

Abstract
Gallbladder cancer (GBC) is the most common malignancy of the biliary tract, with extremely dismal prognosis. Limited therapeutic options are available for GBC patients. We used whole-exome sequencing of human GBC to identify the ErbB and epigenetic pathways as two vulnerabilities in GBC. We screened two focused small-molecule libraries that target these two pathways using GBC cell lines and identified the mTOR inhibitor INK-128 and the histone deacetylase (HDAC) inhibitor JNJ-26481585 as compounds that inhibited proliferation at low concentrations. Both significantly suppressed tumor growth and metastases in mouse models. Both synergized with the standard of care chemotherapeutic agent gemcitabine in cell lines and in mouse models. Furthermore, the activation of the mTOR pathway, measured by immunostaining for phosphorylated mTOR and downstream effector S6K1, is correlated with poor prognosis in GBC. Phosphorylated mTOR or p-S6K1 in clinical samples is an independent indicator for overall survival in GBC patients. Taken together, our findings suggest that mTOR inhibitors and HDAC inhibitors can serve as potential therapeutics for GBC, and the phosphorylation of mTOR and S6K1 may serve as biomarkers for GBC.
Graphical Abstract
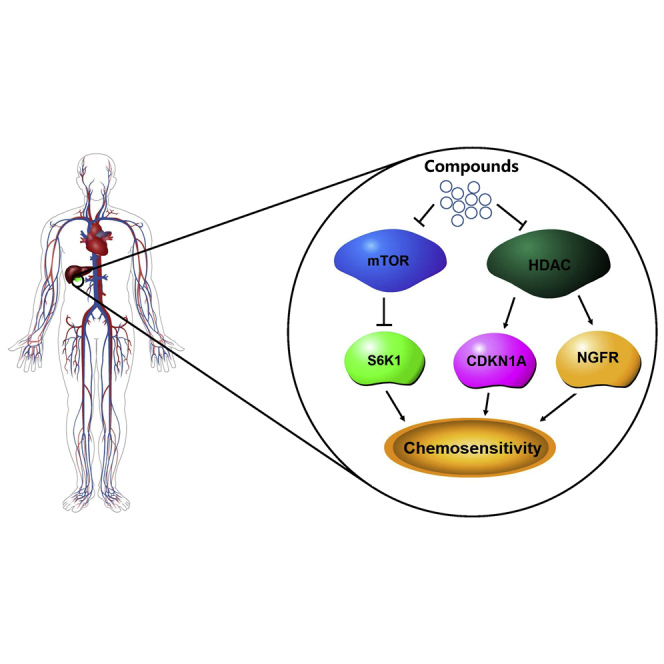
Gallbladder cancer is the most common malignancy of biliary tract, with dismal prognosis. We identified mTOR and HDAC as two potential therapeutic targets and proved p-mTOR and p-S6K1 may serve as biomarkers, which opens opportunities for the development of new targeted therapeutics for gallbladder cancer patients.
Introduction
Gallbladder cancer (GBC) is the most aggressive cancer of the biliary tract with the shortest median survival once diagnosis is confirmed.1 Although radical surgical resection has been the only possible cure for GBC, over 90% of patients are clinically unresectable, and nearly 50% have lymph node metastasis.2 Gemcitabine is the first-line therapy for GBC, bile duct cancer, and pancreatic cancer.3,4 However, the response rate of biliary tract tumors to gemcitabine is only 10%–30%,5 and therefore chemoresistance is one major challenge in the treatment of GBC. Identification of novel therapeutics for GBC is critical for the management of this disease.
The ErbB family, including EGFR (ERBB1 or HER1), ERBB2 (HER2), ERBB3 (HER3), and ERBB4 (HER4), has been strongly implicated in proliferative malignancy.6, 7, 8 ErbB family members control downstream pathways such as phosphatidylinositol 3-kinase (PI3K)-Akt and mitogen-activated protein kinase (MAPK) that participate in cell growth and survival.9,10 Targeting EGFR/HER2 pathways has been shown to synergize with gemcitabine in treating GBC and biliary tract cancer.11 Downstream of the ErbB-PI3K-AKT signaling pathway is mTOR, a ser/thr kinase that plays an important role in PI3K/AKT-mediated cell proliferation, survival, and tumor growth.12,13 Inhibition of mTOR signaling is able to suppress the invasiveness and metastasis of malignancies.13,14 Signaling molecules of the mTOR pathway have been shown to be involved in the mammalian cell size change,15 and their expression is upregulated in osteosarcoma16 and breast cancer.17 mTOR inhibition shows potential anti-proliferative effects in GBC cell lines.18,19 However, whether the anti-proliferative effect of mTOR inhibitors extends to GBC is not known.
The activity of proteins controlled by histone acetylation/deacetylation has been linked to various biological functions, including gene expression, cell cycle progression, and cell death.20 The deacetylation of histones is controlled by histone deacetylases (HDACs).21,22 HDAC inhibitors have been shown to have anti-proliferative effects in various cancer types, including gastric and breast cancer.23 In GBC, the HDAC inhibitors TSA (trichostatin A) and SAHA (vorinostat) showed pro-apoptotic properties in cancer cell lines.24 However, whether HDAC inhibitors can suppress tumor growth and metastasis in GBC is not known.
In the current study, we identified mTOR and epigenetic pathways as therapeutic targets in human GBC using screens of small-molecule libraries. We found that the dual mTOR inhibitor INK-128 and HDAC inhibitor JNJ-26481585 suppress cancer cell growth in vitro and in animal models. Both compounds synergize with the standard of care chemotherapeutic drug gemcitabine to suppress gallbladder tumor growth and metastases. These results suggest that mTOR inhibitors and HDAC inhibitors can serve as potential therapeutics for GBC.
Results
Whole-exome sequencing of human GBC
Whole-exome sequencing was performed on 11 pairs of GBC tissues and their corresponding normal gallbladder tissues. All 11 cancer tissues were pathologically confirmed as GBC. The characteristics of tumor samples for whole-exome sequencing in 11 GBC patients were shown in Table 1. We found that cancer tissues have approximately 69 single nucleotide polymorphism (SNPs) for each sample except sample G22, which was highly mutated, with 3,399 SNPs. For the other 10 samples excluding G22, a non-synonymous (missense, nonsense, and splice) mutation percentage was calculated for each gene (Figure 1A). TP53 is extensively mutated (6 out of 10 samples), followed by ERBB2, OBSCN, and RIN2 (3 out of 10 samples) (Figure 1A). C > T/G > A alteration represents the most frequently mutated pattern in the 10 samples and the trinucleotide signature dominated at T_A sites (Figure 1B), consistent with the data from Li et al.7 In sample G22 (Figure 1C), both C > T/G > A and C > A/G > T were frequent mutation signatures. Genes that are involved in DNA repair pathways such as ATM in sample G22 are shown in Figure 1D. It is possible that the DNA repair mechanism was impaired in G22, which provides an explanation for the extremely high mutation rate in G22. Although it is not common, GBCs with such extremely high mutation rates have been observed in other cohorts.7 The treatment options for gallbladder patients with DNA repair deficiency may be different from those for other patients.
Table 1.
Characteristics of tumor samples for whole-exome sequencing in gallbladder cancer patients
| Sample | Gender | Age | Treatment | Tumor size (cm) | Pathology |
|---|---|---|---|---|---|
| 01 | male | 63 | surgery | 3 × 2.5 × 1.2 | adenocarcinoma |
| 02 | female | 60 | surgery | 2 × 1 × 0.5 | adenocarcinoma |
| 03 | female | 60 | surgery | 6 × 3 × 1.5 | adenocarcinoma |
| 04 | female | 74 | surgery | 4.5 × 2 × 2 | adenocarcinoma |
| 05 | male | 60 | surgery | 6 × 5 × 4 | adenocarcinoma |
| 06 | female | 78 | surgery | 6 × 5 × 4 | adenocarcinoma |
| 07 | female | 57 | surgery | 4 × 3 × 2 | adenocarcinoma |
| 08 | female | 48 | surgery | 3 × 2 × 1.5 | adenocarcinoma |
| 09 | female | 59 | surgery | 4 × 2.5 × 2 | adenocarcinoma |
| 10 | male | 75 | surgery | 8 × 4 × 4 | adenocarcinoma |
| 11 | female | 79 | surgery | 5 × 4 × 1.5 | adenocarcinoma |
Figure 1.

Gene mutation patterns in human gallbladder cancer samples
(A) Non-synonymous gene mutations in 10 cancer samples. TP53 is the most frequently mutated gene (60%) followed by ERBB2, OBSCN, and RIN2 (30% for each). (B) Nucleotide mutation pattern for 10 cancer samples. C > T/G > A and C > G/G > C mutation pattern represent the most frequently mutated type. (C) Nucleotide mutation pattern for hyper-mutated G22 sample. C > T/G > A and C > A/G > T mutation patterns represent the most frequently mutated type. (D) Mutation of genes involved in DNA repair in G22 sample.
Genes in ErbB and epigenetic pathways are highly mutated in GBC
Because GBC is not a common cancer type, we performed a comprehensive analysis that included 79 GBC samples from all the studies to date for which mutation information was available (Li et al.,7 Jiao et al.,25 Nakamura et al.,8 and our study) (Figure 2A). An evaluation of the types of genes that were mutated revealed two major mutational pathways in GBC: genes involved in epigenetic regulation and genes of the ErbB pathway. Mutations in genes that regulate epigenetic changes include histone acetylation, methylation, and phosphorylation, DNA methylation, and chromatin remodeling; ErbB pathway genes include EGFR, ERBB2, ERBB3, and ERBB4. The mutation rates are 63.3% in epigenetic pathway genes (Figure 2B) and 36.7% in ErbB pathway genes (Figure 2C). These data indicate that these two pathways are critical for GBC development and serve as potential therapeutic targets.
Figure 2.

Comprehensive mutation analysis from four whole-exome sequencing in gallbladder cancer
(A) Different mutation types (synonymous, missense, nonsense, frameshift, or splice) and the number of samples with mutation for each gene are indicated. (B) Number of samples containing the mutated genes involved in epigenetic regulation. (C) Number of samples containing the mutated genes of ErbB pathway.
Screening of small molecules targeting ErbB and epigenetic pathways
To identify compounds that have anti-proliferative effects in GBC, we screened two focused libraries of compounds that target ErbB and epigenetic pathways (Table S1), using three GBC cell lines SGC-996, GBC-SD, and NOZ. Most of the compounds in the libraries are either FDA approved or in clinical trials for cancer (Figures 3A and 3B). Among the compounds targeting epigenetic pathways and ErbB pathway, the HDAC inhibitors JNJ-26481585 and LAQ824 (Figure 3A) and the mTOR inhibitor INK-128 (Figure 3B) significantly suppressed cell growth in all three cancer cell lines. We also noticed that apart from INK-128, other compounds such as XL388, AZD8055, and OSI027, which targets mTORC1/2, not just mTORC1, also had a significant anti-proliferative effect in at least two GBC cell lines. Because JNJ-26481585 has superior pharmacokinetics in human than LAQ824, it was selected along with INK-128 for further validation. JNJ-26481585 and INK-128 inhibited cell growth in a dose-dependent manner (Figure 3C), with IC50 ranging from 1.78 nM to 4.12 nM and from 18 nM to 41 nM, respectively, consistent with the screening results. Both compounds inhibited cell growth better than standard of care chemotherapy gemcitabine (IC50 55 nM–292 nM) (Figure 3C). The results indicate that HDAC inhibitors JNJ-26481585 and mTOR inhibitor INK-128 are potential therapeutics for GBC.
Figure 3.

IC50 value of compounds targeting ErbB and epigenetic pathways in three gallbladder cancer cell lines
(A) Heatmap of IC50 of compounds targeting epigenetic regulation. Compound names and their targets are indicated. (B) Heatmap of IC50 of compounds targeting ErbB pathway. Compound names and their targets are indicated. (C) Validation of anti-proliferative effects and IC50s of JNJ-26481585, INK-128, and gemcitabine in three gallbladder cancer lines. (D) Synergistic effects of gemcitabine and INK-128 in three gallbladder cancer cells. (E) Synergistic effects of gemcitabine and JNJ-26481585 in three gallbladder cancer cells. (F) Synergistic effects of INK-128 and JNJ-26481585 in three gallbladder cancer cells. (G) HDAC1 and HDAC2 mRNA expression in three gallbladder cancer cells treated with DMSO control, JNJ-26481585, gemcitabine, or gemcitabine and JNJ-26481585. (H) Protein level of AKT (Ser-473), AKT (Thr-308), and p70S6K1 (Thr-389) in GBC-SD cells administered with DMSO control, INK-128, gemcitabine, or gemcitabine and INK-128.
To determine whether JNJ-26481585 and INK-128 might synergize with gemcitabine in GBC cells, we treated SGC-996, GBC-SD, and NOZ cells with combinations of various doses of the compounds. We used a dose-response surface model based on Bliss independence principle. The interaction index () and its 95% confidence interval (CI) were calculated to evaluate a two-drug combination effect.26,27 An index score of less than 1 indicates synergy. Combinations of two compounds (INK-128 and gemcitabine, JNJ-26481585 and gemcitabine, INK-128 and JNJ-26481585) are highly synergistic in all three cell lines, SGC-996, GBC-SD, and NOZ (Figures 3D–3F). Synergism was not observed in NOZ cells treated with gemcitabine at 500 nM because all the cells died when treated with gemcitabine alone at that concentration. For further validation, we performed real-time PCR and western blot analysis in these cell lines. Our results demonstrated that, as compared with control group, either JNJ-26481585 or gemcitabine alone significantly decreased the levels of HDAC1 and HDAC2, and co-treatment of JNJ-26481585 and gemcitabine showed a synergistic inhibition effect (Figures 3G and 3H). In addition, mTORC1 and mTORC2 activation was reported to be reflected by phosphorylation of p70S6K1 (Thr-389) and AKT (Ser-473), respectively.28 As expected, the expression level of both p70S6K1 (Thr-389) and AKT (Ser-473) in GBC-SD cells was downregulated in INK-128 or gemcitabine treatment group, and an additive inhibition effect was demonstrated when the two drugs were administrated together. However, phosphorylation of AKT (Thr-308) was not changed, showing that AKT activity was not completely blocked by INK-128.
Taken together, these results indicate that JNJ-26481585 and INK-128 synergize with gemcitabine in suppressing cell proliferation in GBC, and combinatorial therapies are potentially more effective.
INK-128 and JNJ-26481585 inhibit GBC proliferation and metastasis in mouse models
To determine the efficacy of INK-128 and JNJ-26481585 in vivo, we subcutaneously transplanted luciferase-tagged GBC-SD cells in mice. Mice were then treated with gemcitabine, JNJ-26481585, INK-128, gemcitabine + JNJ-26481585, gemcitabine + INK-128, or DMSO control. All the mice treated with control developed lung metastasis (Figure 4A). When treated with gemcitabine, 80% of mice developed lung metastasis (Figure 4A), although tumor growth was significantly suppressed (Figure 4B). However, JNJ-26481585 treatment significantly suppressed both the primary tumor growth and metastasis compared to controls (Figures 4A and 4B). Interestingly, although the primary tumor growth in mice treated with gemcitabine or JNJ-26481585 was similar (Figure 4B), the number of mice that developed metastasis was significantly less in the JNJ-26481585 treatment group than in the gemcitabine treatment group (Figure 4A), suggesting that HDAC inhibitors suppress metastasis in GBC. The combination of gemcitabine + JNJ-26481585 suppressed primary tumor growth more than either drug alone (Figure 4B). Treatment with INK-128 alone suppressed primary tumor and metastasis growth compared to control and to gemcitabine alone (Figures 4A and 4C). The most effective combination was gemcitabine + INK-128: the primary tumor showed little growth (Figure 4C), and no mice developed metastasis (Figure 4A). No significant difference of body weight was observed among all six groups mice at week 6 (Figure 4D), indicating all remedies are tolerable at the concentrations listed in the Materials and methods section. Partially consistent with our in vitro result, mRNA expression and protein level of HDAC1 and HDAC2 in xenograft tumor were partially inhibited by JNJ-26481585 or combination of gemcitabine + JNJ-26481585. Moreover, western blot assay of tumor tissues showed that activation of either mTORC1 (p70S6K1 Thr-389) or mTORC2 (AKT Ser-473) was inhibited by INK-128, and the anti-proliferative effect was augmented by co-administration of INK-128 and gemcitabine (Figure 4F). Not surprisingly, AKT Thr-308 phosphorylation was not significantly affected by INK-128 treatment (Figure 4F). Together, these results suggest that combining gemcitabine with JNJ-26481585 or INK-128 inhibits primary tumor growth and metastasis in mice to a greater extent than any agent alone.
Figure 4.

INK-128 and JNJ-26481585 suppress gallbladder cancer growth and metastasis in mouse models
(A) Luciferase-tagged GBC-SD cells were transplanted subcutaneously in mice, and tumor growth and metastasis were measured by the luminescence Xenogen system. Representative images of primary tumor and lung metastasis from 1 of 6 mice per group were shown in the upper panel and lower panel. The number of mice that developed lung metastasis is also indicated. (B) Primary tumor growth in mice treated with gemcitabine, JNJ-26481585, gemcitabine and JNJ-26481585, or DMSO control was measured by the luminescence Xenogen system. (C) Primary tumor growth in mice treated with gemcitabine, INK-128, gemcitabine and INK-128, or DMSO control was measured by the luminescence Xenogen system. (D) Body weight were monitored at week 6 for all six groups of mice. (E) HDAC1 and HDAC2 mRNA expression in xenograft tumor tissues treated with DMSO control, JNJ-26481585, gemcitabine, or gemcitabine and JNJ-26481585. (F) Protein level of AKT (Ser-473), AKT (Thr-308), and p70S6K1 (Thr-389) in xenograft tumor tissues administrated with DMSO control, INK-128, gemcitabine, or gemcitabine and INK-128.
p-mTOR and p-S6K1 are inversely associated with postoperative prognosis in GBC patients
We have shown that inhibition of the mTOR pathway by INK-128 significantly suppressed GBC cell growth in vitro and in vivo, which led us to hypothesize that mTOR activation is inversely associated with survival and can be used to select patients for treatment with mTOR inhibitors. We determined the expression of phosphorylated mTOR (active form) and its downstream target phosphorylated S6K1 in human GBC samples (Figures 5A–5D). High expression of p-mTOR was inversely correlated with survival (Figure 5B). Similarly, high expression of p-S6K1 was inversely correlated with survival (Figure 5D). Analysis of the characteristics of GBC indicated that p-mTOR expression was highly correlated with p-S6K1 expression (Table 2). Multivariable analysis showed that in addition to stage, the expression of phosphorylated mTOR and S6K1 can serve as independent markers of survival (Table 3). These results suggest that activation of the mTOR pathway is highly significant in the (poor) prognosis of GBC patients.
Figure 5.

mTOR activation is correlated with poor prognosis and is an independent marker to survival in gallbladder cancer
(A) Immunohistochemistry staining of phosphorylated mTOR in gallbladder cancer samples. (B) Kaplan-Meier curve of survival in gallbladder cancer patients with high or low expression of phosphorylated mTOR. (C) Immunohistochemistry staining of phosphorylated S6K1 in gallbladder cancer samples. (D) Kaplan-Meier curve of survival in gallbladder cancer patients with high or low expression of phosphorylated S6K1.
Table 2.
Correlation analysis of characteristics in gallbladder cancer patients (*p < 0.05; **p < 0.01; ***p < 0.001)
| Spearman r (p value) | Gender | Age | p-mTOR | p-S6K1 | Tumor differentiation | Tumor size | TNM stage |
|---|---|---|---|---|---|---|---|
| Gender (female versus male) | −0.03 (0.750) | −0.07 (0.413) | −0.18 (0.037)∗ | 0.14 (0.116) | 0.06 (0.517) | 0.08 (0.357) | |
| Age (≥60 years versus <60 years) | −0.03 (0.750) | −0.14 (0.117) | −0.12 (0.175) | −0.02 (0.818) | −0.07 (0.442) | −0.06 (0.503) | |
| p-mTOR (low versus high) | −0.07 (0.413) | −0.14 (0.117) | 0.24 (0.006)∗∗ | −0.02 (0.803) | −0.01 (0.926) | 0.01 (0.937) | |
| p-S6K1 (low versus high) | −0.18 (0.037)∗ | −0.12 (0.175) | 0.24 (0.006)∗∗ | −0.04 (0.675) | 0.06 (0.499) | 0.02 (0.867) | |
| Tumor differentiation (well versus moderate, poor) | 0.14 (0.116) | −0.02 (0.818) | −0.02 (0.803) | −0.04 (0.675) | 0.17 (0.047)∗ | 0.16 (0.066) | |
| Tumor size (≥4 cm versus <4 cm) | 0.06 (0.517) | −0.07 (0.442) | −0.01 (0.926) | 0.06 (0.499) | 0.17 (0.047)∗ | 0.37 (< 0.001)∗∗∗ | |
| TNM stage (0–II versus III–IV) | 0.08 (0.357) | −0.06 (0.503) | 0.01 (0.937) | 0.02 (0.867) | 0.16 (0.066) | 0.37 (< 0.001)∗∗∗ |
Table 3.
Univariate and multivariable analysis of clinical variables influencing overall survival in gallbladder cancer patients
| Characteristics | Univariate analysis |
Multivariate analysis |
||
|---|---|---|---|---|
| p value | HR (95% CI) | p value | HR (95% CI) | |
| Gender (female versus male) | 0.120 | 1.40 (0.91–2.30) | 0.331 | 0.80 (0.50–1.26) |
| Age (≥60 years versus <60 years) | 0.326 | 1.25 (0.79-2.07) | 0.795 | 1.07 (0.66–1.71) |
| Tumor differentiation (well versus moderate, poor, or undifferentiated) | 0.019 | 0.63 (0.40–0.92) | 0.234 | 0.78 (0.52–1.18) |
| Tumor size (≥4 cm versus <4 cm) | <0.001 | 0.50 (0.32–0.71) | 0.132 | 0.71 (0.45–1.11) |
| TNM stage (0–II versus III–IV) | <0.001 | 0.39 (0.25–0.56) | 0.002 | 0.48 (0.30-0.76) |
| p-mTOR (low versus high) | 0.012 | 0.61 (0.40–0.89) | 0.010 | 0.57 (0.37–0.87) |
| p-S6K1 (low versus high) | 0.009 | 0.60 (0.39–0.87) | 0.044 | 0.65 (0.43–0.99) |
Discussion
The goal of this study was to identify new therapeutic targets for GBC. The comprehensive analysis, including 79 GBC samples from 4 studies, indicates that, not only in our sequence data, missense gene mutation also represents the top mutation type in 3 other studies. Interestingly, G22, a special sample that was highly mutated with 3,399 SNPs, was proved to be universally mutated with missense mutation, leading us to the speculation that missense mutation might somehow be connected to a DNA repair mechanism in these particular GBC patients. The precise mechanism remains to be further investigated.
Furthermore, we found that two major pathways were highly mutated in GBC: genes involved in epigenetic regulation and genes of the ErbB pathway. A screen of small-molecule inhibitor libraries identified two compounds, INK-128 and JNJ-26481585, an mTOR and an HDAC inhibitor, respectively, that suppress tumor growth and metastases better than the standard of care gemcitabine. Both INK-128 and JNJ-26481585 synergize with gemcitabine in mouse models to suppress tumor growth and metastases. However, from the compound screening data, we also noticed that single inhibition of mTORC1 with rapamycin is unable to induce cell death in all three GBC cell lines, suggesting mTORC2 or both mTORC1 and mTORC2 are critical targets in GBC. In addition, the screening data shows that the anti-proliferative effect of targeting AKT, which is upstream of mTOR, is unsatisfactory in GBC cell lines, indicating that the well-acknowledged PI3K/AKT/mTOR pathway may not play a key role in the development of ErbB-mutation-induced GBC. In contrast to PF05212384, which mainly targets PI3K and inhibits mTOR activity as a non-dominant effect, INK-128, which targets both mTORC1/2 and PI3Kα/β/γ/δ, exhibited a significant anti-proliferative effect, suggesting that inhibition of the mTOR pathway rather than PI3K activity plays a pivotal role in suppressing GBC cell growth. As compared with other mTOR compounds, INK-128 targeting both mTOR and PI3K yields more extensive and satisfactory anti-proliferative effects in all three cancer cell lines, indicating a potential synergy between mTOR and PI3K inhibition in GBC treatment. Furthermore, we compared the chemical structure of all mTOR compounds and identify INK-128 is the only one presenting with 2 aminos in 2 different benzene rings, which led us to the speculation that chemical properties and biological functions of compounds are critical in the development of new therapeutics in GBC.
The synergism between gemcitabine and mTOR inhibitors or HDAC inhibitors suggests that a combination approach may improve treatment of GBC. The response rate for gemcitabine is low. Our animal data also showed that gemcitabine suppresses primary tumor growth but has no effects on metastasis. Adding mTOR or HDAC inhibitors potentially enhances the efficacy of gemcitabine and reduces the toxicity of both drugs because lower doses are required. In our pilot experiment, we also administrated mTOR and HDAC inhibitors along with gemcitabine to 5 mice to see if this triple-drug remedy outscores the dual-drug remedy. However, all 5 mice demonstrated symptoms of asitia and low body weight, and 4 out of 5 mice died between week 3 and 4 (data not shown). Hence, we conclude that this triple-drug program is not tolerable in mice, and further study is required to determine the optimal dosage of these drugs. Although JNJ-26481585 fails to exhibit similar powerful anti-tumor effects in mice as compared to INK-128, which is in contrast to our cell line experiments, we still consider HDAC a potential biomarker for GBC. First, the genomic mutational profiles of GBC cell lines and clinical tumor samples might not be 100% identical, suggesting the possibility of diverse effects of these compounds. Second, it was reported that epigenetic enzymes often synergize in vivo, and combination of HDAC inhibitor and other epigenetic agents like EZH2 repression is an effective therapeutic strategy.29,30 Hence, further evidences are required for confirming HDAC as a biomarker GBC, either alone or in combination with other epigenetic candidates. Using RNA-seq analysis, we demostrated that the anti-proliferative effect of JNJ-26481585 is at least partially mediated by knockdown of two potential genes, CDKN1A and NGFR in gallbladder cancer (Figures S1 and S2). Our study suggests that activation of mTOR is correlated with poor prognosis and is an independent marker for poor survival in GBC patients. mTOR activation therefore may be used not only as prognostic biomarkers but also for the selection of patients who potentially could benefit from mTOR inhibitors. These findings will be validated in larger sample sizes and future clinical trials.
One of the critical questions that remains is whether these findings in cell lines and mouse models can be translated into the clinic. Both INK-12831,32 and JNJ-2648158533, 34, 35 are in several clinical trials for cancer treatment. Safety profiles and tolerant dosages of these two drugs are known. It is possible to stratify patients based on high and low expression of activated mTOR and its downstream signaling molecule S6K1 through biopsy or new technologies such as the isolation of circulating tumor cells. The effectiveness of these drugs as therapeutics in clinic will open new opportunities for the development of new compounds targeting these two pathways.
Materials and methods
Ethics statement and patient tissue specimens
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of the Wistar Institute, USA. Animal procedures were conducted in compliance with the IACUC. 142 GBC tissues (including 11 samples subject to whole-exome sequencing) with follow-up information and 11 matched adjacent normal gallbladder tissues were obtained from patients who underwent surgery in the Department of Biliary-Pancreatic Surgery, Renji Hospital, between 2014 and 2016. The use of human GBC tissues was approved by the Ethics Committee of Renji Hospital, School of Medicine, Shanghai Jiao Tong University. Written informed consent was obtained from all patients involved in this study.
Whole-exome sequencing in GBC samples
A total of 11 pairs of GBC samples from Renji Hospital were subject to whole-exome sequencing. Genomic DNA libraries were prepared using protocols recommended by Illumina. Whole-exome enrichment was performed using the TruSeq exome enrichment kit (Illumina). Captured DNA libraries were sequenced with the Illumina HiSeq 2500 genome analyzer, yielding 200 (2 × 100) base pairs from the final library fragments. Sequencing reads were trimmed and filtered with Trimmomatic.36 The resulting reads were aligned to the hg19 reference genome using Burrows-Wheeler aligner (BWA),37 and the genome analysis toolkit (GATK)38 was used for base quality score recalibration, insertion or deletion (indel) realignment, and duplicate removal. The MuTect39 algorithm was used to identify somatic SNVs in whole-exome gene sequencing data. MuTect identifies candidate somatic SNVs by Bayesian statistical analysis of bases and their qualities in the tumor and normal BAM files at a given genomic locus. We required a minimum of ten reads covering a site in the tumor and eight reads in the normal sample to declare that a site was adequately covered for mutation calling. Default parameters were used for exome data. To filter out likely false positives, we performed a further filtration step requiring that the alternative allele proportion in the normal sample be less than 0.30 of that in the tumor sample. The Pindel40 algorithm was used to identify indels with default parameters. Inversions and large indels (>100 bp) were excluded. Somatic SNV and indel results were then combined and compared to the COSMIC database. Mutation functions were predicted using SnpEff,41 PolyPhen,42 PROVEAN,43 and SIFT.43 The four algorithms showed little difference in predicting non-silent mutations (altering protein sequence) but showed considerable differences in predicting the effects of amino acid changes. To maintain data integrity, we retained all the non-silent mutations predicted by SnpEff and did not perform further filtering. All somatic mutations were stored in a MAF file and analyzed by MutSigCV,44 with default covariates tables, to calculate gene mutational significance. Genes with q (false discovery rate [FDR]) ≤0.1 were considered to be significantly mutated.
Cell culture and chemicals
The human embryonic kidney 293 cells (HEK293FT) and two human GBC cell lines, NOZ and GBC-SD, were maintained in Dulbecco’s modified Eagle’s medium (DMEM). Another human GBC cell line, SGC-996, was maintained in RPMI-1640 medium, with all media containing 10% fetal bovine serum (FBS) and antibiotics (Gibco, Grand Island, NY, USA). Cells were maintained at 37°C in a humidified atmosphere consisting of 5% CO2. NOZ was purchased from the Health Science Research Resources Bank (Osaka, Japan). GBC-SD and SGC-996 cells were provided by the Academy of Life Sciences, Tongji University (Shanghai, China). HEK293FT cells were used for lentivirus amplification. Gemcitabine was dissolved in DMSO. Gemcitabine was obtained from Department of Biliary-Pancreatic Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University (Shanghai, China). Both INK-128 and JNJ-26481585 were purchased from ApexBio (Houston, TX, USA). All antibodies utilized in the study were purchased from Cell Signaling Technology (Shanghai, China).
RNA isolation, reverse transcription (RT), and real-time PCR analysis
Total RNA was extracted from GBC cell lines using Trizol total RNA isolation reagent (Invitrogen) according to the manufacturer’s instructions and treated with DNase. cDNA was synthesized from total RNA using random hexamers with a TaqMan cDNA reverse transcription kit (Applied Biosystems). To determine the expression levels of CDKN1A and NGFR, primers for each gene were designed using Primer Express v3.0 software, and real-time PCR was performed using SYBR green Jumpstart Taq ReadyMix (Sigma) with the Applied Biosystems 7500 Fast Real-Time PCR system. The relative amount of expression was calculated by 2−ΔΔCT method.45 The average of three independent analyses for each gene and sample was calculated and was normalized to the endogenous reference control gene GAPDH.
Screening of small-molecule inhibitors library and cell viability assay
To identify ErbB pathway inhibitors and epigenetic modulators that suppressed the growth of GBC in vitro, two separate libraries of 26 epigenetic and 55 ErbB pathway small-molecule inhibitors from the Wistar Institute were screened in human GBC SGC-996, GBC-SD, and NOZ cells (compound names and CIDs on PubMed are listed in Table S1). Cell viability was determined following 5 days’ incubation in the presence of compounds. For each compound in the screen, a serial dilution (20 μM, 4 μM, 0.8 μM, 0.16 μM, 0.032 μM, 6.4 nM, 1.28 nM, 0.256 nM, 0.0512 nM, and 0.01024 nM) was used. Afterward, cell viability was determined using an MTS assay (MTS; Promega, Madison, WI, USA). In brief, the MTS reagent (20 μL) was added to each well, followed by incubation at 37°C in a humidified, 5% CO2 atmosphere for 2 h. Finally, the absorbance was read at 490 nm by using a Synergy 2 (BioTek, VT, USA) plate reader. The cell viability was calculated as a percentage relative to control. Using cell viability percentage data under a series of drug concentrations, an individual IC50 value was calculated for each compound in cell lines. For a combination with two drugs, with a dose-response surface model based on the Bliss independence principle, interaction index (τ) and its 95% CI were calculated to evaluate the two-drug combination effect. When the τ <1 and the upper limit of its 95% CI is also less than 1, the combination effect of the two drugs will be considered as significant synergism.26,27 This MTS assay was also used for validation experiments.
Lentivirus transfection and transduction
To generate GBC-SD cells stably overexpressing luciferin, luciferin was cloned into lentiviral vector pLu. Lentivirus was produced by co-transfecting subconfluent HEK293T cells with CDKN1A or NGFR expression plasmid and packaging plasmids pMDLg/pRRE and RSV-Rev using Lipofectamine 2000. Infectious lentiviruses were collected 48 h after transfection, centrifuged to remove cell debris, and filtered through 0.45-mm filters (Millipore). GBC-SD cells were transduced with the lentivirus. Efficiency of overexpression was determined by real-time PCR. GBC-SD cells stably expressing CDKN1A short hairpin RNA (shRNA), NGFR shRNA, or control shRNA were established using shRNA technique. The lentiviruses were processed as described above and transduced into GBC-SD cells, respectively. The knockdown efficiency was also determined by real-time PCR.
In vivo experiments
A total of 5∗105 luciferase-tagged GBC-SD human GBC cells stably expressing the firefly luciferase gene were subcutaneously transplanted into NOD/SCID mice (4–6 weeks old) of each group: group 1, control; group 2, gemcitabine (Gem, 75mg/kg, i.p., once weekly); group 3, JNJ-26481585 (JNJ, 10 mg/kg, intraperitoneally [i.p.], every other day46); group 4, gemcitabine + JNJ-26481585 (Gem + JNJ); group 5, INK-128 (INK, 1 mg/kg, p.o., once daily47); group 6, gemcitabine + INK-128 (Gem + INK); n = 6/group. Mice bearing luciferase-positive tumors were imaged by an IVIS 200 imaging system (Xenogen, Hopkinton, MA, USA). Bioluminescent flux (photons/s/sr/cm2) was determined for the primary tumors or lung metastasis. At week 6, xenograft tumors and internal organs of 6 groups were carefully isolated and subjected to subsequent experiments.
Western blot analysis
Cells and freshly isolated xenograft tissues were lysed using protein extraction reagent (Biyuntian, Wuxi, China) supplemented with protease inhibitor cocktail (Calbiochem). Protein concentrations were determined with a BCA assay (Sigma). Equal amount of the extracts (30 mg/sample) were loaded and subjected to SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes. Afterward, the membranes were blocked with blocking buffer, incubated overnight at 4°C with indicated primary antibodies, and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies. The detection was performed by ECL Supersignal West Pico Chemiluminescent Substrate.
RNA sequencing (RNA-seq) analysis
Four groups of GBC-SD cells treated with DMSO, low-concentration, high-concentration JNJ-26481585, or high-concentration UNC1999 separately were scraped and poled into separate tubes. These pooled cells were then subjected to RNA isolation. A RNeasy kit (QIAGEN, Valencia, CA, USA) was used for total RNA preparation. RNA samples were converted into cDNA libraries using a TruSeq stranded total RNA sample preparation kit (Illumina, San Diego, CA, USA). In brief, total RNA samples were concentration normalized, and ribosomal RNA was removed using biotinylated probes that selectively bind ribosomal RNA species. This preserved messenger RNA and other noncoding RNA species, including long noncoding RNA, small nuclear RNA, and small nucleolar RNA. The resulting ribosomal RNA-depleted RNA was fragmented using heat in the presence of divalent cations, with fragmentation times varying according to input RNA degradation. Fragmented RNA was converted into double-stranded cDNA, with dUTP used in place of dTTP in a second-strand master mix. A single base was added to the cDNA, and forked adaptors that included index, or barcode, sequences were attached via ligation. The resulting molecules were amplified via PCR for 15 cycles. During PCR, the polymerase stalled when a dUTP base was encountered in the template. Final libraries were quantified via PCR, normalized to 2 nM, and pooled. Pooled libraries were bound to the surface of a flow cell, and each bound template molecule was clonally amplified up to 1,000-fold to create individual clusters. Four fluorescently labeled nucleotides were then flowed over the surface of the flow cell and incorporated into each nucleic acid chain. Each nucleotide label acted as a terminator for polymerization. The fluorescence of each cluster was measured during the base identification. Dye was then enzymatically removed to allow for incorporation of the next nucleotide during the next cycle.
Immunohistochemistry
A total of 131 GBC tissue samples were used for immunohistochemistry in our present study. All specimens from patients fixed in 10% buffered formalin were embedded in paraffin blocks. One slide from each specimen had been stained with hematoxylin and eosin and marked by a pathologist to ensure that the tissue section contained more than 80% tumor cells for macrodissection. Consecutive 4-μm-thick sections were analyzed using a standard immunohistochemistry protocol and stained by antibodies of phospho-mTOR (Ser-2448) (1:50, Cell Signaling) and phospho-S6K1 kinase (Thr-389) (1:100, Sigma). The scoring of immunohistochemistry is based upon the staining intensity (I) and the proportion of stained quantity (q) of tumor cells to obtain a final score (Q) defined as the product of I × q and was performed by two independent pathologists. The scoring system for I was 0 = negative, 1 = low, 2 = moderate, 3 = intense immunostaining; and for q was 0 = negative, 1 = 1%–9% positive, 2 = 10%–39% positive, 3 = 40%–69% positive, and 4 = 70%–100% positive cells.
Statistical analyses
All data are expressed as mean ± SEM. Two-group comparisons were performed with unpaired two-tailed Student’s t test. Repeated-measures analysis of variance (ANOVA) was used to determine tumor differences at various time points within one group of mice. One-way ANOVA was used to determine tumor differences at W6 between all four groups. Survival probabilities were determined using Kaplan-Meier analyses and compared by the log-rank test. Each experiment consisted of at least three replicates per condition. SPSS 19.0 software was used for all statistical analysis. p <0.05 was considered statistically significant.
Acknowledgments
This work was supported by the scholarships from the Chinese Scholarship Council (CSC) awarded to D.Y. (no. 201406230237); the Foundation of Shanghai Shen Kang Hospital Development Center (nos. 16CR2002A and 16CR3028A); the National Science Foundation of China (nos. 81472240, 81773184, 81272748, 81872358, and 81572834); the High-Level Collaborative Innovation Team Incentive Program of Shanghai Municipal Education Commission (2018, J.W.); and the Shanghai Outstanding Academic Leaders Plan (2016, J.W.).
Author contributions
J.W. and Q.H. conceived of the study and carried out its design. D.Y., T.C., M.Z., and S.X. performed the experiments. W.C., YH.Z., D.J., and JC.Y. provided materials. D.Y., T.C., M.Z., XF.Y., Q.L., J.W., and Q.H. analyzed the data. D.Y., T.C., M.Z., Q.H., and J.W. wrote and revised the manuscript with input from all authors. All authors read and approved the final manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.11.007.
Contributor Information
Qihong Huang, Email: huang19104@yahoo.com.
Jian Wang, Email: prof_wangjian@163.com.
Supplemental information
References
- 1.Kanthan R., Senger J.L., Ahmed S., Kanthan S.C. Gallbladder Cancer in the 21st Century. J. Oncol. 2015;2015:967472. doi: 10.1155/2015/967472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rakić M., Patrlj L., Kopljar M., Kliček R., Kolovrat M., Loncar B., Busic Z. Gallbladder cancer. Hepatobiliary Surg. Nutr. 2014;3:221–226. doi: 10.3978/j.issn.2304-3881.2014.09.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gelibter A., Malaguti P., Di Cosimo S., Bria E., Ruggeri E.M., Carlini P., Carboni F., Ettorre G.M., Pellicciotta M., Giannarelli D. Fixed dose-rate gemcitabine infusion as first-line treatment for advanced-stage carcinoma of the pancreas and biliary tree. Cancer. 2005;104:1237–1245. doi: 10.1002/cncr.21286. [DOI] [PubMed] [Google Scholar]
- 4.Thongprasert S., Napapan S., Charoentum C., Moonprakan S. Phase II study of gemcitabine and cisplatin as first-line chemotherapy in inoperable biliary tract carcinoma. Ann. Oncol. 2005;16:279–281. doi: 10.1093/annonc/mdi046. [DOI] [PubMed] [Google Scholar]
- 5.Kim M.J., Oh D.Y., Lee S.H., Kim D.W., Im S.A., Kim T.Y., Heo D.S., Bang Y.J. Gemcitabine-based versus fluoropyrimidine-based chemotherapy with or without platinum in unresectable biliary tract cancer: a retrospective study. BMC Cancer. 2008;8:374. doi: 10.1186/1471-2407-8-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tebbutt N., Pedersen M.W., Johns T.G. Targeting the ERBB family in cancer: couples therapy. Nat. Rev. Cancer. 2013;13:663–673. doi: 10.1038/nrc3559. [DOI] [PubMed] [Google Scholar]
- 7.Li M., Zhang Z., Li X., Ye J., Wu X., Tan Z., Liu C., Shen B., Wang X.A., Wu W. Whole-exome and targeted gene sequencing of gallbladder carcinoma identifies recurrent mutations in the ErbB pathway. Nat. Genet. 2014;46:872–876. doi: 10.1038/ng.3030. [DOI] [PubMed] [Google Scholar]
- 8.Nakamura H., Arai Y., Totoki Y., Shirota T., Elzawahry A., Kato M., Hama N., Hosoda F., Urushidate T., Ohashi S. Genomic spectra of biliary tract cancer. Nat. Genet. 2015;47:1003–1010. doi: 10.1038/ng.3375. [DOI] [PubMed] [Google Scholar]
- 9.Yarden Y., Sliwkowski M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 10.Schlessinger J. Common and distinct elements in cellular signaling via EGF and FGF receptors. Science. 2004;306:1506–1507. doi: 10.1126/science.1105396. [DOI] [PubMed] [Google Scholar]
- 11.Pignochino Y., Sarotto I., Peraldo-Neia C., Penachioni J.Y., Cavalloni G., Migliardi G., Casorzo L., Chiorino G., Risio M., Bardelli A. Targeting EGFR/HER2 pathways enhances the antiproliferative effect of gemcitabine in biliary tract and gallbladder carcinomas. BMC Cancer. 2010;10:631. doi: 10.1186/1471-2407-10-631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hossan M.S., Chan Z.Y., Collins H.M., Shipton F.N., Butler M.S., Rahmatullah M., Lee J.B., Gershkovich P., Kagan L., Khoo T.J. Cardiac glycoside cerberin exerts anticancer activity through PI3K/AKT/mTOR signal transduction inhibition. Cancer Lett. 2019;453:57–73. doi: 10.1016/j.canlet.2019.03.034. [DOI] [PubMed] [Google Scholar]
- 13.Yu T., Li J., Yan M., Liu L., Lin H., Zhao F., Sun L., Zhang Y., Cui Y., Zhang F. MicroRNA-193a-3p and -5p suppress the metastasis of human non-small-cell lung cancer by downregulating the ERBB4/PIK3R3/mTOR/S6K2 signaling pathway. Oncogene. 2015;34:413–423. doi: 10.1038/onc.2013.574. [DOI] [PubMed] [Google Scholar]
- 14.Liu T., Sun Q., Li Q., Yang H., Zhang Y., Wang R., Lin X., Xiao D., Yuan Y., Chen L., Wang W. Dual PI3K/mTOR inhibitors, GSK2126458 and PKI-587, suppress tumor progression and increase radiosensitivity in nasopharyngeal carcinoma. Mol. Cancer Ther. 2015;14:429–439. doi: 10.1158/1535-7163.MCT-14-0548. [DOI] [PubMed] [Google Scholar]
- 15.Fingar D.C., Salama S., Tsou C., Harlow E., Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002;16:1472–1487. doi: 10.1101/gad.995802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu K., Dai H.B., Qiu Z.L. mTOR signaling in osteosarcoma: Oncogenesis and therapeutic aspects (Review) Oncol. Rep. 2016;36:1219–1225. doi: 10.3892/or.2016.4922. [DOI] [PubMed] [Google Scholar]
- 17.Karlsson E., Pérez-Tenorio G., Amin R., Bostner J., Skoog L., Fornander T., Sgroi D.C., Nordenskjöld B., Hallbeck A.L., Stål O. The mTOR effectors 4EBP1 and S6K2 are frequently coexpressed, and associated with a poor prognosis and endocrine resistance in breast cancer: a retrospective study including patients from the randomised Stockholm tamoxifen trials. Breast Cancer Res. 2013;15:R96. doi: 10.1186/bcr3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Y., Huang X., Huang Z., Feng J. Phenoxodiol enhances the antitumor activity of gemcitabine in gallbladder cancer through suppressing Akt/mTOR pathway. Cell Biochem. Biophys. 2014;70:1337–1342. doi: 10.1007/s12013-014-0061-y. [DOI] [PubMed] [Google Scholar]
- 19.Wu W.D., Hu Z.M., Shang M.J., Zhao D.J., Zhang C.W., Hong D.F., Huang D.S. Cordycepin down-regulates multiple drug resistant (MDR)/HIF-1α through regulating AMPK/mTORC1 signaling in GBC-SD gallbladder cancer cells. Int. J. Mol. Sci. 2014;15:12778–12790. doi: 10.3390/ijms150712778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dokmanovic M., Marks P.A. Prospects: histone deacetylase inhibitors. J. Cell. Biochem. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 21.Verdin E., Ott M. 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015;16:258–264. doi: 10.1038/nrm3931. [DOI] [PubMed] [Google Scholar]
- 22.Lomberk G.A., Iovanna J., Urrutia R. The promise of epigenomic therapeutics in pancreatic cancer. Epigenomics. 2016;8:831–842. doi: 10.2217/epi-2015-0016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Han R.F., Li K., Yang Z.S., Chen Z.G., Yang W.C. Trichostatin A induces mesenchymal-like morphological change and gene expression but inhibits migration and colony formation in human cancer cells. Mol. Med. Rep. 2014;10:3211–3216. doi: 10.3892/mmr.2014.2594. [DOI] [PubMed] [Google Scholar]
- 24.Chen M.Y., Liao W.S., Lu Z., Bornmann W.G., Hennessey V., Washington M.N., Rosner G.L., Yu Y., Ahmed A.A., Bast R.C., Jr. Decitabine and suberoylanilide hydroxamic acid (SAHA) inhibit growth of ovarian cancer cell lines and xenografts while inducing expression of imprinted tumor suppressor genes, apoptosis, G2/M arrest, and autophagy. Cancer. 2011;117:4424–4438. doi: 10.1002/cncr.26073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiao Y., Pawlik T.M., Anders R.A., Selaru F.M., Streppel M.M., Lucas D.J., Niknafs N., Guthrie V.B., Maitra A., Argani P. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013;45:1470–1473. doi: 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harbron C. A flexible unified approach to the analysis of pre-clinical combination studies. Stat. Med. 2010;29:1746–1756. doi: 10.1002/sim.3916. [DOI] [PubMed] [Google Scholar]
- 27.Liu Q., Yin X., Languino L.R., Altieri D.C. Evaluation of drug combination effect using a Bliss independence dose-response surface model. Stat. Biopharm. Res. 2018;10:112–122. doi: 10.1080/19466315.2018.1437071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sabatini D.M. mTOR and cancer: insights into a complex relationship. Nat. Rev. Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 29.Yamaguchi J., Sasaki M., Sato Y., Itatsu K., Harada K., Zen Y., Ikeda H., Nimura Y., Nagino M., Nakanuma Y. Histone deacetylase inhibitor (SAHA) and repression of EZH2 synergistically inhibit proliferation of gallbladder carcinoma. Cancer Sci. 2010;101:355–362. doi: 10.1111/j.1349-7006.2009.01387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang Y., Chen S.Y., Colborne S., Lambert G., Shin C.Y., Dos Santos N., Orlando K.A., Lang J.D., Hendricks W.P.D., Bally M.B. Histone deacetylase inhibitors synergize with catalytic inhibitors of EZH2 to exhibit anti-tumor activity in small cell carcinoma of the ovary, hypercalcemic type. Mol. Cancer Ther. 2018;17:2767–2779. doi: 10.1158/1535-7163.MCT-18-0348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.García-García C., Ibrahim Y.H., Serra V., Calvo M.T., Guzmán M., Grueso J., Aura C., Pérez J., Jessen K., Liu Y. Dual mTORC1/2 and HER2 blockade results in antitumor activity in preclinical models of breast cancer resistant to anti-HER2 therapy. Clin. Cancer Res. 2012;18:2603–2612. doi: 10.1158/1078-0432.CCR-11-2750. [DOI] [PubMed] [Google Scholar]
- 32.García-Echeverría C. Allosteric and ATP-competitive kinase inhibitors of mTOR for cancer treatment. Bioorg. Med. Chem. Lett. 2010;20:4308–4312. doi: 10.1016/j.bmcl.2010.05.099. [DOI] [PubMed] [Google Scholar]
- 33.Child F., Ortiz-Romero P.L., Alvarez R., Bagot M., Stadler R., Weichenthal M., Alves R., Quaglino P., Beylot-Barry M., Cowan R. Phase II multicentre trial of oral quisinostat, a histone deacetylase inhibitor, in patients with previously treated stage IB-IVA mycosis fungoides/Sézary syndrome. Br. J. Dermatol. 2016;175:80–88. doi: 10.1111/bjd.14427. [DOI] [PubMed] [Google Scholar]
- 34.Venugopal B., Baird R., Kristeleit R.S., Plummer R., Cowan R., Stewart A., Fourneau N., Hellemans P., Elsayed Y., McClue S. A phase I study of quisinostat (JNJ-26481585), an oral hydroxamate histone deacetylase inhibitor with evidence of target modulation and antitumor activity, in patients with advanced solid tumors. Clin. Cancer Res. 2013;19:4262–4272. doi: 10.1158/1078-0432.CCR-13-0312. [DOI] [PubMed] [Google Scholar]
- 35.Moreau P., Facon T., Touzeau C., Benboubker L., Delain M., Badamo-Dotzis J., Phelps C., Doty C., Smit H., Fourneau N. Quisinostat, bortezomib, and dexamethasone combination therapy for relapsed multiple myeloma. Leuk. Lymphoma. 2016;57:1546–1559. doi: 10.3109/10428194.2015.1117611. [DOI] [PubMed] [Google Scholar]
- 36.Lohse M., Bolger A.M., Nagel A., Fernie A.R., Lunn J.E., Stitt M., Usadel B. RobiNA: a user-friendly, integrated software solution for RNA-Seq-based transcriptomics. Nucleic Acids Res. 2012;40:W622–W627. doi: 10.1093/nar/gks540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cibulskis K., Lawrence M.S., Carter S.L., Sivachenko A., Jaffe D., Sougnez C., Gabriel S., Meyerson M., Lander E.S., Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat. Biotechnol. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ye K., Schulz M.H., Long Q., Apweiler R., Ning Z. Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009;25:2865–2871. doi: 10.1093/bioinformatics/btp394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cingolani P., Platts A., Wang L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Choi Y., Sims G.E., Murphy S., Miller J.R., Chan A.P. Predicting the functional effect of amino acid substitutions and indels. PLoS ONE. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lawrence M.S., Stojanov P., Polak P., Kryukov G.V., Cibulskis K., Sivachenko A., Carter S.L., Stewart C., Mermel C.H., Roberts S.A. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 46.Arts J., King P., Marien A., Floren W., Belien A., Janssen L., Pilatte I., Roux B., Decrane L., Gilissen R. JNJ-26481585, a novel “second-generation” oral histone deacetylase inhibitor, shows broad-spectrum preclinical antitumoral activity. Clin. Cancer Res. 2009;15:6841–6851. doi: 10.1158/1078-0432.CCR-09-0547. [DOI] [PubMed] [Google Scholar]
- 47.Jiang S.J., Wang S. Dual targeting of mTORC1 and mTORC2 by INK-128 potently inhibits human prostate cancer cell growth in vitro and in vivo. Tumour Biol. 2015;36:8177–8184. doi: 10.1007/s13277-015-3536-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.