

Certain stressful stimuli activate the glucocorticoid receptor (GR) and increase the incidence of herpes simplex virus 1 (HSV-1) reactivation from latency. For example, a corticosteroid antagonist impairs productive infection and virus shedding following explant of trigeminal ganglia from latently infected mice.
KEYWORDS: ICP4 promoter/enhancer, KLF4, SLUG, glucocorticoid receptor, stress response
ABSTRACT
Following acute infection, herpes simplex virus 1 (HSV-1) lytic cycle viral gene expression is silenced; consequently, lifelong latency in neurons is established. Certain external stimuli that trigger reactivation from latency also activate the glucocorticoid receptor (GR). The synthetic corticosteroid dexamethasone, but not a GR-specific antagonist, increases the frequency of explant-induced reactivation from latency and stimulates productive infection. Furthermore, dexamethasone increases expression of cellular transcription factors in trigeminal ganglionic neurons: for example, SLUG and three Krüppel-like transcription factor (KLF) family members, KLF4, KLF15, and promyelocytic leukemia zinc finger protein (PLZF). Consequently, we hypothesized that stress-induced transcription factors stimulate expression of ICP4, a viral transcriptional regulator required for productive infection. New studies demonstrated that GR and KLF4, PLZF, or SLUG cooperatively transactivate the ICP4 enhancer upstream of a minimal promoter in monkey kidney cells (Vero) and mouse neuroblastoma cells (Neuro-2A). Strikingly, mutagenesis of two KLF4/Sp1 binding sites reduced GR- plus KLF4-, PLZF-, or SLUG-mediated transactivation to basal levels. A consensus enhancer (E)-Box adjacent to a KLF4/Sp1 binding site was also required for GR- and SLUG-, but not KLF family member-, mediated transactivation of the ICP4 promoter. Chromatin immunoprecipitation studies (ChIP) revealed GR and stress-induced transcription factors occupy ICP4 enhancer sequences. Conversely, specific binding was generally reduced in the KLF4/Sp1 mutant. Furthermore, GR and SLUG occupancy of ICP4 enhancer sequences was reduced in the E-Box mutant. Based on these studies, we suggest stressful stimuli can trigger productive infection because GR and specific stress-induced transcription factors activate ICP4 expression.
IMPORTANCE Certain stressful stimuli activate the glucocorticoid receptor (GR) and increase the incidence of herpes simplex virus 1 (HSV-1) reactivation from latency. For example, a corticosteroid antagonist impairs productive infection and virus shedding following explant of trigeminal ganglia from latently infected mice. Infected cell protein 4 (ICP4) is the only immediate early viral transcriptional regulator required for productive infection, suggesting stressful stimuli stimulate ICP4 expression. New studies revealed GR and stress-induced transcription factors identified during reactivation from latency, SLUG and three Krüppel-like transcription factor family members (KLF4, KLF15, and promyelocytic leukemia zinc finger protein), cooperatively transactivate the ICP4 enhancer. Two KLF4 consensus binding sites were crucial for cooperative transactivation of the ICP4 enhancer. A consensus enhancer-box also mediated cooperative transactivation of the ICP4 enhancer by GR and SLUG. The ability of GR and stress-induced transcription factors to transactivate ICP4 enhancer activity is predicted to trigger productive infection following stressful stimuli.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) establishes lifelong latent infections in sensory neurons as well as other neuronal types after acute infection (1, 2). Periodically, reactivation from latency occurs, which leads to virus shedding and transmission. Infection can result in serious recurrent eye infections, including herpetic stromal keratitis (HSK) (3). HSK is characterized by corneal scarring and neovascularization, which can ultimately lead to blindness. Long-term oral acyclovir treatment reduces HSK recurrences by only 41% because many cases are the result of reactivation from latency (1, 2). Many cases of HSV-1-induced recurrent encephalitis are also due to reactivation from latency; consequently, acyclovir or related drugs do not prevent encephalitis (4). A better understanding of early stages of reactivation from latency may lead to improved therapeutics.
During productive infection, five immediate early (IE) viral mRNAs are expressed in the absence of de novo protein synthesis: ICP0, ICP4, ICP22, ICP27, and ICP47 (5). The ICP4 protein, a 175-kDa phosphoprotein, is the only viral transcriptional activator that is essential for productive infection (6) because it activates E and L genes (7). ICP4 specifically binds many sites on the viral genome (8) and interacts with the TATA-binding protein plus TFIIB to stimulate early and late viral gene expression (9). There is also a correlation between the abilities of ICP4 to stimulate viral transcription and to increase histone dynamics (10). ICP0 is a promiscuous activator of promoters and contains an E3 ubiquitin ligase near its amino terminus (reviewed in references 11 and 12). The viral tegument protein VP16 interacts with two cellular transcription factors, Oct 1 and host cellular factor 1, to specifically activate IE gene expression (reviewed in references 13 and 14). In contrast to productive infection, lytic cycle viral gene expression is not readily detected during latency because the genome exists as silent chromatin (15, 16). The only viral gene abundantly expressed during latency is the latency-associated transcript (2, 17).
Several lines of evidence point toward stress triggering HSV-1 productive infection and reactivation from latency in humans (18–20). Support for this premise comes from studies demonstrating that a glucocorticoid receptor (GR)-specific antagonist (CORT-108297) reduces trigeminal ganglion (TG) explant-induced reactivation in latently infected mice (21). Second, the synthetic corticosteroid dexamethasone (DEX) accelerates reactivation and increases the number of TG neurons that express ICP0, ICP4, and VP16 following explant (21, 22). Third, treatment of human gingival fibroblasts with glucocorticoids enhances HSV-1 replication (23) and DEX stimulates productive infection in Neuro-2A cells (24). Stressful stimuli generally increase levels of corticosteroid, which specifically bind GR or mineralocorticoid receptor (MR) (reviewed in reference 25). A GR or MR homodimer bound to corticosteroids enters the nucleus, remodels chromatin, and induces transcription in the absence of de novo protein synthesis; these steps are referred to as ligand-dependent induction of gene expression (25, 26). Nuclear GR or MR dimers specifically regulate transcription by binding consensus glucocorticoid response elements (GREs; 5′-GGTACANNNTGTTCT-3′) (27, 28). GR activation can also occur via corticosteroid-independent mechanisms (unliganded GR activation) (reviewed in reference 29). While the details of ligand-independent GR activation are not well understood, unliganded GR activation stimulates a different subset of cellular genes than those stimulated by ligand-dependent GR activation (30), including the tumor suppressor BRCA1 (31).
Approximately 50% of TG sensory neurons express GR (32), indicating these neurons can directly respond to stressful stimuli. Support for this prediction includes the finding that DEX induces expression of cellular genes in TG neurons during early stages of bovine herpesvirus 1 (BoHV-1) reactivation from latency (33). A subset of these cellular genes consists of transcription factors, including four Krüppel-like transcription factors (KLFs), KLF4, KLF6, KLF15, and PZLF (promyelocytic leukemia zinc finger), also referred to as ZBTB16 (zinc finger and BTB domain-containing protein 16). The transcription factor SLUG (Snail superfamily of C2H2-type zinc finger transcription factors) is also stimulated during early stages of reactivation. Several of these transcription factors are also induced by DEX following explant of mouse TGs (34). KLF family members and specificity protein 1 (Sp1) belong to the same superfamily of transcription factors, and these family members bind GC- and/or CACC-rich sequences (35, 36). Certain KLF family members can activate promoters via Sp1 binding sites, which are present in many HSV-1 promoters. Recent studies demonstrated KLF15 and GR cooperate to transactivate the HSV-1 ICP0 promoter (24) and the BoHV-1 immediate early transcription unit 1 (IEtu1) promoter that drives IE expression of bICP0 and bICP4 (37). GR and KLF15 regulate gene expression dynamics and integrate signals via a positive feed-forward loop (38–40). KLF4 and GR cooperatively transactivate the BoHV-1 bICP0 early promoter even though there is not a consensus GRE in this promoter (41).
ICP4, ICP0, or VP16 expression can induce reactivation in latently infected cells derived from trigeminal ganglia (TG) (42). Since these viral transcriptional regulators are not abundantly expressed during latency, we hypothesized GR and/or stress-induced cellular transcription factors transactivate promoters of these key viral transcriptional regulators during early stages of reactivation from latency. Consequently, we tested whether GR and stress-induced transcription factors can stimulate ICP4 promoter activity. These studies demonstrated GR and specific stress-induced transcription factors cooperatively activate the ICP4 enhancer by directly interacting with KLF4/Sp1 binding sites.
RESULTS
Analysis of ICP4 enhancer sequences in Neuro-2A cells.
A construct that contains ICP4 enhancer sequences within the HSV-1 genome (Fig. 1A) spanning −330 to −110 cloned upstream of the minimal promoter in the pGL4.24[luc2/minP] vector (pα4R; Fig. 1B and C) was used to examine the effects that stress-induced transcription factors have on ICP4 expression because it lacks cryptic transcription factor binding sites. The ICP4 promoter/enhancer construct was initially cloned into a luciferase reporter (pGL3 Basic), which contains numerous cryptic transcription factor binding sites, suggesting this is why stress had only modest effects on ICP4 promoter activity (reference 34 and data not shown).
FIG 1.

Location of ICP4 gene, ICP4 enhancer, adjacent genes, and oriS within HSV-1 genome. (A) The prototypic HSV-1 genomic structure is shown. The viral repeat regions are shown as open rectangles. TRL is the terminal long repeat. IRL is the internal (or inverted) long repeat. TRS is the terminal short repeat. IRS is the internal (or inverted) short repeat. The unique long (UL) and the unique short (US) regions are each represented by a solid line. (B) Location of ICP4 coding regions and flanking genes in IRS (ICP22) or TRS (ICP47). The thin lines in ICP22 denote the location of an intron. Location of the origin of replication (oriS; denoted by black oval) and ICP4 enhancer sequences (−330 to −110; denoted by gray triangle). (C) Schematic of ICP4 enhancer fragment (pα4R) used in these studies. This fragment was inserted upstream of the minimal promoter of the firefly luciferase reporter plasmid, pGL4.24[luc2/minP]. Nucleotide position is given relative to the ICP4 transcription initiation site. Positions of consensus transcription factor binding sites are shown, and the key for these sites is below the schematic.
The pα4R construct was cotransfected with plasmids that express stress-induced transcription factors. After transfection, cultures were incubated in minimal essential medium (MEM) plus 2% charcoal-stripped fetal bovine serum (FBS) in the presence or absence of DEX as previously described (24). Relative to the empty vector (pGL4.24[luc2/minP] vector), pα4R basal promoter activity was more than 350-fold higher in Neuro-2A cells (Fig. 2A, white columns), indicating the ICP4 enhancer was active in Neuro-2A cells. KLF4, but not KLF15, stimulated promoter activity nearly 700-fold when cotransfected with the enhancer element (Fig. 2A, blue columns). When pα4R was cotransfected with GR plus KLF4 and cultures were treated with DEX, promoter activity was approximately 1,100-fold higher than with the minimal promoter (yellow column). Without DEX treatment (black column), or when adding RU486 (gray column), a GR antagonist (43, 44), promoter activity was similar to that with KLF4 alone. Notably, GR did not increase the promoter activity alone or with DEX, suggesting a synergistic interaction between KLF4, GR, and DEX treatment.
FIG 2.

Transactivation of ICP4 enhancer element by GR and stress-induced transcription factors. (A) Neuro-2A cells were grown in MEM containing 2% charcoal-stripped FBS and transfected with the pα4R construct (0.5 μg DNA), Renilla luciferase expression plasmid (0.05 μg DNA), GR expression plasmid (1 μg DNA), and, where denoted, KLF4, KLF15, PLZF, or SLUG (0.5 μg DNA). To maintain the same amount of DNA in each sample, empty vector was included in certain samples. At 24 h after transfection, designated cultures were treated with DEX (10 μM) or DEX and RU486 (10 μM each). (B) Vero cells were transfected with the denoted plasmids as described in the legend to panel A. Cells were harvested at 48 h after transfection, and protein lysate was subjected to dual-luciferase assay as described in Materials and Methods. Promoter activity was calculated as firefly luciferase activity compared to the transfection control, Renilla luciferase. For panels A and B, fold activation is presented as fold increase in promoter activity versus the empty minimal promoter reporter plasmid, pGL4.24[luc2/minP]. For panels C (Neuro-2A cells) and D (Vero cells), fold activation was presented as the fold increase in pα4R activity when cotransfected with the designated expression constructs relative to basal levels of pα4R promoter activity. The results are the mean from three separate experiments. Asterisks denote significant differences between the indicated treatments (*, >0.05; **, >0.01; ***, >0.001). Student’s t test was performed to determine significance.
PLZF and SLUG also transactivated the pα4R construct between 900- and 1,100-fold, but only when cotransfected with GR (Fig. 2A, black column). Addition of DEX significantly decreased GR-dependent transactivation by PLZF but significantly increased GR-dependent transactivation by SLUG (Fig. 2A, yellow column). Addition of RU486 abolished the effect of GR and GR+DEX for PLZF- and SLUG-mediated transactivation (Fig. 2A, gray column), suggesting transactivation was mediated via a GR ligand-dependent mechanism. By itself, GR, PLZF, or SLUG did not significantly increase pα4R promoter activity (Fig. 2A, blue column), providing additional support for the idea that synergistic transactivation of pα4R promoter activity occurred.
As a comparison to the results in Neuro-2A cells, the effects of GR and these stress-induced transcription factors were examined in Vero cells (Fig. 2B). In contrast to Neuro-2A cells, GR+KLF15 significantly stimulated pα4R promoter activity in Vero cells relative to basal promoter activity (Fig. 2B, black column). GR+KLF15-mediated transactivation appeared to occur in a ligand-independent manner because neither RU486 nor DEX significantly changed pα4R promoter activity (Fig. 2B, gray and yellow columns). As in Neuro-2A cells, PLZF and KLF4 significantly transactivated pα4R promoter activity relative to basal activity. SLUG did not significantly stimulate pα4R promoter activity alone or with GR relative to the effects of GR.
Because basal promoter activity of pα4R appeared to be higher in Neuro-2A cells, the results from both cell lines were reassessed by setting pα4R activity as 1 and then comparing levels of transactivation by GR and stress-induced transcription factors plus DEX and/or RU486 treatment (Fig. 2C, Neuro-2A cells, and Fig. 2D, Vero cells). Several marked differences were noted between the two cell lines. The most obvious difference was that GR+KLF15 transactivated the pα4R construct approximately 3-fold in Vero cells but not in Neuro-2A cells. Second, KLF4 and GR cooperatively transactivated the pα4R construct in Vero cells in the absence of DEX treatment; however, this was not the case in Neuro-2A cells. Third, PLZF-mediated transactivation in Neuro-2A, but not Vero, cells was ligand dependent because RU486 treatment reduced promoter activity to a value lower than basal levels of pα4R promoter activity. Finally, GR, SLUG, and DEX treatment significantly stimulated pα4R promoter activity (more than 2-fold) in Neuro-2A cells but not in Vero cells. In summary, GR and stress-induced transcription factors exhibited cell-type-specific effects on ICP4 enhancer activity.
Transactivation of the ICP4 enhancer by GR and stress-induced transcription factor requires KLF4/Sp1 binding sites.
Three consensus KLF4 binding sites (45) were identified in the ICP4 enhancer fragment: two sites are frequently detected as KLF4 binding sites (Fig. 3A; denoted by a blue oval) whereas the third site is bound by KLF4 less frequently (green oval). Each putative KLF4 binding site is contained within a consensus Sp1 binding site; thus, mutating these sites also disrupts the Sp1 binding site. Our studies with ICP4 enhancer deletion constructs indicated that the KLF4 variant binding site, which is not frequently bound by KLF4, was not important for GR- and stress-induced transcription factor-mediated transactivation in Neuro-2A or Vero cells (data not shown). These studies also suggested that the two perfect matches for KLF4 appeared to be important.
FIG 3.

KLF4 binding sites in ICP4 enhancer are crucial for cooperative transactivation. (A) Schematic of wt ICP4 enhancer element (pα4R) and mutants that lack one or both of the consensus KLF4/Sp1 binding sites, denoted by blue ovals (KLF4) or black triangles (Sp1), yellow ovals (TAATGARAT), green ovals (KLF4 variant), and red ovals (E-Box). Nucleotide number shown is relative to the transcription initiation site. The pα4RΔ5′KLF4 and pα4RΔ3′KLF4 are the respective single KLF4/Sp1 binding site mutants. The pα4RΔKLF4 construct is the double KLF4 binding site mutant. Positions of consensus transcription factor binding sites are shown, and the key for these sites is to the right of the schematic. (B) Neuro-2A cells were transfected with the wt pα4R construct or the designated mutant constructs (0.5 μg DNA), Renilla luciferase expression plasmid (0.05 μg DNA), GR expression plasmid (1 μg DNA), and, where denoted, KLF4, KLF15, PLZF, or SLUG (0.5 μg DNA). To maintain the same amount of DNA in each sample, empty vector was included in certain samples. Following transfection, cells were cultured in medium containing 2% charcoal-stripped FBS. At 24 h after transfection, designated cultures were treated with DEX (10 μM). (C) Vero cells were transfected with the denoted plasmids as described in the legend to panel B. At 48 h after transfection, cells were harvested and protein lysate was subjected to dual-luciferase assay as described in Materials and Methods. Promoter activity was calculated as firefly luciferase activity compared to the transfection control, Renilla luciferase. Fold activation is presented as fold increase in promoter activity versus the empty minimal promoter reporter plasmid, pGL4.24[luc2/minP]. The results are the mean from three separate experiments. Asterisks denote significant differences between the indicated treatments (*, >0.05; **, >0.01; ***, >0.001). Student’s t test was performed to determine significance.
Mutating the 3′ KLF4 binding site (pα4RΔ3′KLF4), but not the 5′ KLF4 binding site (pα4RΔ5′KLF4), significantly reduced GR+KLF4+DEX-mediated transactivation in Neuro-2A (Fig. 3B) and Vero (Fig. 3C) cells. Mutating both sites (pαRΔKLF4) essentially eliminated GR+KLF4+DEX-mediated transactivation. In Vero cells, a similar trend was observed for GR+KLF15+DEX transactivation. Mutating the 3′ KLF4 binding site also significantly reduced PLZF- and SLUG-mediated transactivation as well as basal promoter activity in both cell lines. Furthermore, mutating both KLF4 binding sites essentially eliminated KLF15-, PLZF-, or SLUG- and GR+DEX-mediated transactivation of pα4R compared to basal promoter activity in both cell lines. The biological effects of PLZF are primarily associated with repressing transcription because it stably interacts with the SMRT-Sin3-HDAC-Ncor and Polycomb group complexes (46–48); hence, it was surprising to find that PLZF cooperated with GR to transactivate the ICP4 enhancer. Interestingly, a recent study demonstrated that PLZF activates transcription by interacting with a histone methyltransferase (EZH2) (49), which supports the finding that GR and PLZF stimulated ICP4 enhancer activity. Finally, these studies revealed that basal enhancer activity of pαRΔKLF4 and pα4RΔ3′KLF4 was reduced compared to the pα4R construct in both cell lines. In summary, these studies revealed that two KLF4 binding sites, in particular, the 3′ binding site, were required for GR and stress-induced transactivation of the ICP4 enhancer.
A consensus E-Box in the ICP4 enhancer is important for GR- and SLUG-mediated transcription.
ICP4 enhancer sequences also contain a consensus enhancer (E)-Box, CANNTG, that is 8 nucleotides upstream of the 3′ KLF4 binding site (Fig. 4A). Although SLUG is generally considered to be a repressor of transcription (reviewed in reference 50), it was reported to stimulate transcription by directly interacting with an E-Box in the ZEB1 (zinc finger E-Box binding homeobox 1) promoter (51). To test whether the E-Box was important for GR- and SLUG-mediated transactivation of the ICP4 enhancer, an E-Box mutant (pα4RΔE-Box; Fig. 4A) was examined in Neuro-2A cells. Transactivation of pα4RΔE-Box by GR and SLUG was significantly reduced compared to that of wild-type (wt) pα4R (Fig. 4B). Furthermore, synergistic activation by GR and DEX was reduced to basal levels. Conversely, the pα4RΔE-Box and wt pα4R constructs were transactivated at similar levels by KLF4 (Fig. 4C) and PLZF regardless of GR transactivation in Neuro-2A cells (Fig. 4D). In summary, these studies provided evidence that the E-Box and KLF4 binding sites were essential for GR- and SLUG-mediated transactivation of the ICP4 enhancer.
FIG 4.

Role of E-Box binding site on ICP4 enhancer element transactivation. (A) Schematic of ICP4 enhancer construct (pαR) and mutant that lacks the consensus E-Box binding site (pα4RΔE-Box); see summary of consensus binding sites for transcription factors below. Positions of consensus transcription factor binding sites are shown, and the key for these sites is to the right of the schematic. (B) Neuro-2A cells were transfected with the wt pα4R construct or pα4RΔE-Box construct (0.5 μg DNA), Renilla luciferase expression plasmid (0.05 μg DNA), GR expression plasmid (1 μg DNA), and, where denoted, SLUG (0.5 μg DNA). (C) Neuro-2A cells were transfected with the wt pα4R construct or pα4RΔE-Box construct (0.5 μg DNA), Renilla luciferase expression plasmid (0.05 μg DNA), GR expression plasmid (1 μg DNA), and, where denoted, KLF4 (0.5 μg DNA). (D) Neuro-2A cells were transfected with the wt pα4R construct or pα4RΔE-Box construct (0.5 μg DNA), Renilla luciferase expression plasmid (0.05 μg DNA), GR expression plasmid (1 μg DNA), and, where denoted, PLZF (0.5 μg DNA). To maintain the same amount of DNA in each sample, empty vector was included in certain samples. Following transfection, cells were cultured in MEM containing 2% charcoal-stripped FBS. Indicated samples were treated with DEX (10 μM) 24 h following transfection. At 48 h after transfection, cells were harvested and protein lysate was subjected to dual-luciferase assay as described in Materials and Methods. Promoter activity was calculated as firefly luciferase activity compared to the transfection control, Renilla luciferase. Fold activation is presented as fold increase in promoter activity versus the empty minimal promoter reporter plasmid, pGL4.24[luc2/minP]. Data are the means from three separate experiments. Asterisks denote significant differences between pα4RΔE-Box construct and wild-type pα4R construct (*, >0.05; **, >0.01; ***, >0.001). Student’s t test was performed to determine significance.
Stress-induced transcription factors enhance GR association with wt ICP4 enhancer sequences in transfected Neuro-2A cells.
To test whether GR occupies ICP4 enhancer sequences, Neuro-2A cells were cotransfected with the wt pα4R construct or pα4RΔKLF mutant plus GR and a stress-induced transcription factor. Chromatin immunoprecipitation (ChIP) studies using specific primers (Fig. 5A) were performed using the GR antibody or an isotype control antibody. GR and DEX treatment significantly increased occupancy of ICP4 sequences in the wt pα4R construct (Fig. 5B) but not the pα4RΔKLF mutant. Strikingly, cotransfection of the wt pα4R construct with GR, a stress-induced transcription factor (KLF4, KLF15, SLUG, and PLZF), and DEX treatment significantly increased GR occupancy relative to GR alone. In sharp contrast, increased GR occupancy was not observed when the pα4RΔKLF mutant was cotransfected with GR and KLF4, KLF15, SLUG, or PLZF. In summary, these studies revealed a correlation between GR occupancy of pα4R sequences and a significant increase in promoter activity mediated by GR and KLF4, PLZF, or SLUG (Table 1; blue + or − signs). In accordance with this finding, decreased GR occupancy of pα4RΔKLF sequences correlated with reduced promoter activity by GR and a stress-induced transcription factor.
FIG 5.

Chromatin immunoprecipitation with GR-specific antibodies in transfected Neuro-2A cells. (A) Schematic of ICP4 enhancer (pα4R construct, black line) cloned into pGL4.24 [luc2/minP] (red lines) at unique KpnI/BglII restriction sites. Relative positions of primers used to amplify ChIP target DNA (red arrows) are shown. This primer pair generates a 274-bp PCR product. Positions of consensus transcription factor binding sites are shown and are the same as in Fig. 1. (B) Neuro-2A cells were grown in MEM containing 2% charcoal-stripped FBS following transfection with plasmid containing wild-type pα4R construct or the double KLF4 binding site mutant pα4RΔKLF4 construct (1.5 μg DNA). Neuro-2A cells were cotransfected with a GR expression plasmid (3 μg DNA) and/or KLF4, KLF15, or SLUG expression plasmids (1.5 μg DNA). In order to maintain an equal quantity of DNA in each reaction mixture, empty vector was added as necessary. At 24 h posttransfection, the designated samples were treated with DEX (10 μM). At 48 h after transfection, cells were cross-linked with formaldehyde and harvested. ChIP was performed as described in Materials and Methods using GR-specific antibodies (5 μg) or with IgG (5 μg) as an isotype control. Target DNA was amplified using PCR with ICP4 forward and pGL4.24[luc2/minP] reverse primers (see panel A), which generated a 274-bp product on agarose gels stained with ethidium bromide. Individual bands were quantified using Image Lab software and presented as a percentage of the input sample, representing 13.3% of the cell lysate used for each sample. Data presented are the means from three separate transfection studies performed on different days. Asterisks denote a significant difference between the GR-specific antibody and the respective isotype control (*, >0.05; **, >0.01; ***, >0.001). Student’s t test was performed to determine significance.
TABLE 1.
Summary of transactivation and ChIP studies of ICP4 enhancera

Summary of effects of wt ICP4 enhancer (pα4R), KLF4 binding site mutant (pα4RΔKLF4), and E-Box binding site mutant (pα4RΔE-Box) on basal promoter activity (Fig. 2 and 4 show original data obtained in Neuro-2A cells) and ChIP results (Fig. 5 and 7). For promoter activity, “+++” denotes significant transactivation relative to basal promoter activity; conversely, “−” denotes that there was no significant difference. For ChIP studies, “+” denotes a significant increase in occupancy of the denoted transcription factor with the ICP4 enhancer construct relative to the isotype control antibody. Conversely, “−” indicates no significant difference. ChIP results using a GR-specific antibody are noted in blue, while data from antibodies directed against the indicated stress-induced transcription factor are in red. For clarity, results from Neuro-2A cells transfected with the denoted constructs are shown. The question mark in the GR+DEX+PLZF studies denotes that the PLZF antibody yielded high background in ChIP studies. ChIP studies with the pα4RΔE-Box construct were performed only with GR or SLUG antibody.
Occupancy of stress-induced transcription factors with the ICP4 enhancer was altered when KLF4/Sp1 binding sites were mutated.
Additional studies tested whether occupancy of KLF4, KLF15, PLZF, and SLUG was influenced when the KLF4/Sp1 binding sites were mutated. As described for studies in Fig. 4, Neuro-2A cells were transfected with the wt pα4R construct or double KLF4 binding site mutant (pα4RΔKLF4), GR expression plasmid, and one of the stress-induced transcription factors; where indicated, cultures were treated with DEX. Significantly more KLF4 was associated with pα4R or pα4RΔKLF4 following transfection of these promoter constructs with GR, KLF4, and cultures treated with DEX (Fig. 6A, black columns). Compared to wt ICP4 sequences, similar levels of KLF4 occupied pα4RΔKLF4 sequences when Neuro-2A cells were cotransfected with GR plus KLF4 and cultures were treated with DEX (Fig. 6A). Occupancy of KLF15 to wt ICP4 sequences (Fig. 6B, black columns) was significantly higher than that to ΔKLF4/Sp1 ICP4 sequences (blue columns) when transfected with just KLF15. When wt ICP4 or the ΔKLF4/Sp1 construct was cotransfected with GR+DEX or GR+DEX+KLF15, there was not a significant difference in KLF15 occupancy, which correlated with negligible transactivation by GR and KLF15 or GR, KLF15, and DEX treatment in Neuro-2A cells.
FIG 6.

Chromatin immunoprecipitation with KLF4, KLF15, PLZF, and SLUG in Neuro-2A cells transfected with wild type or ΔKLF mutant. (A to C) Neuro-2A cells were grown in MEM containing 2% charcoal-stripped FBS and transfected with wild-type pα4R construct or pα4RΔKLF4 construct (1.5 μg DNA). As denoted, samples were cotransfected with the GR expression plasmid (3 μg DNA) and/or a plasmid expressing KLF4, KLF15, or SLUG (1.5 μg DNA). To maintain the same DNA concentrations in each sample, empty vector was added to samples as necessary. At 24 h posttransfection, cells were treated with DEX (10 μM) where indicated. At 48 h posttransfection, cells were formaldehyde cross-linked and harvested. ChIP was performed as described in Materials and Methods using KLF4 (A)-, KLF15 (B)-, or SLUG (C)-specific antibodies or the isotype IgG control. Target DNA was amplified by PCR using ICP4 forward and pGL4.24[luc2/minP] reverse primers (Fig. 5A), generating a 274-bp product. DNA was detected in agarose gels stained with ethidium bromide. Individual bands were quantified using Image Lab software and presented as a percentage of the input sample, which represents approximately 13% of the cell lysate used for each sample. Data presented are the means from three independent transfection experiments. Asterisks denote a significant difference between the protein-specific antibody and the respective isotype control (*, >0.05; **, >0.01; ***, >0.001). Student’s t test was performed to determine significance.
SLUG occupancy of ICP4 sequences in the wt pα4R construct, but not pα4RΔKLF4, was significantly higher when Neuro-2A cells were cotransfected with just SLUG (no DEX treatment) or GR plus SLUG and cultures were treated with DEX (Fig. 6C). We attempted to assess PLZF occupancy of the ICP4 enhancer construct; however, high background levels of PLZF were amplified following ChIP. This suggested the PLZF antibody nonspecifically interacts with other proteins after formalin fixation, leading to high background PCR amplification. Consequently, we were unable to definitively compare PLZF occupancy with the wt ICP4 enhancer or KLF4 binding site mutants (data not shown). These studies revealed there was not a correlation between KLF4 occupancy of pα4R versus pα4RΔKLF4 and GR+KLF4+DEX-mediated transactivation (Table 1). In contrast, there was a correlation between SLUG occupancy of pα4R but not pα4RΔKLF4 and transactivation by GR+KLF4+DEX in Neuro-2A cells.
E-Box mutation abolishes SLUG association with the ICP4 enhancer.
Since mutating the E-Box significantly reduced GR+SLUG-mediated transactivation of the ICP4 enhancer regardless of DEX treatment, ChIP studies were performed following transfection of Neuro-2A cells with wt pα4R or pα4RΔE-Box mutant, GR, and/or SLUG. Consistent with the results in Fig. 5, GR occupied ICP4 enhancer sequences when Neuro-2A cells were transfected with the wt pα4R construct, GR, and SLUG and cultures were treated with DEX (Fig. 7A, black columns). In sharp contrast, transfection of the pα4RΔE-Box mutant with GR+SLUG and treatment with DEX (blue columns) did not yield significantly higher levels of occupancy relative to the isotype control antibody. SLUG occupancy of wt pα4R sequences was also significantly higher than the E-Box mutant following cotransfection with SLUG or GR+SLUG and DEX treatment (Fig. 7B, black columns versus blue columns). In summary, these studies revealed that mutating the E-Box significantly reduced GR and SLUG occupancy in transfected Neuro-2A cells, which correlated with high levels of transactivation of wt pα4R relative to the pα4RΔE-Box construct (summarized in Table 1).
FIG 7.

Chromatin immunoprecipitation of ICP4 enhancer element with the E-Box binding site mutation. (A and B) Neuro-2A cells were grown in MEM containing 2% charcoal-stripped FBS following transfection with wild-type pα4R construct (1.5 μg DNA) or mutant lacking the putative E-Box binding site (pα4RΔE-Box, 1.5 μg DNA). Samples were cotransfected with the GR expression plasmid (1 μg DNA) and/or SLUG expression plasmid (0.5 μg DNA) as denoted. At 24 h postinfection, designated samples were treated with DEX (10 μM). At 48 h after transfection, cells were cross-linked with formaldehyde and harvested in PBS. ChIP was performed using 5 μg of antibody directed against GR, SLUG, or isotype control IgG. Target DNA was amplified by PCR using ICP4 forward and pGL4.24[luc2/minP] reverse primers (Fig. 5A), generating a 274-bp product and imaged in agarose gels containing ethidium bromide. Individual bands were quantified using Image Lab software and presented as a percentage of input, which represents approximately 13% of the cell lysate for each sample. Data represent the means from three separate experiments. Asterisks denote a significant difference between the protein-specific antibody and the respective isotype control (*, >0.05; **, >0.01; ***, >0.001). Student’s t test was performed to determine significance.
GR and stress-induced transcription factors associate with the HSV-1 ICP4 promoter during productive infection.
To assess whether GR and the stress-induced transcription factors associate with ICP4 promoter/enhancer sequences during productive infection (Fig. 8A), Vero cells were infected with HSV-1 at a multiplicity of infection (MOI) of 1. Infected cells were cultured in medium containing 2% charcoal-stripped serum with or without DEX. At the indicated time points, cells were cross-linked with paraformaldehyde and harvested. ChIP was performed using isotype IgG, GR, KLF4, KLF15, or SLUG antibodies, as described in Materials and Methods. All four transcription factors occupied ICP4 promoter sequences at 8 h postinfection with DEX treatment (Fig. 8B). KLF4 and SLUG also occupied ICP4 promoter/enhancer sequences at 8 h in the absence of DEX, but to a lesser extent than with DEX treatment. None of the transcription factors associated with the promoter at 4 h after infection, and only KLF4 occupied the ICP4 promoter at 16 h after infection, regardless of DEX treatment. Occupancy of ICP4 enhancer sequences by GR and stress-induced transcription factors was different from GR and KLF15 occupancy of the ICP0 promoter, which occurred at 4 h postinfection but not at 8 or 16 h (24).
FIG 8.

Occupancy of GR, KLF4, KLF15, and SLUG with ICP4 enhancer sequences during productive infection. (A) Schematic of ICP4 promoter within the HSV-1 genome. Nucleotide positions are numbered relative to the transcription initiation site. ICP4 forward and reverse primer positions are denoted (red arrows), which result in a 247-bp PCR fragment. Positions of consensus transcription factor binding sites are the same as in Fig. 1. (B) Vero cells were cultured in MEM containing 2% charcoal-stripped FBS, following infection with HSV-1. Immediately following infection, cells were paraformaldehyde cross-linked and harvested for ChIP for the 0-h-after-infection time point. Samples treated with DEX (10 μM) were cultured in MEM containing 2% charcoal-stripped FBS. ChIP was performed as described in Materials and Methods using antibodies specific to IgG isotype (negative control, white columns), GR (black columns), KLF4 (yellow columns), KLF15 (blue columns), or SLUG (orange columns). Target DNA was amplified by PCR using ICP4 forward and reverse primers (A). Individual bands were quantified using Image Lab software and presented as a percentage of the input sample, which represents 13% of the harvested cell lysate for each sample. Data represent the means from three separate experiments with each antibody. Asterisks denote a significant difference between the protein-specific antibody and the respective isotype control (*, >0.05; **, >0.01; ***, >0.001). Student’s t test was performed to determine significance.
DISCUSSION
These studies provide new evidence that two KLF4/Sp1 binding sites were critical for cooperative transactivation of the ICP4 enhancer by GR and KLF4, PLZF, or SLUG in Neuro-2A and Vero cells in the absence of viral transcriptional regulatory proteins. ICP4 enhancer sequences do not contain a consensus GRE or a 1/2 GRE, suggesting that direct or indirect interactions between GR and a respective stress-induced transcription factor were important for stimulating ICP4 enhancer activation. GR and KLF4 may be particularly important for the ability of HSV-1 to respond to stressful stimuli because GR (52, 53) and KLF4 (54) are pioneer transcription factors that can specifically bind silent chromatin and stimulate transcription (25, 45, 55–57). The BoHV-1 ICP0 early promoter also lacks consensus GREs and is cooperatively transactivated by GR and KLF4 (41). Strikingly, bICP0 E promoter sequences contain consensus KLF4 binding sites and nucleosome-enriched KLF4 binding sites. Studies designed to prove that these binding sites are crucial for GR- and KLF4-mediated transactivation of the bICP0 E promoter are in progress. Of further note, increased KLF4 expression promotes lytic cycle Epstein-Barr virus gene expression and productive infection in latently infected epithelial cells and Burkitt lymphoma cells (58). The effects of KLF4 correlate with its ability to transactivate the Epstein-Barr virus immediate early promoter BRLF1 via two motifs (CACCCC and GGGGTG). Several KLF4 binding sites have been reported, (reviewed in reference 54), including those in the BRLF1 promoter, suggesting transcriptional coactivators influence KLF4 binding specificity. Collectively, these observations suggest KLF4 is an important cellular transcription factor that transactivates key herpesvirus promoters and productive infection.
The KLF4/Sp1 binding sites in the ICP4 enhancer (GGGCGGGGC) are identical and match a consensus KLF4 binding site plus a preferred nucleosome-depleted KLF4 binding site (45). ChIP studies indicated that GR occupied wt ICP4 enhancer sequences at higher levels than the pα4RΔKLF4 mutant (Fig. 5), which correlated with increased promoter activity of wt pα4R (Fig. 4B and C and Table 1). Conversely, KLF4 occupancy of the ΔKLF4/Sp1 binding site contained significantly higher KLF4 levels relative to wt ICP4 enhancer when transfected with GR and KLF4 but without DEX treatment (Fig. 6A), suggesting that KLF4 interactions with the mutant occurred but did not lead to a complex that stimulated promoter activity. This result implied that DEX treatment induced GR and KLF4 recruitment to wt ICP4 enhancer sequences, which correlated with transcriptional activation (Fig. 9A shows a schematic of these steps). It is also likely that additional transcriptional coactivators were directly or indirectly associated with GR or KLF4 (denoted by the X in Fig. 9A). Since the E-Box was not important for GR- and KLF4- or PLZF-mediated transactivation (Fig. 4), specific binding to the E-Box by GR, KLF4, PLZF, or other transcriptional coactivators does not appear to be important for transcriptional activation.
FIG 9.
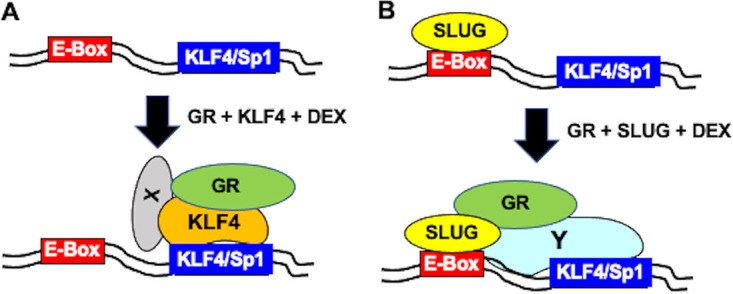
Model comparing potential steps that occur during cooperatively mediated transactivation of ICP4 enhancer by GR and KLF4 versus GR and SLUG. (A) Schematic describing how GR and KLF4 may cooperatively transactivate the ICP4 enhancer via a consensus KLF4/Sp1 binding site independent of an E-Box and in the absence of a consensus GRE. (B) Schematic describing how SLUG may cooperatively transactivate the ICP4 enhancer via a consensus E-Box and KLF4/Sp1 binding site, which lacks a consensus GRE. Additional details are presented in Discussion.
In contrast to KLF4, SLUG interactions with wt ICP4 enhancer sequences, but not the ΔKLF4/Sp1 mutant, occurred in the absence of GR and DEX. Cotransfection with GR and DEX treatment led to recruitment of GR to wt ICP4 enhancer sequences, but not to the ΔKLF4/Sp1 mutant, which correlates with efficient transactivation of wt pα4R but not pα4RΔKLF4 (Fig. 9B shows a schematic of these steps). We suggest that a transcriptional cofactor, perhaps Sp1, or a complex of transcriptional coactivators needs to interact with the KLF4/Sp1 binding site for GR+SLUG+DEX-mediated transactivation (denoted by Y in Fig. 9B). In general, there was a correlation between GR recruitment to wt ICP4 enhancer sequences, but not to the ΔKLF4/Sp1 mutant, when cotransfected with KLF4, PLZF, or SLUG. SLUG expression is stimulated in TG more than 10-fold at 90 min after DEX treatment of calves latently infected with BoHV-1 (33). Following explant of mouse TG, more neurons express the SLUG protein relative to controls (34). SLUG expression is also induced by UV light (59) and, like PLZF, can negatively or positively regulate transcription. Since KLF4, SLUG, and PLZF can repress transcription, occupancy of the ICP4 promoter by these transcription factors during latency may impair ICP4 promoter activity. Conversely, GR activation may stimulate ICP4 promoter activity via interactions with stress-induced transcription factors.
UV light, heat stress (fever), trauma, and increased corticosteroid levels are associated with increasing the incidence of reactivation from latency in humans (17, 19, 20, 60–63). These stimuli also increase the incidence of reactivation from latency in mouse models of infection and/or stimulate productive infection in cultured cells (21). Remarkably, all of these dissimilar reactivation stimuli activate GR. For example, GR phosphorylation is induced by UV light, which correlates with GR-mediated transcriptional activation and GR-mediated induction of certain enzymes (30, 64, 65). Cyanoketone, a glucocorticoid synthesis inhibitor, reduces the incidence of heat-stress-induced reactivation from latency in a mouse ocular model of HSV-1 infection (66). Furthermore, heat stress increases the frequency of reactivation from latency in cultured TG cells (67). KLF4 expression is also increased by heat stress (68) and DEX in bovine TG (33). GR activation (ligand dependent or independent) may not always be required for HSV-1 reactivation from latency; however, it may be more important than expected.
Transactivation of the ICP4 enhancer and ICP0 promoter (24) by GR and stress-induced transcription factors may not be the only impact that stressful stimuli have on viral gene expression and productive infection. For example, the ICP4 enhancer fragment that we examined in this study is within 1 to 2 kb of oriS and ICP22 or ICP47 proximal promoters, suggesting that GR and the respective stress-induced transcription factors can influence oriS and/or these promoters (Fig. 1A and B). Furthermore, an HSV-1 origin of replication (oriL) contains a functional GRE and viral replication is stimulated by DEX in neuronal cells (69). Point mutations in the oriL GRE impair acute infection and reactivation in mice (70). Third, GR can transactivate a subset of GREs that are more than 5 kb from the nearest promoter (71). Since the HSV-1 genome contains many consensus GREs (unpublished data), additional viral genes may be stimulated following a stressful stimulus. In addition to directly influencing viral gene expression and replication, the anti-inflammatory and immunosuppressive properties of corticosteroids (26, 72) are likely to promote virus spread following stressful stimuli. Finally, KLF4 interferes with antiviral responses by inhibiting ISRE (interferon-stimulated response element) and IRF3 binding to the beta-interferon (IFN-β) promoter (73). In summary, GR activation may have numerous effects (direct or indirect) on productive infection.
MATERIALS AND METHODS
Cells and virus.
Vero and Neuro-2A cells were grown in MEM supplemented with l-glutamine (2 mM), streptomycin (100 µg/ml), penicillin (10 U/ml), and 10% FBS. Where denoted, 2% charcoal-stripped FBS was used in place of 10% FBS.
Virus infection.
A virulent strain of HSV-1 (McKrae) was obtained from Steve Wechsler. Virus was cultured as previously described (21). Vero cells were washed with phosphate-buffered saline (PBS) and infected for ChIP studies at an MOI of 1 for 1 h, with periodic shaking. At the end of the infection period, cells were again washed with PBS, fresh medium containing 2% stripped FBS was added, and cells were cultured for the indicated times.
Primers.
Primers used for amplifying the ICP4 enhancer fragment in plasmid constructs included the ICP4 forward primer 5′-CGGAACGGAAGCGGAAAC-3′ (spanning −293 to −275 of the ICP4 enhancer). The reverse primer was within pGL4.24[luc2/minP] sequences: 5′-ACAGTACCGGATTGCCAAG-3′ (spanning nucleotides 127 to 109 of pGL4.24[luc2/minP]). These sequences are located between the minimal promoter and luciferase coding sequence. The two primers were used as a pair to amplify ChIP target DNA in transfected cells that resulted in a 274-bp PCR product (Fig. 5A).
To assess ICP4 occupancy during productive infection, a different reverse primer was used, AGGAGGAGCAGCGGAGGC, which spans nucleotides −63 to −46 of the ICP4 promoter. The ICP4 forward and reverse primers were used to amplify ChIP target DNA from HSV-1-infected cells and create a 247-bp PCR product (Fig. 8A).
Plasmids.
A plasmid containing the ICP4 promoter enhancer sequence (α4R) was obtained from Thomas Kristie (NIH) and was cloned into pGL3-Promoter plasmid (Promega) at SmaI-BglII sites, which are upstream of the minimal promoter that drives firefly luciferase expression. The ICP4 enhancer fragment was released from pGL3-Promoter using SmaI and BglII restriction enzymes. The released fragment was cloned into the unique KpnI and BglII sites of pGL4.24[luc2/minP] (Promega), which are upstream of the minimal promoter that drives firefly luciferase gene expression. The KLF4 binding site mutants contain an EcoRI restriction site (GAATCC) at the denoted sites (Δ5′ KLF4, Δ3′ KLF4, and ΔKLF4) as shown in Fig. 3A. These constructs were synthesized by GenScript and cloned into pGL4.24[luc2/minP] at SacI and HindIII sites of pGL4.24[luc2/minP]. The E-Box mutant was also synthesized by GenScript and contains a BamHI restriction site (GGATCC) in place of wt sequences (Fig. 4A). The E-Box mutant ICP4 fragment was inserted between the SacI and HindIII sites in pGL4.24[luc2/minP].
The mouse GR expression plasmid was obtained from Joseph Cidlowski, NIH. The human KLF4 expression vector was obtained from Jonathan Katz (University of Pennsylvania). The human KLF15 expression plasmid was obtained from Deborah Otteson (University of Houston). The mouse PLZF expression vector was obtained from Derek Sant’Angelo (Sloan-Kettering Cancer Center). The snail family transcriptional repressor 2 (SLUG) expression vector was obtained from Paul Wade (NIEHS, Research Triangle Park, NC).
Transfection and measurement of luciferase activity in transfected cells.
Neuro-2A and Vero cells were grown to ≥80% confluence in 60-mm plates and washed with PBS. Antibiotic-free MEM with 2% charcoal-stripped FBS was added to plates. Cells were transfected with either a plasmid expressing a firefly luciferase gene from pGL4.24[luc2/minP] (0.5 μg DNA) or the same plasmid with the indicated ICP4 enhancer construct (pα4R) cloned upstream of the minimal promoter using TransIT-X2 (Mirus, Madison, WI) transfection reagent, following the manufacturer’s guidelines.
Cells used for the dual-luciferase assay were also transfected with a plasmid expressing Renilla luciferase (0.05 μg DNA) from a minimal thymidine kinase (TK) promoter as a transfection control. Indicated samples were cotransfected with a GR expression plasmid (1 μg DNA) and/or an expression plasmid containing one of the stress-induced transcription factors (KLF4, KLF15, PLZF, or SLUG; 0.5 μg DNA). To maintain equivalent amounts of DNA for every sample, empty vector was added as needed. Following transfection, cells were incubated in medium containing 2% charcoal-stripped FBS. At 24 h following transfection, designated cultures were treated with water-soluble DEX (Sigma; D2915; 10 µM final concentration) or RU486 (Sigma; M8046; 10 µM final concentration). At 48 h following transfection, cells were washed with PBS, harvested, and stored at −80°C.
Transfected Neuro-2A and Vero cells for dual-luciferase assay were harvested using passive lysis buffer. Luciferase activity in each cell lysate was measured using a commercially available kit (Promega; E1910) and a GloMax 20/20 luminometer (Promega E5331). Promoter activity was measured as firefly luciferase activity compared to Renilla luciferase activity, the transfection control. Firefly luciferase activity from pGL4.24[luc2/minP] was used as the baseline promoter activity, and enhancer activity by the various ICP4 constructs was denoted as fold activation over baseline levels. Transactivation by the indicated transcription factors was also measured as fold activity over basal promoter or fold activity over the pα4R construct transfected alone.
Chromatin immunoprecipitation.
Neuro-2A cells for ChIP were cultured in 100-mm plates with antibiotic-free medium containing 2% charcoal-stripped FBS and transfected with the pGL4.24[luc2/minP] plasmid containing the pα4R construct (1.5 μg DNA). Indicated samples were cotransfected with expression plasmids expressing GR (3 μg DNA) and/or a stress-induced transcription factor (KLF4, KLF15, PLZF, or SLUG; 0.5 μg DNA). At 24 h after transfection, designated samples were treated with DEX (10 μM). At 48 h posttransfection, Neuro-2A cells were formaldehyde cross-linked and harvested in PBS. Chromatin immunoprecipitation was performed as previously described (24, 37, 41) using 5 µg of GR (Cell Signaling; 3660S), KLF15 (Abcam; ab2647), KLF4 (Abcam; ab106629), PLZF (Active Motif; 39987), or SLUG (Santa Cruz; sc166476) antibody. Five micrograms of IgG served as an isotype control. PCR was performed using ICP4 forward and pGL4.24[luc2/minP] reverse primers, generating a 274-bp product, and run on a 1% agarose gel with ethidium bromide for visualization. Individual bands were quantified using Image Lab software, with isotype and antibody samples presented as a percentage of input, representing approximately 13% of input cell lysate for each sample.
ACKNOWLEDGMENTS
We thank Thomas Kristie and Jodi Vogel for ICP4 promoter and enhancer constructs as well as their insightful comments regarding these studies.
This research was supported by a grant from the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number R01NS111167, by the Oklahoma Center for Respiratory and Infectious Diseases (National Institutes of Health Centers for Biomedical Research Excellence grant number P20GM103648), and by funds from the Sitlington Endowment. J. Ostler was partially supported by a postdoctoral fellowship from the USDA NIFA (2019-07214).
REFERENCES
- 1.Shimeld C, Efstathiou S, Hill T. 2001. Tracking the spread of a lacZ-tagged herpes simplex virus type 1 between the eye and the nervous system of the mouse: comparison of primary and recurrent infection. J Virol 75:5252–5262. doi: 10.1128/JVI.75.11.5252-5262.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Phelan D, Barrozo ER, Bloom DC. 2017. HSV1 latent transcription and non-coding RNA: a critical retrospective. J Neuroimmunol 308:65–101. doi: 10.1016/j.jneuroim.2017.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Liesegang TJ. 1999. Herpes simplex virus epidemiology and ocular importance. Cornea 18:127–143. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Yamada S, Kameyama T, Nagaya S, Hashizume Y, Yoshida M. 2003. Relapsing herpes simplex encephalitis: pathological confirmation of viral reactivation. J Neurol Neurosurg Psychiatry 74:262–264. doi: 10.1136/jnnp.74.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roizmann B, Knipe DM. 2001. Herpes simplex viruses and their replication, p 2399–2459. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, vol 4. Lippincott-Williams and Wilkins, Philadelphia, PA. [Google Scholar]
- 6.DeLuca NA, McCarthy AM, Schaffer PA. 1985. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J Virol 56:558–570. doi: 10.1128/JVI.56.2.558-570.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.DeLuca NA. 2011. Functions and mechanism of action of the herpes simplex virus regulatory protein, ICP4, p 17–38. In Weller SK (ed), Alphaherpesviruses: molecular virology. Caister Academic Press, Poole, United Kingdom. [Google Scholar]
- 8.Kristie TM, Roizman B. 1986. Alpha 4, the major regulatory protein of herpes simplex virus type 1, is stably and specifically associated with promoter-regulatory domains of alpha genes and of selected other viral genes. Proc Natl Acad Sci U S A 83:3218–3222. doi: 10.1073/pnas.83.10.3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smith CA, Bates P, Rivera-Gonzalez R, Gu B, DeLuca NA. 1993. ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J Virol 67:4676–4687. doi: 10.1128/JVI.67.8.4676-4687.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibeault R, Conn KL, Bildersheim MD, Schang LM. 2016. An essential viral transcription activator modulates chromatin dynamics. PLoS Pathog 12:e1005842. doi: 10.1371/journal.ppat.1005842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boutell C, Everett RD. 2013. Regulation of alphaherpesvirus infections by the ICP0 family of proteins. J Gen Virol 94:465–481. doi: 10.1099/vir.0.048900-0. [DOI] [PubMed] [Google Scholar]
- 12.Everett RD. 2000. ICP0, a regulator of herpes simplex virus during lytic and latent infection. Bioessays 22:761–770. doi:. [DOI] [PubMed] [Google Scholar]
- 13.Kristie TM. 2007. Early pre-initiation of alphaherpesvirus viral gene expression, p 112–127. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Whitley R, Yamanishi K (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis, vol 1. Cambridge University Press, Cambridge, United Kingdom. [PubMed] [Google Scholar]
- 14.Kristie TM. 2015. Dynamic modulation of HSV chromatin drives initiation of infection and provides targets for epigenetic therapies. Virology 479-480:555–561. doi: 10.1016/j.virol.2015.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bloom DC, Giordani NV, Kwiatkowski DL. 2010. Epigenetic regulation of latent HSV-1 gene expression. Biochim Biophys Acta 1799:246–256. doi: 10.1016/j.bbagrm.2009.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Knipe DM, Cliffe A. 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Microbiol 6:211–221. doi: 10.1038/nrmicro1794. [DOI] [PubMed] [Google Scholar]
- 17.Perng G-C, Jones C. 2010. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis 2010:262415–262418. doi: 10.1155/2010/262415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Glaser R, Kiecolt-Glaser JK. 2005. Stress-induced immune dysfunction: implications for health. Nat Rev Immunol 5:243–251. doi: 10.1038/nri1571. [DOI] [PubMed] [Google Scholar]
- 19.Padgett DA, Sherida J, Dorne FJ, Berntson GG, Candelora J, Glaser R. 1998. Social stress and the reactivation of latent herpes simplex virus type 1. Proc Natl Acad Sci U S A 95:7231–7235. doi: 10.1073/pnas.95.12.7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rooney JT, Straus SE, Mannix ML, Wohlenberg CR, Banks S, Jagannath S, Brauer JE, Notkins AL. 1992. UV light-induced reactivation of herpes simplex virus type 2 and prevention by acyclovir. J Infect Dis 166:500–506. doi: 10.1093/infdis/166.3.500. [DOI] [PubMed] [Google Scholar]
- 21.Harrison K, Zhu L, Thunuguntla P, Jones C. 2019. Antagonizing the glucocorticoid receptor impairs explant-induced reactivation in mice latently infected with herpes simplex virus 1. J Virol 93:e00418-19. doi: 10.1128/JVI.00418-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Du T, Zhou G, Roizman B. 2012. Induction of apoptosis accelerates reactivation from latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc Natl Acad Sci U S A 109:14616–14621. doi: 10.1073/pnas.1212661109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erlandsson AC, Bladh L-G, Stierna P, Yucel-Lindberg T, Hammarsten O, Modéer T, Harmenberg J, Wikström A-C. 2002. Herpes simplex virus type 1 infection and glucocorticoid treatment regulate viral yield, glucocorticoid receptor and NF-kB levels. J Endocrinol 175:165–176. doi: 10.1677/joe.0.1750165. [DOI] [PubMed] [Google Scholar]
- 24.Ostler J, Harrison KS, Schroeder K, Thunuguntla P, Jones C. 2019. The glucocorticoid receptor (GR) stimulates herpes simplex virus 1 productive infection, in part because the infected cell protein 0 (ICP0) promoter is cooperatively transactivated by the GR and Krüppel-like transcription factor 15. J Virol 93:e02063-18. doi: 10.1128/JVI.02063-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oakley RH, Cidlowski JA. 2013. The biology of the glucocorticoid receptor: new signaling mechanisms in health and disease. J Allergy Clin Immunol 132:1033–1044. doi: 10.1016/j.jaci.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smoak KL, Cidlowski JA. 2004. Mechanisms of glucocorticoid receptor signaling during inflammation. Mech Ageing Dev 125:697–706. doi: 10.1016/j.mad.2004.06.010. [DOI] [PubMed] [Google Scholar]
- 27.Wang J-C, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR. 2004. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci U S A 101:15603–15608. doi: 10.1073/pnas.0407008101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giguere V, Hollenberg SM, Rosenfeld MG, Evans RM. 1986. Functional domains of the human glucocorticoid receptor. Cell 46:645–652. doi: 10.1016/0092-8674(86)90339-9. [DOI] [PubMed] [Google Scholar]
- 29.Hapgood J, Avenant C, Moliki JM. 2016. Glucocorticoid-independent modulation of GR activity: implications for immunotherapy. Pharmacol Ther 165:93–113. doi: 10.1016/j.pharmthera.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galliher-Beckley A, Williams JG, Cidlowski JA. 2011. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol Cell Biol 31:4663–4675. doi: 10.1128/MCB.05866-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ritter H, Antonova L, Mueller CR. 2012. The unliganded glucocorticoid receptor positively regulates the tumor suppressor gene BRCA1 through GABP beta. Mol Cancer Res 10:558–569. doi: 10.1158/1541-7786.MCR-11-0423-T. [DOI] [PubMed] [Google Scholar]
- 32.DeLeón M, Coveñas R, Chadi G, Narváez JA, Fuxe K, Cintra A. 1994. Subpopulations of primary sensory neurons show coexistence of neuropeptides and glucocorticoid receptors in the rat spinal and trigeminal ganglia. Brain Res 636:338–342. doi: 10.1016/0006-8993(94)91034-0. [DOI] [PubMed] [Google Scholar]
- 33.Workman A, Eudy J, Smith L, Frizzo da Silva L, Sinani D, Bricker H, Cook E, Doster A, Jones C. 2012. Cellular transcription factors induced in trigeminal ganglia during dexamethasone-induced reactivation from latency stimulate bovine herpesvirus 1 productive infection and certain viral promoters. J Virol 86:2459–2473. doi: 10.1128/JVI.06143-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinani D, Cordes E, Workman A, Thunuguntia P, Jones C. 2013. Stress induced cellular transcription factors expressed in trigeminal ganglionic neurons stimulate the herpes simplex virus type 1 (HSV-1) infected cell protein 0 (ICP0) promoter. J Virol 87:1183–1192. doi: 10.1128/JVI.02783-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Black AR, Black JD, Azizkhan-Clifford J. 2001. Sp1 and Kruppel-like transcription factor family of transcription factors in cell growth and cancer. J Cell Physiol 188:143–160. doi: 10.1002/jcp.1111. [DOI] [PubMed] [Google Scholar]
- 36.Kaczynski J, Cook T, Urrutia R. 2003. Sp1- and Kruppel-like transcription factors. Genome Biol 4:206–208. doi: 10.1186/gb-2003-4-2-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.El-Mayet FS, Sawant L, Thunuguntla P, Jones C. 2017. Combinatorial effects of the glucocorticoid receptor and Krüppel-like transcription factor 15 on bovine herpesvirus 1 transcription and productive infection. J Virol 91:e00904-17. doi: 10.1128/JVI.00904-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mangan S, Alon U. 2003. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A 100:11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sasse S, Zuo Z, Kadiyala V, Zhang L, Pufall MA, Jain MK, Phang TL, Stormo GD, Gerber AN. 2015. Response element composition governs correlations between binding site affinity and transcription in glucocorticoid receptor feed-forward loops. J Biol Chem 290:19756–19769. doi: 10.1074/jbc.M115.668558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sasse SK, Mailloux CM, Barczak AJ, Wang Q, Altonsy MO, Jain MK, Haldar SM, Gerber AN. 2013. The glucocorticoid receptor and KLF15 regulate gene expression dynamics and integrate signals through feed-forward circuitry. Mol Cell Biol 33:2104–2115. doi: 10.1128/MCB.01474-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.El-Mayet F, Sawant L, Thunuguntla P, Zhao J, Jones C. 2019. Two pioneer transcription factors, Krüppel-like transcription factor 4 and glucocorticoid receptor, cooperatively transactivate the bovine herpesvirus 1 ICP0 early promoter and stimulate productive infection. J Virol 94:e01670-19. doi: 10.1128/JVI.01670-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halford WP, Kemp CD, Isler JA, Davido DJ, Schaffer PA. 2001. ICP0, ICP4, or VP16 expressed from adenovirus vectors induces reactivation of latent herpes simplex virus type 1 in primary cultures of latently infected trigeminal ganglion cells. J Virol 75:6143–6153. doi: 10.1128/JVI.75.13.6143-6153.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bardon S, Vignon F, Chalbos D, Rochefort H. 1985. RU486, a progestin and glucocorticoid antagonist, inhibits the growth of breast cancer cells via the progesterone receptor. J Clin Endocrinol Metab 60:692–697. doi: 10.1210/jcem-60-4-692. [DOI] [PubMed] [Google Scholar]
- 44.Pandit S, Geissler W, Harris G, Sitlani A. 2002. Allosteric effects of dexamethasone and RU486 on glucocorticoid receptor-DNA interactions. J Biol Chem 277:1538–1543. doi: 10.1074/jbc.M105438200. [DOI] [PubMed] [Google Scholar]
- 45.Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, Zaret KS. 2015. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161:555–568. doi: 10.1016/j.cell.2015.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.David G, Alland L, Hong SH, Wong CW, DePinho RA, Dejean A. 1998. Histone deacetylase associated with mSin3A mediates repression by the acute promyelocytic leukemia-associated PLZF protein. Oncogene 16:2549–2556. doi: 10.1038/sj.onc.1202043. [DOI] [PubMed] [Google Scholar]
- 47.Hong SH, David G, Wong CW, Dejean A, Privalsky ML. 1997. SMRT corepressor interacts with PLZF and with the PML-retinoic acid receptor alpha (RARalpha) and PLZF-RARalpha oncoproteins associated with acute promyelocytic leukemia. Proc Natl Acad Sci U S A 94:9028–9033. doi: 10.1073/pnas.94.17.9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Boukarabila H, Saurin AJ, Batsche E, Mossadegh N, van Lohuizen M, Otte AP, Pradel J, Muchardt C, Sieweke M, Duprez E. 2009. The PRC1 Polycomb group complex interacts with PLZF/RARA to mediate leukemic transformation. Genes Dev 23:1195–1206. doi: 10.1101/gad.512009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koubi M, Poplineau M, Vernerey J, N’Guyen L, Tiberi G, Garciaz S, El-Kaoutari A, Maqbool MA, Andrau J-C, Guillouf C, Saurin AJ, Duprez E. 2018. Regulation of the positive transcriptional effect of PLZF through a non-canonical EZH2 activity. Nucleic Acids Res 46:3339–3350. doi: 10.1093/nar/gky080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hemavathy K, Ashraf SI, Ip IT. 2000. Snail/slug family of repressors: slowly going into the fast lane of development and cancer. Gene 257:1–12. doi: 10.1016/S0378-1119(00)00371-1. [DOI] [PubMed] [Google Scholar]
- 51.Wels C, Joshi S, Koefinger P, Bergler H, Schaider H. 2011. Transcriptional activation of ZEB1 by Slug leads to cooperative regulation of the EMT like phenotype in melanoma. J Invest Dermatol 131:1877–1885. doi: 10.1038/jid.2011.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.John S, Sabo PJ, Thurman RE, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. 2011. Chromatin accessibility pre-determined glucocorticoid receptor binding patterns. Nat Genet 43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perlman T. 1992. Glucocorticoid receptor DNA-binding specificity is increased by the organization of DNA in nucleosomes. Proc Natl Acad Sci U S A 89:3884–3888. doi: 10.1073/pnas.89.9.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ghaleb A, Yang VW. 2017. Krüppel-like factor 4 (KLF4): what we currently know. Gene 611:27–37. doi: 10.1016/j.gene.2017.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mayran A, Drouin J. 2018. Pioneer transcription factors shape the epigenetic landscape. J Biol Chem 293:13795–13804. doi: 10.1074/jbc.R117.001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Iwafuchi-Doi M, Zaret KS. 2014. Pioneer transcription factors in cell reprogramming. Genes Dev 28:2679–2692. doi: 10.1101/gad.253443.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zaret KS, Carroll JS. 2011. Pioneer transcription factors: establishing competence for gene expression. Genes Dev 25:2227–2241. doi: 10.1101/gad.176826.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nawandar DM, Wang A, Makielski K, Lee D, Ma S, Barlow E, Reusch J, Jiang R, Wille CK, Greenspan D, Greenspan JS, Mertz JE, Hutt-Fletcher L, Johannsen EC, Lambert PF, Kenney SC. 2015. Differentiation-dependent KLF4 expression promotes lytic Epstein-Barr virus infection in epithelial cells. PLoS Pathog 11:e1005195. doi: 10.1371/journal.ppat.1005195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hudson LG, Choi C, Newkirk KM, Parkhani J, Cooper KL, Lu P, Kusewitt DJ. 2007. Ultraviolet radiation stimulates expression of Snail family transcription factors in keratinocytes. Mol Carcinog 46:257–268. doi: 10.1002/mc.20257. [DOI] [PubMed] [Google Scholar]
- 60.Cassidy L, Meadows J, Catalan J, Barton S. 1997. Are stress and coping style associated with frequent recurrence of genital herpes? Genitourin Med 73:263–266. doi: 10.1136/sti.73.4.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glaser R, Kiecolt-Glaser JK, Speicher CE, Holliday JE. 1985. Stress, loneliness, and changes in herpesvirus latency. J Behav Med 8:249–260. doi: 10.1007/BF00870312. [DOI] [PubMed] [Google Scholar]
- 62.Jones C. 1998. Alphaherpesvirus latency: its role in disease and survival of the virus in nature. Adv Virus Res 51:81–133. doi: 10.1016/s0065-3527(08)60784-8. [DOI] [PubMed] [Google Scholar]
- 63.Jones C. 2003. Herpes simplex virus type 1 and bovine herpesvirus 1 latency. Clin Microbiol Rev 16:79–95. doi: 10.1128/cmr.16.1.79-95.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Davies L, Karthikeyan N, Lynch JT, Sial E-A, Gkourtsa A, Demonacos C, Krstic-Demonacos M. 2008. Cross talk of signaling pathways in the regulation of the glucocorticoid receptor function. Mol Endocrinol 22:1331–1344. doi: 10.1210/me.2007-0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Skobowiat C, Sayre RM, Dowdy JC, Slominski AT. 2013. Ultraviolet radiation regulates cortisol activity in a waveband dependent manner in human skin ex-vivo. Br J Dermatol 168:595–601. doi: 10.1111/bjd.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Noisakran S, Halford WP, Veress L, Carr DJJ. 1998. Role of the hypothalamic pituitary adrenal axis and IL-6 in stress-induced reactivation of latent herpes simplex virus type 1. J Immunol 160:5441–5447. [PubMed] [Google Scholar]
- 67.Halford WP, Gebhardt BM, Carr DJJ. 1996. Mechanisms of herpes simplex virus type 1 reactivation from latency. J Virol 70:5051–5060. doi: 10.1128/JVI.70.8.5051-5060.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu Y, Wang J, Yi Y, Zhang H, Liu J, Liu M, Yuan C, Tang D, Benjamin IJ, Xiao X. 2006. Induction of KLF4 in response to heat stress. Cell Stress Chaperones 11:379–389. doi: 10.1379/csc-210.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hardwicke MA, Schaffer PA. 1997. Differential effects of nerve growth factor and dexamethasone on herpes simplex virus type 1 oriL- and oriS-dependent DNA replication in PC12 cells. J Virol 71:3580–3587. doi: 10.1128/JVI.71.5.3580-3587.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Balliet JW, Schaffer PA. 2006. Point mutations in herpes simplex virus type 1 oriL, but not in oriS, reduce pathogenesis during acute infection of mice and impair reactivation from latency. J Virol 80:440–450. doi: 10.1128/JVI.80.1.440-450.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Polman JAE, Welten JE, Bosch DS, de Jonge RT, Balog J, van der Maarel SM, de Kloet ER, Datson NA. 2012. A genome-wide signature of glucocorticoid receptor binding in neuronal PC12 cells. BMC Neurosci 13:118–125. doi: 10.1186/1471-2202-13-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rhen T, Cidlowski JA. 2005. Antiinflammatory action of glucocorticoids—new mechanisms of old drugs. N Engl J Med 353:1711–1723. doi: 10.1056/NEJMra050541. [DOI] [PubMed] [Google Scholar]
- 73.Wei-Wei L, Lian H, Zhong B, Shu H-B, Li S. 2016. Kruppel-like factor 4 negatively regulates cellular antiviral immune response. Cell Mol Immunol 13:65–72. doi: 10.1038/cmi.2014.125. [DOI] [PMC free article] [PubMed] [Google Scholar]