which is available to authorized users.
Keywords: Dexamethasone, ischemic stroke, inflammation, intranasal administration, MCAO
Introduction
The World Health Organization estimates that strokes affect 15 million people worldwide every year and that more than 80% of stroke cases are ischemic [1]. Of these, 5 million patients die, whereas survivors remain affected with different degrees of disabilities depending on the area and the extent of damage to the brain [2, 3].
Brain tissue injury after an ischemic stroke results in the expression of damage-associated molecular patterns, which release inflammatory cytokines and activate the microglia that migrate to the damaged area to remove cellular debris and restore tissue homeostasis [4]. The sustained neuroinflammation (NI) exacerbates the neuronal injury, favors edema, and disrupts the blood–brain barrier (BBB), leading to clinical deterioration, and eventually death [5].
Currently, restoring blood flow to ischemic tissues by using the tissue plasminogen activator (rtPA) remains the only available pharmacologic therapy to improve the outcome of ischemic stroke. This treatment is costly and only modestly effective [6].
One alternative is to focus the treatment on controlling NI. Synthetic glucocorticoids (GC) stand out as the most potent anti-inflammatory therapeutic agents commonly used in the treatment for an array of inflammatory disorders [7]. More specifically, dexamethasone (DX) is known as one of the most potent prescribed GC, which also boasts minor side effects at low doses for brief periods. In addition, DX restores the overall structure and improves the tightness of the BBB [8]. However, their beneficial or detrimental effects in strokes remain uncertain. Indeed, GC administered before patients’ hospital admission has been associated with increased mortality, suggesting the protective role of acute NI in stroke [9]. The use of dexamethasone in stroke patients has been evaluated in different studies. In a review including a set of 8 studies comparing corticosteroids versus placebo or a control group in people with acute ischemic stroke, a great imprecision was observed [10]. The treatment could increase the odds of death by as much as 34% or reduce it by as much as 43%. The time window in which the treatment was applied as well as the magnitude of the damage induced by the stroke could underlie these differences. Indeed, corticosteroids could be beneficial in high-risk patients with large infarcts and much vasogenic edema [11], and ineffective or harmful among those with smaller infarcts and less vasogenic edema. Furthermore, these studies included patients within 48 h after the infarction, a time range long enough to be associated with an acute nondamage or harmful subacute inflammatory response. Another variable that can critically affect the efficacy of the treatment is the glucocorticoid dose and the administration regimen used. High and sustained doses can be counterproductive in old patients that frequently have other comorbidities. This was the case of a large randomized controlled trial that included over 10,000 patients treated with a high dose of corticosteroids for 14 days. Corticosteroid treatment was associated with a higher number of deaths from all causes and no reduction in the disability of survivors [12]. In summary, the time in which the treatment is applied, the dose and regimen used, and the degree of harm of the affected patients are fundamental variables for corticosteroid success. Finally, it is important to consider the route of administration. In all studies carried out to date, corticosteroids are administered intravenously, intramuscularly, or orally, all routes that required high doses to reach a therapeutic concentration in the central nervous system (CNS) with the negative side effects that this implies [7]. Compelling evidence suggests that this can be overcome by using the intranasal (IN) route of administration for DX treatment, which we observed to be more efficient than the intravenous option in the treatment of NI [13, 14].
The present study aimed to evaluate the effectiveness of the intranasal administration of DX in morbidity and mortality induced by middle cerebral artery occlusion (MCAO).
Materials and Methods
Mice
C57BL/6 male mice of 8 to 10 weeks of age were purchased from Charles River Laboratories (Wilmington, MA) and bred at the Instituto de Investigaciones Biomédicas. Mice were grown at 22 ± 3 °C with a 12/12-h light–dark cycle and free access to water and food. Animal handling and experimental procedures followed the Guidelines for Care and Use of Laboratory Animals published by the USA National Institutes of Health and the Guidelines of the Mexican Law for Animal Protection (Norma Oficial Mexicana NOM-062-ZOO-1999). All experimental procedures were approved by the Committee for the Use and Care of Laboratory Animals of the Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México (approval number ID: 231).
Middle Cerebral Artery Occlusion
Brain ischemia was induced using the intraluminal MCAO model [15, 16]. Initially, mice were anesthetized with 5% isoflurane in oxygen for 3 min. During surgery, anesthesia was maintained with 1.5 to 2.5% isoflurane in oxygen. A midline ventral neck incision was made, and the left common carotid artery and its proximal branches were isolated. A silicone-coated 6-0 nylon suture (Doccol Corp, Redlands, CA) was introduced via the arteriotomy in the external carotid artery and slowly advanced through the left internal carotid artery to reach the origin of the middle cerebral artery. The nylon suture was removed after a 60-min occlusion, and the external carotid artery was ligated permanently. Sham-operated animals underwent the same surgical procedure, with the exception that the filament was not advanced beyond the middle cerebral artery. After surgery, all mice received intraperitoneal (IP) buprenorphine as analgesia (0.03 mg/kg). Animals that died of anesthetic, procedural problems during surgeries, or without neurological deficit after operation were excluded from the study.
Experimental Groups and Drug Administration
Mice were randomly assigned to the experimental groups and quantified by investigators blinded to treatment. The first 3 experiments were performed to identify the most promising conditions to treat MCAO mice, e.g., a side-by-side comparison using IN-DX versus IP-DX, a timecourse including immediate (0 h) and 4 and 12 h poststroke and 1 versus 4 IN daily doses of DX beginning 12 h after MCAO.
Based on the results of the above experiments, MCAO mice were randomly divided into 2 groups: MCAO + 1 IN dose of DX (0.25 mg/kg) in 20 μl saline 12 h after artery occlusion or MCAO + IN-saline in 20 μl at the same timepoint. Sham-operated mice did not receive any DX or saline after surgery. Mice were caged in polysulfone boxes with food and water ad libitum before and during the experiments. The body weight, mortality, and neurological deficits were evaluated daily 7 days after surgery. The surviving animals were euthanized with a lethal intraperitoneal injection of 90 to 100 mg/kg ketamine and 10 mg/kg xylazine. Similar groups of mice were sacrificed at 24 h to evaluate the effect of IN-DX in a subacute phase of the ischemic stroke or evaluated weekly up to 42 days after MCAO.
Neurological Test
Neurological deficit was assessed daily for 7 days, or weekly up to 42 days after MCAO using a modified 7-point neuroscore scale [17]. The neurological severity score was graded as follows:
0, no observable deficit
1, failure to extend the contralateral forepaw
2, mild circling behavior when picked up by the tail, < 50% attempts to rotate to the contralateral side
3, mild consistent circling, > 50% attempts to rotate to the contralateral side
4, consistent and strong circling, the mouse holds a rotation position for more than 1 to 2 s, with its nose almost reaching its tail
5, severe rotation with falling in a direction contralateral to the infarct, loss of walking or righting reflex, and
6, depressed level of consciousness, comatose, or moribund
Cerebral Infarct Volume Evaluation
Seven days after MCAO, 4 mice randomly selected from the DX- or saline-treated groups and 3 sham mice were anesthetized with a lethal IP injection of 90 to 100 mg/kg ketamine and 10 mg/kg xylazine, and perfused transcardially with physiological saline solution (0.9%). Brains were extracted and cut into 4 coronal sections (2 mm thick each) using a mouse brain slicer (Stoelting, Wood Dale, IL) and stained with a 2% solution of 2,3,5-triphenyl tetrazolium chloride (TTC) (Sigma, St. Louis, MO) in phosphate-buffered saline at 37 °C for 30 min. Images of brain sections were obtained using a digital camera (Nikon Digital Sight DS-Ri1, Nikon, Tokyo, Japan). The infarct volume was quantified by the ImageJ digital analysis software using the Freehand selection tool (National Institutes of Health, Bethesda, MD). The infarct volumes corrected by edema were calculated (percentage of corrected infarct volume = (normal hemisphere volume − noninfarct volume of infarct side)/normal hemispheric volume) × 100 [18].
Blood–Brain Barrier Permeability Quantification
Randomly selected mice from the corresponding group were euthanized with a lethal IP injection of 90 to 100 mg/kg ketamine and 10 mg/kg xylazine. Briefly, 0.2 mL/100 g body weight of Evans blue was administrated in the left heart ventricle and circulated for 10 min [19]. After that, the animals were perfused with saline solution (0.9%), followed by 4% paraformaldehyde. Brains were postfixed overnight by immersion in 4% paraformaldehyde at 4 °C. Two-millimeter coronal sections were obtained, and slices were photographed without magnification with a digital camera (Nikon D3200). Evans blue extravasation was analyzed by measuring the mean optical density of the somatosensory cortex and striatum using ImageJ software. The quantification of the optical density using the calibrated optical density step tablet and the Rodbard function provided by ImageJ software, has been used before to study the BBB integrity [19, 20], and is based on previously validated studies in which the empirical relationship between the amount of Evans blue per unit area and the optical density was obtained [21].
Tissue Processing for Histopathological Analysis
At 24 h and 7 days, 3 mice of each group were anesthetized and perfused with physiological saline followed by 4% paraformaldehyde solution previously cooled to 4 °C. The brains were removed and postfixed in 4% formaldehyde solution for 24 h at 4 °C. After being dehydrated and embedded with paraffin, 10- and 20-μm coronal sections were taken for hematoxylin–eosin (H&E) and Nissl staining, respectively. The paraffin sections were deparaffined in xylene and rehydrated in gradient ethanol from 100 to 96% and stained with H&E to characterize tissue damage. 20 μm coronal sections were stained with Nissl staining to estimate neuronal density. Slides were examined and micrographs were taken in a digital camera attached to a light microscope (Olympus Microscope BX51 W1). Neuronal density was estimated from the striatum and the somatosensory cortex of ipsilateral and contralateral sides in 4 adjacent histological sections of each region (800 μm total distance). The number of viable cells was manually quantified with the Multi-point tool of the ImageJ software based on pathological changes. Well-rounded neurons with normal-looking nuclei were counted in areas of 0.2 mm2 in the 4 20-μm-thick coronal sections and were multiplied by 40, which corresponds to the number of slices that comprise 800 μm of depth. The value obtained is equivalent to the number of living cells in 0.16 mm3. The above estimation of the number or density of neurons was performed according to a previously described method [22].
Immunofluorescence Analysis
Immunohistological studies were performed by immunofluorescence following the protocol previously described [13]. More details are available in the Supplementary Materials and Methods.
Statistical Analysis
A 2-way ANOVA followed by Bonferroni’s multiple comparisons test was performed to determine significant differences between groups. Neuronal density was analyzed by Kruskal–Wallis, followed by Dunn’s multiple-comparisons test. A 1-way ANOVA, followed by Tukey’s multiple-comparisons test, was performed to determine significant differences for the blood–brain permeability and peaks from the area under the curve for the neuroscore scale. Mortality was analyzed performing a Mantel–Cox log–rank test. A 2-tailed unpaired t test analyzed the infarct volume. Tests were carried out using GraphPad Prism® 7.0 (GraphPad Software, San Diego, CA). Statistical significance for all tests was considered when p values were lower than 0.05. Data supporting the results are available to readers upon request to the corresponding author.
Results
One Single Dose of Dexamethasone Administered Intranasally 12 h after Stroke Improves Survival and Neurological Performance in MCAO Mice
The first three experiments were performed to assess the effectiveness of the intranasal route, the number of doses, the most appropriate administration time of DX treatment in MCAO mice on the survival, neurological deficit, and body weight. In the first experiment, we examined the comparative effectiveness in dose (IN vs IP); in the second, the number of doses (1 vs 4); and in the third, the effect of an intranasal dose of DX administered 12 h after occlusion versus 4 doses administered over 4 days beginning at 12 h after MCAO to improve survival, neurological deficit (neuroscore), and the body weight (an indicator of mice health) during the first 7 days after the stroke. As shown in Fig. 1A, no significant difference in percentage of survival was observed in the IN-DX group when compared to the IP-DX group (p = 0.0706). However, a statistically significant weight recovery at days 4, 5, and 7 (p = 0.0154, p = 0.0153, and p = 0.0138 respectively) and lower neurological deficits (p < 0.0001) were observed in those IN-DX versus IP-DX-treated mice. Statistical analysis also revealed significant differences in neurological deficits between DX and saline groups (Fig. 1A, p < 0.05). We next evaluated the effectiveness of a single dose compared to 4 doses of DX using these same metrics. We found that 4 doses administered every 24 h beginning 12 h after MCAO significantly increased mortality (p = 0.0033) and neurological deficits (p < 0.0001) and reduced the bodyweight compared to those that received only 1 dose (Fig. 1B). Next, we investigated whether timing of DX administration affected outcome measures, and we found that the treatment administered immediately (zero) or 4 h after MCAO did not modify the outcome of the stroke (Fig. 1C). Considering that the best results were obtained using 1 dose of IN-DX 12 h after MCAO, another series of experiments were performed to study long-term weight and survival, medium-term infarct volume, BBB measurements, and histological assessments (Fig. 2A). The follow-up work found a higher percentage of survival in DX-treated (69%) versus saline-treated mice (36%) 42 days after MCAO (Fig. 2B, p = 0.011). As expected, all mice lost body mass during the 3 days postsurgery. Sham animals gained body mass at a significantly faster rate compared to MCAO mice, but DX-treated MCAO mice more effectively recovered weight compared to those that were saline-treated (Fig. 2C). There was no sign of neurological deficit observed in sham-operated mice. DX-treated MCAO mice showed lower neurological deficits compared to saline-treated mice (Fig. 2D, p < 0.0001).
Fig. 1.
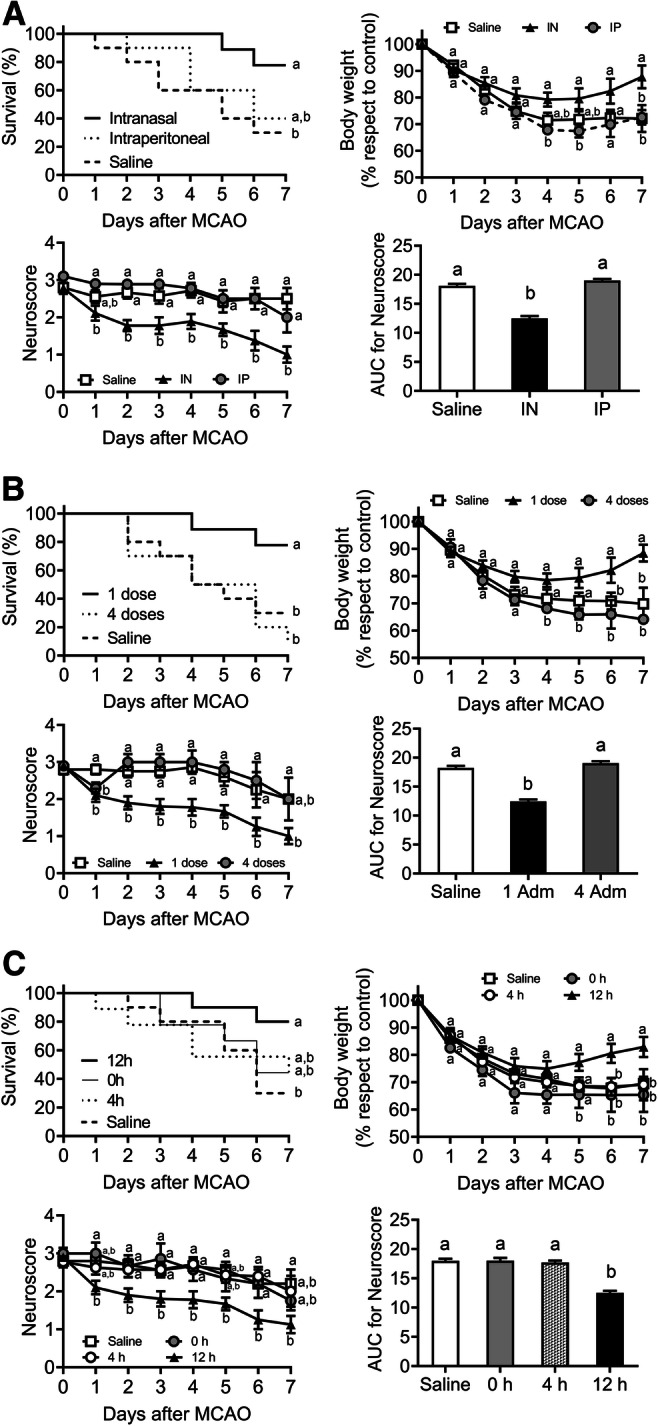
One single dose of dexamethasone (DX) intranasally administered 12 h after stroke improves survival and neurological performance in MCAO mice. Intranasal (IN) versus intraperitoneal (IP) DX routes of administration were compared at 12 h after MCAO. Each mouse was treated with 0.25 mg/kg of DX by the IN or IP route or received 20 μl IN saline solution (A). A single dose of 0.25 mg/kg of DX administered 12 h after MCAO was compared with 4 repeated administrations every 24 h starting at 12 h (B), and with single administrations at 0 and 4 h after the insult (C) (n = 10 per group). The percentage of survival, body weight, and the area under the curve (AUC) for neuroscore were determined daily for 7 days after MCAO. Data represent the mean ± SEM. Survival was analyzed by the Mantel–Cox log–rank test; the percentage of body weight loss was analyzed by 2-way ANOVA, followed by Bonferroni’s multiple-comparisons test. The AUC was calculated averaging neuroscore scale for 7 days and peaks were compared by 1-way ANOVA, followed by Tukey’s multiple-comparisons test. Different letters (a and b) indicate significant differences between groups (p < 0.05)
Fig. 2.

Dexamethasone (DX) treatment decreased mortality and improved the neuroscore and weight at 7 days and promotes long-term recovery after MCAO. Experimental design line (A). Twelve hours after MCAO, each mouse was treated with 0.25 mg/kg DX or 20 μl saline solution by the intranasal route. The percentage of survival (B), body weight (C), and area under the curve (AUC) for neuroscore (D) were determined daily for 7 days after MCAO. From mice that survived 7 days after the MCAO, a group of animals (n = 10 per group) were evaluated for an extra period of 42 days (6 weeks) (A, B, and C). Data represent the mean ± SEM. Survival was analyzed by the Mantel–Cox log–rank test; the percentage of body weight loss was analyzed by 2-way ANOVA, followed by Bonferroni’s multiple-comparisons test. The AUC was calculated averaging the neuroscore scale for 7 days, and peaks were compared by 1-way ANOVA, followed by Tukey’s multiple-comparisons test. Different letters (a, b, and c) indicate significant differences in the survival, body weight, and neuroscore between sham, treated, and nontreated mice (p < 0.05)
Dexamethasone Treatment Reduces Brain Damage Induced by Ischemic Stroke
To correlate if the clinical improvement with DX-treated mice was related to a decrease in brain damage, we evaluated brain infarct volume and BBB permeability changes 7 days after MCAO. In the brains of sham mice, ischemic areas were not observed. The brains of MCAO mice showed extensive areas of ischemic damage in the somatosensory cortex and the striatum, areas of the brain that are supplied by the middle cerebral artery (Fig. 3A). As observed in Fig. 3, intranasal dexamethasone treatment reduced the total volume of infarction (Fig. 3B, p = 0.0198). This reduction is caused mainly by the effect observed in the striatum (Fig. 3D, p = 0.0049) and not in the cortex (Fig. 3C, p = 0.2326).
Fig. 3.

Intranasal dexamethasone (DX) reduced blood–brain barrier permeability and infarct volume 7 days after the MCAO. Coronal brain sections of the sham-, saline-, and DX-treated groups are shown (A). Images correspond to the 4 saline- and DX-treated mice presented in vertical columns separated by pointed lines. The total infarction volume (B), and infarct volume in the cortex (C) and striatum (D) were quantified using the Image J software. Representative coronal brain slices of Evans blue extravasation evaluated in the cortex and striatum (marked with dotted lines) of the sham, saline, and DX groups are shown (E). Quantification of Evans blue is shown as relative units of optical density in the cortex (F) and striatum (G) from sham-, saline-, or DX-treated mice. Each point represents the data of an individual mouse (n = 4 per group). Data represent the mean ± SEM. Data were analyzed by a 2-tailed unpaired t test (infarct volume) and 1-way ANOVA, followed by Tukey’s multiple-comparisons test (BBB permeability). Different letters (a, b, and c) indicate significant differences in the infarct volume and BBD permeability between treated and nontreated mice (p < 0.05)
As far as the BBB, assessed by Evans blue extravasation (Fig. 3E), the permeability was increased in the somatosensory cortex and the striatum of the saline-treated group (Fig. 3F, G p = 0.0006, p = 0.0040, respectively). In comparison with the saline-treated group, the IN-DX administration significantly reduced the BBB permeability in the somatosensory cortex (Fig. 3F, p = 0.0136), but no significant differences were found in the striatum (Fig. 3G). Additionally, in the DX-treated group, Evans blue extravasation was not significant in the somatosensory cortex and striatum areas when compared with the sham group (Fig. 3F, G).
In addition, we carried out a histopathological analysis of the brain damage induced by MCAO and the therapeutic effects of IN-DX at 24 h and 7 days after MCAO with H&E-stained paraffin sections (Fig. 4). A group of sham mice and 2 groups of mice subjected to MCAO were included. One group received an IN saline solution and the other 0.25 mg/kg of IN-DX. Sham mice presented no abnormalities of the somatosensory cortex and the striatum and the microscopic appearance of neurons, glial cells, gemistocytes, and astrocytes was unremarkable at 24 h and 7 d. (Fig. 4). In the somatosensory cortex and striatum of 24 h MCAO mice that were treated with IN saline, diffuse edema and increased cellularity were apparent (Fig. 4). The majority of neurons presented ischemic changes with eosinophilic cytoplasm and pyknotic and fragmented nuclei (Fig. 4, arrows). At 7 days, the interstitial edema was more intense, and areas of tissue disruption were observed in the cortex (Fig. 4, asterisk). Additionally, at this time, the nuclei of many cells were pyknotic and fragmented. Occasional inflammatory cells, such as macrophages and polymorphonuclear leukocytes, were observed in the ischemic brain. The IN-DX treatment markedly prevented the changes described above in both cortex and striatum more clearly on day 7 after MCAO (Fig. 4). The interstitial edema was no longer present, although neurons with pyknotic nuclei (Fig. 4, arrows) were still observed in the cortex and striatum. Glial cells with abundant vacuolated cytoplasm were also observed (Fig. 4, arrowheads).
Fig. 4.
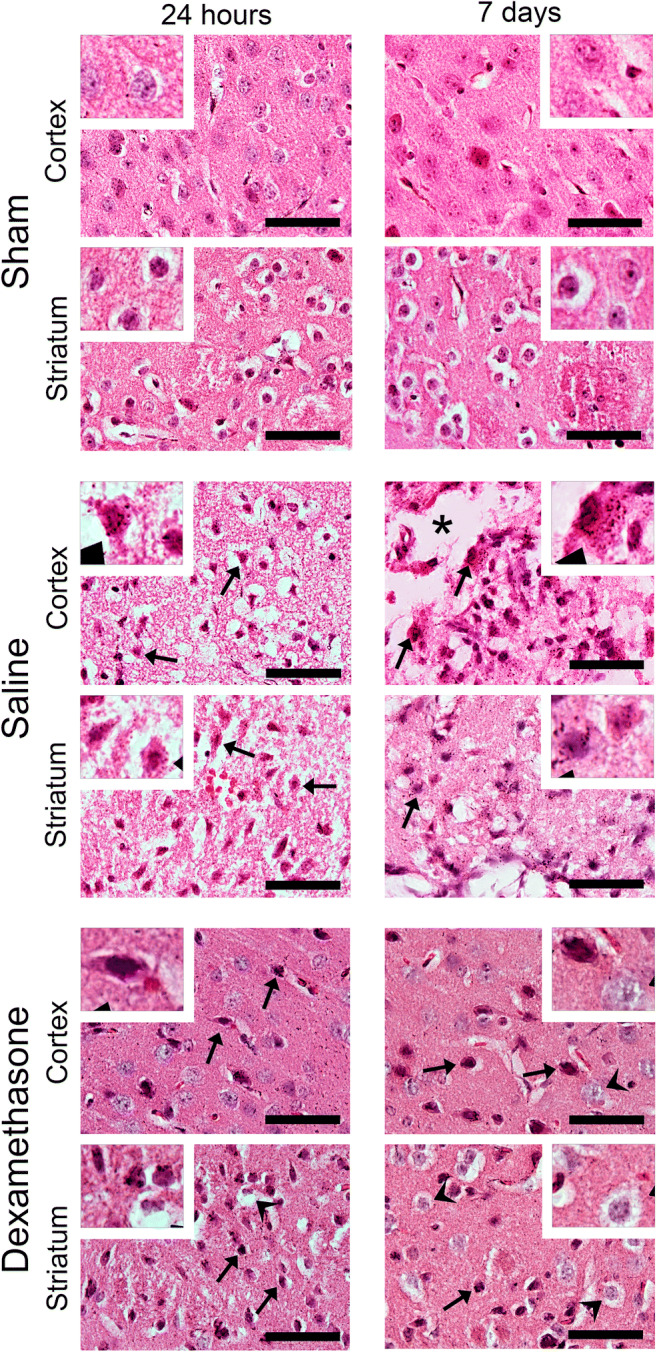
Intranasal dexamethasone reduces markedly the ischemic brain tissue damage in an MCAO model. Representative images of a brain section (ipsilateral hemisphere) stained with hematoxylin and eosin (H&E) of the sham-, saline-, or DX-treated groups are shown. Coronal sections of whole brains of sham mice and mice subjected to MCAO for 24 h and 7 days are observed. One control group was treated by the intranasal route with saline solution, and the other group received 0.25 mg/kg intranasal dexamethasone. For histologic examination, low (× 60)-magnification H&E-stained micrographs of the somatosensory cortex and striatum were examined for each animal (n = 3 per group). High-magnification images are shown in white boxes. Scale bar, 50 μm
Considering that neuronal death is observed during a stroke, the density of neurons in the cortex and the striatum at 24 h after MCAO was quantified (Fig. 5). There were no changes in neuronal density in the hemisphere contralateral to the arterial occlusion (Fig. 5B, C). In the ipsilateral hemisphere of the saline-treated mice, a significant reduction in neuronal density was observed in the somatosensory cortex and striatum compared to the sham group (Fig. 5B, Cp = 0.022, p = 0.034, respectively). In contrast, in the DX-treated mice, the neuronal density in the somatosensory cortex and striatum was similar to those of the saline and sham mice (Fig. 5B, C). After 7 days, neuronal density was quantified and histologic differences between groups were observed (Fig. S1). In the cortex and striatum of MCAO mice treated with saline, neuron depletion was observed, and the remaining neurons showed degenerative changes, including pyknotic nuclei. These changes were much less evident in mice treated with DX (Fig. S1).
Fig. 5.

Increased neuronal density in the somatosensory cortex and the striatum after intranasal dexamethasone (DX) treatment in the MCAO model. Representative image of a brain section stained with Nissl to illustrate where neuronal quantifications were analyzed (A). Representative images of Nissl staining of the somatosensory cortex and striatum from the sham-, saline-, and DX-treated mice after 24 h of performing MCAO (B). Quantification of the neuronal density of 4 adjacent histological sections of 20 μm (800 μm total distance) stained with Nissl in the cortex and striatum from the ipsilateral (Ips) and contralateral (Cont) hemisphere of the sham, saline, or DX animals (C). Each point represents the data of an individual mouse (n = 3 per group). Data represent the mean ± SEM. Data were analyzed by a Kruskal–Wallis, followed by Dunn’s multiple-comparisons test. Different letters (a, b, and c) indicate significant differences in the neuronal density between groups (p < 0.05). Scale bar, 50 μm
Dexamethasone Treatment Diminishes the Glial Response 7 Days after MCAO
During a stroke, microglia/macrophages and astrocytes are activated [4]. To test if IN-DX administration reduced the activation of these cells, we quantified Iba-1 (whose overexpression allows differentiation between microglial cells engaged in routine surveillance and those which are activated in response to injury) and GFAP (a marker of activated astrocytes) after artery occlusion. DX treatment did not change the astrocytes or microglia response at 24 h significantly (Fig. S2). Nevertheless, at 7 days post-MCAO, a more pronounced Iba-1 and GFAP expression was observed compared to 24 h (Fig. 6 and S2). Both MCAO saline- and DX-treated groups presented a significantly higher microglia and astrocyte response in both affected areas (Fig. 6, p < 0.0001). The saline group displayed more reactive astrocytes and ameboid cells in both affected structures when compared with the DX group (Fig. 6A); also, the intensity of the astrocyte response seemed to be higher in the striatum than in the cortex. The DX group showed more ramified microglia in the somatosensory cortex and striatum when compared to the saline group (Fig. 6A). Furthermore, the DX-treated group presented significantly fewer microglia cells (Fig. 6B, cortex p = 0.0033, striatum p = 0.0001) and astrocytes (Fig. 6C, cortex and striatum p < 0.0001) than the saline-treated group. Interestingly, in a parallel immunofluorescence study, we found that IN-DX accumulated in the meninges of healthy mice in both hemispheres, but the GC was directed towards the area affected by cerebral infarction in MCAO mice; notably, it was absent in the contralateral hemisphere (Fig. S3).
Fig. 6.

Analysis of brain GFAP (green) and Iba-1 (red) expression in treated and nontreated mice 7 days after the MCAO. Nuclei were stained with DAPI (blue). Representative mouse brain sections of the sham-, saline-, and dexamethasone (DX)-treated mice (A). Left images show a low-magnification reconstruction of the ipsilateral hemisphere. The mean fluorescence intensity (MFI) of Iba-1 (B) and GFAP (C) were quantified using the Image J software. Each point represents the data from 1 of 3 photographs analyzed in both the cortex and striatum (n = 3 animals per group). Data represent the mean ± SEM. Data were analyzed with 2-way ANOVA followed by Bonferroni’s test. Different letters (a, b, and c) indicate significant differences in the MFI between groups (p < 0.05). Scale bar, 200 μm
Discussion
In this study, we explore the use of DX, a potent anti-inflammatory synthetic GC [13], IN versus IP administered and at different times and doses in the murine MCAO model. As Fig. 1A shows, a better outcome of the stroke was observed in those IN-DX-treated mice, which could reflect that the intranasal administration route provides a more direct delivery of the drug than the systemic route, allowing it to reach higher central and lower systemic concentrations than the oral or parenteral routes [23, 24] and greater efficiency in transport glucocorticoids directly through the olfactory and the trigeminus nerves [14, 25] overpassing the BBB. A worse performance was observed in those mice that received 4 versus 1 dose of DX, which may be the result of neural damage due to an overexposure of GC [26]. No significant effects were observed when DX was employed immediately or 4 h after MCAO, which may indicate that suppression of inflammation during the repair phase is significant and that suppression of acute inflammation does not affect outcome. This is important because NI participates with active intrinsic repair responses [27]. Available evidence suggests that early NI can restore CNS homeostasis in stroke [28–30]. Thus, the use of GC to alleviate NI should be considered in the subacute phase of the stroke, as it has been reported by Fu and colleagues [31] and carried out in this study. In accordance, a single low dose of IN-DX administered 12 h after stroke significantly reduced mortality and morbidity 42 days after MCAO. The highest mortality rate in non-IN DX groups was observed during the first 7 days after MCAO. Indeed, this high mortality is similar to that reported by other authors in which the same model was used [32, 33]. Other long-term experiments show that the largest number of animals also dies during the first week after MCAO, and that mortality is lower thereafter in the following weeks, as in our work [34, 35]. The treatment also decreased the infarct volume significantly. A unique dose of 0.25 mg/kg was employed since it is known that low doses of GC are more beneficial, preserving the physiological metabolism of the neurons and the HPA axis [26]. This is the proposed dose for the treatment of the acute respiratory distress syndrome (ARDS) [36], to reduce postsurgical pain in children [37] and to improve the pulmonary outcome in intubated very low birth weight infants [38]. Lower doses are used to treat acute asthma exacerbations in children [39] and much higher for systemic lupus [40]. IN-DX administered at 0.25 mg/kg has higher effectiveness than intravenous to control NI [13]. Moreover, we demonstrated that the IN administration gives a much higher brain or cerebrovascular load of the drug than intravenous [14]. The dose of 0.25 mg/kg is extensively employed intravenously to control NI in different pathologies in humans [36]. Thus, this dose has been also employed in this study.
Disruption of the BBB plays an important role in the development of neurological dysfunction in ischemic stroke, triggering edema, and it can be secondary to the activation of microglia and astrocytes by promoting tissue damage into the ischemic brain [41]. Postischemic breakdown of the BBB can last for hours and is a central event of postischemic inflammation [42]. DX treatment may improve the BBB integrity by preventing the infiltration of peripheral inflammatory cells, probably by maintaining tight junctions between endothelial cells [43]. DX has a known effect on cells of the immune system [44], and is also known to decrease endothelial permeability [45]. The decreases in the BBB leakiness may be the consequence of the reduction of the infarct volume or the direct effect of DX on the integrity of the endothelial barrier. Both mechanisms could be involved which are not distinguished in this study.
The effect of a single low dose of DX was remarkable. Histologically, it preserved the brain tissue and decreased the neuronal damage in the somatosensory cortex and striatum. It also markedly reduced the activation of astrocytes and microglia, the main inflammatory cells in the CNS. Indeed, activation of astrocytes and microglia, the major inflammatory cell types in the CNS, was reduced 7 days after stroke in DX-treated mice, possibly by the recognition of nuclear receptors through the mechanisms that have been extensively reported [4]. GC such as DX have been reported to exert significant effects on macrophage function and differentiation [46–48], promoting M2 macrophages which may play a critical role in tissue recovery after cerebral vascular event [49]. In this sense, it would be very useful to study markers of immune cell polarization in the brains to assess its relevance in the recovery of treated mice. The high effectiveness of the treatment could also be associated with an increased uptake of DX into ischemic brain areas revealed by immunohistochemistry. A similar distribution has been observed after intravenous injection of DX in experimental edema using more massive ischemic procedures such as the occlusion of the right carotid artery and cold injury of the right temporal lobe [50]. The accumulation of DX in the ischemic brain tissue could be an essential factor in the better neurological outcome of treated mice.
Conclusion
A single IN low dose of DX in mice with ischemic damage due to MCAO has beneficial effects attenuating the inflammatory responses, improving neurological performance, and significantly reducing mortality. Altogether, our results provide experimental evidence that supports a novel, noninvasive approach for the treatment of patients with ischemic stroke.
Electronic Supplementary Material
(PDF 1224 kb)
(PDF 553 kb)
Acknowledgments
The authors thank Omar Noel Medina-Campos, Alonso Reyes, Pedro Medina, Dunia Rassy, Patricia Espinosa, Marisela Hernández, and Manuel Giraldo for their technical support, and Daniel Garzón, Jorge Omar García, and Georgina Díaz for the assistance with animal care.
Required Author Forms
Disclosure forms provided by the authors are available with the online version of this article.
Funding Information
Alejandro Espinosa is a doctoral student from Programa de Doctorado en Ciencias Biológicas, Universidad Nacional Autónoma de México, and received a fellowship CVU699520 from CONACYT. This work was supported by DGAPA-UNAM (IN207720 to E.S.) and DGAPA-UNAM (IN211917 to E.S.) Mexico. This study was also supported by the institutional program “Programa de Investigación para el Desarrollo y la Optimización de Vacunas, Inmunomoduladores y Métodos Diagnósticos del IIB” (PROVACADI).
Compliance with Ethical Standards
Conflict of Interest
The authors declare that they have no conflict of interest.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Institute for Health Metrics and Evaluation (IHME) Findings from the Global Burden of Disease Study 2017. Seattle, WA: IHME; 2018. [Google Scholar]
- 2.López-Espuela F, Pedrera-Zamorano JD, Jiménez-Caballero PE, et al. Functional status and disability in patients after acute stroke: a longitudinal study. Am J Crit Care. 2016;25:144–151. doi: 10.4037/ajcc2016215. [DOI] [PubMed] [Google Scholar]
- 3.Noe-Sebastian E, Balasch-Bernat M, Colomer-Font C, et al. Disability after stroke: a longitudinal study in moderate and severe stroke patients included in a multidisciplinary rehabilitation program. Rev Neurol. 2017;64:385–392. [PubMed] [Google Scholar]
- 4.Gülke E, Gelderblom M, Magnus T. Danger signals in stroke and their role on microglia activation after ischemia. Ther Adv Neurol Disord. 2018;11:1756286418774254. doi: 10.1177/1756286418774254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Patel AR, Ritzel R, McCullough LD, Liu F. Microglia and ischemic stroke: a double-edged sword. Int J Physiol Pathophysiol Pharmacol. 2013;5:73–90. [PMC free article] [PubMed] [Google Scholar]
- 6.Xian Y, Federspiel JJ, Grau-Sepulveda M, et al. Risks and benefits associated with prestroke antiplatelet therapy among patients with acute ischemic stroke treated with intravenous tissue plasminogen activator. JAMA Neurol. 2016;73:50–59. doi: 10.1001/jamaneurol.2015.3106. [DOI] [PubMed] [Google Scholar]
- 7.Tuckermann JP, Kleiman A, McPherson KG, Reichardt HM. Molecular mechanisms of glucocorticoids in the control of inflammation and lymphocyte apoptosis. Crit Rev Clin Lab Sci. 2005;42:71–104. doi: 10.1080/10408360590888983. [DOI] [PubMed] [Google Scholar]
- 8.Blecharz KG, Drenckhahn D, Förster CY. Glucocorticoids increase VE-cadherin expression and cause cytoskeletal rearrangements in murine brain endothelial cEND cells. J Cereb Blood Flow Metab. 2008;28:1139–1149. doi: 10.1038/jcbfm.2008.2. [DOI] [PubMed] [Google Scholar]
- 9.Sundbøll J, Horváth-Puhó E, Schmidt M, et al. Preadmission use of glucocorticoids and 30-day mortality after stroke. Stroke. 2016;47:829–835. doi: 10.1161/STROKEAHA.115.012231. [DOI] [PubMed] [Google Scholar]
- 10.Sandercock PA, Soane T. Corticosteroids for acute ischaemic stroke. Cochrane Database Syst Rev. 2011;2011(9):CD000064. doi: 10.1002/14651858.CD000064.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulley G, Wilcox RG, Mitchel JR. Dexamethasone in acute stroke. Br Med J. 1979;1:753–754. doi: 10.1136/bmj.1.6165.753-e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Edwards P, Arango M, Balica L, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury - outcomes at 6 months. Lancet. 2005;365:1957–1959. doi: 10.1016/S0140-6736(05)66552-X. [DOI] [PubMed] [Google Scholar]
- 13.Meneses G, Gevorkian G, Florentino A, et al. Intranasal delivery of dexamethasone efficiently controls LPS-induced murine neuroinflammation. Clin Exp Immunol. 2017;190:304–314. doi: 10.1111/cei.13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Meneses G, Cárdenas G, Espinosa A, et al. Sepsis: developing new alternatives to reduce neuroinflammation and attenuate brain injury. Ann N Y Acad Sci. 2019;1437:43–56. doi: 10.1111/nyas.13985. [DOI] [PubMed] [Google Scholar]
- 15.Koizumi J, Yoshida Y, Nakazawa T, Ooneda G. Experimental studies of ischemic brain edema. I. A new experimental model of cerebral embolism in rats in which recirculation can be introduced in the ischemic area. Jpn J Stroke. 1986;8:1–8. [Google Scholar]
- 16.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 17.Rousselet E, Kriz J, Seidah NG. Mouse model of intraluminal MCAO: cerebral infarct evaluation by cresyl violet staining. J Vis Exp. 2012;69:e4038. doi: 10.3791/4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McBride DW, Klebe D, Tang J, Zhang JH. Correcting for brain swelling’s effects on infarct volume calculation after middle cerebral artery occlusion in rats. Transl Stroke Res. 2015;6:323–338. doi: 10.1007/s12975-015-0400-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hurtado-Alvarado G, Domínguez-Salazar E, Velázquez-Moctezuma J, Gómez-González B. A2AAdenosine receptor antagonism reverts the blood-brain barrier dysfunction induced by sleep restriction. PLoS One. 2016;11:e0167236. doi: 10.1371/journal.pone.0167236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gómez-González B, Hurtado-Alvarado G, Esqueda-León E, Santana-Miranda R, Rojas-Zamorano JA, Velázquez-Moctezuma J. REM sleep loss and recovery regulates blood-brain barrier function. Curr Neurovasc Res. 2013;10:197–207. doi: 10.2174/15672026113109990002. [DOI] [PubMed] [Google Scholar]
- 21.Fry DL. Aortic Evans blue dye accumulation: its measurement and interpretation. Amer J Physiol. 1977;232:H204–H222. doi: 10.1152/ajpheart.1977.232.2.H204. [DOI] [PubMed] [Google Scholar]
- 22.García-Cabezas MA, John YJ, Barbas H, Zikopoulos B. Distinction of neurons, glia and endothelial cells in the cerebral cortex: an algorithm based on cytological features. Front Neuroanat. 2016;10:107. doi: 10.3389/fnana.2016.00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ducharme N, Banks WA, Morley JE, et al. Brain distribution and behavioral effects of progesterone and pregnenolone after intranasal or intravenous administration. Eur J Pharmacol. 2010;641:128–34. doi: 10.1016/j.ejphar.2010.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen H, Chen CC, Acosta C, Wu SY, Sun T, Konofagou EE. A new brain drug delivery strategy: focused ultrasound-enhanced intranasal drug delivery. PLOS ONE. 2014;9:e108880. doi: 10.1371/journal.pone.0108880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lochhead JJ, Thorne RG. Intranasal delivery of biologics to the central nervous system. Adv Drug Deliv Rev. 2012;64:614–628. doi: 10.1016/j.addr.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 26.Abrahám IM, Meerlo P, Luiten PG. Concentration dependent actions of glucocorticoids on neuronal viability and survival. Dose Response. 2006;4:38–54. doi: 10.2203/dose-response.004.01.004.Abraham. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DiSabato D, Quan N, Godbout JP. Neuroinflammation: the devil is in the details. J Neurochem. 2016;139:136–153. doi: 10.1111/jnc.13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cramer SC. Treatments to promote neural repair after stroke. J Stroke. 2018;20:57–70. doi: 10.5853/jos.2017.02796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin R, Yang G, Li G. Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J Leukoc Biol. 2010;87:779–789. doi: 10.1189/jlb.1109766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vidale S, Consoli A, Arnaboldi M, Consoli D. Postischemic inflammation in acute stroke. J Clin Neurol. 2017;13:1–9. doi: 10.3988/jcn.2017.13.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fu Y, Zhang N, Ren L, et al. Impact of an immune modulator fingolimod on acute ischemic stroke. Proc Natl Acad Sci U S A. 2014;111:18315–18320. doi: 10.1073/pnas.1416166111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Groger M, Lebesgue D, Pruneau D, et al. Release of bradykinin and expression of kinin B2 receptors in the brain: role for cell death and brain edema formation after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2005;25:978–989. doi: 10.1038/sj.jcbfm.9600096. [DOI] [PubMed] [Google Scholar]
- 33.Ma T, Shi YL, Wang YL. Forsythiaside A protects against focal cerebral ischemic injury by mediating the activation of the Nrf2 and endoplasmic reticulum stress pathways. Mol Med Rep. 2019;20:1313–1320. doi: 10.3892/mmr.2019.10312. [DOI] [PubMed] [Google Scholar]
- 34.Hase Y, Okamoto Y, Fujita Y, et al. Cilostazol, a phosphodiesterase inhibitor, prevents no-reflow and hemorrhage in mice with focal cerebral ischemia. Exp Neurol. 2012;233:523–533. doi: 10.1016/j.expneurol.2011.11.038. [DOI] [PubMed] [Google Scholar]
- 35.Romer C, Engel O, Winek K, et al. Blocking stroke-induced immunodeficiency increases CNS antigen-specific autoreactivity but does not worsen functional outcome after experimental stroke. J Neurosci. 2015;35(20):7777–7794. doi: 10.1523/JNEUROSCI.1532-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Villar J, Ferrando C, Martínez D, et al. Dexamethasone in ARDS network. Dexamethasone treatment for the acute respiratory distress syndrome: a multicentre, randomised controlled trial. Lancet Respir Med. 2020;8:267–276. doi: 10.1016/S2213-2600(19)30417-5. [DOI] [PubMed] [Google Scholar]
- 37.Salami OF. Amanor-Boadu SD, Eyelade OR, Olateju SO. Effects of low-dose intravenous dexamethasone combined with caudal analgesia on post-herniotomy pain. Niger Postgrad Med J. 2017;24:230–235. doi: 10.4103/npmj.npmj_120_17. [DOI] [PubMed] [Google Scholar]
- 38.McEvoy C, Bowling S, Williamson K, McGaw P, Durand M. Randomized, double-blinded trial of low-dose dexamethasone: II. Functional residual capacity and pulmonary outcome in very low birth weight infants at risk for bronchopulmonary dysplasia. Pediatr Pulmonol. 2004;38:55–63. doi: 10.1002/ppul.20037. [DOI] [PubMed] [Google Scholar]
- 39.Keeney GE, Gray MP, Morrison AK, et al. Dexamethasone for acute asthma exacerbations in children: a meta-analysis. Pediatrics. 2014;133:493–499. doi: 10.1542/peds.2013-2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Waselchuk EA, Hildrew DM, Winters RD, Ellis MS. Intractable epistaxis and systemic lupus: high-dose intravenous pulse steroids. Am J Otolaryngol. 2014;35:236–238. doi: 10.1016/j.amjoto.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 41.Yang C, Hawkins KE, Dore S, Candelario-Jalil E. Neuroinflammatory mechanisms of blood-brain barrier damage in ischemic stroke. Am J Physiol Cell Physiol. 2018;316:C135–C153. doi: 10.1152/ajpcell.00136.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim JY, Kawabori M, Yenari MA. Innate inflammatory responses in stroke: mechanisms and potential therapeutic targets. Curr Med Chem. 2014;21:2076–2097. doi: 10.2174/0929867321666131228205146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hue CD, Cho FS, Cao S, Dale Bass CR, Meaney DF, Morrison B., 3rd Dexamethasone potentiates in vitro blood-brain barrier recovery after primary blast injury by glucocorticoid receptor-mediated upregulation of ZO-1 tight junction protein. J Cereb Blood Flow Metab. 2015;35:1191–1198. doi: 10.1038/jcbfm.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Giles AJ, Hutchinson MND, Sonnemann HM, et al. Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J Immunother Cancer. 2018;6:51. doi: 10.1186/s40425-018-0371-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oberleithner H, Reithmüller C, Ludwig T, et al. Differential action of steroid hormones on human endothelium. J Cell Sci. 2006;119:1926–1932. doi: 10.1242/jcs.02886. [DOI] [PubMed] [Google Scholar]
- 46.Lim HY, Müller N, Reichhardt HM. Glucocorticoids exert opposing effects on macrophage function dependent on their concentration. Immunology. 2007;122:47–53. doi: 10.1111/j.1365-2567.2007.02611.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xie Y, Tolmeijer S, Oskam JM, Tonkens T, Meijer AH, Schaaf MJM. Glucocorticoids inhibit macrophage differentiation towards a pro-inflammatory phenotype upon wounding without affecting their migration. Dis Model Mech. 2019;12:dmm037887. doi: 10.1242/dmm.037887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Desgeorges T, Caratti G, Mounier R, Tuckerman J, Chazaud B. Glucocorticoids shape macrophage phenotype for tissue repair. Front Inmunol. 2019;10:1591. doi: 10.3389/fimmu.2019.01591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kanazawa M, Ninomiya I, Hatakeyama M, Takahashi T, Shimohata T. Microglia and monocytes/macrophages polarization reveal novel therapeutic mechanism against stroke. Int J Mol Sci. 2017;18:2135. doi: 10.3390/ijms18102135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kostron H, Fischer J. Regional, cellular, and subcellular distribution of [3H]dexamethasone in rat brain edema. Surg Neurol. 1983;20:48–54. doi: 10.1016/0090-3019(83)90105-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 1224 kb)
(PDF 553 kb)