

Abstract
Aberrant expression of long non-coding RNAs (lncRNA) is associated with altered DNA methylation and histone modifications during carcinogenesis. However, identifying epigenetically dysregulated lncRNAs and characterizing their functional mechanisms in different cancer subtypes are still major challenges for cancer studies. In this study, we systematically analyzed the epigenetic alterations of lncRNAs at important regulatory elements in three breast cancer subtypes. We identified 87, 691, and 1,197 epigenetically dysregulated lncRNAs in luminal, basal, and claudin-low subtypes of breast cancer, respectively. The landscape of epigenetically dysregulated lncRNAs at enhancer elements revealed that epigenetic changes of the majority of lncRNAs occurred in a subtype-specific manner and contributed to subtype-specific biological functions. We identified six acetylation of lysine 27 on histone H3 (H3K27ac)-dysregulated lncRNAs and three DNA methylation-dysregulated lncRNAs (CTC-303L1.2, RP11-738B7.1, and SLC26A4-AS1) as prognostic biomarkers of basal subtype. These lncRNAs were involved in immune response-related biological functions. Treatment of the basal breast cancer cell line MDA-MB-468 with CREBBP/EP300 bromodomain inhibitors downregulated H3K27 acetylation levels and caused a decrease in the expression of five H3K27ac-dysregulated lncRNAs (LINC00393, KB-1836B5.1, RP1-140K8.5, AC005162.1, and AC020916.2) and inhibition of the growth of breast cancer cells. One epigenetically dysregulated lncRNA (LINC01983) and four lncRNA regulators (UCA1, RP11-221J22.2, RP11-221J22.1, and RP1-212P9.3) were identified as prognostic biomarkers of the luminal molecular subtype of breast cancer by controlling the tumor necrosis factor (TNF) signaling pathway, T helper (Th)17 cell differentiation, and T cell migration. Finally, our results highlighted a profound role of enhancer-related H3K27ac-dysregulated lncRNAs, DNA methylation-dysregulated lncRNAs, and lncRNA regulators in breast cancer subtype carcinogenesis and their potential prognostic value.
Keywords: epigenetics, long non-coding RNAs, enhancer, breast cancer subtypes, prognostic biomarkers
Graphical Abstract
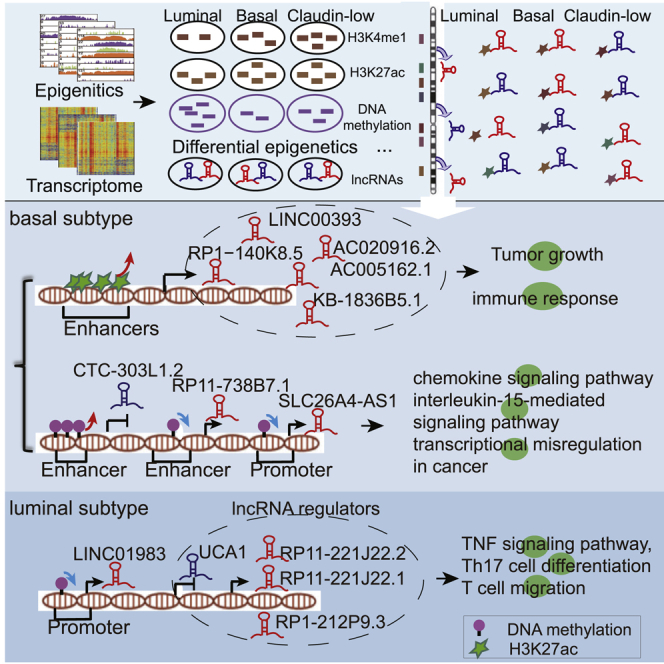
The corresponding authors and colleagues identified H3K27ac-dysregulated lncRNAs (LINC00393, KB-1836B5.1, RP1-140K8.5, AC005162.1, and AC020916.2) and DNA methylation-dysregulated lncRNAs (CTC-303L1.2, RP11-738B7.1, and SLC26A4-AS1) as prognostic biomarkers of basal subtypes. LINC01983, UCA1, RP11-221J22.2, RP11-221J22.1, and RP1-212P9.3 act as prognostic biomarkers of the luminal subtype. The inhibition of enhancer-related lncRNAs could suppress breast cancer cell growth.
Introduction
Long non-coding RNA (lncRNA) has been linked to many human cancers, including genetically heterogeneous breast cancers.1,2 Distinct breast cancer molecular subtypes (luminal, basal, human epidermal growth factor receptor 2 [HER2], and claudin-low) have been characterized based on gene expression, clinical, and prognostic features of breast cancer.3 lncRNAs are frequently differentially expressed in breast cancer subtypes.4 Aberrant lncRNA expression could be caused by alteration of epigenetic patterns, such as changes in DNA methylation and post-translational histone modifications. For example, loss of expression of lncRNA LOC554202 in triple-negative breast cancer (TNBC) can be explained by increased DNA methylation of its promoter-associated CpG island.5 lncRNA Esrp2-as, which is regulated by DNA methylation at enhancers, acts as a prognostic biomarker for breast cancer.6 Analyses of associations between expression of lncRNAs and their promoter histone modifications showed an important role of trimethylation of lysine 27 on histone H3 (H3K27me3) in lncRNA silencing.7 Although global epigenetic abnormalities have been identified as prominent cancer hallmarks, attempts to characterize the relationship between epigenetic alterations and expression of lncRNAs and their prognostic value in breast cancer subtypes have been limited.
This study aims to analyze the relationship between lncRNA expression and epigenetic alterations. It also explores the prognostic value of these findings in patients with breast cancer subtypes. We identified a large number of epigenetic-dysregulated lncRNAs in luminal, basal, and claudin-low subtypes of breast cancer. This was achieved by comparing the alterations of the following histone modifications at the promoter and enhancer elements of lncRNAs: lysine H3K27 acetylation (H3K27ac), H3K27me3, trimethylation of Lys36 in histone H3 (H3K36me3), monomethylation of histone H3 at lysine 4 (H3K4me1), trimethylation of histone H3 at lysine 4 (H3K4me3), and histone H3 lysine 9 trimethylation (H3K9me3). Epigenetic-dysregulated lncRNAs at enhancer elements play an important role in controlling subtype-specific biological functions. We identified six lncRNAs with differential histone modifications at enhancers (LINC00393, KB-1836B5.1, CASC11, RP1-140K8.5, AC005162.1, and AC020916.2) and three lncRNAs with differential DNA methylation at enhancers or promoters (CTC-303L1.2, RP11-738B7.1, and SLC26A4-AS1) as prognostic biomarkers of the basal subtype of breast cancer. DNA methylation-dysregulated lncRNA LINC01983 and four lncRNA regulators (UCA1, RP11-221J22.2, RP11-221J22.1, and RP1-212P9.3) were identified as prognostic biomarkers of luminal subtype. Our results help provide a better understanding of the aberrant epigenetic regulation of lncRNA expression in breast cancer subtypes.
Results
Identifying epigenetically dysregulated lncRNAs and protein-coding genes (PCGs) in breast cancer subtypes
Our goal was to analyze the relationship between lncRNA expression and epigenetic alterations in breast cancer subtypes. We focused on differentially expressed lncRNAs and looked for differences in three breast cancer subtypes compared with normal breast tissue or cell lines using DESeq2. 318 (2.50%), 2,493 (19.59%), and 3,414 (24.62%) lncRNAs were differentially expressed in luminal, basal, and claudin-low subtypes, respectively (false discovery rate [FDR] < 0.05). We also identified 2,141 (11.54%), 7,582 (40.85%), and 9,851 (51.87%) differentially expressed PCGs in luminal, basal, and claudin-low subtypes, respectively. Epigenetic changes have been reported in a number of PCGs in breast cancer subtypes.8 We hypothesized that aberrant epigenetic modifications might play a crucial role in regulating lncRNAs in breast cancer. We systematically analyzed differential histone modification regions (DHMRs; H3K4me1, H3K4me3, H3K36me3, H3K27ac, H3K27me3, and H3K9me3) and DNA methylation between breast cancer subtypes and normal samples to determine whether epigenetic changes are involved in differential expression of lncRNAs. The genome-wide epigenetic alterations within the lncRNA promoter and enhancer regions were dissected. An epigenetically dysregulated lncRNA (termed epi-lncRNA) was identified when at least one DHMR or differentially methylated region (DMR) was located in the promoter or enhancer of differentially expressed lncRNAs and when the dysregulated pattern of active (or repressive) epigenetic marks contributed to transcriptional dysregulation in the same (or reverse) direction (Figure 1A). As a result, we identified 65, 553, and 1,197 epi-lncRNAs with differential histone modification in luminal, basal, and claudin-low subtypes, respectively. There were 196 and 36 epi-lncRNAs with differential DNA methylation in basal and luminal subtypes, respectively. Similarly, 370, 884, and 4,139 epi-PCGs were identified in luminal, basal, and claudin-low subtypes, respectively. lncRNAs (12.16%) exhibited a much lower aberrant frequency than did PCGs (26.23%) in breast cancer (Figure 1B). This result is consistent with previous studies that have shown that overall occupancy of histone marks across the transcriptional start sites of protein-coding and lncRNA genes is in the range 65%–73% and 27%–38%, respectively, for a particular tissue or cell type.9 Notably, epigenetically dysregulated lncRNAs were significantly enriched for known cancer lncRNAs when analyzed using Lnc2Cancer v2.0 (p = 0.02; hypergeometric test).10 More specifically, 13 (p = 7.53 × 10−4), 69 (p = 1.80 × 10−5), and 96 (p = 3.47 × 10−5) epigenetically dysregulated lncRNAs in luminal, basal, and claudin-low subtypes, respectively, are known lncRNAs associated with human cancers (Figure 1C). These lncRNAs include RMST, CASC8, and RP11-65J3.1 in the luminal subtype, SNHG12, CASC11, and HOTAIRM1 in the basal subtype, and GAS5, HOTTIP, HAS2-AS1, and HOXD-AS1 in the claudin-low subtype. For example, LINC01016 in the luminal subtype has been reported to be a known prognostic marker of breast cancer.11 We observed significant increases in H3K27ac, H3K4me1, and H3K4me3 levels in the LINC01016 proximal enhancer region, which resulted in a significant upregulation of LINC01016 (fold change [FC] = 3.68, FDR = 5.74 × 10−5; Figures 1D and 1E). As another example, a significant enhancement of the H3K4me3 signal was observed in the enhancer region of SNHG12 in the basal subtype, which resulted in a significant upregulation of SNHG12 (FC = 1.74, FDR = 5.61 × 10−6; Figures 1D and 1E). The overexpression of SNHG12 has been previously reported to be involved in the proliferation, apoptosis, and invasion of triple-negative breast cancer.12 These observations suggested that several breast cancer lncRNAs are subject to epigenetic dysregulation in their enhancer regions.
Figure 1.

Epigenetically dysregulated lncRNAs in breast cancer subtypes
(A) Identifying epigenetically dysregulated lncRNAs in different breast cancer subtypes. (B) Percentage of epi-lncRNAs and epi-PCGs among all lncRNAs and PCGs on the genome. (C) Overlapping of epi-lncRNAs in three breast cancer subtypes with cancer-lncRNAs from Lnc2Cancer 2.0. (D) Expression distribution of LINC01016 (left) in the claudin-low subtype and HMEC and expression distribution of SNHG12 (right) in the basal subtype and normal samples. (E) Histone modification profile of LINC01016 (left) and SNHG12 (right). Histone modification data were visualized using the Integrative Genomics Viewer (IGV) tool.
Additionally, we explored the regulation of lncRNA to lncRNA expression using partial least-squares regression. We identified 98, 25, and 87 lncRNA regulators in luminal, basal, and claudin-low subtypes, respectively. Several lncRNA regulators are known cancer genes. For example, taurine-upregulated gene 1 (TUG1) is significantly upregulated in the basal subtype (FC = 1.21, FDR = 3.18 × 10−2). TUG1 knockdown was reported to be significantly associated with decreased cell proliferation and promoted apoptosis of breast cancer cells.13 A lncRNA regulator MALAT1 was significantly downregulated in the claudin-low subtype (FC = 0.20, FDR = 1.22 × 10−3). It has been reported that MALAT1 suppresses breast cancer progression and metastasis.14
Characterizing genomic signatures of epigenetically dysregulated lncRNAs
We compared the number and length of genes, exons, introns, and isoforms of epi-lncRNAs and other lncRNAs (non-epi-lncRNAs) to characterize genomic signatures of epigenetically dysregulated lncRNAs. epi-lncRNAs harbored longer exon (p = 1.1 × 10−11), intron (p = 1.3 × 10−4), and gene lengths (p = 1.3 × 10−14), and higher exon, intron, and isoform numbers (p < 2.2 × 10−16, Wilcoxon rank sum test; Figure S1A), regardless of different aberrant epigenetic modifications. epi-PCGs exhibited similar structural features to epi-lncRNAs with the exception of exon and intron length. Furthermore, we found that H3K4me1-dysregulated lncRNAs had a higher exon number and longer intron and gene length (p < 0.05; Figure S1B) compared with lncRNAs that have different aberrant epigenetic modifications. epi-lncRNAs were classified into five categories on the basis of their relationship with PCGs: intergenic, overlapping, partially overlapping, intronic, or exonic.15,16 859 (53.1%) and 27 (1.6%) epi-lncRNAs belonged to intergenic lncRNAs and overlapping lncRNAs, respectively. Structural analysis revealed that epigenetic-dysregulated overlapping lncRNAs had higher exon, intron. and isoform number, longer intron and gene length, and shorter exon length (p < 0.05; Figure S2A). These results suggest that lncRNAs with complex splicing patterns are more likely regulated by aberrant epigenetic modifications.
In addition, we compared the expression level and expression variation of epi-lncRNAs with other lncRNAs to characterize the expression pattern of epigenetically dysregulated lncRNAs. The coefficient of variation (CV) was used to assess expression variation of genes among patients with breast cancer. lncRNAs showed substantially lower expression levels than did PCGs in all three breast cancer subtypes, which has been described in previous studies.17 Interestingly, epigenetically dysregulated lncRNAs displayed substantially higher expression levels and lower expression variation compared with other lncRNAs in all three breast cancer subtypes (p < 0.05). Consistently, we observed higher expression levels and lower expression variation of epigenetically dysregulated PCGs compared with other PCGs in the basal and claudin-low subtypes (p < 0.05; Figure S2B). These results suggest that epigenetically dysregulated lncRNAs are stably expressed in breast cancer subtypes and may be suitable as potential biomarkers for breast cancer.18
Epigenetically dysregulated lncRNAs at enhancer elements contributing to cancer subtype-specific functions
The genomic distribution of epi-lncRNAs showed that abnormal histone modifications H3K27ac (47.91%), H3K4me3 (25.95%), and H3K4me1 (15.11%) within enhancers (77.77%) mainly contribute to dysregulation of lncRNAs in the basal subtype. Both H3K27ac-dysregulated lncRNAs and H3K4me1-dysregulated lncRNAs in the enhancer region are associated with regulation of cell cycle, immune system process, and cell communication based on guilt by association (Figure 2A). epi-lncRNAs in the luminal subtype were mainly associated with H3K27ac (26.41%), H3K36me3 (25.90%), and H3K4me3 (19.49%) dysregulation at enhancer elements (53.46%), which mainly affected ripoptosome assembly involved in the necroptotic process (Figure 2B). epi-lncRNAs in the claudin-low subtype were mainly regulated by H3K27ac (78.27%) and H3K27me3 (14.98%) at enhancer elements (60.85%), which affected a large number of basic biological functions, including macromolecule metabolism, cellular macromolecule biosynthesis, and cellular protein modification processes (Figure 2C). Overall, epi-lncRNAs in breast cancer subtypes were mainly associated with H3K27ac dysregulation (67.16%) within enhancer elements (Figure 2D). However, lncRNAs with H3K27ac dysregulation affected different biological functions in different breast cancer subtypes, such as the necroptotic process in the luminal subtype, cell communication, cell proliferation, and immune system processes in the basal subtype, and macromolecule biosynthetic and protein modification processes in the claudin-low subtype (Figure 2D).
Figure 2.

Subtype-specific functions of epi-lncRNAs
(A–C) Distribution and function analysis of epi-lncRNAs in the basal (A), luminal (B), and claudin-low (C) subtypes. Distribution and function analysis of epi-lncRNAs in breast cancer subtypes are shown. (D) Distribution of epi-lncRNAs of all three breast cancer subtypes and function analysis of epi-lncRNAs, which are associated with H3K27ac dysregulation. (E) Unsupervised clustering was performed using epigenetically dysregulated lncRNAs at enhancer elements. PAM50 classification, ER, PR, and HER2 status are shown above the heatmap.
We performed unsupervised hierarchical clustering analysis using Euclidean distance for breast cancer samples based on the expression of these lncRNAs to further characterize the important role of epigenetically dysregulated lncRNAs at enhancer elements. We found that expression of these lncRNAs, especially for H3K4me1-, H3K4me3-, and H3K27me3-dysregulated lncRNAs, can be used to distinguish the breast cancer samples by PAM50 subtypes, which suggests that epi-lncRNAs at enhancer elements may contribute to the distinct biology of these subtypes. These epi-lncRNAs could not be used to distinguish between estrogen receptor (ER)-driven luminal A and luminal B subtypes. However, H3K4me1-, H3K4me3-, and H3K27me3-dysregulated lncRNAs at enhancer elements are able to distinguish luminal subtypes from basal and claudin-low subtypes (Figure 2E). In summary, epigenetically dysregulated lncRNAs at enhancer elements play an important role in the pathogenesis of breast cancer subtypes by affecting distinct subtype-specific biological functions.
The landscape of epigenetically dysregulated lncRNAs reveals a subtype-specific pattern for cancer
Next, we systematically analyzed the epigenetically dysregulated lncRNAs in different breast cancer subtypes and revealed the landscape of enhancer-associated epigenetically dysregulated lncRNAs. 92.59% of epi-lncRNAs showed a subtype-specific pattern (Figures 3A and 3B; Figure S3). Specifically, 75.38% of epi-lncRNAs in the luminal subtype showed a subtype-specific pattern. For example, the known breast cancer lncRNA RMST showed elevated H3K27ac, H3K4me1, and H3K4me3 and decreased H3K27me3 at distal enhancer elements (Figure 3C), which contributed to its upregulation (FC = 3.87, FDR = 1.05 × 10−4). RMST is reported to be involved in cell proliferation and apoptosis of breast cancer.19 79.02% of epi-lncRNAs in the basal subtype showed a subtype-specific pattern. For example, known breast cancer lncRNAs TUG1, SNHG12, and SOX2-OT were specifically epigenetically dysregulated in the basal subtype. A significant enhancement of the H3K4me3 signal was observed at the enhancer element of SNHG12, which contributed to its upregulation (FC = 1.74, FDR = 5.61 × 10−6). SNHG12 has been reported to regulate triple-negative breast cancer cell proliferation, apoptosis, and migration.12 We observed decreased H3K36me3 in the enhancer region of SOX2-OT, which resulted in its significant downregulation (FC = 4.34, FDR = 3.99 × 10−9). A previous study has shown that ectopic expression of SOX2-OT could reduce proliferation and increase breast cancer cell growth.20 89.81% of epi-lncRNAs in the claudin-low subtype showed a subtype-specific pattern. lncRNAs LINC00152, AFAP1-AS1, NNT-AS1, and SNHG15, which are known breast cancer lncRNAs, were specifically epigenetically dysregulated in the claudin-low subtype. We observed elevated H3K27ac at distal enhancer elements of LINC00152 (FC = 4.07, FDR = 3.78 × 10−12). LINC00152 was reported to be significantly upregulated in MDA-MB-231 and MCF-7 cell lines, and it is thought to be involved in the epithelial-to-mesenchymal transition (EMT) and chemoresistance in breast cancer cells.21
Figure 3.

Landscape of lncRNAs with differential histone modifications reveals a highly cancer subtype-specific pattern
(A) Subtype specificity distribution of epi-lncRNAs. Some epi-lncRNAs that are cancer-lncRNAs are shown on the left. (B) Landscape of lncRNAs with differential histone modifications at enhancer elements in three breast cancer subtypes. (C) Histone modification profile of RMST in the luminal subtype and LINC00152 in the claudin-low subtype.
Additionally, a small number of lncRNAs are epigenetically dysregulated in multiple breast cancer subtypes, which suggests that some molecular mechanisms are shared by these three subtypes. However, the same lncRNAs in the three subtypes were altered by different types of epigenetic regulations in a subtype-specific manner. The basal and claudin-low subtypes had the most epi-lncRNAs in common (Figure 3A). This result is consistent with previous studies that the claudin-low subtype is similar to basal-like breast cancer. For example, lncRNAs AC006262.5, LINC00704, and LUCAT1 are shared by basal and claudin-low subtypes. AC006262.5 is an lncRNA associated with the risk of breast cancer. It was downregulated in the claudin-low subtype, and it regulated cell migration by downregulating IGFL1. In the present study, we observed elevated H3K27me3 in its promoter region, which results in its significant downregulation in the claudin-low subtype (FC = 19.61, FDR = 3.37 × 10−14; Figures S4A and S4B). However, we observed elevated H3K27ac and decreased H3K27me3 in its enhancer region in the basal subtype, resulting in its upregulation (FC = 2.58, FDR = 1.10 × 10−2). In another example, RP11-395G23.3, CASC8, APCDD1L-AS1, and RP11-154D17.1 are common lncRNAs in three cancer subtypes. We observed downregulated expression and decreased H3K27ac and H3K36me3 at the promoter element in the luminal subtype, but upregulated expression and elevated H3K27ac, H3K4me1, and H3K4me3 at enhancer elements in the basal subtype, and upregulated expression and elevated H3K27ac and decreased H3K27me3 at enhancer elements in the claudin-low subtype (Figures S4B and S4C). Although the role of CASC8 in breast cancer has not been studied, CASC8 is a known protective factor of bladder cancer.22 CASC8 was reported to be significantly downregulated in bladder cancer, and it reduced glycolysis and inhibited cell proliferation.22 Taken together, a large number of epigenetic-dysregulated lncRNAs, including many known breast cancer lncRNAs, show distinct epigenetically dysregulated patterns in different breast cancer subtypes. Investigation of these lncRNAs might provide approaches for the subtype-specific treatment of breast cancer.
Epigenetically dysregulated lncRNAs at enhancer elements are associated with disease prognosis in breast cancer subtypes
To gain insights into the potential prognostic value of dysregulated H3K27ac at enhancer elements, DNA methylation-associated lncRNAs and lncRNA regulators were analyzed using multivariate Cox regression analysis (see Materials and methods). We found six enhancer-related epi-lncRNAs with differential histone modification in the basal subtype (LINC00393, KB-1836B5.1, CASC11, RP1-140K8.5, AC005162.1, and AC020916.2) and were able to significantly distinguish patients in high-risk groups from those in low-risk groups in terms of overall survival (Table 1). We observed elevated H3K27ac and H3K4me1 at enhancer elements of LINC00393 and KB-1836B5.1, which contributed to their upregulated expression (FC = 20.49, FDR = 8.07 × 10−11 for LINC00393; FC = 2.46, FDR = 1.02 × 10−4 for KB-1836B5.1; Figures 4A–4D). High KB-1836B5.1 expression was associated with advanced tumor stage (p = 0.01; Fisher’s exact test). The higher expression group of LINC00393 or KB-1836B5.1 had significantly shorter survival times in the basal subtype (FDR = 3.87 × 10−2 for LINC00393; FDR = 3.87 × 10−2 for KB-1836B5.1; Figure 4C). These results are consistent with those of previous studies where upregulation of LINC00393 was proposed as a biomarker of the basal subtype.23 KB-1836B5.1 was reported to be significantly upregulated in glioma.24 Significant enhancement of H3K27ac, H3K4me1, and H3K4me3 signals in the enhancer region of CASC11 were observed, which resulted in significant CASC11 upregulation (FC = 2.21, FDR = 6.84 × 10−4; Figures 4B–4D). The higher expression group of CASC11 had shorter survival time than did the lower expression group (FDR = 4.15 × 10−2; Figure 4C). Previous evidence has shown that CASC11 is a risk lncRNA of colorectal cancer and hepatocellular carcinoma. In colorectal cancer, c-MYC binds to the promoter of CASC11 and increases the H3K27ac signal to enhance CASC11 expression. Increased CASC11 expression in colorectal cancer is associated with tumor size, serosal invasion, lymph metastasis, and the tumor-node-metastasis (TNM) stage.25 In hepatocellular carcinoma, higher expression of CASC11 is associated with a worse prognosis in hepatocellular carcinoma patients.26 In addition, we identified three biomarkers for the basal subtype, which include protective factor RP1-140K8.5, AC005162.1, and risk factor AC020916.2. We observed elevated H3K27ac and H3K4me1 at enhancer elements of RP1-140K8.5 and AC005162.1, which contributed to their upregulation (FC = 5.82, FDR = 4.92 × 10−9 for RP1-140K8.5; FC = 3.89, FDR = 2.00 × 10−6 for AC005162.1; Figures 4B–4D). Lower expression of RP1-140K8.5 or AC005162.1 was associated with poor prognosis in the basal subtype (FDR = 3.87 × 10−2; Figure 4C). Functional analysis showed that RP1-140K8.5 was involved in the T cell-mediated immune response and defense response by regulating interleukin (IL)-12A, ADAM15, and IL-5. AC005162.1 was involved in the regulation of the immune system process, the T cell receptor signaling pathway, and tumor necrosis factor (TNF) production by regulating CD6, CD47, and GSTP1. We observed elevated H3K27ac and H3K4me3 in the enhancer region of AC020916.2, which resulted in its significant upregulation (FC = 2.63, FDR = 7.49 × 10−4; Figures 4B–4D). A log-rank test showed that higher expression of AC020916.2 indicated poorer prognosis (FDR = 4.15 × 10−2; Figure 4C). Functional analysis showed that AC020916.2 was involved in the immune system process and apoptotic process by regulating, for example, PARP1, CD44, and CD48. These observations may suggest a heavy immune infiltration in basal breast cancer. This is supported by previous studies that showed that a considerable number of immune response-related genes exhibit significant variable expression across the basal cell subtype. Targeting these enhancer-related epi-lncRNAs in the basal subtype may help to achieve a more favorable response to immune therapy. We further investigated the effects of C646 treatment in the expression of six H3K27ac-dysregulated lncRNAs in human breast cancer (MDA-MB-468) cells. C646 is a histone acetyltransferase CREBBP/EP300 inhibitor. As expected, inhibition of CREBBP/EP300 histone acetyltransferase activity resulted in a significant loss of active enhancer mark H3K27ac (Figure 4E). The results of real-time polymerase chain reaction (PCR) showed that treatment with 20 μM C646 in MDA-MB-468 cells for 24 h significantly decreased expression levels of LINC00393, KB-1836B5.1, RP1-140K8.5, AC005162.1, and AC020916.2 compared with untreated cells (p < 0.01, Student’s t test; Figure 4F). CREBBP/EP300 knockdown resulted in significant inhibition of the growth of MDA-MB-468 cells in a dose-dependent manner (Figure 4G; p < 0.001). These results indicate that active enhancer mark H3K27ac takes part in modulating the enhancer-related lncRNA transcription of LINC00393, KB-1836B5.1, RP1-140K8.5, AC005162.1, and AC020916.2, which have potential tumor-promoting activities in the breast cancer basal subtype. Additionally, we identified three epi-lncRNAs (SLC26A4-AS1, CTC-303L1.2, and RP11-738B7.1) with differential DNA methylation that can significantly affect the survival of patients with basal subtype breast cancer (Table 1; Figures 4B and 4C). Upregulation of SLC26A4-AS1 is associated with its promoter hypermethylation (FC = 2.36, FDR = 0.02), and high SLC26A4-AS1 expression had significantly longer overall survival (FDR = 7.14 × 10−4). SLC26A4-AS1 has been reported to be significantly associated with papillary thyroid cancer patient disease-free survival and overall survival for gastric cancer patients.27 RP11-738B7.1 enhancer hypomethylation increases its expression (FC = 2.13, FDR = 1.2 × 10−3), and high levels of RP11-738B7.1 expression were correlated with poor outcome (FDR = 0.02). DNA hypermethylation at CTC-303L1.2 enhancer regions decreased its expression (FC = 0.53, FDR = 7.3 × 10−3), and low CTC-303L1.2 expression had significantly shorter overall survival (FDR = 0.03). Functional analysis showed that SLC26A4-AS1, CTC-303L1.2, and RP11-738B7.1 were related to the chemokine signaling pathway, IL-15-mediated signaling pathway, and transcriptional misregulation in cancer, respectively. We obtained an RNA sequencing (RNA-seq) dataset of 42 triple-negative breast cancer clinical samples and 21 adjacent non-tumor tissues (Gene Expression Omnibus [GEO]: GSE58135) to further validate the pattern of lncRNA expression in independent clinical samples. The expression changes for 77.8% (seven out of nine) epi-lncRNAs were validated (FDR < 0.05; Figure S5A).
Table 1.
The information of epi-lncRNAs associated with survival in breast cancer subtypes
| Subtype | lncRNA name | Alteration of epigenetics | Regulatory elements | Epigenetic alteration | Expression | Multiple Cox HR model(p) | Log-rank test(FDR) |
|---|---|---|---|---|---|---|---|
| Basal | LINC00393 | H3K27ac, H3K4me1 | enhancer | up | Up | 7.61 × 10–3 | 3.87 × 10–2 |
| Basal | KB-1836B5.1 | H3K27ac, H3K4me1 | enhancer | up | Up | 1.86 × 10–3 | 3.87 × 10–2 |
| Basal | CASC11 | H3K27ac, H3K4me1, H3K4me3 | enhancer | up | Up | 7.50 × 10–3 | 4.15 × 10–2 |
| Basal | RP1-140K8.5 | H3K27ac, H3K4me1 | enhancer | up | Up | 3.74 × 10–2 | 3.87 × 10–2 |
| Basal | AC005162.1 | H3K27ac, H3K4me1 | enhancer | up | Up | 1.80 × 10–2 | 3.87 × 10–2 |
| Basal | AC020916.2 | H3K27ac, H3K4me1, H3K4me3 | enhancer | up | Up | 3.96 × 10–3 | 4.15 × 10–2 |
| Basal | CTC-303L1.2 | DNA methylation | enhancer | up | down | 2.45 × 10–2 | 3.14 × 10–2 |
| Basal | RP11-738B7.1 | DNA methylation | enhancer | down | Up | 6.25 × 10–4 | 2.45 × 10–2 |
| Basal | SLC26A4-AS1 | DNA methylation | promoter | down | Up | 0 × 100 | 7.15 × 10–4 |
| Luminal | LINC01983 | DNA methylation | promoter | down | Up | 2.53 × 10–2 | 3.25 × 10–2 |
| Luminal | UCA1 | lncRNA regulator | – | – | down | 5.14 × 10–3 | 7.70 × 10–3 |
| Luminal | RP11-221J22.2 | lncRNA regulator | – | – | Up | 4.81 × 10–2 | 1.94 × 10–2 |
| Luminal | RP11-221J22.1 | lncRNA regulator | – | – | Up | 4.66 × 10–2 | 2.56 × 10–2 |
| Luminal | RP1-212P9.3 | lncRNA regulator | – | – | up | 2.25 × 10–2 | 2.56 × 10–2 |
Figure 4.

Epigenetically dysregulated lncRNAs at enhancer elements are associated with prognosis in basal subtypes
(A) Forest plot according to the result of multivariate Cox regression analysis of epigenetically dysregulated lncRNAs at enhancer elements. (B) Expression distribution of epigenetically dysregulated lncRNAs in basal and normal samples. (C) Kaplan-Meier survival curves according to the expression of epigenetically dysregulated lncRNAs. (D) Histone modification profile of epigenetically dysregulated lncRNAs in basal subtype. (E) Western blot analysis with H3K27ac after adding CREBBP/EP300 bromodomain inhibitor C646 (20 μM) to the MDA-MB-468 cell line. Cells were treated with C646 for 12 h. (F) qRT-PCR analyses of H3K27ac-dysregulated lncRNAs following its suppression. n = 6; mean ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 by Student’s t test. (G) CCK-8 assays of the viability of the MDA-MB-468 cell line following C646 treatment (5, 10, 20, and 40 μM) for 12 h.
In the luminal subtype, we identified one epi-lncRNA (LINC01983) with differential DNA methylation and four lncRNA regulators (UCA1, RP11-221J22.2, RP11-221J22.1, and RP1-212P9.3) that significantly affected the survival of patients (Table 1; Figures 5A and 5B). Upregulation of LINC01983 is associated with its promoter hypomethylation (FC = 2.56, FDR = 0.04), and high LINC01983 expression had a significantly poorer overall survival (FDR = 0.03). LINC01983 was involved in lymphocyte chemotaxis, T cell migration, and the chemokine-mediated signaling pathway. UCA1 was downregulated in the luminal subtype (FC = 0.18; FDR = 3.51 × 10−8) and associated with the TNF signaling pathway through regulating CXCL6 and MAP3K8, which can mediate the inflammatory immune response. Low UCA1 expression is an independent prognostic factor for poor overall survival (FDR = 7.70 × 10−3). UCA1 has been found to be differentially expressed in many cancer types, such as breast cancer.4 Low expression of RP11-221J22.1 and RP11-221J22.2 is independently correlated with decreased overall survival (FDR = 2.56 × 10−2 and FDR = 1.94 × 10−2, respectively). Functional analysis showed that both RP11-221J22.1 and RP11-221J22.2 were related to T helper (Th)1 and Th2 cell differentiation, the mitogen-activated protein kinase (MAPK) signaling pathway, and Th17 cell differentiation by regulating STAT6, MAPK10, and STAT3. High expression of RP1-212P9.3 is independently correlated with decreased overall survival (FDR = 2.56 × 10−2). Additionally, in the luminal subtype, we did not identify any survival-related epi-lncRNAs with differential histone modification. We set out to explore the prognostic values of these epi-lncRNA-related modules. We identified three modules, including epi-lncRNAs (KCNC4-AS1 and APCDD1L-AS1), as prognostic biomarkers of the luminal subtype (Table 2). Module 1 consists of two epi-lncRNAs (KCNC4-AS1 and APCDD1L-AS1) and 33 PCGs. Module 2 consists of KCNC4-AS1 and 34 PCGs. Module 3 consists of APCDD1L-AS1 and 74 PCGs. Functional analysis showed that these two lncRNAs are involved in the regulation of inflammatory response, apoptosis, cell division, cell communication, and nuclear factor κB (NF-κB) signaling by regulating genes such as STAG2, JAG2, VAMP8, HMGB1, and LITAF (Figure 5C). APCCD1L-AS1 is reported to significantly distinguish the high-expression group from the low-expression group in lung squamous cell carcinoma.28 Each of these modules was able to significantly distinguish patients in high-risk groups from those in low-risk groups in terms of overall survival in the luminal subtype (Figure 5D). We obtained an RNA-seq dataset of 42 ER-positive (ER+) breast cancer primary tumors and 30 adjacent breast tissues (GEO: GSE58135) to further validate the pattern of lncRNA expression in independent clinical samples. The expression changes for 66.7% (four out of six) detected lncRNAs were validated (FDR < 0.05; Figure S5B). Taken together, these results confirm the reproducibility of lncRNA biomarkers in independent samples.
Figure 5.

Epigenetically dysregulated lncRNAs at enhancer elements are associated with prognosis in luminal subtypes
(A) Expression distribution and Kaplan-Meier survival curves of lncRNA regulators in the luminal subtype. (B) Kaplan-Meier survival curves of an lncRNA with differential DNA methylation and epi-lncRNA-related modules in the luminal subtype. (C) The lncRNA-PCG co-expression networks showed the epi-lncRNAs-related modules in the luminal subtype.
Table 2.
Modules of epi-lncRNAs associated with survival in the luminal subtype
| Modules | lncRNAs | PCGs |
|---|---|---|
| 1 | KCNC4-AS1, APCDD1L-AS1 | IRS1, ABHD2, HMGB1, CALML3, SLC4A7, |
| ADAM23, TBXA2R, CFB, VDAC3, EVI2A, | ||
| BUB1B, GPR65, ADAMTS2, TNFRSF10A, | ||
| RIN1, TPP1, ABCD2, IFIT3, SCN4B, ZNF256, | ||
| CPB1, RBM4, NPIPA1, LIG4, DGKQ, GRINA, | ||
| LITAF, ADAM20, VAMP8, CHRNA3, URI1, | ||
| STXBP5L, ZKSCAN1 | ||
| 2 | KCNC4-AS1 | IRS1, ABHD2, HMGB1, CALML3, SLC4A7, |
| ADAM23, TBXA2R, CFB, VDAC3, EVI2A, | ||
| BUB1B, GPR65, ADAMTS2, TNFRSF10A, | ||
| RIN1, TPP1, ABCD2, IFIT3, SCN4B, ZNF256, | ||
| CPB1, RBM4, NPIPA1, LIG4, DGKQ, GRINA, | ||
| LITAF, ADAM20, VAMP8, CHRNA3, URI1, | ||
| STXBP5L, ZKSCAN1, GBF1 | ||
| 3 | APCDD1L-AS1 | IRS1, ABHD2, HMGB1, CALML3, SLC4A7, |
| ADAM23, TBXA2R, CFB, VDAC3, EVI2A, | ||
| BUB1B, GPR65, ADAMTS2, TNFRSF10A, | ||
| RIN1, TPP1, ABCD2, IFIT3, SCN4B, ZNF256, | ||
| CPB1, RBM4, NPIPA1, LIG4, DGKQ, GRINA, | ||
| LITAF, ADAM20, VAMP8, CHRNA3, URI1, | ||
| STXBP5L, ZKSCAN1, KCND2, ETV6, ITPR2, | ||
| JAG2, APOA1, LIG3, SNRPG, WASF3, MC3R, | ||
| LAMA3, NPC1, NDUFA10, TCEA2, C11orf58, | ||
| SCGB1D1, B2M, GPX5, SSTR2, SSTR5, C8A, | ||
| SLC16A6, NKTR, TNFRSF1A, ABCA2, MADD, | ||
| NOL4, GEM, CCNB2, COL8A2, MGAM, ORC5, | ||
| IDH3G, PPP1R10, KIF20B, DHX16, TSHR, STAG2, | ||
| RPS13, FGF3, HIST1H1C, GRIN2A |
Collectively, our findings underline the crucial roles of H3K27ac-dysregulated lncRNAs, DNA methylation-dysregulated lncRNAs, and lncRNA regulators in basal and luminal breast cancer carcinogenesis and their potential prognostic value. In addition, the inhibition of enhancer-related H3K27ac-dysregulated lncRNAs by histone acetyltransferase CREBBP/EP300 inhibitors offers a potential way to inhibit breast cancer growth and warrants further investigation.
Discussion
In this study, we systematically analyzed epigenetically dysregulated lncRNAs in luminal, basal, and claudin-low breast cancer subtypes. A large number of epigenetically dysregulated lncRNAs were identified in each of the three breast cancer subtypes, which included many known cancer lncRNAs. Epigenetically dysregulated lncRNAs showed complex splicing patterns and stable expression in breast cancer subtypes. Epigenetic alterations in a subtype-specific manner were shown by 92.6% of lncRNAs. A small number of common epi-lncRNAs shared by different subtypes showed differences in epigenetic dysregulation patterns. Functional analysis revealed that epigenetic-dysregulated lncRNAs at enhancer elements could control breast cancer subtype-specific biological functions. For the same histone modification mark H3K27ac, which accounts for 67.16% epi-lncRNAs in the three subtypes, H3K27ac-dysregulated lncRNAs at enhancer elements contributed to breast cancer subtype-specific biological functions. We thus present a landscape of epigenetic-dysregulated lncRNAs at enhancer elements as a valuable resource for studies of breast cancer subtypes and highlight a crucial role of enhancer-related epi-lncRNAs in the development of breast cancer subtypes.
We identified six H3K27ac-dysregulated lncRNAs at enhancer elements and three DNA methylated-dysregulated lncRNAs as independent prognostic biomarkers of the basal subtype of breast cancer. Three of these lncRNAs (LINC00393, KB-1836B5.1, and CASC11) have been identified as risk factors in many cancers, such as glioma, liver cancer, and colorectal cancer. SLC26A4-AS1 has been reported to be a protective factor of gastric cancer and papillary thyroid cancer. Five lncRNAs (RP1-140K8.5, AC005162.1, AC020916.2, CTC-303L1.2, and RP11-738B7.1) were identified as biomarkers in the basal subtype. RP1-140K8.5, AC005162.1, and CTC-303L1.2 act as protective factors, while AC020916.2 and RP11-738B7.1 are risk factors in the basal subtype. In the breast cancer MDA-MB-468 cell line, CREBBP/EP300 bromodomain inhibitors downregulated the expression of lncRNAs associated with enhancers and inhibition of tumor growth. This observation underlined that LINC00393, KB-1836B5.1, RP1-140K8.5, AC005162.1, and AC020916.2 were modulated by the active enhancer mark H3K27ac and that they have potential tumor-promoting activities. They may therefore be candidates for gene therapy approaches to breast cancer. In the luminal subtype, DNA methylation-deregulated lncRNA LINC01983 and lncRNA regulators (UCA1, RP11-221J22.2, RP11-221J22.1, and RP1-212P9.3) were identified as prognostic biomarkers. UCA1 has been reported to promote the EMT of breast cancer. LINC01983, RP11-221J22.2, RP11-221J22.1, and RP1-212P9.3 were identified as biomarkers in the luminal subtype. Currently, large-scale multi-dimensional omics and clinical data on breast cancer subtypes, especially for the claudin-low subtype, are scarce. The predictive capacities of our approach could be further improved as more large-scale multi-dimensional omics and clinical data of breast cancer subtypes become available.
In summary, our study analyzes the relationship between lncRNA expression and epigenetic alterations and explores their prognostic value in patients with breast cancer subtypes. We report that six enhancer-related H3K27ac-dysregulated lncRNAs, four DNA methylation-dysregulated lncRNAs, and four lncRNA regulators could serve as prognostic biomarkers for breast cancer subtypes. The expression levels of enhancer-related H3K27ac-dysregulated lncRNAs were downregulated after inhibition of histone acetyltransferases CREBBP and EP300. The inhibition of the expression of these lncRNAs has potential as a long-term treatment for breast cancer, but further studies are needed in this direction.
Materials and methods
Histone modification and expression data on breast cancer subtypes
We downloaded six types of histone modification data (including H3K27ac, H3K27me3, H3K36me3, H3K4me1, H3K4me3, and H3K9me3) in breast cancer cell lines (MCF-7, HCC1954 and MDA-MB-231) from the Encyclopedia of DNA Elements (ENCODE) and the GEO from the National Center for Biotechnology Information (GEO: GSE29069 and GSE85158). These cell lines represent luminal (MCF-7), basal (HCC1954), and claudin-low (MDA-MB-231) subtypes. Human mammary epithelial cells (HMECs) were used as a control cell line. Transcriptomic data for breast cancer were obtained from The Cancer Genome Atlas (TCGA). Samples of breast cancer subtypes were classified by PAM50 subtyping as described by Niknafs et al.29 We obtained 351 luminal subtype samples, 93 basal subtype samples, and 29 normal samples. We collected RNA-seq data for 23 claudin-low subtype samples (MDA-MB-231 cell line) and 25 control samples (HMEC cell line) from the GEO (GEO: GSE37918, GSE58135, GSE72141, GSE59335, GSE68248, GSE72524, GSE80409, GSE84577, GSE85579, and GSE91395). Raw reads for RNA-seq data were uniquely mapped to human reference genome versions of hg19/GRCh37 using Bowtie 2 (version 2.0.6). The value of reads per kilobase transcript per million mapped reads (RPKM) was calculated as the expression of the University of California Santa Cruz (UCSC) known genes and lncRNAs of GENCODE (release 35). DNA methylation data from 565 luminal subtype samples, 137 basal breast cancers, and 34 normal tissues using the Illumina human methylation 450K array were obtained from TCGA. We also obtained survival data of luminal and basal subtype patients from TCGA.
Identification of epigenetically dysregulated lncRNAs and PCGs
First, we identified differentially expressed lncRNAs and PCGs by comparing gene expression of three breast cancer subtypes with normal cells using DESeq2.30 The p values were determined by the Benjamini-Hochberg method. lncRNAs or PCGs with an FC greater than 2 and an FDR less than 0.05 were considered significant. Second, reads from chromatin immunoprecipitation combined with high-throughput sequencing (ChIP-seq) data were mapped to hg19/GRCh37 assembly of the human genome using Bowtie 2.31 Peak detection was run using MACS2 with a threshold of q less than 0.05.32 H3K27ac peaks observed for breast cancer subtypes are considered to be active or potentially active enhancer regions. The promoter of a gene was defined as 0.5 kb upstream and downstream of the transcriptional start site (TSS).33,34 DHMRs of different histone modifications were identified using MACS2 bdgdiff using default parameters in three breast cancer cell lines; HMECs were used as a control. We used the DMRcate R package to identify DMRs.35 A region was called significant when the Benjamini-Hochberg adjusted p value was less than 1 × 10−3. Finally, lncRNAs and PCGs were defined as epigenetically dysregulated lncRNAs or PCGs according to the following criteria: (1) lncRNAs and PCGs were differentially expressed in the breast cancer subtype; (2) their promoters or enhancers overlapped at least one DHMR or DMR; and (3) the histone modification and DNA methylation alteration at regulatory elements corresponded with expression changes of genes.
Identification of lncRNA regulators
We used partial least-squares regression to determine the regulation of lncRNA to lncRNA expression. For each differentially expressed lncRNA, a partial least-squares regression model was constructed using the pls package to explain lncRNA expression changes using other differentially expressed lncRNAs as covariates. lncRNAs with significant coefficients (FDR < 0.05) were identified as lncRNA regulators.
Co-expression networks and functional enrichment analysis of epigenetic-dysregulated lncRNAs
A lncRNA-PCG co-expression network was constructed for each breast cancer subtype using the normalized signal intensity of individual genes based on guilt by association.29,36 For each lncRNA-PCG pair, we calculated the Pearson correlation coefficient (PCC). We chose lncRNA-PCG pairs with significant correlations (PCC > 0.7 and FDR < 0.05) to construct the co-expression network. Functional enrichment analysis based on Gene Ontology (GO) annotation terms was carried out for each of the epigenetic-dysregulated lncRNAs using its co-expressed PCGs with the R package clusterProfiler.37 The significance threshold was FDR less than 0.05.
Identification of lncRNA biomarkers
Multivariable Cox proportional hazard regression analysis was performed considering the pathologic stage, ER status, progesterone receptor (PR) status, HER2 status, and lymph node number as covariates. We estimated the expression median of the selected lncRNAs (p < 0.05) and divided the patients with a specific breast cancer subtype into two groups. A log-rank test was used to assess the differences in survival times between different groups of patients.38
We identified modules as lncRNA-related modules based on lncRNA-PCG co-expressed networks using the R package biclique.39 Univariate Cox regression analyses were carried out for individual PCGs and lncRNAs in the modules with their expression values as variables. A risk score was computed for every patient according to a linear combination of the expression values weighted by coefficients from univariate Cox regression analysis. The risk score was computed as follows:
| (1) |
where n is the number of genes in the module, βi represents the coefficient of univariate Cox regression analysis of the ith gene in the module, and Expi represents the expression of the ith gene in the module. All patients were classified into two groups for each module according to the median of the risk scores. The Kaplan-Meier method and log-rank test were used to estimate the difference in survival time between the two groups.
Cell culture and treatment
The human breast cancer cell line MDA-MB-468 was obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were cultured in Roswell Park Memorial Institute 1640 (RPMI 1640) medium (Gibco, USA), which contained 10% fetal bovine serum (Gibco) and 1% penicillin-streptomycin (100 U/mL penicillin, 0.1 mg/mL streptomycin; C0222, Beyotime, China) at 37°C with 95% air and 5% carbon dioxide. The cells were seeded into 6-, 12-, and 96-well plates and incubated in RPMI 1640 medium for 12 h followed by incubation with different concentrations of C646 or dimethyl sulfoxide (DMSO, Sigma-Aldrich) as a negative control (NC). C646 was purchased from MedChemExpress (HY-13823, MedChemExpress, China).
Cell Counting Kit-8 (CCK-8) assay
The cells were seeded into 96-well plates for 24 h at a density of 1 × 103 cells/well. C646 was diluted to different concentrations (0, 5, 10, 20, and 40 μM) and subsequently added to the plates for another 24-h treatment. After 24 h, the cell viability was measured using a CCK-8 proliferation assay (Beijing Solarbio Science & Technology, Beijing, China). The cells were incubated in 100 μL of RPMI 1640 medium mixed with 10 μL of CCK-8 solution/well for an additional 2 h at 37°C. The optical density (OD) of each well was measured at 450 nm with a microplate reader (Bio-Rad, USA) to determine the cell viability of MDA-MB-468.
Total RNA extraction and quantitative reverse-transcriptase PCR (qRT-PCR)
Total RNA from different groups of the MDA-MB-468 cell line was extracted with TRIzol reagent (Invitrogen, USA) according to the instructions of the manufacturer. The concentration and purity of RNA were determined using a NanoDrop 8000 spectrophotometer (Thermo Fisher Scientific, USA). cDNA was prepared using a high-capacity cDNA reverse transcription kit (Applied Biosystems, USA) following the protocol of the manufacturer. qRT-PCR was performed on an ABI 7500 Fast real-time PCR system (Applied Biosystems, USA). The conditions used were as follows: hold stage was 95°C for 10 min, and cycling was 40 cycles of 95°C for 30 s, 60°C for 30 s, and 72°C for 30 s. Expression levels of the following lncRNAs were examined: AC005162, AC020916, RP1-140K8.5, KB-1836B5.1, LINC00393, and CASC11. The expression level of the β-actin gene served as the reference. The forward and reverse primer sequences used are listed in Table S1. Each qRT-PCR was processed in triplicate. The threshold cycle (Ct) value was subsequently determined, and the relative quantification of lncRNA expression was calculated using the comparative Ct method.
Western blot
The cells in six-well plates were washed with phosphate-buffered saline (PBS, pH 7.4) three times on ice. Cells were subsequently lysed for 10 min using radioimmunoprecipitation assay (RIPA) buffer (Sigma-Aldrich) and centrifuged at 10,000 × g for 10 min at 4°C. The supernatants were collected. The total protein in lysates was measured using the bicinchoninic acid (BCA) protein assay kit (Beyotime, China). Cell lysates (60 μg) were electrophoresed using SDS-PAGE on a 12.5% polyacrylamide gel and transferred onto nitrocellulose membranes by semi-dry electroblotting using the Trans-Blot Turbo system (Bio-Rad). Membranes were blocked with 5% non-fat milk and 0.1% Tween 20 (Thermo Fisher Scientific, USA) for 60 min at room temperature, and subsequently probed at 4°C for 12 h with the following primary antibodies: anti-histone H3 (1:500, YT2163, Immunoway, China), acetyl-histone H3 (1:500, YK0010, Immunoway, China), and β-actin (1:1,000, Abcam, UK). The membranes were washed and incubated with horseradish peroxidase (HRP)-labeled secondary antibodies (goat anti-rabbit-HRP or goat anti-mouse-HRP) (Abcam) and diluted 1:10,000 for 1 h at room temperature on a rocker. Lastly, membranes were washed three times with PBST (PBS with 0.05% Tween 20, pH 7.4) followed by scanning and analysis using Odyssey version 1.2 software (LI-COR Biosciences, USA). The β-actin was used as the internal control. All blots were performed in three or more biological replicates.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (31801115 and 61803129); the China Postdoctoral Science Foundation (2018M631943 and 2018M641860), the China Postdoctoral Science Special Foundation (2019T120280 and 2020T130161); the Hei Long Jiang Postdoctoral Special Foundation (LBH-TZ1018); the Hei Long Jiang Postdoctoral Foundation (LBH-Z17110, and LBH-Z17218); and by the Heilongjiang Provincial Planning Office Key Subjects (GBB1318066).
Author contributions
H.Z., X.L., and L.W. designed the study, implemented the algorithm, and performed the analysis. H.Z., X.L., J.L., and L.W. wrote and revised the manuscript. L.Y., S.L., C.Z., H.X., Z.L., W.H., J.L., and T.L. help to collect the data and prepare the figures and tables. Y.F. helped to perform some experiments. All authors read, reviewed, and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.12.024.
Contributor Information
Jing Li, Email: ljcross2008@126.com.
Fan Yang, Email: yangfan@hrbmu.edu.cn.
Li Wang, Email: wangli@hrbmu.edu.cn.
Supplemental information
References
- 1.Zhao H., Zhang G., Pang L., Lan Y., Wang L., Yu F., Hu J., Li F., Zhao T., Xiao Y., Li X. “Traffic light rules”: chromatin states direct miRNA-mediated network motifs running by integrating epigenome and regulatome. Biochim. Biophys. Acta. 2016;1860:1475–1488. doi: 10.1016/j.bbagen.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Wang L., Zhao H., Xu Y., Li J., Deng C., Deng Y., Bai J., Li X., Xiao Y., Zhang Y. Systematic identification of lincRNA-based prognostic biomarkers by integrating lincRNA expression and copy number variation in lung adenocarcinoma. Int. J. Cancer. 2019;144:1723–1734. doi: 10.1002/ijc.31865. [DOI] [PubMed] [Google Scholar]
- 3.Prat A., Parker J.S., Karginova O., Fan C., Livasy C., Herschkowitz J.I., He X., Perou C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010;12:R68. doi: 10.1186/bcr2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao H., Shi J., Zhang Y., Xie A., Yu L., Zhang C., Lei J., Xu H., Leng Z., Li T. LncTarD: a manually-curated database of experimentally-supported functional lncRNA-target regulations in human diseases. Nucleic Acids Res. 2020;48(D1):D118–D126. doi: 10.1093/nar/gkz985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Augoff K., McCue B., Plow E.F., Sossey-Alaoui K. miR-31 and its host gene lncRNA LOC554202 are regulated by promoter hypermethylation in triple-negative breast cancer. Mol. Cancer. 2012;11:5. doi: 10.1186/1476-4598-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heilmann K., Toth R., Bossmann C., Klimo K., Plass C., Gerhauser C. Genome-wide screen for differentially methylated long noncoding RNAs identifies Esrp2 and lncRNA Esrp2-as regulated by enhancer DNA methylation with prognostic relevance for human breast cancer. Oncogene. 2017;36:6446–6461. doi: 10.1038/onc.2017.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wu S.C., Kallin E.M., Zhang Y. Role of H3K27 methylation in the regulation of lncRNA expression. Cell Res. 2010;20:1109–1116. doi: 10.1038/cr.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y., Li S., Chen J., Shao T., Jiang C., Wang Y., Chen H., Xu J., Li X. Comparative epigenetic analyses reveal distinct patterns of oncogenic pathways activation in breast cancer subtypes. Hum. Mol. Genet. 2014;23:5378–5393. doi: 10.1093/hmg/ddu256. [DOI] [PubMed] [Google Scholar]
- 9.Sati S., Ghosh S., Jain V., Scaria V., Sengupta S. Genome-wide analysis reveals distinct patterns of epigenetic features in long non-coding RNA loci. Nucleic Acids Res. 2012;40:10018–10031. doi: 10.1093/nar/gks776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao Y., Wang P., Wang Y., Ma X., Zhi H., Zhou D., Li X., Fang Y., Shen W., Xu Y. Lnc2Cancer v2.0: updated database of experimentally supported long non-coding RNAs in human cancers. Nucleic Acids Res. 2019;47(D1):D1028–D1033. doi: 10.1093/nar/gky1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jonsson P., Coarfa C., Mesmar F., Raz T., Rajapakshe K., Thompson J.F., Gunaratne P.H., Williams C. Single-molecule sequencing reveals estrogen-regulated clinically relevant lncRNAs in breast cancer. Mol. Endocrinol. 2015;29:1634–1645. doi: 10.1210/me.2015-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang O., Yang F., Liu Y., Lv L., Ma R., Chen C., Wang J., Tan Q., Cheng Y., Xia E. c-MYC-induced upregulation of lncRNA SNHG12 regulates cell proliferation, apoptosis and migration in triple-negative breast cancer. Am. J. Transl. Res. 2017;9:533–545. [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao X.B., Ren G.S. lncRNA taurine-upregulated gene 1 promotes cell proliferation by inhibiting microRNA-9 in MCF-7 cells. J. Breast Cancer. 2016;19:349–357. doi: 10.4048/jbc.2016.19.4.349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J., Piao H.L., Kim B.J., Yao F., Han Z., Wang Y., Xiao Z., Siverly A.N., Lawhon S.E., Ton B.N. Long noncoding RNA MALAT1 suppresses breast cancer metastasis. Nat. Genet. 2018;50:1705–1715. doi: 10.1038/s41588-018-0252-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frankish A., Diekhans M., Ferreira A.M., Johnson R., Jungreis I., Loveland J., Mudge J.M., Sisu C., Wright J., Armstrong J. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47(D1):D766–D773. doi: 10.1093/nar/gky955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Du Z., Fei T., Verhaak R.G., Su Z., Zhang Y., Brown M., Chen Y., Liu X.S. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nat. Struct. Mol. Biol. 2013;20:908–913. doi: 10.1038/nsmb.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Derrien T., Johnson R., Bussotti G., Tanzer A., Djebali S., Tilgner H., Guernec G., Martin D., Merkel A., Knowles D.G. The GENCODE v7 catalog of human long noncoding RNAs: analysis of their gene structure, evolution, and expression. Genome Res. 2012;22:1775–1789. doi: 10.1101/gr.132159.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y., Zheng Q., Bao C., Li S., Guo W., Zhao J., Chen D., Gu J., He X., Huang S. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 2015;25:981–984. doi: 10.1038/cr.2015.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang L., Liu D., Wu X., Zeng Y., Li L., Hou Y., Li W., Liu Z. Long non-coding RNA (lncRNA) RMST in triple-negative breast cancer (TNBC): expression analysis and biological roles research. J. Cell. Physiol. 2018;233:6603–6612. doi: 10.1002/jcp.26311. [DOI] [PubMed] [Google Scholar]
- 20.Askarian-Amiri M.E., Seyfoddin V., Smart C.E., Wang J., Kim J.E., Hansji H., Baguley B.C., Finlay G.J., Leung E.Y. Emerging role of long non-coding RNA SOX2OT in SOX2 regulation in breast cancer. PLoS ONE. 2014;9:e102140. doi: 10.1371/journal.pone.0102140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu X.L., Wang J., He W., Zhao P., Wu W.Q. Down-regulation of lncRNA Linc00152 suppressed cell viability, invasion, migration, and epithelial to mesenchymal transition, and reversed chemo-resistance in breast cancer cells. Eur. Rev. Med. Pharmacol. Sci. 2018;22:3074–3084. doi: 10.26355/eurrev_201805_15067. [DOI] [PubMed] [Google Scholar]
- 22.Hu R., Zhong P., Xiong L., Duan L. Long noncoding RNA cancer susceptibility candidate 8 suppresses the proliferation of bladder cancer cells via regulating glycolysis. DNA Cell Biol. 2017;36:767–774. doi: 10.1089/dna.2017.3785. [DOI] [PubMed] [Google Scholar]
- 23.Bradford J.R., Cox A., Bernard P., Camp N.J. Consensus analysis of whole transcriptome profiles from two breast cancer patient cohorts reveals long non-coding RNAs associated with intrinsic subtype and the tumour microenvironment. PLoS ONE. 2016;11:e0163238. doi: 10.1371/journal.pone.0163238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matjasic A., Popovic M., Matos B., Glavac D. Expression of LOC285758, a potential long non-coding biomarker, is methylation-dependent and correlates with glioma malignancy grade. Radiol. Oncol. 2017;51:331–341. doi: 10.1515/raon-2017-0004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Z., Zhou C., Chang Y., Zhang Z., Hu Y., Zhang F., Lu Y., Zheng L., Zhang W., Li X., Li X. Long non-coding RNA CASC11 interacts with hnRNP-K and activates the WNT/β-catenin pathway to promote growth and metastasis in colorectal cancer. Cancer Lett. 2016;376:62–73. doi: 10.1016/j.canlet.2016.03.022. [DOI] [PubMed] [Google Scholar]
- 26.Yin X., Zheng S.S., Zhang L., Xie X.Y., Wang Y., Zhang B.H., Wu W., Qiu S., Ren Z.G. Identification of long noncoding RNA expression profile in oxaliplatin-resistant hepatocellular carcinoma cells. Gene. 2017;596:53–88. doi: 10.1016/j.gene.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 27.Li C.Y., Liang G.Y., Yao W.Z., Sui J., Shen X., Zhang Y.Q., Peng H., Hong W.W., Ye Y.C., Zhang Z.Y. Integrated analysis of long non-coding RNA competing interactions reveals the potential role in progression of human gastric cancer. Int. J. Oncol. 2016;48:1965–1976. doi: 10.3892/ijo.2016.3407. [DOI] [PubMed] [Google Scholar]
- 28.Luo Y., Xuan Z., Zhu X., Zhan P., Wang Z. Long non-coding RNAs RP5-821D11.7, APCDD1L-AS1 and RP11-277P12.9 were associated with the prognosis of lung squamous cell carcinoma. Mol. Med. Rep. 2018;17:7238–7248. doi: 10.3892/mmr.2018.8770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niknafs Y.S., Han S., Ma T., Speers C., Zhang C., Wilder-Romans K., Iyer M.K., Pitchiaya S., Malik R., Hosono Y. The lncRNA landscape of breast cancer reveals a role for DSCAM-AS1 in breast cancer progression. Nat. Commun. 2016;7:12791. doi: 10.1038/ncomms12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vernimmen D., Bickmore W.A. The hierarchy of transcriptional activation: from enhancer to promoter. Trends Genet. 2015;31:696–708. doi: 10.1016/j.tig.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 34.Wang L., Zhao H., Li J., Xu Y., Lan Y., Yin W., Liu X., Yu L., Lin S., Du M.Y. Identifying functions and prognostic biomarkers of network motifs marked by diverse chromatin states in human cell lines. Oncogene. 2020;39:677–689. doi: 10.1038/s41388-019-1005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peters T.J., Buckley M.J., Statham A.L., Pidsley R., Samaras K., V Lord R., Clark S.J., Molloy P.L. De novo identification of differentially methylated regions in the human genome. Epigenetics Chromatin. 2015;8:6. doi: 10.1186/1756-8935-8-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guttman M., Amit I., Garber M., French C., Lin M.F., Feldser D., Huarte M., Zuk O., Carey B.W., Cassady J.P. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature. 2009;458:223–227. doi: 10.1038/nature07672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yu G., Wang L.G., Yan G.R., He Q.Y. DOSE: an R/Bioconductor package for disease ontology semantic and enrichment analysis. Bioinformatics. 2015;31:608–609. doi: 10.1093/bioinformatics/btu684. [DOI] [PubMed] [Google Scholar]
- 38.Jung M., Russell A.J., Liu B., George J., Liu P.Y., Liu T., DeFazio A., Bowtell D.D., Oberthuer A., London W.B. A Myc activity signature predicts poor clinical outcomes in Myc-Associated Cancers. Cancer Res. 2017;77:971–981. doi: 10.1158/0008-5472.CAN-15-2906. [DOI] [PubMed] [Google Scholar]
- 39.Xu J., Wang Z., Li S., Chen J., Zhang J., Jiang C., Zhao Z., Li J., Li Y., Li X. Combinatorial epigenetic regulation of non-coding RNAs has profound effects on oncogenic pathways in breast cancer subtypes. Brief. Bioinform. 2018;19:52–64. doi: 10.1093/bib/bbw099. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.