

Abstract
Oncolytic virotherapy (OVT) has been suggested to be effective. However, the suppressive effects of checkpoints and insufficient costimulatory signals limit OVT-induced antitumor immune responses. In this study, we constructed a replicative adenovirus, Ad5sPVR, that expresses the soluble extracellular domain of poliovirus receptor (sPVR). We showed that sPVR can bind to both T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT) and CD226, and the binding affinity of sPVR to TIGIT is stronger than that of PVR to CD226. In the H22 hepatocellular carcinoma (HCC) ascites model, Ad5sPVR treatment increased the infiltration of CD8+ T cells and the release of interferon (IFN)-γ, exhibiting an antitumor effect with long-term tumor-specific immune surveillance. In line with this, Ad5sPVR also effectively improved antitumor outcomes in solid tumors. In conclusion, while Ad5sPVR plays a role in oncolysis and transforms cold tumors into hot tumors, sPVR expressed by Ad5sPVR can block the PVR/TIGIT checkpoint and activate CD226, thereby greatly improving the efficacy of OVT. This study provides a new way to develop potential oncolytic viral drugs.
Keywords: oncolytic virus, PVR/CD155, T cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain, TIGIT, immune checkpoints, CD226, cancer therapy
Graphical Abstract
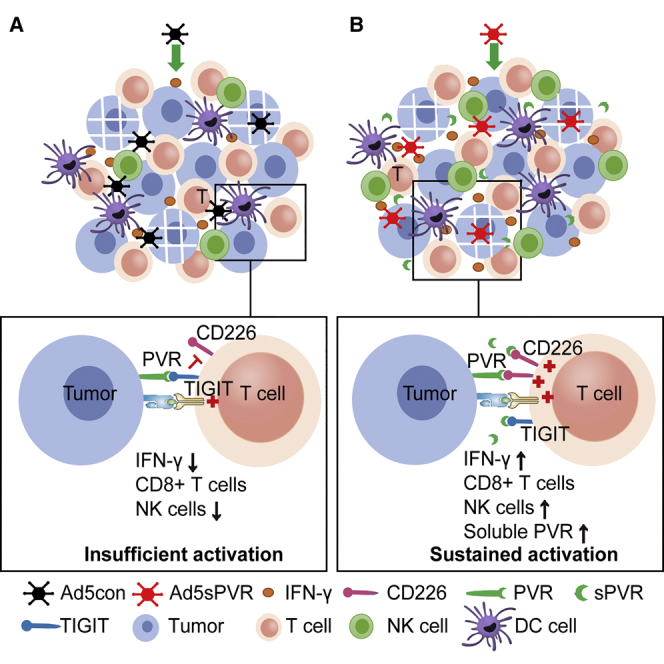
Zhang et al. constructed a replicative oncolytic adenovirus Ad5sPVR that not only enhances infiltration of lymphocytes, but also blocks the PVR/TIGIT checkpoint and activates CD226. This design effectively overcomes the hurdles of viro-immunotherapies: the inducible immune checkpoints and insufficient costimulatory signals.
Introduction
Since the approval of talimogene laherparepvec (T-VEC, Imlygic) by the US Food and Drug Administration (FDA) in 2015, oncolytic virus-mediated antitumor immunotherapy has received increasing attention.1 As a foreign invader, an oncolytic virus can rapidly induce type I interferon (IFN)-mediated immune activation and upregulate local lymphocyte chemokines to enhance the recruitment and infiltration of immune cells into the tumor microenvironment (TME),1, 2, 3 ultimately transforming “cold” tumors into “hot” tumors.4 Oncolytic adenovirus, a double-stranded DNA virus, is an important member of the oncolytic virus family. Oncolytic adenovirus with deletion of E1B-55kDa possesses the ability to selectively replicate in tumors. The oncolytic adenovirus H101 with E1B-55kDa deleted was approved for the treatment of head and neck squamous cell carcinoma in 2005.5 There are currently multiple oncolytic adenoviruses being studied in clinical6, 7, 8, 9, 10 and preclinical research,11, 12, 13, 14 and these oncolytic adenoviruses have shown some application prospects and have characteristics of oncolytic adenovirus-mediated immunotherapy. First, an oncolytic adenovirus can cause immunogenic cell death (ICD) of tumor cells, leading to the release of damage-associated molecular patterns (DAMPs) and the activation of innate immunity.2,15 Second, an oncolytic adenovirus can recruit lymphocytes and activate the adaptive immune response mediated by CD8+ T cells.16 Third, an oncolytic adenovirus is easy to modify and is an ideal foreign gene expression vector because it can express foreign genes in infecting tumor cells.15,17 Finally, oncolytic adenoviruses can selectively infect tumor cells with high safety, thus restricting the encoded immune checkpoints or cytokines to local expression in tumors and reducing autoimmune-like damage caused by off-target effects.16,18,19 However, while oncolytic adenoviruses are used in tumor immunotherapy, the tumor immune microenvironment can accordingly induce immunosuppression through the activation of immune checkpoint pathways and restriction of immune costimulatory pathways, thereby greatly limiting the antitumor immune response of these oncolytic adenoviruses.20,21
TIGIT was recently discovered as an immune checkpoint molecule expressed on lymphocytes.20,22,23 TIGIT has been found to be involved in the negative regulation of activated T cells and natural killer (NK) cells.20,22,23 As an immune checkpoint molecule, TIGIT regulates T cells mainly in the following ways. First, TIGIT on the surface of T cells can bind to PVR (CD155) on tumor cell or antigen-presenting cell (APC) surfaces. After binding, signaling is transduced into tumor cells or APCs to achieve immune suppression by upregulating interleukin (IL)-10 expression and downregulating IL-12 expression.20 Second, TIGIT on T cells can also directly bind to the immune costimulatory molecule CD226, blocking the formation of the CD226 homodimer and thereby inhibiting the immune costimulatory signals transduced by CD226. In this way, TIGIT plays another type of immunosuppressive role.23 Third, PVR is a ligand of both TIGIT and CD226.24 PVR can be used as a ligand to activate the immune costimulatory pathway mediated by CD226. The affinity of PVR and TIGIT has been found to be much greater than that of PVR and CD226. Therefore, TIGIT can be used to reduce the binding of PVR and CD226, thereby reducing the immune costimulatory signaling pathway mediated by PVR/CD226 and playing a novel immunosuppressive role.22,23,25
Clinical studies have shown that TIGIT is related to the poor clinical prognosis of acute myeloid leukemia. Blocking TIGIT can restore T cell function and antitumor immunity and is a novel and effective treatment for leukemia.26,27 It was reported that blocking TIGIT can prevent the exhaustion of NK cells and induce a strong antitumor immune response.28 TIGIT is highly expressed on the surface of CD8+ T cells, and blocking TIGIT can enhance the antitumor immune response mediated by CD8+ T cells.23 Blocking TIGIT or PVR can enhance the lysis of breast cancer cells mediated by immune cells. Targeting the TIGIT/PVR immune checkpoint pathway is therefore a novel treatment for breast cancer.29 These results indicate that the PVR/TIGIT pathway is an effective target for tumor immunotherapy. If the soluble extracellular domain of PVR (sPVR) is used for tumor immunotherapy, then sPVR can block the immunosuppressive effect mediated by the immune checkpoint PVR/TIGIT pathway by binding to TIGIT and also directly and indirectly activate the immunostimulatory pathway mediated by CD226, thus inducing an antitumor immune response via multiple routes.
In this study, we successfully constructed a recombinant oncolytic adenovirus expressing sPVR with E1B-55kDa, called Ad5sPVR, using the ViraPower pAd/PL-DEST adenovirus expression system. We speculate that Ad5sPVR, as a replication-type oncolytic adenovirus, has a direct oncolytic effect and also increases lymphocyte infiltration, thereby transforming cold tumors into hot tumors. Simultaneously, sPVR directly blocks the PVR/TIGIT immune checkpoint pathway and activates the CD226-mediated immune costimulatory pathway. In the present study, we determined whether Ad5sPVR could promote the infiltration and activation of immune cells in the TME and whether the expressed sPVR could enhance antitumor immune responses by blocking the PVR/TIGIT inhibitory pathway and activating the PVR/CD226 pathway to achieve potent antitumor effects.
Results
Generation of a novel recombinant adenovirus that expresses sPVR
To elicit a therapeutic antitumor immune response, a novel recombinant adenovirus, Ad5sPVR, was designed to express sPVR, which blocks PVR/TIGIT immune checkpoint signaling and provides a costimulatory signal to effector immune cells. Fragments for expression were inserted into the genome of a replicative adenovirus (Figure 1A). First, we confirmed that sPVR could be secreted into and accumulate in the supernatants of virus-infected tumor cells (Figure 1B). To further confirm whether sPVR bound to TIGIT and CD226, we used the Octet RED96 protein interaction analysis system to determine the affinity.30,31 The affinity between free sPVR and TIGIT or CD226, as indicated by the equilibrium dissociation constant (Kd), was 33 nM and 6.13 μM, respectively (Figure 1C). The results demonstrated that the affinity between sPVR and TIGIT was stronger than that between sPVR and CD226, a result that is consistent with previous research.22 Therefore, we constructed the recombinant adenovirus Ad5sPVR.
Figure 1.

Generation of a novel recombinant adenovirus, Ad5sPVR, expressing the soluble extracellular domain of PVR (sPVR)
(A) Recombinant adenovirus constructs. EXO, extracellular domain; Ad5sPVR, recombinant adenovirus encoding sPVR. (B) H22 cells and B16/F10 cells were infected with the recombinant adenovirus (multiplicity of infection [MOI] of 20 and 10, respectively) for 72 h, and the levels of sPVR in the supernatant were determined by western blotting. The molecular weights were estimated using a molecular weight marker. (C) 293T cells were infected with Ad5sPVR, and soluble PVR in the cell culture supernatant was harvested and purified by a nickel column. The affinity of sPVR/TIGIT and sPVR/CD226 was detected using an Octet RED96 protein interaction analysis system.
The replication capacity and oncolytic activities of the recombinant adenovirus are not impaired by recombination of the sPVR gene
Next, we explored whether recombination of the sPVR gene affected the replication capacity and oncolytic activities of the recombinant adenovirus. Compared with the replication and oncolysis of the control virus (Ad5con), Ad5sPVR replication (Figure 2A; Figure S1) and oncolysis (Figure 2B) did not change significantly. In conclusion, in vitro analysis demonstrated that recombination of the sPVR gene did not alter the replication capacity or oncolytic activities of the recombinant adenovirus in various cell lines. These data suggest that the recombinant adenovirus retained its replication and oncolysis capabilities while expressing and secreting the sPVR protein.
Figure 2.

Replication and oncolytic activities of the recombinant adenovirus Ad5sPVR
(A) Hepa1-6, H22, LM3, B16/F10, 4T1, and LLC1 cells were infected with recombinant adenoviruses at an MOI of 5, 20, 2, 10, 5, and 10 and were harvested at various time points. DNA was extracted and the viral copy number was determined by RT-PCR. (B) Hepa1-6, H22, LM3, B16/F10, 4T1, and LLC1 cells were infected with recombinant adenovirus at the indicated MOIs for 72 h, and cell viability was determined by the MTT assay. Virus replication and oncolysis are presented using one-way ANOVA with repeated measures (SPSS). The data are shown as the means ± SD. ns, not significant.
Recombinant Ad5sPVR significantly increases IFN-γ and lymphocyte infiltration within the TME
We further investigated whether the amount of IFN-γ in the TME could be modulated by Ad5sPVR. In immune-competent C57BL/6 mice, we established an intraperitoneal (i.p.) murine H22 model and monitored tumor growth during the intervention. The tumor model and the therapeutic regimen are depicted in Figure 3A. On days 12 and 16, before i.p. injection of saline or virus, ascites were collected for the follow-up study. We monitored viral load (Figure 3B) and soluble protein levels (Figure 3C) in the ascites during treatment at earlier stages by an enzyme-linked immunosorbent assay (ELISA) on day 16.
Figure 3.

Effect of recombinant adenovirus Ad5sPVR on antitumor immune activation and lymphocyte infiltration
(A) Schematic diagram of the experimental setup for adenovirus therapy in H22-challenged mice. On day 0, male C57BL/6 mice were injected intraperitoneally with 5 × 106 H22 cells, and the mice were randomly divided into three groups: saline group, Ad5con group, and Ad5sPVR group. On days 8, 12, and 16, mice in the saline group, Ad5con group, and Ad5sPVR group were injected intraperitoneally with saline, the control virus Ad5con (5 × 108 PFU), and Ad5sPVR (5 × 108 PFU), respectively. On days 12 and 16, before intraperitoneal injection of saline or virus, ascites were collected. (B) Virus replication was measured in the ascites of mice infected with recombinant adenovirus. On day 16, before intraperitoneal injection of saline or virus, ascites were collected and the viral copy number was determined by RT-PCR. (C) On day 16, before intraperitoneal injection of saline or virus, ascites were collected and the concentration of sPVR in ascites was determined by ELISA. (D) On day 16, before intraperitoneal injection of saline or virus, ascites were collected, and CD4+ T cell, CD8+ T cell, and NK cell frequencies in ascites were determined by flow cytometry. (E) On day 16, before intraperitoneal injection of saline or virus, ascites were collected, and immune activity in the TME was detected using a mouse IFN-γ ELISpot assay, and the number of IFN-γ spots in each group was calculated. The data are shown as the means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. i.p., intraperitoneal; s.c., subcutaneous; i.t., intratumoral.
Compared with saline and Ad5con, there were no significant changes in the level of CD4+ T cells in the ascites of mice infected with Ad5sPVR; however, increased levels of CD8+ T cell and NK cell infiltration were observed in the ascites of mice infected with virus, and the proportion of NK cells was highest in the ascites of mice infected with Ad5sPVR (Figure 3D). Then, using an enzyme-linked immunosorbent spot (ELISpot) assay, we found that the amount of IFN-γ was robustly upregulated within the TME in mice treated with Ad5sPVR compared with Ad5con (Figure 3E). Using IFN-γ as the index of immune activation,32 the results indicated that, compared with Ad5con, Ad5sPVR possessed significantly enhanced immune activation ability against tumors in the TME. In the H22 ascites mouse model, we investigated lymphocyte infiltration. Therefore, Ad5sPVR robustly induced lymphocyte infiltration and an activated immune response to eradicate tumor cells.
Ad5sPVR induces an antitumor immune response and results in a durable cure rate in the H22 ascites model
The H22 mouse ascites model and the therapeutic regimen are depicted in Figure 4A. On days 12 and 16, before i.p. injection of saline or virus, ascites were collected for the upcoming study. We monitored viral load (Figure 4B) and sPVR levels (Figure 4C) in the ascites during treatment at earlier stages on day 16. In vivo analysis further demonstrated that there were no differences in viral load between the two groups. The survival of mice treated with Ad5con was similar to that of mice that received saline. However, Ad5sPVR treatment significantly prolonged mouse survival, resulting in a 20% cure rate (Figure 4D). To elucidate whether Ad5sPVR-mediated tumor clearance was tumor-specific, cured H22-bearing mice were challenged twice with i.p. injections of H22 cells, but no tumor burden (ascites) was observed; all naive mice were susceptible to H22 cells (Figure 4E). These results indicate that Ad5sPVR elicits long-term immune memory. In summary, recombinant Ad5sPVR induces a robust antitumor immune response and results in a durable cure rate.
Figure 4.

Effect of recombinant adenovirus Ad5sPVR on the antitumor immune response and cure rate in the H22 ascites model
(A) Schematic diagram of the experimental setup for adenovirus therapy. On day 0, male C57BL/6 mice were injected intraperitoneally with 5 × 106 H22 cells, and the mice were randomly divided into three groups: saline group, Ad5con group, and Ad5sPVR group. On days 8, 12, and 16, mice in the saline group, Ad5con group, and Ad5sPVR group were injected intraperitoneally with saline, the control virus Ad5con (5 × 108 PFU), and Ad5sPVR (5 × 108 PFU), respectively. On days 12 and 16, before intraperitoneal injection of saline or virus, ascites were collected for further study. (B) Virus replication in the ascites of mice infected with recombinant adenovirus. On day 16, before intraperitoneal injection of saline or virus, ascites were collected and the viral copy number was determined by RT-PCR. (C) On day 16, before intraperitoneal injection of saline or virus, ascites were collected and the concentration of sPVR in ascites was determined by ELISA. (D) Survival curves for mice treated with recombinant adenovirus. (E) Mice that had been cured were challenged with 5 × 106 H22 cells on day 90 and day 150 post-inoculation. Naive mice were used as a control. The day of challenge was regarded as day 0 in survival curves. The data are shown as the means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Recombinant Ad5sPVR induces an antitumor immune response in B16/F10 solid tumor models
To prove that Ad5sPVR possesses a significant antitumor effect in both the ascites and solid tumor models, we used the B16/F10 solid tumor model. The tumor models and therapeutic regimens are provided in Figures 5A and 5C. Then, using an ELISpot assay, we found that the number of IFN-γ-producing cells was robustly increased within the TME of mice treated with Ad5sPVR compared with Ad5con on day 13 (Figure 5B). The results indicated that, compared with Ad5con, Ad5sPVR possesses significantly enhanced immune activation ability against tumors in the TME in B16/F10 solid tumor models. Treatment with Ad5sPVR significantly inhibited solid tumor growth (Figures 4E and 4F). In solid tumor models, Ad5sPVR had no clear influence on body weight (Figure 4D), which reflects the safety of Ad5sPVR.
Figure 5.

Effect of recombinant adenovirus Ad5sPVR on the antitumor immune response in B16/F10 solid tumor models
(A) Schematic diagram of the experimental setup for adenovirus therapy in B16/F10-challenged mice. On day 0, male C57BL/6 mice were injected subcutaneously with 2 × 106 B16/F10 cells in the right flank, and the mice were randomly divided into three groups: saline group, Ad5con group, and Ad5sPVR group. On days 8, 9, and 10, mice in the saline group, Ad5con group, and Ad5sPVR group were injected intratumorally with saline, the control virus Ad5con (5 × 108 PFU), and Ad5sPVR (5 × 108 PFU), respectively. On day 13, tumors were dissected. Single-cell suspensions were obtained from the tumor tissue, and cells were counted after trypan blue staining using CountStar. (B) Immune activity in the TME was detected using a mouse IFN-γ ELISpot assay, and the number of IFN-γ spots in each group was calculated. (C) Schematic diagram of the experimental setup for adenovirus therapy. Male C57BL/6 mice received a subcutaneous injection of 2 × 106 B16/F10 cells in the right flank. When the tumor size reached approximately 4–6 mm in diameter, the mice were randomly divided into three groups. On days 8, 9, 10, and 11, mice in the saline group, Ad5con group, and Ad5sPVR group were injected intratumorally with saline, the control virus Ad5con (5 × 108 PFU), and Ad5sPVR (5 × 108 PFU), respectively. (D and E) Tumor volumes were measured using a caliper, and body weight was monitored every day during treatment. (F) Volume of each tumor in the group of mice treated with saline, Ad5con, and Ad5sPVR. The data are shown as the means ± SD. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Discussion
Oncolytic viruses have unique advantages in activating immune responses, such as inducing ICD, recruiting lymphocytes, and encoding genes that regulate the immune microenvironment.10 Nevertheless, oncolytic virus immunotherapy also faces challenges. The application of oncolytic viruses will induce immunosuppressive negative feedback signaling pathways.14,33,34 Moreover, in most cases, oncolytic virus therapy lacks sufficient immune costimulatory activation.35,36 These pathways may disable infiltrating lymphocytes, including T cells. In this study, we constructed a novel replicative oncolytic adenovirus, Ad5sPVR, expressing the soluble protein sPVR. As expected, Ad5sPVR significantly induced lymphocyte infiltration and turned cold tumors into hot tumors, with a significantly enhanced antitumor immune response and antitumor effect. This effect was confirmed in both ascites and solid tumors. We also observed that the antitumor immune response induced by Ad5sPVR has immune memory. The present study provides a new, highly effective, and safe weapon for cancer treatment; it provides a theoretical and technical basis for the development of a new generation of antitumor drugs and methods; and it has strong clinical translation prospects.
In the present study, in the H22 hepatocellular carcinoma (HCC) ascites tumor model, we confirmed that Ad5con significantly induced the infiltration of CD8+ T cells and NK cells and robustly upregulated the amount of IFN-γ in the TME, indicating that the oncolytic adenovirus itself can induce lymphocyte infiltration. These results are consistent with the studies by Kanaya et al.12 and Zhang et al.14 However, in the present study, the massive infiltration of lymphocytes did not induce a significant antitumor immune response, and the survival of mice was not prolonged, which suggested that there are potential factors that suppress antitumor immunity and hinder the antitumor effect of the oncolytic virus Ad5con; there may also be insufficient activation of immune costimulatory signals, which may disable infiltrating lymphocytes, including T cells.
To solve the problem of possibly inducing an immunosuppressive negative feedback pathway, researchers combined oncolytic virotherapy with PD-1/PD-L1 immune checkpoint blockade therapy and showed a significantly improved therapeutic effect compared with oncolytic virus therapy alone.8,33 These studies indicate that the oncolytic adenovirus can induce negative immune feedback signaling, and blocking these negative immune feedback signaling pathways can significantly enhance the therapeutic effect of the immunotherapy. In this study, we also adopted the strategy of combining immune checkpoint blockade with oncolytic adenovirus treatment, but we cleverly used an oncolytic adenovirus to express sPVR, which can block the PVR/TIGIT immune negative regulatory pathway. The combination of oncolytic virotherapy and PVR/TIGIT blocking therapy has been proven to be feasible in many studies.21,37 The difference among these studies and ours is that we used an oncolytic virus to express the blocking agent, while other studies have used additional PVR/TIGIT pathway blockers.
To solve the problem of insufficient activation of immune costimulatory signals upon oncolytic virus treatment, researchers constructed viruses expressing activating proteins of the immune costimulatory signaling pathway, and these recombinant oncolytic viruses exhibited significantly improved therapeutic effects.35,38,39 The activated immune costimulatory signaling pathway is mainly mediated by OX4035 and CD137.38,39 Our previous study found that the lack of costimulatory immune activation instead of immune checkpoint, such as PD-L1/PD-1, seriously affected the activation of cytotoxic T lymphocyte (CTLs). Indeed, by encoding the secretory protein CD137L, the recombinant Ad5sCD137L had a significantly enhanced effect of CD8+ T cell infiltration and IFN-γ release compared with the control Ad5con and Ad5sPD1 virus, significantly prolonging the survival of tumor-burdened mice.14 In this study, we also adopted a similar strategy. The sPVR encoded and expressed by Ad5sPVR also activated the immune costimulatory pathway, but the CD226-mediated immune costimulatory pathway was activated.
Many clinical studies have indicated that systemic administration of immune checkpoint blockers can cause off-target effects and lead to autoimmune-like damage.40, 41, 42 Oncolytic viruses are easy to genetically modify and can selectively replicate in tumor cells. Oncolytic viruses encoding immune checkpoint blockers express and secrete molecules that block immune checkpoints in the tumor to avoid side effects caused by off-target effects, and the shortcomings (off-target effects) of immune checkpoint blockade therapy can be overcome to some extent by this strategy.15,18,37,43,44 Consistently, in our study, the Ad5sPVR we constructed allows Ad5sPVR to selectively infect tumor cells, replicate in tumor cells, and express sPVR, which can reduce the possibility of systemic damage by sPVR due to off-target effects. In the B16/F10 melanoma subcutaneous tumor model, intratumoral (i.t.) injection of Ad5sPVR significantly inhibited tumor growth and prolonged the survival of mice, and there were no significant changes in the body weight of mice, which proved its safety.
In terms of the antitumor immune mechanism, compared with the saline group, the control Ad5con virus significantly increased the ratio of CD8+ T cells. Blocking TIGIT can enhance the antitumor immunity of CD8+ T cells.23 Therefore, we speculate that the sPVR expressed by Ad5sPVR in our study has a stronger activation effect on CD8+ T cells than on NK cells. This claim is supported by evidence that the Ad5sPVR-cured mice were resistant to rechallenge with the same tumor cell line because only memory T cells exert long-term tumor-specific immune surveillance. Since the proportion of NK cells in the Ad5sPVR group was significantly higher than that in the Ad5con group, we cannot exclude the possibility that Ad5sPVR may also promote the activation and proliferation of NK cells by blocking PVR/TIGIT. Indeed, a recent study showed that oncolytic adenovirus combined with anti-TIGIT antibody significantly increased the antitumor effect of NK cells.21 Nevertheless, in this study, we observed a significant increase in NK and CD8+ T cell numbers in the Ad5sPVR group. The same results have been observed in other oncolytic virus studies, but these studies have shown that, among these two kinds of cells, CD8+ T cells play a crucial role in antitumor responses.14,33,44,45 NK cells may play a role in the antitumor immune response, but this effect may be limited, and NK cells do not play a major role.46, 47, 48 Therefore, we postulate that CD8+ T cells play a crucial role in the Ad5sPVR-induced antitumor effect.
To the best of our knowledge, few studies have examined the combination of oncolytic virus therapy using oncolytic viruses to express proteins that simultaneously block immune checkpoints and activate immune costimulatory pathways. Zhang et al.14 constructed the oncolytic adenovirus Ad5sPD1CD137L, which expresses sPD1CD137L, and the expressed sPD1CD137L can simultaneously block the PD-1/PD-L1 immune checkpoint pathway and activate the CD137-mediated immune costimulatory pathway. Compared with sPD1CD137L, in the present study, sPVR simultaneously blocked the PVR/TIGIT immune checkpoint pathway and activated the CD226-mediated immune costimulatory pathway, but the pathways that were blocked and activated by sPVR were different. Therefore, sPVR is a single protein that can perform multiple functions.
In summary, the recombinant oncolytic adenovirus Ad5sPVR blocks the negative immune checkpoints and also provides sufficient costimulatory signals through the introduction of sPVR. By local administration, Ad5sPVR overcomes the shortcomings of low universality and off-target effects concerning immune checkpoint therapy. This strategy deserves further study in more tumor models by using Ad5sPVR alone or in combination.
Materials and methods
Cell culture
The human HCC cell line HCC-LM3 and the mouse HCC cell line H22 were obtained from the China Center for Type Culture Collection. The mouse HCC cell line Hepa1-6 was obtained from the Cell Bank of the Type Culture Collection of the Chinese Academy of Sciences. A549 human lung adenocarcinoma cells (CCL-185), B16-F10 mouse melanoma cells, LLC1 mouse Lewis lung carcinoma cells, and 293T human embryonic kidney cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). These cell lines were authenticated by short tandem repeat (STR) analysis and tested for mycoplasma contamination. H22 cells were cultured in Roswell Park Memorial Institute 1640 (RPMI 1640) medium supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin (all from Thermo Fisher Scientific, Gibco, Grand Island, NY, USA). All other cell types were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin (all from Thermo Fisher Scientific, Gibco, Grand Island, NY, USA). All cells were kept at 37°C in a humidified atmosphere in a 5% CO2 incubator.
Recombinant adenovirus construction
The recombinant adenovirus was generated as previously described.49 E1A cDNA was obtained from 293T cells. The cDNAs encoding the extracellular domains of PVR were purchased from Sino Biological. The gene sequences were generated by PCR amplification using the following specific primer pairs. E1A, forward, 5′-ATCCCGGCCCTACCGGAATGAGACATATTATCTGCCAC-3′, reverse, 5′-AGCTTATCGATAGGTGTTATGGCCTGGGGCGTTTACAG-3′; PVR, forward, 5′-GGCGGTGGCGGATCGGATATACGTGTGCTG-3′, reverse, 5′-GTGGTGGTGGTGGTGGTGTAATCTTGTATTTTGCTG-3′. For the secretion of sPVR, the CD33 signal peptide (MPLLLLLPLLWAGALAM) was designed upstream of the PVR sequence. The prepared sequences were cloned into the adenovirus shuttle plasmid pENTR using the AgeI and XhoI restriction sites. The recombinant adenoviral vectors expressing soluble proteins were obtained via homologous recombination between the shuttle plasmid and the adenovirus backbone pAD/PL-DEST (Thermo Fisher Scientific, Invitrogen, Carlsbad, CA, USA). After digestion with the restriction enzyme PacI, the recombinant adenovirus was generated by transfecting 293T cells. The virus was then amplified in 293T cells and purified using iodixanol gradient ultracentrifugation. Virus titration was then performed (10,000 cells/well). Cells were cultured for 4 days, and fluorescence was evaluated via microscopy. The viral titer was calculated according to the following formula: TCID50 = 102+(S/N−0.5)/mL, PFU/mL = 0.7 × TCID50/mL, where TCID50 is the 50% tissue culture infective dose, S is the total number of fluorescence-positive wells, N is the number of replicates, and PFU indicates plaque-forming units.
Mice and tumor inoculation
Male C57BL/6 mice were obtained from Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China). All mice were acclimatized for at least 1 week prior to the initiation of each experiment. Mice were 6–8 weeks of age in the experiments. To study the therapeutic efficacy of our virus constructs, male C57BL/6 mice were injected i.p. with 5 × 106 H22 ascites cells (diluted in 100 μL of saline). On days 8, 12, and 16, mice were injected i.p. with recombinant adenovirus (5 × 108 PFU/mouse), and saline was used as a control. The ascites were collected on day 16, and subsequent analyses were performed immediately. The mice that had been cured were challenged with 5 × 106 H22 cells on days 90 and 150, and naive mice were used as a negative control.
To establish B16/F10 xenograft models, male C57BL/6 mice were injected subcutaneously with 2 × 106 B16/F10 melanoma cells (diluted in 100 μL of saline) in the right flank. Eight days post-challenge, mice were injected i.t. with 5 × 108 PFU of virus in 200 μL of saline or with saline alone. Following vaccination, tumor growth was measured with calipers every day, and the tumor volume was calculated as tumor length × tumor width2 × 0.5. Mice bearing a tumor with a volume ≥2 cm3 were sacrificed as required by national experimental animal care regulations.
All animal experimental procedures were approved by the Animal Care Committee of Nanjing University in accordance with the Institutional Animal Care and Use Committee guidelines.
Western blot analysis
Proteins in the supernatant were separated by SDS-PAGE and electrophoretically transferred to a polyvinylidene fluoride membrane (03010040001, Roche). After blocking in 5% nonfat milk in Tris-buffered saline, the membrane was incubated with specific primary antibodies, followed by incubation in appropriate horseradish peroxidase (HRP)-conjugated secondary antibodies. Signals were detected using enhanced chemiluminescence reagent (WBKLS0500, Millipore, Billerica, MA, USA) and imaged using a chemiluminescent imaging system (ChampChemi 610, Sage Creation Science, Beijing, China). The following antibodies were used: anti-His antibody (A00186-100, GenScript, Nanjing, China) and HRP-conjugated rabbit anti-mouse immunoglobulin G (IgG) secondary antibody (31430, Thermo Fisher Scientific, Pierce; 1:2,000 diluted).
Quantitative RT-PCR analysis of virus copies
Hepa1-6, H22, LM3, B16/F10, 4T1, and LLC1 cells were infected with recombinant adenoviruses at multiplicities of infection (MOIs) of 5, 20, 2, 10, 5, and 10 and were harvested at various time points in a 24-well plate. DNA was extracted, and the viral copy number was determined by RT-PCR. Total adenovirus genomic DNA was extracted with a TIANamp genomic DNA kit (DP304-03, TIANGEN). Then, SYBR Green PCR master mix (04913914001, Roche, Switzerland) was used according to the manufacturer’s protocol, and PCR was performed using a real-time PCR system (Viia7, Thermo Fisher Scientific, USA). Virus copies were calculated using the comparative threshold cycle (CT) method and normalized to the endogenous levels of GAPDH. The following specific primer pairs were used: E1A, forward, 5′-CCTTCTAACACACCTCCTGAGATACA-3′, reverse, 5′-CAGGCTCGTTAAGCAAGTCCTC-3′; GAPDH, forward, 5′-CGCTGAGTACGTCGTGGAGTC-3′, reverse, 5′-CCTTTGCAGGGCTGAGTCAG-3′.
MTT assay
Cells were seeded into a 96-well plate, and adenovirus was added at the indicated MOI. Then, 72 h later, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, M5655, Sigma, USA) (5 mg/mL, 100 μL) was added to each well of a 96-well plate and incubated for 3 h at 37°C. Then, the supernatant was discarded. Isopropyl alcohol (100 μL; 12090611516, Nanjing Chemical Reagent, China) was then added, and the plates were agitated for 20 min to dissolve the formed crystals. The absorbance was measured using a multimode reader (SMP500-13497-JWYK, Molecular Devices, USA) at 570 nm. The cell viability was calculated as the ratio of the absorbance of treated cells to the absorbance of control cells.
Flow cytometry analysis
Ascites and blood were collected at predefined time points, and cells were obtained by centrifugation. After washing or red blood cell lysis, the harvested cells were stained with anti-CD3-allophycocyanin (APC) (17-0032082, eBioscience, USA), anti-CD4-fluorescein isothiocyanate (FITC) (557307, BD Biosciences, USA), anti-CD8a-peridinin chlorophyll protein (PerCP)-Cy5.5 (45-0081-82, eBioscience, USA), or anti-NK1.1-FITC (553164, BD Biosciences, USA). The fluorescence intensity of the cells was then detected using a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA).
IFN-γ ELISpot assay
Immune activity in the TME was measured with a mouse IFN-γ ELISpot assay kit (3321-2A, Mabtech, Sweden), according to the kit protocol. In brief, the plate was coated with mouse IFN-γ capture antibodies and kept overnight at 4°C in the dark. Cells isolated from tumor masses were seeded at a density of 1.5 × 105 cells/well. After 24 h of incubation at 37°C in a humidified atmosphere in a 5% CO2 incubator, cells were removed, and the wells were rinsed with PBS. Biotinylated anti-IFN-γ antibodies were then added, and the plate was incubated for 2 h at room temperature (RT). Then, the wells were rinsed with PBS and then incubated with streptavidin-alkaline phosphatase (ALP) for 1 h. The reaction was stopped by washing the wells with tap water when spots emerged. Spot number and activity were determined using an ELISpot reader system (Autoimmune Diagnostika, Germany). Spot activity was characterized as the average spot size and intensity per well.
ELISA
For the quantification of soluble proteins, a polystyrene microplate was precoated with a His-tag antibody (A00186-100, GenScript Biotech, Nanjing, China). One hundred microliters of supernatant or ascites was added and incubated for 2 h at 37°C. After washing, the anti-CD155 antibody (50259-R001, Sino Biological, Beijing, China) and HRP-conjugated streptavidin were added, and the plate was incubated for another 2 h at 37°C. Tetramethyl benzidine (TMB) was used as the substrate, and the absorbance was read at 450 nm. The concentration of IFN-γ in the plasma or ascites was quantified using a mouse IFN-γ ELISA kit according to the manufacturer’s instructions (BD Biosciences, Franklin Lakes, NJ, USA).
Expression, purification, and affinity analysis of free PVR
293T cells were infected with Ad5sPVR, and the cell culture supernatant was passed through a nickel column (Beyotime Biotechnology). Washing solution was added to wash away unbound protein, elution solution was added, and the protein was then collected. The collected protein was identified by SDS-PAGE and Coomassie brilliant blue staining, and its purity was greater than 85%. The purified sPVR protein was used in the Octet RED96 protein interaction analysis system (ForteBio) for affinity analysis. Purchased mouse TIGIT and CD226 proteins (200 nM) were prepared with PBS. The receptor used by Octet RED96 has activity against other proteins and can therefore bind to the purified sPVR protein. The Octet RED96 receptor undergoes a process of self-equilibration for 5 min, binding to the sPVR protein for 10 min, equilibration for 3 min, binding to TIGIT or CD226 for 5 min, and dissociation for 3 min. The affinity between proteins was calculated by dynamically monitoring the binding process between PVR and TIGIT or between PVR and CD226.
Statistical analysis
The results are presented as the means ± SD. Statistical analyses were performed using a Student’s t test. Virus replication and oncolysis are presented using one-way ANOVA with repeated measures (SPSS). Animal survival is presented using Kaplan-Meier survival curves and was statistically analyzed using the log-rank test (GraphPad Prism version 6). p <0.05 was considered to be statistically significant.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81773255, 81972888, 81700037, and 81472820) and by the Primary Research and Development Plan of Jiangsu Province (BE2018701).
Author contributions
J. Wei conceived the study, J. Wei and J. Wu designed the experiments and supervised the project. H.Z., Y.Z., and J.D. performed the experiments and analyzed the data. H.Z., J. Wu, and J. Wei wrote the manuscript. All authors critically reviewed and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.11.001.
Contributor Information
Junhua Wu, Email: wujunhua@nju.edu.cn.
Jiwu Wei, Email: wjw@nju.edu.cn.
Supplemental information
References
- 1.Keller B.A., Bell J.C. Oncolytic viruses—immunotherapeutics on the rise. J. Mol. Med. (Berl.) 2016;94:979–991. doi: 10.1007/s00109-016-1453-9. [DOI] [PubMed] [Google Scholar]
- 2.Lichty B.D., Breitbach C.J., Stojdl D.F., Bell J.C. Going viral with cancer immunotherapy. Nat. Rev. Cancer. 2014;14:559–567. doi: 10.1038/nrc3770. [DOI] [PubMed] [Google Scholar]
- 3.Zamarin D., Holmgaard R.B., Subudhi S.K., Park J.S., Mansour M., Palese P., Merghoub T., Wolchok J.D., Allison J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014;6:226ra32. doi: 10.1126/scitranslmed.3008095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galon J., Bruni D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019;18:197–218. doi: 10.1038/s41573-018-0007-y. [DOI] [PubMed] [Google Scholar]
- 5.Niemann J., Kuhnel F. Oncolytic viruses: adenoviruses. Virus Genes. 2017;53:700–706. doi: 10.1007/s11262-017-1488-1. [DOI] [PubMed] [Google Scholar]
- 6.Garcia-Carbonero R., Salazar R., Duran I., Osman-Garcia I., Paz-Ares L., Bozada J.M., Boni V., Blanc C., Seymour L., Beadle J. Phase 1 study of intravenous administration of the chimeric adenovirus enadenotucirev in patients undergoing primary tumor resection. J. Immunother. Cancer. 2017;5:71. doi: 10.1186/s40425-017-0277-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Machiels J.P., Salazar R., Rottey S., Duran I., Dirix L., Geboes K., Wilkinson-Blanc C., Pover G., Alvis S., Champion B. A phase 1 dose escalation study of the oncolytic adenovirus enadenotucirev, administered intravenously to patients with epithelial solid tumors (EVOLVE) J. Immunother. Cancer. 2019;7:20. doi: 10.1186/s40425-019-0510-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuryk L., Møller A.W., Jaderberg M. Combination of immunogenic oncolytic adenovirus ONCOS-102 with anti-PD-1 pembrolizumab exhibits synergistic antitumor effect in humanized A2058 melanoma huNOG mouse model. OncoImmunology. 2018;8:e1532763. doi: 10.1080/2162402X.2018.1532763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuryk L., Møller A.W., Garofalo M., Cerullo V., Pesonen S., Alemany R., Jaderberg M. Antitumor-specific T-cell responses induced by oncolytic adenovirus ONCOS-102 (AdV5/3-D24-GM-CSF) in peritoneal mesothelioma mouse model. J. Med. Virol. 2018;90:1669–1673. doi: 10.1002/jmv.25229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaufman H.L., Kohlhapp F.J., Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2015;14:642–662. doi: 10.1038/nrd4663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heiniö C., Havunen R., Santos J., de Lint K., Cervera-Carrascon V., Kanerva A., Hemminki A. TNFa and IL2 encoding oncolytic adenovirus activates pathogen and danger-associated immunological signaling. Cells. 2020;9:798. doi: 10.3390/cells9040798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kanaya N., Kuroda S., Kakiuchi Y., Kumon K., Tsumura T., Hashimoto M., Morihiro T., Kubota T., Aoyama K., Kikuchi S. Immune modulation by telomerase-specific oncolytic adenovirus synergistically enhances antitumor efficacy with Anti-PD1 antibody. Mol. Ther. 2020;28:794–804. doi: 10.1016/j.ymthe.2020.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter C.E., Rosewell Shaw A., Jung Y., Yip T., Castro P.D., Sandulache V.C., Sikora A., Gottschalk S., Ittman M.M., Brenner M.K., Suzuki M. Oncolytic adenovirus armed with BiTE, cytokine, and checkpoint inhibitor enables CAR T cells to control the growth of heterogeneous tumors. Mol. Ther. 2020;28:1251–1262. doi: 10.1016/j.ymthe.2020.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y., Zhang H., Wei M., Mou T., Shi T., Ma Y., Cai X., Li Y., Dong J., Wei J. Recombinant adenovirus expressing a soluble fusion protein PD-1/CD137L subverts the suppression of CD8+ T cells in HCC. Mol. Ther. 2019;27:1906–1918. doi: 10.1016/j.ymthe.2019.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Gruijl T.D., Janssen A.B., van Beusechem V.W. Arming oncolytic viruses to leverage antitumor immunity. Expert Opin. Biol. Ther. 2015;15:959–971. doi: 10.1517/14712598.2015.1044433. [DOI] [PubMed] [Google Scholar]
- 16.Harrington K., Freeman D.J., Kelly B., Harper J., Soria J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019;18:689–706. doi: 10.1038/s41573-019-0029-0. [DOI] [PubMed] [Google Scholar]
- 17.Guo Z.S., Liu Z., Bartlett D.L. Oncolytic immunotherapy: dying the right way is a key to eliciting potent antitumor immunity. Front. Oncol. 2014;4:74. doi: 10.3389/fonc.2014.00074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ribas A., Dummer R., Puzanov I., VanderWalde A., Andtbacka R.H.I., Michielin O., Olszanski A.J., Malvehy J., Cebon J., Fernandez E. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell. 2017;170:1109–1119.e10. doi: 10.1016/j.cell.2017.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Twumasi-Boateng K., Pettigrew J.L., Kwok Y.Y.E., Bell J.C., Nelson B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer. 2018;18:419–432. doi: 10.1038/s41568-018-0009-4. [DOI] [PubMed] [Google Scholar]
- 20.Yu X., Harden K., Gonzalez L.C., Francesco M., Chiang E., Irving B., Tom I., Ivelja S., Refino C.J., Clark H. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat. Immunol. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- 21.Leung E.Y.L., Ennis D.P., Kennedy P.R., Hansell C., Dowson S., Farquharson M., Spiliopoulou P., Nautiyal J., McNamara S., Carlin L.M. NK cells augment oncolytic adenovirus cytotoxicity in ovarian cancer. Mol. Ther. Oncolytics. 2020;16:289–301. doi: 10.1016/j.omto.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manieri N.A., Chiang E.Y., Grogan J.L. TIGIT: a key inhibitor of the cancer immunity cycle. Trends Immunol. 2017;38:20–28. doi: 10.1016/j.it.2016.10.002. [DOI] [PubMed] [Google Scholar]
- 23.Johnston R.J., Comps-Agrar L., Hackney J., Yu X., Huseni M., Yang Y., Park S., Javinal V., Chiu H., Irving B. The immunoreceptor TIGIT regulates antitumor and antiviral CD8+ T cell effector function. Cancer Cell. 2014;26:923–937. doi: 10.1016/j.ccell.2014.10.018. [DOI] [PubMed] [Google Scholar]
- 24.Bottino C., Castriconi R., Pende D., Rivera P., Nanni M., Carnemolla B., Cantoni C., Grassi J., Marcenaro S., Reymond N. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003;198:557–567. doi: 10.1084/jem.20030788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pauken K.E., Wherry E.J. TIGIT and CD226: tipping the balance between costimulatory and coinhibitory molecules to augment the cancer immunotherapy toolkit. Cancer Cell. 2014;26:785–787. doi: 10.1016/j.ccell.2014.11.016. [DOI] [PubMed] [Google Scholar]
- 26.Kong Y., Zhu L., Schell T.D., Zhang J., Claxton D.F., Ehmann W.C., Rybka W.B., George M.R., Zeng H., Zheng H. T-cell immunoglobulin and ITIM domain (TIGIT) associates with CD8+ T-cell exhaustion and poor clinical outcome in AML patients. Clin. Cancer Res. 2016;22:3057–3066. doi: 10.1158/1078-0432.CCR-15-2626. [DOI] [PubMed] [Google Scholar]
- 27.Stamm H., Klingler F., Grossjohann E.M., Muschhammer J., Vettorazzi E., Heuser M., Mock U., Thol F., Vohwinkel G., Latuske E. Immune checkpoints PVR and PVRL2 are prognostic markers in AML and their blockade represents a new therapeutic option. Oncogene. 2018;37:5269–5280. doi: 10.1038/s41388-018-0288-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Q., Bi J., Zheng X., Chen Y., Wang H., Wu W., Wang Z., Wu Q., Peng H., Wei H. Blockade of the checkpoint receptor TIGIT prevents NK cell exhaustion and elicits potent anti-tumor immunity. Nat. Immunol. 2018;19:723–732. doi: 10.1038/s41590-018-0132-0. [DOI] [PubMed] [Google Scholar]
- 29.Stamm H., Oliveira-Ferrer L., Grossjohann E.M., Muschhammer J., Thaden V., Brauneck F., Kischel R., Müller V., Bokemeyer C., Fiedler W., Wellbrock J. Targeting the TIGIT-PVR immune checkpoint axis as novel therapeutic option in breast cancer. OncoImmunology. 2019;8:e1674605. doi: 10.1080/2162402X.2019.1674605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cairns T.M., Huang Z.Y., Gallagher J.R., Lin Y., Lou H., Whitbeck J.C., Wald A., Cohen G.H., Eisenberg R.J. Patient-specific neutralizing antibody responses to herpes simplex virus are attributed to epitopes on gD, gB, or both and can be type specific. J. Virol. 2015;89:9213–9231. doi: 10.1128/JVI.01213-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang L., Wang P., Sun Y.J., Wu Y.J. Ivermectin reverses the drug resistance in cancer cells through EGFR/ERK/Akt/NF-κB pathway. J. Exp. Clin. Cancer Res. 2019;38:265. doi: 10.1186/s13046-019-1251-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kursunel M.A., Esendagli G. The untold story of IFN-γ in cancer biology. Cytokine Growth Factor Rev. 2016;31:73–81. doi: 10.1016/j.cytogfr.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 33.Liu Z., Ravindranathan R., Kalinski P., Guo Z.S., Bartlett D.L. Rational combination of oncolytic vaccinia virus and PD-L1 blockade works synergistically to enhance therapeutic efficacy. Nat. Commun. 2017;8:14754. doi: 10.1038/ncomms14754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zamarin D., Ricca J.M., Sadekova S., Oseledchyk A., Yu Y., Blumenschein W.M., Wong J., Gigoux M., Merghoub T., Wolchok J.D. PD-L1 in tumor microenvironment mediates resistance to oncolytic immunotherapy. J. Clin. Invest. 2018;128:1413–1428. doi: 10.1172/JCI98047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang H., Dwyer K., Bover L., Lang F., Gomez-Manzano C., Fueyo J. Potentiation of anti-glioma immunity induced by oncolytic adenovirus Delta-24-RGD through viral expression of immune co-stimulator OX40 ligand. J. Immunother. Cancer. 2015;3(Suppl 2):P337. [Google Scholar]
- 36.John L.B., Howland L.J., Flynn J.K., West A.C., Devaud C., Duong C.P., Stewart T.J., Westwood J.A., Guo Z.S., Bartlett D.L. Oncolytic virus and anti-4-1BB combination therapy elicits strong antitumor immunity against established cancer. Cancer Res. 2012;72:1651–1660. doi: 10.1158/0008-5472.CAN-11-2788. [DOI] [PubMed] [Google Scholar]
- 37.Lin C., Ren W., Luo Y., Li S., Chang Y., Li L., Xiong D., Huang X., Xu Z., Yu Z. Intratumoral delivery of a PD-1-blocking scFv encoded in oncolytic HSV-1 promotes antitumor immunity and synergizes with TIGIT blockade. Cancer Immunol. Res. 2020;8:632–647. doi: 10.1158/2326-6066.CIR-19-0628. [DOI] [PubMed] [Google Scholar]
- 38.Kim H.S., Kim-Schulze S., Kim D.W., Kaufman H.L. Host lymphodepletion enhances the therapeutic activity of an oncolytic vaccinia virus expressing 4-1BB ligand. Cancer Res. 2009;69:8516–8525. doi: 10.1158/0008-5472.CAN-09-2522. [DOI] [PubMed] [Google Scholar]
- 39.Ragonnaud E., Andersson A.M., Pedersen A.E., Laursen H., Holst P.J. An adenoviral cancer vaccine co-encoding a tumor associated antigen together with secreted 4-1BBL leads to delayed tumor progression. Vaccine. 2016;34:2147–2156. doi: 10.1016/j.vaccine.2015.06.087. [DOI] [PubMed] [Google Scholar]
- 40.Postow M.A., Sidlow R., Hellmann M.D. Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 2018;378:158–168. doi: 10.1056/NEJMra1703481. [DOI] [PubMed] [Google Scholar]
- 41.Pollack M.H., Betof A., Dearden H., Rapazzo K., Valentine I., Brohl A.S., Ancell K.K., Long G.V., Menzies A.M., Eroglu Z. Safety of resuming anti-PD-1 in patients with immune-related adverse events (irAEs) during combined anti-CTLA-4 and anti-PD1 in metastatic melanoma. Ann. Oncol. 2018;29:250–255. doi: 10.1093/annonc/mdx642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martins F., Sofiya L., Sykiotis G.P., Lamine F., Maillard M., Fraga M., Shabafrouz K., Ribi C., Cairoli A., Guex-Crosier Y. Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 2019;16:563–580. doi: 10.1038/s41571-019-0218-0. [DOI] [PubMed] [Google Scholar]
- 43.Twumasi-Boateng K., Pettigrew J.L., Kwok Y.Y.E., Bell J.C., Nelson B.H. Oncolytic viruses as engineering platforms for combination immunotherapy. Nat. Rev. Cancer. 2018;18:419–432. doi: 10.1038/s41568-018-0009-4. [DOI] [PubMed] [Google Scholar]
- 44.Bartee M.Y., Dunlap K.M., Bartee E. Tumor-localized secretion of soluble PD1 enhances oncolytic virotherapy. Cancer Res. 2017;77:2952–2963. doi: 10.1158/0008-5472.CAN-16-1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shin S.P., Seo H.H., Shin J.H., Park H.B., Lim D.P., Eom H.S., Bae Y.S., Kim I.H., Choi K., Lee S.J. Adenovirus expressing both thymidine kinase and soluble PD1 enhances antitumor immunity by strengthening CD8 T-cell response. Mol. Ther. 2013;21:688–695. doi: 10.1038/mt.2012.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ricca J.M., Oseledchyk A., Walther T., Liu C., Mangarin L., Merghoub T., Wolchok J.D., Zamarin D. Pre-existing immunity to oncolytic virus potentiates its immunotherapeutic efficacy. Mol. Ther. 2018;26:1008–1019. doi: 10.1016/j.ymthe.2018.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rojas J.J., Sampath P., Hou W., Thorne S.H. Defining effective combinations of immune checkpoint blockade and oncolytic virotherapy. Clin. Cancer Res. 2015;21:5543–5551. doi: 10.1158/1078-0432.CCR-14-2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kowalsky S.J., Liu Z., Feist M., Berkey S.E., Ma C., Ravindranathan R., Dai E., Roy E.J., Guo Z.S., Bartlett D.L. Superagonist IL-15-armed oncolytic virus elicits potent antitumor immunity and therapy that are enhanced with PD-1 blockade. Mol. Ther. 2018;26:2476–2486. doi: 10.1016/j.ymthe.2018.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu D.P., Sauter B.V., Huang T.G., Meseck M., Woo S.L., Chen S.H. The systemic administration of Ig-4-1BB ligand in combination with IL-12 gene transfer eradicates hepatic colon carcinoma. Gene Ther. 2005;12:1526–1533. doi: 10.1038/sj.gt.3302556. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.