

Abstract
Mitochondrial DNA (mtDNA) mutations are closely implicated in the pathogenesis of multiple cancers, making circulating cell-free mtDNA (ccf-mtDNA) as a potential non-invasive tumor biomarker. However, an effective approach to comprehensively profile ccf-mtDNA mutations is still lacking. In this study, we first characterized ccf-mtDNA by low-depth whole-genome sequencing (WGS) and found that plasma DNA samples exhibited a dramatic decrease in mtDNA copy number when compared with fresh tumor tissues. Further analysis revealed that plasma ccf-mtDNA had a biased distribution of fragment size with a peak around 90 bp. Based on these insights, we developed a robust captured-based mtDNA deep-sequencing approach that enables accurate and efficient detection of plasma ccf-mtDNA mutations by systematic optimization of probe quantity and length, hybridization temperature, and PCR amplification cycles. Moreover, we found that placement of isolated plasma for 6 h at both 4°C and room temperature (RT) led to a dramatic decrease of ccf-mtDNA stability, highlighting the importance of proper plasma sample processing. We further showed that the optimized approach can successfully detect a substantial fraction of tumor-specific mtDNA mutations in plasma ccf-mtDNA specifically from hepatocellular carcinoma (HCC) patients but not from colorectal cancer (CRC) patients, suggesting the presence of a potential cancer-specific difference in the abundance of tumor-derived mtDNA in plasma.
Graphical Abstract
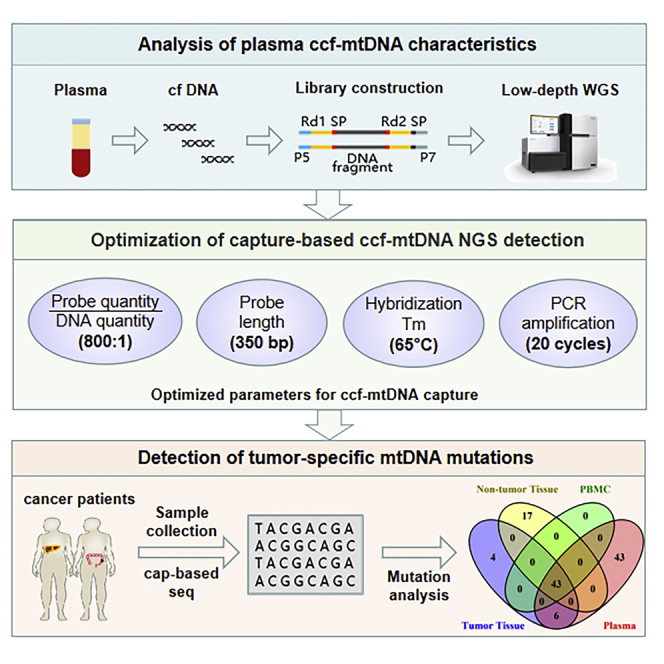
Based on the comprehensive characterization of plasma ccf-mtDNA, a robust capture-based mtDNA deep-sequencing approach was developed by optimization of probe quantity and length, hybridization temperature, and PCR amplification cycles. Applying this approach, abundant tumor-specific mtDNA mutations were successfully detected in plasma samples from HCC but not CRC patients.
Introduction
Human mitochondrial DNA (mtDNA) is a maternally inherited 16-kb genome, which is characterized as double-stranded circular DNA that encodes 13 essential components of the mitochondrial electron transport chain and ATP synthase.1 Due to the lack of protective histones and an inefficient DNA repair system, mtDNA exhibits a much higher mutation rate than nuclear DNA.2 As opposed to diploid nuclear DNA, the number of mtDNA copies fluctuates from several hundred to more than 10,000 according to cell types and pathological conditions.3 Consequently, the common coexistence of wild-type and mutant mtDNA in the same cell is defined as mitochondrial heteroplasmy.4 Mitochondrial dysfunctions, mainly mtDNA mutations occurring in the heteroplasmic state and mtDNA copy number variations, have been demonstrated to be strongly associated with tumor development and metastasis.5,6
The discovery of circulating cell-free DNA (ccf-DNA) in the plasma of cancer patients has opened up an exciting opportunity for non-invasive diagnostic and prognostic evaluation of cancer.7 Empowered by the rapid advances of next-generation sequencing (NGS), extensive studies have been attempted to identify tumor-specific mutations, copy number variations, chromosomal alterations, and fragmentation patterns in plasma ccf-DNA from cancer patients.8 Notably, most of these investigations have focused on ccf-DNA of nuclear origin (ccf-nDNA), but the abundance of tumor-derived ccf-nDNA is extremely low in many early stage and certain metastatic cancers.9,10 In addition, it has recently been found that leucocyte-derived mutations are unexpectedly pervasive in plasma ccf-nDNA, even accounting for more than 50% of total ccf-nDNA variants, which may greatly hinder its clinical application.11
Because of the high copy number and high mutation rate, circulating cell-free mtDNA (ccf-mtDNA) may hold great promise for a novel tumor biomarker.12,13 Unfortunately, our current understanding of the biological properties of plasma ccf-mtDNA is poor. Meanwhile, an accurate and efficient approach to comprehensively profile ccf-mtDNA mutations and copy numbers is still lacking. Traditional technologies, such as Sanger sequencing and high-resolution melt analysis, face the great limitations of sensitivity and throughput in identifying low heteroplasmic mtDNA mutations.14 Moreover, Weerts et al.15 recently reported limited ability in tracing tumor-specific ccf-mtDNA variants by single-molecule real-time sequencing, showing that most of the variants they identified from tumor tissues were undetectable in matched plasma mtDNA. Alternatively, although both capture and PCR-based strategies have been used to efficiently enrich mtDNA from genomic DNA of tissue samples,16 none of them is directly applicable to enrich ccf-mtDNA from plasma. Specifically, the highly fragmented nature of plasma DNA can impose great prohibition for PCR-based ccf-mtDNA enrichment while the atypical abundance of plasma mtDNA also renders capture-based enrichment suffering from low capture efficiency.17, 18, 19 Thus, it is of great importance to develop an NGS-based approach for accurate and highly efficient detection of plasma ccf-mtDNA, especially in cancer patients.
In the present study, we first comprehensively analyzed the basic characteristics of plasma ccf-mtDNA from both healthy and cancer patients using the whole-genome sequencing (WGS) approach. Based on these insights, we developed an optimized capture-based mtDNA deep-sequencing procedure enabling the accurate and efficient detection of ccf-mtDNA. Finally, we applied this innovative approach into the measurement of paired tumor, tumor-adjacent, peripheral blood and plasma samples, and successfully detected tumor-specific mtDNA mutations from plasma ccf-mtDNA of hepatocellular carcinoma (HCC) patients.
Results
Analysis of ccf-mtDNA copy number, fragment size, and breakpoint by low-depth WGS
To explore the clinical application potential of plasma ccf-mtDNA in various cancers, we first comprehensively analyzed the characteristics of ccf-mtDNA from 40 plasma samples (10 healthy subjects, 10 hepatitis patients, 10 HCC patients, and 10 colorectal cancer [CRC] patients) using low-depth WGS. All sequencing data are summarized in Table S1. For comparison, we also analyzed the mtDNA copy number in matched fresh tumor tissues for HCC and CRC patients. Based on low-depth WGS data, we calculated the mtDNA copy number and found that plasma samples exhibited a dramatic decrease in mtDNA copy number when compared with fresh tumor tissues (p < 0.001), with an average number of 5.05 (ranging from 0.76 to 27.83) and 935.40 (ranging from 139.52 to 3,170.49), respectively (Figure 1A). Also, we found that healthy subjects had significantly higher plasma ccf-mtDNA copy numbers than the other three groups (Figure S1A). Further analysis of fragment size distribution revealed that plasma ccf-mtDNA had a biased distribution of fragment size with peaks around 90 bp, and plasma ccf-DNA of nuclear origin had an approximately normal distribution of fragment size with peaks around 167 bp, consistent with the reported nucleosome length (Figure 1B).20 When median insert size was compared among four groups, we found that hepatitis patients had significantly longer insert size than did healthy subjects and HCC patients, and no difference of insert size was observed between healthy subjects and HCC patients, suggesting that inflammation may play a role in the distribution of ccf-mtDNA fragment size (Figure S1B). In addition, the ccf-mtDNA breakpoint, which is defined as the cleavage position of mtDNA fragments at the mitochondrial genome, seemed to be distributed randomly across the whole mitochondrial genome among all plasma samples (Figure 1C).
Figure 1.

Analysis of ccf-mtDNA copy number, fragment size, and breakpoint by low-depth whole genome sequencing (WGS)
(A) Comparison of mtDNA copy number between plasma (orange) and fresh tissues (purple). The significance was determined by a Student’s t test. (B) Fragment size distribution of ccf-mtDNA (orange) and ccf-DNA (purple) among healthy controls, hepatitis patients, HCC patients, and CRC patients. (C) Proportion of ccf-mtDNA breakpoints across the mitochondrial genome among healthy controls, hepatitis patients, HCC patients, and CRC patients. HC, heathy control; HCC, hepatocellular carcinoma; CRC, colorectal cancer.
Optimization of plasma ccf-mtDNA detection by capture-based mtDNA sequencing
The low abundance and unique size distribution of plasma ccf-mtDNA presented a great challenge for the WGS-based full characterization of ccf-mtDNA. To overcome this obstacle, based on insights gained from low-depth WGS analysis of plasma ccf-mtDNA, we turned to capture-based detection of tissue mtDNA as described in our previous study21 and performed a systematic optimization against probe quantity, probe length, probe-mtDNA hybridization, and post-capture amplification using 10 HCC plasma samples. The mtDNA mapping rate and sequencing depth were used for assessing mtDNA enrichment efficiency. As shown in Figure 2A, the highest mtDNA enrichment efficiency, with an average mtDNA mapping rate of 38% (ranging from 23% to 50%) and average mtDNA sequencing depth of 1,578× (ranging from 868 to 2,036), was achieved when using 0.8 ng of probe per 200 ng of the WGS library. In addition, we found that the enrichment efficiency can further be moderately improved by using gradually increased probe length (150, 250, and 350 bp) (p < 0.001, Figure 2B). Although previous reports suggested that hybridization at 55°C is more suitable for capture-based enrichment of ancient DNA,22 we found that hybridization at 65°C performed significantly better for enrichment of ccf-mtDNA from plasma samples (p < 0.001, Figure 2C). Finally, we showed that maximum enrichment of plasma ccf-mtDNA can be achieved by further setting 20 cycles of PCR amplification (all p < 0.001, Figure 2D), which does not affect the detection of homoplasmic and heteroplasmic mtDNA variants in each of 10 HCC samples (Table S2). Interestingly, we also observed a significant positive correlation between mtDNA copy number and enrichment efficiency (Spearman r = 0.952, p < 0.001; Figure S2). Taken together, these results identified probe quantity of 0.8 ng, probe length of 350 bp, probe-mtDNA hybridization at 65°C, and subsequent PCR amplification for 20 cycles as the optimal condition for detection of plasma ccf-mtDNA by captured-based mtDNA sequencing.
Figure 2.

Optimization of plasma ccf-mtDNA detection by capture-based mtDNA sequencing
(A–D) Comparison of mtDNA mapping rate and mtDNA sequencing depth among (A) different probe quantity, (B) different probe length, (C) different hybridization temperature, and (D) different PCR amplification cycles. The significance between different treatments was determined by a paired t test.
Robust detection of plasma ccf-mtDNA variants by optimized captured-based mtDNA sequencing
We next evaluated the performance of our optimized capture procedure for detecting plasma ccf-mtDNA using the same set of samples. Compared with the capture procedure that was routinely used for tissue DNA samples, the optimized procedure greatly improved both the mtDNA mapping rate and the mtDNA sequencing depth for plasma DNA samples (p < 0.001, Figures 3A and 3B). Furthermore, while both procedures detected similar numbers of homoplasmic mtDNA variants, the optimized procedure can detect significantly higher numbers of heteroplasmic mtDNA variants (p < 0.001, Figures 3C and 3D). Further analysis revealed that most of the mtDNA variants (92%) only detectable by the optimized procedure could be confirmed by replicate experiments (Figure 3E). Notably, most mtDNA variants only detectable by the optimized procedure had low heteroplasmic level (≤10%) and exhibited significant transition predominance (Figures 3F and 3G), consistent with the mtDNA mutation signature previously reported.5 Taken together, these data demonstrated that our optimized procedure greatly enhanced the specificity and efficiency of mtDNA capture from plasma DNA samples, thus allowing more accurate and sensitive detection of ccf-mtDNA mutations, especially for those with low heteroplasmic levels.
Figure 3.

Robust detection of plasma ccf-mtDNA variants by optimized captured-based mtDNA sequencing
(A–D) Comparison of (A) mtDNA mapping rate, (B) mtDNA sequencing depth, and (C) homoplasmic and (D) heteroplasmic mtDNA variant numbers between conventional capture-based sequencing procedure and the optimized capture-based sequencing procedure. Significance was determined by a paired t test. (E) Number of repeatable and unrepeatable mtDNA variants only detected by optimized capture-based sequencing. (F) Distribution of mtDNA variants only detected by optimized capture-based sequencing at different heteroplasmy level. (G) Substitution spectrum of mtDNA variants only detected by optimized capture-based sequencing.
Reliability evaluation of optimized capture-based ccf-mtDNA sequencing
We then assessed the reproducibility of the optimized capture-based NGS approach in detecting ccf-mtDNA by independently preparing another replicate sequencing library for each of the 10 plasma DNA samples from HCC patients. We found that all of the homoplasmic variants and the vast majority of the heteroplasmic variants exhibited an almost 100% consistency between two replicate experiments (Figure 4A). Even in four cases where the heteroplasmic variants showed relatively lower consistency, the most heteroplasmic variants could still be confidently reproduced (Figure 4B). Furthermore, the optimized procedure exhibited very high consistency in determining the exact heteroplasmy levels (Spearman r = 0.986; Figure 4C). In addition, the mutation spectrum of heteroplasmic variants in both replicates showed highly similar transition predominance, consistent with the established mtDNA mutation signatures5 (Figure 4D).
Figure 4.

Reproducibility of captured-based mtDNA sequencing in detecting plasma ccf-mtDNA
(A) Consistency of homoplasmic and heteroplasmic mtDNA variants between two repeat experiments using 10 HCC plasma samples. (B) Comparison of heteroplasmic mtDNA variants in HCC plasma samples with lower consistency. (C) Correlation of mtDNA heteroplasmy level of all mtDNA variants between two repeat experiments. The significance of correlation was determined by Spearman’s rank correlation coefficient. (D) Percentage of heteroplasmic mtDNA variants with different substitution category of two repeat experiments. Rep, replicate.
To further analyze whether the optimized procedure may generate potential capture-related bias, we compared the basic features of plasma ccf-mtDNA between data from optimized capture sequencing and WGS. To our satisfaction, both analyses revealed a highly consistent portrait of plasma ccf-mtDNA in terms of mtDNA copy number, fragment size, and distribution of fragment ends across the whole mitochondrial genome (Figure 5). Overall, these data demonstrate that the optimized capture-based sequencing enables comprehensive profiling of plasma ccf-mtDNA with prominent reliability.
Figure 5.

Consistency between capture-based approach and low-depth WGS for profiling ccf-mtDNA characteristics
(A) Correlation of mtDNA copy number between optimized capture-based sequencing and low-depth WGS. The significance of correlation was determined by Spearman’s rank correlation coefficient. (B) Distribution of ccf-mtDNA fragment size detected by optimized capture-based sequencing (orange) and low-depth WGS (purple). (C) Distribution of ccf-mtDNA breakpoints across the mitochondrial genome detected by optimized capture-based sequencing (orange) and low-depth WGS (purple).
Suitable sample processing and placement are critical for accurate detection of circulating mtDNA
To further investigate the suitable sample processing for accurate profiling of ccf-mtDNA, we further evaluated the impact of different sample placement times and temperatures on downstream ccf-mtDNA detection. mtDNA copy number and fragment size were used as indicators of ccf-mtDNA stability. Intriguingly, placement of peripheral blood for up to 24 h at either 4°C or room temperature (RT) had no discernible impact on ccf-mtDNA stability (Figures 6A and 6B), whereas placement of isolated plasma for 6 h at both 4°C and RT led to a dramatic decrease of ccf-mtDNA copy number and fragment size (Figures 6C and 6D), suggesting that plasma ccf-mtDNA is susceptible to degradation. Although the reason underlying this distinct ccf-mtDNA behavior between whole blood and plasma requires further investigation, these findings highlight the importance of proper plasma sample processing.
Figure 6.

Suitable sample processing and placement are critical for accurate detection of circulating mtDNA
(A) Comparison of mtDNA copy number between 4°C (orange) and room temperature (RT, purple) at different placement times of peripheral blood. (B) Comparison of median mtDNA fragment size between 4°C (orange) and RT (purple) at different placement times of peripheral blood. (C) Comparison of mtDNA copy number between 4°C (orange) and RT (purple) at different placement times of plasma. (D) Comparison of median mtDNA fragment size between 4°C (orange) and RT (purple) at different placement times of plasma. The significance between groups was determined by a paired t test. ∗p < 0.05, ∗∗p < 0.01.
Tumor-specific mtDNA mutations can be effectively detected in plasma samples based on an optimized NGS approach
To explore the clinical application potential of plasma ccf-mtDNA, we simultaneously detected mtDNA variants in matched tumor tissue, adjacent non-tumor tissue, peripheral blood mononuclear cells (PBMCs), and plasma samples from 5 HCC and 10 CRC patients (Figure 7; Figures S3 and S4; see Table S3 for detailed patient information). On average, we were able to detect 64.2 mtDNA variants (range, 51–92) in plasma from HCC patients and 50.6 mtDNA variants (range, 31–112) in plasma from CRC patients by the optimized procedure, when the heteroplasmy level was set at 1%. In addition, the repertoire of germline mtDNA variants (defined as variants that were shared by tumor tissue and adjacent non-tumor tissue) can be fully recovered from the plasma sample of each patient. Remarkably, a fraction of tumor-specific mutations (defined as variants presented only in tumor tissue but absent in both adjacent non-tumor tissue and PBMCs) was recovered in each of 5 HCC plasma samples, but such mutations were completely undetectable in all 10 CRC plasma samples. However, there still existed a substantial amount of plasma-specific variants with unknown originations (VUOs) in both HCC and CRC patients.
Figure 7.

Tumor-specific mtDNA mutations can be effectively detected in plasma samples based on optimized NGS approach
(A) Representative Venn diagrams of mtDNA variants detected by single-spot sequencing of matched tumor tissue, adjacent non-tumor tissue, PBMCs and plasma samples from HCC and CRC patients. (B) Representative Venn diagrams of mtDNA variants detected by multiple-spot sequencing of matched tumor tissue, adjacent non-tumor tissue, PBMCs, and plasma samples from HCC and CRC patients.
To examine whether intratumor heterogeneity may constitute a source for plasma VUOs, we further applied multiple-spot mtDNA sequencing of tumor and adjacent non-tumor tissues (10 sampling spots for each tumor tissue and 5 spots for each adjacent non-tumor tissue). Multiple-spot sequencing of HCC samples revealed a considerable increase of tumor-specific mutations (range, 1–4), among which a substantial fraction can find identical counterparts in the plasma VUOs (Table S4). Furthermore, we revealed that the heteroplasmy levels of tumor-specific mutations was highly correlated (spearman r = 0.761, p < 0.001) between HCC tumor tissue and plasma samples (Figure S5). Surprisingly, multiple-spot sequencing of CRC samples identified much greater tumor-specific mutations (range, 2–22), suggesting the presence of tremendous CRC intratumor heterogeneity. However, the vast majority of them were still undetectable in the corresponding plasma samples. These results strongly suggest that the abundance of tumor-derived ccf-mtDNA in plasma may be cancer-type specific.
Discussion
In the present study, we first comprehensively profiled the characteristics of plasma ccf-mtDNA in both healthy controls and cancer patients. Based on the insight of ccf-mtDNA properties, we successfully optimized the capture-based NGS approach, which remarkably increased the capture efficiency of mtDNA from plasma DNA samples and thus greatly improved in the detection of ccf-mtDNA mutations. More importantly, the substantial fraction of tumor-derived ccf-mtDNA mutations was successfully detected in plasma samples from HCC patients but not CRC patients by the optimized approach. Our study provides a more robust means to identify tumorous ccf-mtDNA in the noninvasive diagnosis of specific cancer patients.
Plasma mtDNA has long been considered as a promising liquid biopsy candidate because of its high copy number and high mutation rate. Moreover, several studies have reported that the release of mtDNA into plasma is involved in immune responses and is elevated in cancer patients.23,24 Most previous studies have used the quantitative PCR (qPCR)-based approach to measure plasma mtDNA content in cancer patients.13 Despite the great importance of ccf-mtDNA content as the tumor biomarker, there are still a number of inconsistent findings. Studies have reported a decreased ccf-mtDNA copy number in the plasma of HCC patients.20 However, others have observed the opposite trend.25 This discrepancy may be partially due to the different detection methods, including massively parallel sequencing and qPCR. In our previous study, we directly compared these approaches in the same sample set and demonstrated that qPCR is not appropriate for the detection of mtDNA content in plasma DNA samples, due to their low quality. In addition, because alterations of ccf-mtDNA content have also been reported in multiple non-malignant pathologies, such as Alzheimer’s disease, Parkinson’s disease, and coronary heart disease,26,27 the usage of plasma mtDNA content alone as a cancer biomarker may be inaccurate and misleading, highlighting the importance of identifying tumor-derived mtDNA mutations in plasma samples. Recently, we developed a novel capture-based NGS approach using both mtDNA and nDNA probes to capture target fragments, enabling simultaneous detection of mtDNA mutation and copy number.28 Nonetheless, considering the complex environment of plasma greatly different from tissue samples, these approaches remain to be comprehensively evaluated and optimized when applied in the detection of circulating mtDNA.
Based on low-depth WGS data, we first comprehensively characterized the circulating mtDNA (ccf-mtDNA), including copy number, fragment distribution, and break points, providing a definitive description of ccf-mtDNA. We found that only around 0.001% of plasma DNA can be mapped to mtDNA, which is almost 180-fold less compared to that of matched fresh tissue samples. The extremely low abundance of mtDNA in plasma samples may prohibit further mutation calling based on capture-free deep sequencing, such as WGS. A few studies have attempted to detect ccf-mtDNA mutations in plasma or other bodily fluids,29,30 although all of these detections are faced with various limitations, such as low sensitivity or high cost. For instance, Weerts et al.15 reported the frustrated detection of tumor-specific mtDNA in plasma using single-molecule real-time sequencing and concluded that tracing tumor-specific mtDNA variants in plasma is of limited value. In our present study, based on novel insights into mtDNA characteristics, we performed a systematic optimization of the conventional capture-based NGS approach against capture probe (length and quantity), capture condition (hybridization temperature), and post-capture amplification. We found that increasing the probe quantity promoted mtDNA capture when the probe quantity is insufficient. However, when the probe quantity is excessive, further increasing the probe quantity may cause non-specific capture, such as the capture of nuclear mtDNA (NUMT). Therefore, the mtDNA mapping rate first increases and then decreases as the quantity of the capture probe increases. These efforts enabled us to develop a highly efficient capture-based plasma mtDNA detection pipeline. Compared to conventional capture-based deep sequencing, our optimized procedure can achieve at least 4-fold higher mtDNA enrichment using plasma DNA samples. In addition, we found that ccf-mtDNA in plasma samples, but not in whole-blood samples, was susceptible to degradation, highlighting the importance of proper plasma sample processing. Recently, Hotz et al.31 reported that red blood cells can homeostatically bind a large proportion of circulating mtDNA through Toll-like receptor 9 (TLR9). Thus, it is possible that whole-blood samples have a more stable level of circulating cell-free mtDNA than do isolated plasma samples due to the dynamic release of ccf-mtDNA from red blood cells.
More importantly, we demonstrated that the optimized procedure could greatly enhance the accurate and sensitive detection of ccf-mtDNA mutations. We analyzed the plasma mtDNA in a few HCC and CRC patients, leading to the recovery of abundant plasma mtDNA variants. Combined analysis of tumor tissue, adjacent non-tumor tissue, and PBMC samples allowed us to partially decipher the biological origins of plasma mtDNA variants. Interestingly, tumor-specific mtDNA mutations were identified in all 5 HCC plasma samples, but no tumor-specific mtDNA mutations were identified in 10 CRC plasma samples. Owing to the intratumor heterogeneity and sampling regional biases, the detection of tumor-specific mtDNA mutations inferred from single biopsy-based sequencing data may be limited.32 Therefore, we further took intratumor heterogeneity into account by using multi-regional sequencing of tissue samples. Our results revealed significantly increased numbers of tumor-specific mtDNA mutations in HCC plasma samples. However, tumor-specific mtDNA mutations were still barely detectable in CRC plasma samples. We thus propose that different types of cancer may have great differences in tumor-derived plasma mtDNA abundance. In line with previous studies, we also observed the high fractions of plasma-specific mtDNA variants that are of unknown origins in both HCC and CRC patients, which are actually tumor-specific mtDNA mutations that originated from intratumor heterogeneity, or possibly owing to the intricate mixture of DNA released from different organs or cell types to plasma.33 Therefore, the comprehensive profiling of ccf-mtDNA in plasma may provide more promising biomarkers to identify patients from cancer-free healthy populations and monitor the tumor progression.
In summary, we successfully optimized a capture-based NGS approach for accurate and highly efficient detection of both plasma ccf-mtDNA copy number and mutations, which provides a great possibility with plasma ccf-mtDNA as an invasive tumor biomarker. Based on our preliminary analysis, plasma mtDNA from HCC patients seems to contain abundant tumor-specific mtDNA mutations that can be attributed to intratumor heterogeneity. The comprehensive profiling of plasma mtDNA in large cohorts of HCC patients deserves special attention in the future.
Materials and methods
Sample collection
Peripheral blood and/or tissue samples were collected from a total of 15 healthy controls, 10 hepatitis patients, 15 HCC patients, and 20 CRC patients in Xijing Hospital, Fourth Military Medical University (FMMU) in Xi’an, China. This study was approved by the Ethical Committees of FMMU (permission no. KY20183331-1; date issued, March 6, 2018), and written consent was obtained from each subject.
Sample processing and DNA extraction
To obtain matched ccf-DNA and leucocyte genomic DNA, we simultaneously isolated plasma and PBMCs from 5 mL of peripheral blood. The peripheral blood was collected in EDTAK2 tubes and processed. Blood was first centrifuged at 4°C at 1,600 rpm for 10 min to separate plasma from blood cells. The plasma supernatant was purified by centrifugation at 4°C at 10,000 rpm for 15 min to remove residual cells or debris. A total of 55 ccf-DNA samples were extracted using 2 mL of plasma from 10 healthy controls, 10 hepatitis patients, 15 HCC patients, and 20 CRC patients by the QIAamp circulating nucleic acid kit (QIAGEN, USA). Also, 15 genomic DNA samples were extracted using PBMCs from 5 HCC and 10 CRC patients by an EZNA DNA kit (Omega, USA). To further identify the source of mtDNA variants in plasma samples, multiple-spot samplings of tumor and adjacent non-tumor tissue were performed for 5 HCC patients and 10 CRC patients, and the genomic DNA was extracted with an EZNA DNA kit (Omega, USA). Qubit 3.0 (Thermo Fisher Scientific, USA) and an Agilent 2100 Bioanalyzer system (Agilent Technologies, USA) were used to evaluate DNA quality and quantity.
Library construction and low-depth WGS
Genomic DNA (1 μg for each sample) was used to construct the WGS library as previously described.34 The plasma ccf-DNA (20 ng for each sample) was used to construct the WGS library by an NEB ultra v2 kit (NEB, USA) as previously described.35 The WGS libraries from 40 plasma samples and 20 tumor tissues were sequenced by the Illumina HiSeq X Ten platform using paired-end runs with 2 × 150 cycles (PE 150).
Capture-based mtDNA sequencing
Capture-based mtDNA sequencing was performed as previously described.21 In brief, 200 ng of WGS library was mixed with 10 ng of home-made biotinylated mtDNA capture probes (average 250 bp in length) and hybridized at 65°C for 24 h. The captured mtDNA library was further PCR-amplified for 16 cycles and sequenced on an Illumina HiSeq X Ten platform using PE 150.
Optimization of mtDNA enrichment for plasma samples
To specifically optimize mtDNA enrichment for plasma samples, we selected 10 plasma samples from HCC patients to compare the impact of different experimental conditions on mtDNA enrichment efficiency. We evaluated probe quantity (0.005, 0.01, 0.02, 0.05, 0.1, 0.2, 0.4, 0.8, 1.6, 3.2, 6.4, and 10 ng), probe length (150, 250, and 350 bp), hybridization temperature (55°C and 65°C), and subsequent mtDNA amplification cycles (16, 20, and 24). The enrichment efficiency of mtDNA was assessed by mtDNA mapping rate and sequencing depth.
Optimized capture-based mtDNA sequencing
Optimization of the mtDNA enrichment procedure enabled us to establish a novel captured-based ccf-mtDNA sequencing pipeline. Briefly, 200 ng of the WGS library from the plasma sample was mixed with mtDNA capture probes (probe length, 350 bp) using probe quantity of 0.8 ng and hybridized at 65°C for 24 h. In addition, the PCR amplification cycle for the mtDNA library was increased from 16 to 20. Finally, the amplified mtDNA libraries were sequenced on an Illumina HiSeq X Ten platform using PE 150. To assess the repeatability of the optimized capture-based ccf-mtDNA sequencing pipeline, WGS libraries constructed from 10 plasma samples were re-captured, amplified, and sequenced on an Illumina HiSeq X Ten platform using PE 150 to compare the consistency of mtDNA mutations.
Assessment of plasma sample processing
To investigate whether the condition of blood sample processing affects downstream ccf-mtDNA detection, we obtained 30 mL of peripheral blood from each of five additional healthy subjects. The acquired peripheral blood was divided into two groups. In group 1, peripheral blood samples were placed for different times (0, 3, 6, 9, 12, and 24 h) at either 4°C or RT before plasma centrifugation and DNA extraction. In group 2, plasma samples were obtained immediately by centrifugation and placed for different time (0, 3, and 6 h) at either 4°C or RT before DNA extraction. The impact of different treatments on ccf-mtDNA was evaluated by optimized capture-based mtDNA sequencing.
Calculation of mtDNA copy number and mtDNA mapping rate
For WGS data, the mtDNA copy number was calculated using the following formula: mtDNA copy number = 2 × average mtDNA sequencing depth/average nuclear DNA sequencing depth. For capture-based mtDNA sequencing data, the relative mtDNA copy number was calculated using the following formula: mtDNA copy number = 2 × average mtDNA depth/average depth of reference gene. The mtDNA mapping rate was calculated as sequencing reads aligned to mtDNA/total sequencing reads after deduplication.
Somatic mtDNA mutation calling and analysis
Raw sequencing data were trimmed using the FASTQ preprocessor fastp software version 0.20.0 (https://github.com/OpenGene/fastp; last accessed July 14, 2019),36 and the trimmed reads were mapped to the revised Cambridge Reference Sequence (rCRS) and whole-genome hg19 with BWA-MEM software version 0.7.1521 to minimize contamination from nuclear DNA of mitochondrial origin (NUMT). Then, Picard Tools software version 1.119 (Broad Institute, Cambridge, MA; https://broadinstitute.github.io/picard; last accessed November 12, 2014) was used for sorting the reads and marking the duplicate reads. Local realignment was performed with IndelRealigner in GATK software version 3.2-2 (Broad Institute; https://gatk.broadinstitute.org/hc/en-us; last accessed September 29, 2014) to reduce the false-positive rate of nearby indel positions. Finally, SAMtools (http://www.htslib.org; last accessed September 19, 2014) was used for selecting reads of high quality. For each site, we first counted the respective read numbers of the major and minor allele and calculated the site-specific minor allele frequency (MAF). Also, the mtDNA-mutation calling was performed according to the following filter conditions: (1) at least three reads on each strand have the mutation site; (2) minimum MAF cutoff 1%; (3) remove heterogeneity sites in rCRS repeat regions (66–71, 303–311, 514–523, 12418–12425, 16184–16193); (4) remove C>A/G>T mutations with low MAF (<1%) and strong sequence context bias (at CpCpN>CpApN, most frequently CpCpG>CpApG),37 which is known to arise from artificial guanine oxidation during sequencing library preparation; and (5) remove mtDNA mutations if the mutant rate and mutant base quality do not pass a binominal test (p > 0.001).38
Statistical analysis
GraphPad Prism 5.0 (GraphPad, USA) was used for statistical analysis. A Student’s t test was used to compare the difference between groups with continuous variables. Spearman’s rank correlation coefficient was performed to measure the correlations between groups. All p values were two-tailed and reported using a significance level of 0.05.
Acknowledgments
The authors thank Huanqin Zhang and Qinqin Mo in the State Key Laboratory of Cancer Biology and Department of Physiology and Pathophysiology for ongoing support and discussion. This work was supported by the National Natural Science Foundation of China (grant 81830070), the Autonomous Project of State Key Laboratory of Cancer Biology, China (grant CBSKL2019ZZ06), the National Science and Technology Major Projects of China (grant 2018ZX10302205-002), and by the Key Research and Development Program of Shaanxi Province, China (grant 2018ZDXM-SF-061).
Author contributions
J.X. and X. Gu conceptualized and designed the study. Y.L. and K.Z. carried out the sample collection, performed the data analysis, and drafted the manuscript. S.G., Q.Y., and L.S. participated in the bioinformatics analyses. X.J. and Y.W. performed the laboratory experiments. X. Guo and X. Gu participated in the design of the study and performed the draft revision. J.X. conceived of the study, participated in its design and coordination, and helped to revise the manuscript. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.12.017.
Contributor Information
Xiwen Gu, Email: xiwen.gu@xjtu.edu.cn.
Jinliang Xing, Email: xingjinliang@163.com.
Supplemental information
References
- 1.Anderson S., Bankier A.T., Barrell B.G., de Bruijn M.H., Coulson A.R., Drouin J., Eperon I.C., Nierlich D.P., Roe B.A., Sanger F. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 2.Ju Y.S., Alexandrov L.B., Gerstung M., Martincorena I., Nik-Zainal S., Ramakrishna M., Davies H.R., Papaemmanuil E., Gundem G., Shlien A., ICGC Breast Cancer Group. ICGC Chronic Myeloid Disorders Group. ICGC Prostate Cancer Group Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. eLife. 2014;3:e02935. doi: 10.7554/eLife.02935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Reznik E., Miller M.L., Şenbabaoğlu Y., Riaz N., Sarungbam J., Tickoo S.K., Al-Ahmadie H.A., Lee W., Seshan V.E., Hakimi A.A., Sander C. Mitochondrial DNA copy number variation across human cancers. eLife. 2016;5:e10769. doi: 10.7554/eLife.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart J.B., Chinnery P.F. The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat. Rev. Genet. 2015;16:530–542. doi: 10.1038/nrg3966. [DOI] [PubMed] [Google Scholar]
- 5.Yuan Y., Ju Y.S., Kim Y., Li J., Wang Y., Yoon C.J., Yang Y., Martincorena I., Creighton C.J., Weinstein J.N., PCAWG Consortium Comprehensive molecular characterization of mitochondrial genomes in human cancers. Nat. Genet. 2020;52:342–352. doi: 10.1038/s41588-019-0557-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He Y., Wu J., Dressman D.C., Iacobuzio-Donahue C., Markowitz S.D., Velculescu V.E., Diaz L.A., Jr., Kinzler K.W., Vogelstein B., Papadopoulos N. Heteroplasmic mitochondrial DNA mutations in normal and tumour cells. Nature. 2010;464:610–614. doi: 10.1038/nature08802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Rubis G., Rajeev Krishnan S., Bebawy M. Liquid Biopsies in Cancer Diagnosis, Monitoring, and Prognosis. Trends Pharmacol. Sci. 2019;40:172–186. doi: 10.1016/j.tips.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 8.Goodwin S., McPherson J.D., McCombie W.R. Coming of age: ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Volik S., Alcaide M., Morin R.D., Collins C. Cell-free DNA (cfDNA): Clinical Significance and Utility in Cancer Shaped By Emerging Technologies. Mol. Cancer Res. 2016;14:898–908. doi: 10.1158/1541-7786.MCR-16-0044. [DOI] [PubMed] [Google Scholar]
- 10.Tang J.C., Feng Y.L., Guo T., Xie A.Y., Cai X.J. Circulating tumor DNA in hepatocellular carcinoma: trends and challenges. Cell Biosci. 2016;6:32. doi: 10.1186/s13578-016-0100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Razavi P., Li B.T., Brown D.N., Jung B., Hubbell E., Shen R., Abida W., Juluru K., De Bruijn I., Hou C. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat. Med. 2019;25:1928–1937. doi: 10.1038/s41591-019-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellinger J., Albers P., Müller S.C., von Ruecker A., Bastian P.J. Circulating mitochondrial DNA in the serum of patients with testicular germ cell cancer as a novel noninvasive diagnostic biomarker. BJU Int. 2009;104:48–52. doi: 10.1111/j.1464-410X.2008.08289.x. [DOI] [PubMed] [Google Scholar]
- 13.Afrifa J., Zhao T., Yu J. Circulating mitochondria DNA, a non-invasive cancer diagnostic biomarker candidate. Mitochondrion. 2019;47:238–243. doi: 10.1016/j.mito.2018.12.003. [DOI] [PubMed] [Google Scholar]
- 14.Rohlin A., Wernersson J., Engwall Y., Wiklund L., Björk J., Nordling M. Parallel sequencing used in detection of mosaic mutations: comparison with four diagnostic DNA screening techniques. Hum. Mutat. 2009;30:1012–1020. doi: 10.1002/humu.20980. [DOI] [PubMed] [Google Scholar]
- 15.Weerts M.J.A., Timmermans E.C., van de Stolpe A., Vossen R.H.A.M., Anvar S.Y., Foekens J.A., Sleijfer S., Martens J.W.M. Tumor-Specific Mitochondrial DNA Variants Are Rarely Detected in Cell-Free DNA. Neoplasia. 2018;20:687–696. doi: 10.1016/j.neo.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ye F., Samuels D.C., Clark T., Guo Y. High-throughput sequencing in mitochondrial DNA research. Mitochondrion. 2014;17:157–163. doi: 10.1016/j.mito.2014.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang R., Nakahira K., Guo X., Choi A.M., Gu Z. Very Short Mitochondrial DNA Fragments and Heteroplasmy in Human Plasma. Sci. Rep. 2016;6:36097. doi: 10.1038/srep36097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y., Guo S., Yin C., Guo X., Liu M., Yuan Z., Zhao Z., Jia Y., Xing J. Optimized PCR-Based Enrichment Improves Coverage Uniformity and Mutation Detection in Mitochondrial DNA Next-Generation Sequencing. J. Mol. Diagn. 2020;22:503–512. doi: 10.1016/j.jmoldx.2020.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Mamanova L., Coffey A.J., Scott C.E., Kozarewa I., Turner E.H., Kumar A., Howard E., Shendure J., Turner D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods. 2010;7:111–118. doi: 10.1038/nmeth.1419. [DOI] [PubMed] [Google Scholar]
- 20.Jiang P., Chan C.W., Chan K.C., Cheng S.H., Wong J., Wong V.W., Wong G.L., Chan S.L., Mok T.S., Chan H.L. Lengthening and shortening of plasma DNA in hepatocellular carcinoma patients. Proc. Natl. Acad. Sci. USA. 2015;112:E1317–E1325. doi: 10.1073/pnas.1500076112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin C., Li D.Y., Guo X., Cao H.Y., Chen Y.B., Zhou F., Ge N.J., Liu Y., Guo S.S., Zhao Z. NGS-based profiling reveals a critical contributing role of somatic D-loop mtDNA mutations in HBV-related hepatocarcinogenesis. Ann. Oncol. 2019;30:953–962. doi: 10.1093/annonc/mdz105. [DOI] [PubMed] [Google Scholar]
- 22.Cruz-Dávalos D.I., Llamas B., Gaunitz C., Fages A., Gamba C., Soubrier J., Librado P., Seguin-Orlando A., Pruvost M., Alfarhan A.H. Experimental conditions improving in-solution target enrichment for ancient DNA. Mol. Ecol. Resour. 2017;17:508–522. doi: 10.1111/1755-0998.12595. [DOI] [PubMed] [Google Scholar]
- 23.Fang C., Wei X., Wei Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell. 2016;7:11–16. doi: 10.1007/s13238-015-0222-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu M. Circulating cell-free mitochondrial DNA as a novel cancer biomarker: opportunities and challenges. Mitochondrial DNA. 2012;23:329–332. doi: 10.3109/19401736.2012.696625. [DOI] [PubMed] [Google Scholar]
- 25.Li L., Hann H.W., Wan S., Hann R.S., Wang C., Lai Y., Ye X., Evans A., Myers R.E., Ye Z. Cell-free circulating mitochondrial DNA content and risk of hepatocellular carcinoma in patients with chronic HBV infection. Sci. Rep. 2016;6:23992. doi: 10.1038/srep23992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lowes H., Pyle A., Santibanez-Koref M., Hudson G. Circulating cell-free mitochondrial DNA levels in Parkinson’s disease are influenced by treatment. Mol. Neurodegener. 2020;15:10. doi: 10.1186/s13024-020-00362-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu J., Cai X., Xie L., Tang Y., Cheng J., Wang J., Wang L., Gong J. Circulating Cell Free Mitochondrial DNA is a Biomarker in the Development of Coronary Heart Disease in the Patients with Type 2 Diabetes. Clin. Lab. 2015;61:661–667. doi: 10.7754/clin.lab.2014.141132. [DOI] [PubMed] [Google Scholar]
- 28.Zhou K., Mo Q., Guo S., Liu Y., Yin C., Ji X., Guo X., Xing J. A Novel Next-Generation Sequencing-Based Approach for Concurrent Detection of Mitochondrial DNA Copy Number and Mutation. J. Mol. Diagn. 2020;22:1408–1418. doi: 10.1016/j.jmoldx.2020.09.005. [DOI] [PubMed] [Google Scholar]
- 29.Uzawa K., Baba T., Uchida F., Yamatoji M., Kasamatsu A., Sakamoto Y., Ogawara K., Shiiba M., Bukawa H., Tanzawa H. Circulating tumor-derived mutant mitochondrial DNA: a predictive biomarker of clinical prognosis in human squamous cell carcinoma. Oncotarget. 2012;3:670–677. doi: 10.18632/oncotarget.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Newell C., Hume S., Greenway S.C., Podemski L., Shearer J., Khan A. Plasma-derived cell-free mitochondrial DNA: A novel non-invasive methodology to identify mitochondrial DNA haplogroups in humans. Mol. Genet. Metab. 2018;125:332–337. doi: 10.1016/j.ymgme.2018.10.002. [DOI] [PubMed] [Google Scholar]
- 31.Hotz M.J., Qing D., Shashaty M.G.S., Zhang P., Faust H., Sondheimer N., Rivella S., Worthen G.S., Mangalmurti N.S. Red Blood Cells Homeostatically Bind Mitochondrial DNA through TLR9 to Maintain Quiescence and to Prevent Lung Injury. Am. J. Respir. Crit. Care Med. 2018;197:470–480. doi: 10.1164/rccm.201706-1161OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gerlinger M., Rowan A.J., Horswell S., Math M., Larkin J., Endesfelder D., Gronroos E., Martinez P., Matthews N., Stewart A. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012;366:883–892. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aucamp J., Bronkhorst A.J., Badenhorst C.P.S., Pretorius P.J. The diverse origins of circulating cell-free DNA in the human body: a critical re-evaluation of the literature. Biol. Rev. Camb. Philos. Soc. 2018;93:1649–1683. doi: 10.1111/brv.12413. [DOI] [PubMed] [Google Scholar]
- 34.Yin C., Liu Y., Guo X., Li D., Fang W., Yang J., Zhou F., Niu W., Jia Y., Yang H., Xing J. An Effective Strategy to Eliminate Inherent Cross-Contamination in mtDNA Next-Generation Sequencing of Multiple Samples. J. Mol. Diagn. 2019;21:593–601. doi: 10.1016/j.jmoldx.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 35.Tao K., Bian Z., Zhang Q., Guo X., Yin C., Wang Y., Zhou K., Wan S., Shi M., Bao D. Machine learning-based genome-wide interrogation of somatic copy number aberrations in circulating tumor DNA for early detection of hepatocellular carcinoma. EBioMedicine. 2020;56:102811. doi: 10.1016/j.ebiom.2020.102811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen S., Zhou Y., Chen Y., Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Costello M., Pugh T.J., Fennell T.J., Stewart C., Lichtenstein L., Meldrim J.C., Fostel J.L., Friedrich D.C., Perrin D., Dionne D. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013;41:e67. doi: 10.1093/nar/gks1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campo D.S., Nayak V., Srinivasamoorthy G., Khudyakov Y. Entropy of mitochondrial DNA circulating in blood is associated with hepatocellular carcinoma. BMC Med. Genomics. 2019;12(Suppl 4):74. doi: 10.1186/s12920-019-0506-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.