

The ongoing pandemic of coronavirus disease 2019 (COVID-19), caused by a newly emerged zoonotic coronavirus (severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2]), highlights the importance of coronaviruses as human and animal pathogens and our knowledge gaps in understanding the cellular mechanisms, especially the mechanisms shared among human and animal coronaviruses, exploited by coronaviruses for optimal replication and enhanced pathogenicity. This study reveals that the upregulation of cFOS, along with other AP-1 transcription factors, as a cell survival strategy is such a mechanism utilized by coronaviruses during their replication cycles.
KEYWORDS: apoptosis, coronavirus, immediate early gene, infectious bronchitis virus, stress response, viral replication
ABSTRACT
Coronaviruses have evolved a variety of strategies to optimize the cellular microenvironment for efficient replication. In this study, we report the induction of AP-1 transcription factors by coronavirus infection based on genome-wide analyses of differentially expressed genes in cells infected with the avian coronavirus infectious bronchitis virus (IBV). Most members of the AP-1 transcription factors were subsequently found to be upregulated during the course of IBV and porcine epidemic diarrhea virus (PEDV) infection of cultured cells as well as in IBV-infected chicken embryos. Further characterization of the induction kinetics and functional roles of cFOS in IBV replication demonstrated that the upregulation of cFOS at early to intermediate phases of IBV replication cycles suppresses IBV-induced apoptosis and promotes viral replication. Blockage of the nuclear translocation of cFOS by the peptide inhibitor nuclear localization signal peptide (NLSP) suppressed IBV replication and apoptosis, ruling out the involvement of the cytoplasmic functions of cFOS in the replication of IBV. Furthermore, knockdown of extracellular signal-regulated kinase 1/2 (ERK1/2) and inhibition of Jun N-terminal protein kinase (JNK) and p38 kinase activities reduced cFOS upregulation and IBV replication. This study reveals an important function of cFOS in the regulation of coronavirus-induced apoptosis, facilitating viral replication.
IMPORTANCE The ongoing pandemic of coronavirus disease 2019 (COVID-19), caused by a newly emerged zoonotic coronavirus (severe acute respiratory syndrome coronavirus 2 [SARS-CoV-2]), highlights the importance of coronaviruses as human and animal pathogens and our knowledge gaps in understanding the cellular mechanisms, especially the mechanisms shared among human and animal coronaviruses, exploited by coronaviruses for optimal replication and enhanced pathogenicity. This study reveals that the upregulation of cFOS, along with other AP-1 transcription factors, as a cell survival strategy is such a mechanism utilized by coronaviruses during their replication cycles. Through the induction and regulation of apoptosis of infected cells at early to intermediate phases of the replication cycles, subtle but appreciable differences in coronavirus replication efficiency were observed when the expression levels of cFOS were manipulated in the infected cells. As the AP-1 transcription factors are multifunctional, further studies of their regulatory roles in proinflammatory responses may provide new insights into the pathogenesis and virus-host interactions during coronavirus infection.
INTRODUCTION
Coronaviruses are important pathogens of humans and animals. In the past 17 years, we have witnessed the emergence of three highly pathogenic coronaviruses of animal origins. These include severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002 to 2003, Middle East respiratory coronavirus (MERS-CoV) in 2012, and SARS-CoV-2, the novel coronavirus that causes the current pandemic, in 2019 to 2020 (coronavirus disease 2019 [COVID-19]) (1–3). Coronaviruses belong to the Coronaviridae family and are classified into four genera, alpha-, beta-, gamma-, and deltacoronaviruses (4). Viruses in this family are enveloped and contain a single-stranded, positive-sense, nonsegmented RNA genome (5). This RNA genome encodes four structural proteins, i.e., spike (S), membrane (M), nucleocapsid (N), and small envelope (E); 15 to 16 nonstructural proteins (nsp’s); as well as a number of accessory proteins (6). The avian coronavirus infectious bronchitis virus (IBV) is a gammacoronavirus that causes an acute, highly contagious infection affecting all ages and types of chickens. IBV infection has caused great losses to the poultry industry worldwide (4, 7, 8). A cell culture-adapted IBV (p65) is used as the main model system in this study. Porcine epidemic diarrhea virus (PEDV), an alphacoronavirus, was first reported in 1978 (9). PEDV causes highly contagious acute enteritis and fatal watery diarrhea in piglets. Clinical characteristics are mainly manifested as vomiting, severe diarrhea, and dehydration of piglets, resulting in huge economic losses to the swine industry (10, 11).
Although different coronaviruses may use unique mechanisms to enter and replicate in specific tissues and cells, they have common strategies to either antagonize or agonize the physiological functions of normal cellular signaling pathways and processes in order to successfully and efficiently complete their infection cycles. Among many cellular pathways and factors, the multifunctional activator protein 1 (AP-1) transcription factors were found to be regulated by coronavirus infection. For example, it was reported that SARS-CoV infection led to the activation of AP-1 in human bronchial epithelial cells (12). The overexpression of SARS-CoV N protein can activate the AP-1 pathway (13). SARS-CoV accessory protein 3b may also induce AP-1 transcriptional activity through the activation of the Jun N-terminal protein kinase (JNK) and extracellular signal-regulated kinase (ERK) pathways (14).
AP-1 transcription factors, composed mainly of JUN and FOS family members, are involved in many cellular functions, including cell proliferation, differentiation, survival, angiogenesis, hematopoiesis, apoptosis, embryo development, inflammation, cancer, and other pathological processes (15, 16). cJUN and cFOS bind in a sequence-specific manner on promoter regions of target genes and form a variety of homo- and heterodimers on a common AP-1 site (14, 17). The Jun protein family includes cJUN, JUNB, and JUND, and the FOS family includes cFOS, FOSB, Fra-1, and Fra-2 (18). AP-1 can be induced by a variety of stimuli such as hormones, growth factors, cytokines, oxygen stress, UV radiation, and overexpressed oncogenes or other forms of cellular stress (19). Activated AP-1 regulates the expression of multiple proinflammatory factors such as interleukin 6 (IL-6), IL-8, and monocyte chemotactic protein 1 (MCP-1) (20). Two prominent functional domains, the basic domain (BD) spanning amino acids 139 to 160 and the leucine zipper domain (LZ) from amino acids 165 to 211, have been characterized in the 380-amino-acid cFOS protein. It was recently reported that cFOS may function as a transcription factor in the nucleus and as an activation factor for lipid synthesis in the cytoplasm (21, 22). BD is the functional domain essential for both the AP-1/DNA-binding activity in the nucleus and phospholipid synthesis activation in the cytoplasm. LZ is a leucine repeat region that allows the heterodimerization of proteins, including other AP-1 transcription factors (23).
In this report, we aim to study the induction of AP-1 transcription factors by IBV and PEDV infections and their regulatory roles in coronavirus replication based on genome-wide analyses of differentially expressed genes in infected cells. We report that most members of the AP-1 transcription factors are upregulated during the course of IBV and PEDV infection of cultured cells as well as in IBV-infected chicken embryos. Characterization of the induction kinetics and functional roles of cFOS in IBV replication reveals that the upregulation of cFOS at early to intermediate phases of the viral replication cycle promotes viral replication by the suppression of apoptosis.
RESULTS
IBV infection of cells upregulates the AP-1 transcription factors as revealed by genome-wide gene expression profiling and RT-qPCR verification.
To gain a general profile of host genes differentially regulated by IBV infection, Vero and H1299 cells were infected with IBV and analyzed by Affymetrix array and transcriptomic analyses, respectively. Among hundreds of differentially upregulated genes, AP-1 transcription factors, including cJUN, JUNB, JUND, cFOS, FOSB, FOSL1, and FOSL2, were found to be upregulated in both cell lines by the two assays. As summarized in Table 1, cJUN and cFOS are the two highly upregulated genes consistently detected in the two infected cell lines by the two assays. To verify these results, IBV-infected H1299 and Vero cells were harvested at 20 h postinfection (hpi) and subjected to reverse transcription-quantitative PCR (RT-qPCR) analysis, confirming that a similar trend of upregulation of these genes at the mRNA level was detected in the infected cells (Table 1).
TABLE 1.
Affymetrix array and transcriptomic analyses of AP-1 transcription factors in Vero and H1299 cells infected with IBVa
| Gene | Fold upregulation by Affymetrix (Vero) | Fold upregulation by mRNA-Seq (H1299) | Mean fold upregulation by RT-qPCR (n = 3) ± SD |
|
|---|---|---|---|---|
| Vero | H1299 | |||
| cJUN | 7.5 | 39.7 | 9.6 ± 1.5 | 99.8 ± 13.6 |
| JUNB | 2.6 | 10.1 | 2.9 ± 0.2 | 33.6 ± 4.9 |
| JUND | 1.3 | 5.9 | 1.2 ± 0.1 | 16.2 ± 1.9 |
| cFOS | 16.1 | 483.3 | 141.7 ± 6.3 | 948.9 ± 63.4 |
| FOSB | ND | 667.3 | 52.7 ± 7.3 | 1,089.9 ± 187.5 |
| FOSL1 | 4.2 | 1.6 | ND | ND |
| FOSL2 | 1.2 | 4.8 | ND | ND |
ND, not determined; mRNA-Seq, mRNA sequencing.
To ensure the reproducibility of these data, in this and all the following studies, all experiments were repeated at least three times, and results from one representative experiment are presented.
Induction kinetics of AP-1 transcription factors by IBV and PEDV infection.
The induction kinetics of these AP-1 transcription factors during the process of IBV infection were then studied with detailed time course experiments in both H1299 and Vero cells. For this purpose, IBV-infected H1299 and Vero cells were harvested at 0, 4, 8, 12, 16, 20, and 24 hpi; total RNA was extracted; and the expressions of cJUN, JUNB, JUND, cFOS, and FOSB at the mRNA level as well as IBV subgenomic RNA2 (sgRNA2) were analyzed by RT-qPCR. The results showed that all five genes were highly upregulated in IBV-infected H1299 cells, reaching their respective peaks at 16 hpi (Fig. 1a). Upregulation of these genes was also observed in IBV-infected Vero cells although at much-reduced levels and with later peaks. As shown in Fig. 1a, the induction peaks of most of the five genes in IBV-infected Vero cells were at or after 24 hpi, the end time point of the experiments.
FIG 1.

Upregulation of AP-1 transcription factors during IBV and PEDV infections. (a) Upregulation of AP-1 transcription factors in IBV- and PEDV-infected H1299 and Vero cells. H1299 and Vero cells were infected with IBV and PEDV at an MOI of approximately 2. Cell lysates were harvested at the indicated time points for RNA extraction. Equal amounts of total RNA were reverse transcribed. The levels of IBV genomic RNA (gRNA) and PEDV gRNA and the mRNA expression levels of AP-1 transcription factors (cJUN/JUNB/JUND/cFOS/FOSB) were determined by quantitative PCR. (b) Ten-day-old SPF chicken embryos were inoculated with 200 μl IBV (500 PFU). At 60 hpi, chicken embryo viscera were collected. Total RNAs were extracted, and the AP-1 transcription factors (cJUN/JUND/cFOS) at the mRNA level as well as IBV gRNA were analyzed by RT-qPCR. Shown are the results of three repeated experiments, as indicated. Significance levels are presented by the P value (***, P < 0.001). (c) H1299 cells were infected with IBV at an MOI of approximately 2 or mock treated with UV-inactivated IBV. Cell lysates were harvested at the indicated time points and subjected to Western blot analysis using cFOS and IBV N antibodies. Beta-actin was included as the loading control. Sizes of protein ladders in kilodaltons are indicated on the left. Significance levels are presented by the P value (**, P < 0.01; ****, P < 0.0001). (d) H1299 cells were infected with PEDV at an MOI of approximately 2 or mock treated with UV-inactivated PEDV. Cell lysates were harvested at the indicated time points and subjected to Western blot analysis using cFOS antibodies and PEDV-positive serum. Beta-actin was included as the loading control. Sizes of protein ladders in kilodaltons are indicated on the left. Significance levels are presented by the P value (***, P < 0.001; ****, P < 0.0001).
To determine if the upregulation of the AP-1 transcription factors was a general phenomenon in coronavirus-infected cells, PEDV-infected H1299 and Vero cells were harvested at 0, 8, 16, 24, 32, and 40 hpi. Total RNAs were extracted, and the mRNA levels of cJUN, JUNB, JUND, cFOS, and FOSB as well as PEDV genomic RNA (gRNA) were analyzed by RT-qPCR, showing upregulation trends similar to the ones observed for IBV infection (Fig. 1a). The five AP-1 transcription factors were significantly upregulated in PEDV-infected H1299 cells, with the induction peaks at or after 32 hpi (Fig. 1a). Upregulation of the five genes was also observed in PEDV-infected Vero cells; their mRNA levels were gradually increased through the course of infection and reached induction peaks at 40 hpi, the end time point of the time course experiments (Fig. 1a).
Next, we investigated the expression of these AP-1 transcription factors in IBV-infected chicken embryos. Ten-day-old specific-pathogen-free (SPF) chicken embryos were inoculated with 200 μl of IBV (500 PFU). At 60 hpi, embryo viscera were collected, and total RNA was extracted. The expressions of cJUN, JUND, and cFOS at the mRNA level as well as IBV gRNA were analyzed by RT-qPCR. As shown in Fig. 1b, the mRNA level of cFOS was approximately 30-fold upregulated in the infected group, and the expression levels of cJUN and JUND were >5-fold increased, confirming that the AP-1 transcription factors were highly upregulated in IBV-infected chicken embryos.
Western blot analysis of the induction kinetics of cFOS at the protein level was then conducted in IBV-infected H1299 cells. As shown in Fig. 1c, a drastic induction of cFOS was observed at 12 and 16 hpi, with a peak at 12 hpi, the onset time point for the largest amount of IBV N protein synthesis. No expression of cFOS and IBV N protein was detected in H1299 cells treated with UV-inactivated IBV (UV-IBV), confirming that the induction of cFOS in IBV-infected H1299 cells is coupled with active viral replication (Fig. 1c).
To validate the expression of cFOS protein in PEDV-infected H1299 cells, H1299 cells were infected with PEDV and harvested at 0, 8, 16, 24, 32, and 40 hpi. The results showed that the expression of cFOS protein was induced at 24, 32, and 40 hpi, along with an increase of the PEDV N protein level at these time points, demonstrating that PEDV infection indeed induces cFOS protein expression in infected cells (Fig. 1d).
Inhibition of cFOS suppresses IBV replication in H1299 cells.
We first used T-5224, an effective cFOS/AP-1 inhibitor, to explore the effect of cFOS inhibition on IBV replication. H1299 cells were infected with IBV for 2 h before being treated with increasing concentrations of T-5224 for 20 h. The effects of different concentrations of T-5224 on cell proliferation are shown in Fig. 2a, revealing no significant difference between 6.25 μM, 12.5 μM, and 25 μM T-5224 in the cell proliferation rates compared with the dimethyl sulfoxide (DMSO) control. Significant differences in the cell proliferation rates were observed at concentrations of between 25 μM and 50 μM (Fig. 2a).
FIG 2.

Suppression of IBV replication by T-5224 in H1299 cells. (a) H1299 cells were plated on a 96-well plate and cultured for 16 to 20 h. Cells were treated with T-5224 at the indicated concentrations or the same volume of DMSO for 24 h. Cell viability was determined by measuring the absorbance value at 450 nm after adding the CCK solution for 1 to 4 h. Significance levels are presented by the P value (ns, nonsignificant; **, P < 0.01). (b) H1299 cells were infected with IBV. After 2 h, cells were treated with T-5224 at the indicated concentrations or the same volume of DMSO for 20 h. Cell lysates were harvested by three freeze-thaw cycles. Virus titers are expressed in units of log TCID50 per milliliter. Significance levels are presented by the P value (**, P < 0.01). (c) Virus titers in the culture supernatants expressed in units of log TCID50 per milliliter. The experiment was repeated three times, and the results of one representative experiment are shown. Significance levels are presented by the P value (*, P < 0.05; **, P < 0.01). (d) H1299 cells were infected and treated with T-5224 as described above for panel b, and cell lysates and supernatants were also subjected to Western blot analysis using IBV N antibodies. Beta-actin was included as the loading control. The sizes of protein ladders in kilodaltons are indicated on the left. Significance levels are presented by the P value (*, P < 0.05; ***, P < 0.001). M, mock. (e) H1299 cells were infected with IBV at an MOI of ∼2. At 2 hpi, cells were treated with T-5224 at the indicated concentrations or the same volume of DMSO, and protein samples were harvested at 16, 24, and 30 hpi and subjected to SDS-PAGE and Western blot analysis using the indicated antisera or antibodies. Beta-actin was included as the loading control. Sizes of protein ladders in kilodaltons are indicated on the left. The percentage of PARP cleavage [PARP Clv.(%)] was calculated as the intensity of cleaved PARP [PARP (Cl)] divided by the total intensities of full-length PARP [PARP (FL)] and cleaved PARP. Significance levels are presented by the P value (ns, nonsignificant; *, P < 0.05; ***, P < 0.001). (f) H1299 cells were treated with T-5224 as described above for panel e. Cell lysates were harvested at 16, 24, and 30 hpi for RNA extraction. Equal amounts of total RNA were reverse transcribed, and the IL-8 mRNA level was determined by qPCR. Significance levels are presented by the P value (ns, nonsignificant; *, P < 0.05; ***, P < 0.001).
To probe the appropriate concentration range of T-5224 that could inhibit IBV-induced cFOS expression but not reduce the cell proliferation rate, H1299 cells were treated with T-5224 and harvested at 24 hpi for titration and Western blotting. Determination of the 50% tissue culture infective doses (TCID50) in the total cell lysates and supernatants showed reduced viral titers when 25 μM and 50 μM T-5224, respectively, were used (Fig. 2b and c). Western blot analysis showed a gradual reduction of IBV N protein synthesis when increasing concentrations of T-5224 were applied (Fig. 2d), confirming the inhibitory effect of this inhibitor on IBV replication.
The effect of T-5224 on IBV replication was then determined in a time course experiment in IBV-infected H1299 cells in the presence of 12.5 μM and 25 μM T-5224. As shown in Fig. 2e, moderate inhibitory effects on IBV replication (23 to 34% lower) were observed when 12.5 μM T-5224 was used at 16, 24, and 30 hpi. As expected, more severe inhibitory effects on IBV replication (80 to 93% lower) were detected when 25 μM T-5224 was used (Fig. 2e).
Cleavage of poly(ADP-ribose) polymerase (PARP), a substrate of active caspase-3, was used as a marker for apoptosis induced by IBV infection. In the presence of 12.5 μM T-5224, IBV-induced apoptosis was almost abolished at 16 hpi, and slightly less induction of apoptosis was observed at the other two later time points (Fig. 2e). IBV-induced apoptosis was almost undetectable when 25 μM T-5224 was used (Fig. 2e). These results demonstrate that inhibition of cFOS by T-5224 significantly reduces IBV replication, and IBV-induced apoptosis is delayed and reduced.
We then explored if the inhibition of IBV-induced cFOS upregulation by T-5224 would also suppress AP-1 activity. In a previous study, we showed that IBV infection upregulates IL-6 and IL-8 (24). As shown in Fig. 2f, the upregulation of IL-8 in IBV-infected H1299 cells was significantly suppressed in the presence of 12.5 μM T-5224 at 24 and 30 hpi.
Knockdown of cFOS increases IBV-induced apoptosis at early to intermediate phases of the IBV replication cycle.
To confirm the involvement of cFOS in the regulation of IBV replication and IBV-induced apoptosis, knockdown of cFOS with small interfering RNA (siRNA) was done in H1299 cells. The knockdown cells were then infected with IBV. As shown in Fig. 3a, the upregulation of total cFOS was significantly reduced in the knockdown cells compared with the enhanced green fluorescent protein siRNA (siEGFP) control. The synthesis of the IBV structural proteins S and N was moderately reduced in the cFOS knockdown cells at later time points (Fig. 3a). The viral titers were reduced at 20 hpi in both the cell lysates and supernatants of cFOS knockdown cells (Fig. 3b and c), confirming the involvement of cFOS in IBV replication in cell culture.
FIG 3.

Increased induction of apoptosis in cFOS knockdown cells infected with IBV. (a) H1299 cells were transfected with siEGFP and sicFOS before being infected with IBV at an MOI of ∼2. Cell lysates were harvested at the indicated time points and subjected to Western blot analysis using the indicated antibodies. Beta-tubulin was included as the loading control. Sizes of protein ladders in kilodaltons are indicated on the left. The percentage of PARP cleavage was determined as described in the legend of Fig. 2e. Significance levels are presented by the P value (ns, nonsignificant; *, P < 0.05; **, P < 0.01; ****, P < 0.0001). (b) H1299 cells were transfected and infected as described above for panel a, and cell lysates were harvested at the indicated time points by three freeze-thaw cycles. Virus titers were determined and are expressed in units of log TCID50 per milliliter. (c) Virus titers in the culture supernatants were determined and are expressed in units of log TCID50 per milliliter.
Interestingly, a significantly higher percentage of PARP cleavage was detected in cFOS knockdown cells than in the control cells at the same time points (Fig. 3a). Whereas IBV infection induced only low levels of apoptosis in the siEGFP control at 24 and 32 hpi, much more pronounced PARP cleavage was detected in the cFOS knockdown cells at these two time points, supporting that cFOS may protect infected cells from apoptosis at early to intermediate phases of the IBV infection cycle.
Inhibition of the nuclear translocation of cFOS by NLSP and its effect on IBV replication and IBV-induced apoptosis.
Nuclear localization signal peptide (NLSP), a peptide carrying an AP-1 nuclear localization sequence, may block the nuclear translocation of cFOS and inhibit its nuclear function. The effects of different concentrations of NLSP on the proliferation of H1299 cells were first tested by the incubation of cells with 25, 50, 75, and 100 μM NLSP for 20 min, and the cells were then cultured in fresh serum-free medium supplemented with (24-h treatment) or without (20-min treatment) the same concentrations of NLSP for 24 h. As shown in Fig. 4a, no significant differences in the cell proliferation rates were observed when 25, 50, and 75 μM NLSP were used for 20 min and when 25 μM NLSP was used for 24 h. Significant differences in the cell proliferation rates were observed with other treatments (Fig. 4a).
FIG 4.

Blockage of the nuclear translocation of cFOS by NLSP and inhibition of IBV replication. (a) H1299 cells treated with 25, 50, 75, and 100 μM NLSP for 20 min and cultured in fresh serum-free medium supplemented with (24-h treatment) or without (20-min treatment) the same concentrations of NLSP for 24 h. The cell proliferation rate was determined by measuring the absorbance value at 450 nm after adding the CCK solution for 2 h. Significance levels are presented by the P value (ns, nonsignificant; ***, P < 0.001). (b) H1299 cells were treated with NLSP at the indicated concentrations or with the same volume of DMSO before being infected with IBV at an MOI of ∼2. Cell lysates and supernatants were harvested at the indicated time points and subjected to SDS-PAGE and Western blot analysis using the indicated antibodies. Beta-actin was included as the loading control. Sizes of protein ladders in kilodaltons are indicated on the left. The degree of cell apoptosis was calculated as described in the legend of Fig. 3a. (c) H1299 cells were treated with 75 μM NLSP or mock treated with the same volume of DMSO for 20 min before being infected with IBV at an MOI of ∼2. After absorption for 2 h, cells were incubated at 37°C in the presence or absence of 75 μM NLSP for 4, 8, 10, 12, 16, and 24 h. The cell proliferation rate was determined by measuring the absorbance value at 450 nm after adding the CCK solution for 2 h. Significance levels are presented by the P value (ns, nonsignificant; ***, P < 0.001). (d) Immunofluorescence staining of cFOS in IBV-infected H1299 cells treated with DMSO, 75 μM NLSP for 20 min, and 75 μM NLSP. Cells were fixed at 8 h and 12 h postinfection, immunostained with anti-cFOS, and nuclear stained with DAPI. The merged images show cFOS/nuclear colocalization.
H1299 cells were then incubated with 75 μM NLSP for 20 min, infected with IBV, cultured in fresh serum-free medium, and harvested at the indicated time points. Compared with control cells incubated with DMSO (mock-treated cells), moderately to slightly low levels of IBV S and N proteins in IBV-infected cells treated with NLSP were detected at 8, 10, 12, and 16 hpi, and comparable levels of the two viral proteins were detected in the total lysates and supernatants at 20 and 24 hpi (Fig. 4b). Higher levels of cFOS were detected in the treated cells at 0 and 4 hpi than in the mock-treated cells (Fig. 4b). Induction of cFOS was delayed at 8 hpi but reached similar levels as those of the mock-treated control at later time points (Fig. 4b). Induction of apoptosis was also significantly delayed in NLSP-treated cells, as judged by the much-reduced levels of PARP cleavage in the treated cells at 16 hpi compared to those in the mock-treated cells (Fig. 4b).
In cells infected with IBV and incubated with 75 μM NLSP throughout the course of the experiment, much more significantly lower levels of viral protein synthesis and a very low level of PARP cleavage were observed in the treated cells at most time points (Fig. 4b). Similarly, the induction of cFOS was much delayed and at much lower levels in the treated cells than in the untreated cells (Fig. 4b). Meanwhile, determination of the proliferation of these cells showed that approximately 75% of the proliferation rates were maintained when cells were incubated with NLSP for up to 12 h; the proliferation rates of the treated cells were reduced to approximately 35% at 16 and 24 hpi (Fig. 4c).
Immunofluorescence staining of the NLSP-treated cells was then performed to check if these treatments affected the nuclear translocation of cFOS. Predominantly cytoplasmic staining of cFOS in the treated cells and no staining of cFOS in the untreated cells were observed at 0 hpi (Fig. 4d), consistent with the levels of cFOS protein in these cells as detected by Western blotting (Fig. 4b). In cells treated with 75 μM NLSP for 20 min, no staining of cFOS was observed at 8 hpi, and nuclear staining of the protein similar to that in the mock-treated cells was observed at 12 hpi (Fig. 4c), revealing that this treatment delayed the induction but did not affect the nuclear translocation of cFOS in IBV-infected H1299 cells. Continuous incubation with 75 μM NLSP demonstrated that a majority of the fluorescence was detected in the cytoplasm at both 8 and 12 hpi (Fig. 4d), showing that this treatment blocked the nuclear translocation of cFOS protein in IBV-infected H1299 cells.
Knockdown of ERK1/2 suppresses IBV-induced cFOS upregulation and increases apoptosis.
To study the upstream signals that may regulate cFOS induction in IBV-infected cells, knockdown of ERK1/2 with siRNA was conducted in H1299 cells. The knockdown cells were then infected with IBV, total lysates were prepared, and the induction of cFOS and IBV replication as well as PARP cleavage were analyzed by Western blotting. As shown in Fig. 5a, the upregulation of total cFOS and cJUN was significantly reduced in the knockdown cells compared with the siEGFP control. Synthesis of IBV N protein was marginally suppressed in the ERK1/2 knockdown cells at all time points (Fig. 5a). Notably, a much higher percentage of PARP cleavage was detected in the knockdown cells (Fig. 5a). Consistently, the mRNA levels of cFOS and cJUN were markedly reduced in ERK1/2 knockdown cells compared with the siEGFP control, whereas IBV genomic RNA synthesis was not significantly affected (Fig. 5b). These results suggest that ERK1 and -2 may be the upstream kinases that activate cFOS in IBV-infected cells.
FIG 5.

Suppression of IBV-induced cFOS upregulation and increased induction of apoptosis in ERK1/2 knockdown cells infected with IBV. (a) H1299 cells were transfected with siEGFP and siERK1/2 before being infected with IBV at an MOI of ∼2. Cell lysates were harvested at the indicated time points and subjected to Western blot analysis using PARP, ERK1/2, cFOS, cJUN, and IBV N antibodies. Beta-tubulin was included as the loading control. Sizes of protein ladders in kilodaltons are indicated on the left. Significance levels are presented by the P value (**, P < 0.01; ***, P < 0.001; ****, P < 0.0001). (b) H1299 cells were transfected and infected as described above for panel a. Cell lysates were harvested at the indicated time points for RNA extraction. Equal amounts of total RNA were reverse transcribed, and the mRNA levels of ERK1/2, cFOS, and cJUN were determined by qPCR. The IBV gRNA levels were determined as an indicator of IBV replication efficiency. Significance levels are presented by the P value (*, P < 0.05; ***, P < 0.001). M, mock.
Inhibition of both JNK and p38 MAPKs suppresses cFOS induction and IBV replication in H1299 cells.
The JNK signal pathway is an important branch of the mitogen-activated protein kinase (MAPK) pathway and plays an important role in various physiological and pathological processes such as the cell cycle, reproduction, apoptosis, and cell stress. SP600125, a widely used JNK inhibitor, was used to investigate whether JNK was involved in IBV-induced cFOS expression. H1299 cells were infected with IBV for 2 h before being treated with either DMSO or 8 μM SP600125 and harvested at 0, 12, 16, 20, and 24 h. Compared with the control cells, the protein levels of cFOS and IBV N were significantly low at each time point (Fig. 6a).
FIG 6.

Suppression of cFOS upregulation and IBV replication by JNK and p38 inhibitors. (a) H1299 cells were infected with IBV at an MOI of ∼2 and treated with SP600125 at the indicated concentrations or with the same volume of DMSO at 2 hpi. Protein samples were harvested at the indicated time points and subjected to Western blot analysis using the indicated antisera. Beta-actin was included as the loading control. Sizes of protein ladders in kilodaltons are indicated on the left. Significance levels are presented by the P value (*, P < 0.05; ***, P < 0.001). (b) H1299 cells were treated with SB-203580 as described above for panel a. Protein samples were harvested at the indicated time points and subjected to Western blot analysis using the indicated antisera. Beta-actin was included as the loading control. Sizes of protein ladders in kilodaltons are indicated on the left. Significance levels are presented by the P value (**, P < 0.01; ****, P < 0.0001).
p38 MAPK participates in a signaling cascade controlling cellular responses to cytokines and stress. SB-203580 is a selective, ATP-competitive p38 MAPK inhibitor. H1299 cells were pretreated with either DMSO or 20 μM SB-203580 after IBV infection. The expression levels of cFOS and IBV N were significantly reduced in SB-203580-treated cells (Fig. 6b). These results indicate that inhibition of both JNK and p38 may suppress cFOS induction and IBV replication.
DISCUSSION
As obligate intracellular parasitic entities, viruses heavily rely on host cell machinery for efficient replication. Over years of coevolution with their respective hosts, viruses have evolved a variety of strategies to modulate cellular processes and pathways in order to create a more conducive microenvironment for the successful completion of the viral replication cycle in a generally hostile cellular setting. In addition to the efforts focusing on dissecting viral strategies antagonizing host antiviral responses, we are now beginning to appreciate the mechanisms exploited by coronaviruses to modulate host cell functions for the benefit of viral replication. In fact, a number of strategies exploited by coronaviruses to optimize the cellular microenvironment for optimal viral replication have been uncovered in recent years. One typical example is to arrest the cell cycle at the S and M/G2 phases through the interaction of SARS-CoV and IBV nsp13 with the p125 subunit of DNA polymerase δ, allowing adequate time and resources for the efficient completion of the viral replication cycle (25, 26). Other examples include the interaction of IBV M protein with β-actin (27), activation of cellular stress responses (28), and promotion and inhibition of apoptosis at different stages of the viral replication cycle (6, 29). In this study, we report that a cell survival strategy mediated by the upregulation of the AP-1 family of transcription factors is exploited by IBV, a gammacoronavirus, and PEDV, an alphacoronavirus, for efficient viral replication. Due to the lack of specific reagents for the detection of cFOS protein levels in avian cells such as DF-1 cells, most experiments described in this study were conducted in human and monkey cells. As the upregulation of the AP-1 transcription factors was also reported in cells infected with SARS-CoV, a betacoronavirus (6, 29), this appears to be a mechanism shared by all coronaviruses. The transcriptional activity of AP-1 was also reported to be stimulated by a wide array of viral infections, including hepatitis B virus, hepatitis C virus, human papillomavirus, Epstein-Barr virus, human T-cell lymphotropic virus type 1, and Kaposi’s sarcoma-associated herpesvirus (30).
Further studies of the induction kinetics and functional characterization showed that the induction of cFOS by IBV infection may reduce IBV-induced apoptotic cell death at early to intermediate phases of the viral replication cycle, thereby creating a more conducive cellular microenvironment for viral replication (Fig. 7). As members of the immediate early gene family, cFOS and cJUN would be synthesized in large quantities immediately after the cell receives an extracellular stress signal, without the requirement for the de novo synthesis of other cellular proteins (31). This would quickly restore cellular homeostasis and normal physiological functions. In this study, the cell survival function of cFOS was found to be exploited by coronaviruses to ensure the efficient completion of the viral replication cycle.
FIG 7.
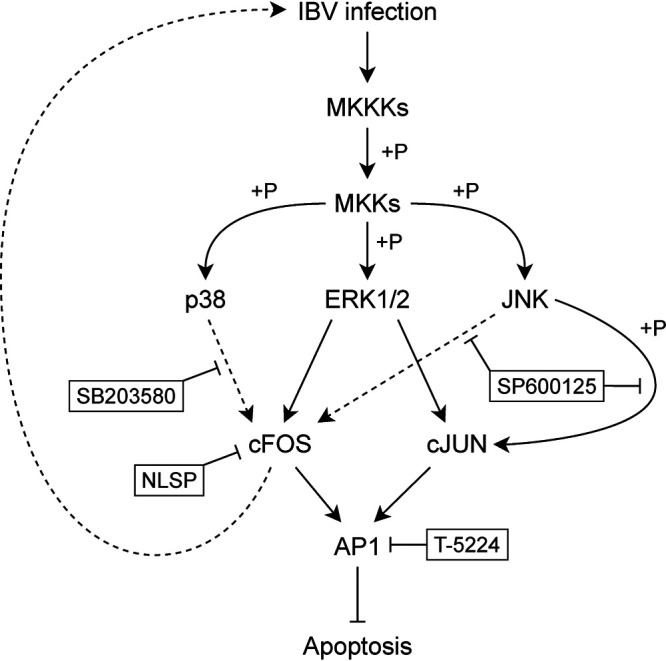
Diagram illustrating the current working model. Pointed and blunt arrows denote activation and suppression, respectively. “+P” denotes phosphorylation. Dotted lines denote processes that are not fully characterized. MKKK, MAPK kinase kinase; MKK, MAPK kinase.
As a member of the AP-1 transcription factor family, cFOS plays multiple, sometimes opposite roles in different cellular settings. Recently, it was found that cFOS mediated the antiviral activity of interferon kappa (IFN-κ) in cultured human lung cells infected with the low-pathogenicity avian influenza A virus H9N2 (32). Acting independent of signal transducer and activator factor of transcription 1 (STAT1), the binding of IFN-κ to interferon alpha and beta receptor (IFNAR) activated p38-cFOS signaling and induced the expression of an antiviral effector protein called chromodomain helicase DNA-binding protein 6 (CHD6) (32).
The role of cFOS in antagonizing apoptosis has been studied mainly in several cancer models. In about 40% of multiple myelomas, a transcription activator of cyclin D2 named musculoaponeurotic fibrosarcoma oncogene homolog (MAF) is overexpressed (33). In these MAF-overexpressing cells, treatment with the MEK1 inhibitor U0126 or cFOS knockdown reduces MAF transcription and activates apoptosis, suggesting that the MEK1/ERK/cFOS/MAF signaling axis promotes survival in these cells (33). This is consistent with our finding that knockdown of either ERK1/2 or cFOS significantly increased IBV-induced apoptosis. A high level of cFOS protein expression has also been associated with poor prognoses in malignant glioma patients, and knockdown of cFOS by short hairpin RNA (shRNA) sensitized glioma cells to radiation-induced apoptosis (34). It was reported previously that microRNAs miR-181b and miR-21 were involved in cFOS-mediated apoptosis suppression and the progression of gliomas (35).
On the other hand, cFOS serves proapoptotic roles in some cancers. In one early study, it was shown that mice with p53 and cFOS double knockout developed proliferative and invasive rhabdomyosarcomas (36). Enhanced apoptosis was observed when cFOS was reexpressed in cell lines established from these tumors, suggesting a proapoptotic tumor-suppressive role of cFOS (36). In prostate cancer cells, cFOS promotes TRAIL-induced apoptosis by repressing the transcription of cellular FLICE-like inhibitory protein (c-FLIP), which is a negative regulator of caspase-8 cleavage in the extrinsic apoptosis pathway (37). Finally, in a mouse model of diethylnitrosamine (DEN)-induced liver cancer initiation, cFOS promoted apoptosis induction by directly activating sirtuin 6 transcription. Sirtuin 6 in turn repressed the transcription of survivin, an inhibitor of apoptosis that binds to and inhibits effector caspase-3 and -7 (38).
Viral replication and host cell apoptosis are two closely intertwined events during the viral infection cycles. Early cell death as a cellular response to viral invasion would result in the abortion of viral replication and therefore can be considered a host antiviral mechanism. Naturally, viruses would have evolved countermeasures to subvert this host antiviral function. In addition, the delicate balance of cell survival and death during the replication of a virus appears to be highly and accurately regulated. We have previously shown that IBV may use a diversity of mechanisms to either induce or suppress apoptosis, including induction of the proapoptotic protein Bak and the antiapoptotic protein Mcl-1, induction of different branches of the unfolded protein response pathways, and activation of the MAPKs p38 and JNK (6, 24, 28, 29, 39, 40). As revealed by this study, some moderate fluctuations in viral protein synthesis in the presence of a cFOS inhibitor or in cFOS knockdown cells would significantly affect the induction of apoptotic cell death, consequently regulating the replication of IBV in treated cells. In this study, we showed that inhibition of JNK and p38 reduced the upregulation of cFOS and the replication of IBV. Knockdown of ERK1/2 was also shown to reduce cFOS expression and IBV replication, consistent with previous reports that cFOS is a downstream molecule in the ERK pathway (41–43). Figure 7 illustrates the involvement of these stress-related kinases in the activation of cFOS, which in turn regulates cell survival and IBV replication.
Recent studies have demonstrated that cFOS may exert two separate functions in different subcellular locations (21, 22). Once translocated to the nucleus, it functions as a transcription factor, whereas the portion of the protein that remains in the cytoplasm may function as an activation factor for lipid synthesis. The addition of NLSP to the culture medium may block the nuclear translocation of cFOS, thereby inhibiting its nuclear function while virtually maintaining its cytoplasmic function (21, 44). This cytoplasmic function may play an enhancement role in IBV replication, as the promotion of lipid biosynthesis would benefit the replication of enveloped viruses, including IBV. However, treatment of infected cells either with 75 μM NLSP for 20 min or by continuous incubation with the same concentration of NLSP, as reported in this study, plays an inhibitory role in IBV replication in general. One obvious reason would be due to the reduced cell proliferation rate by the treatments. However, no detectable effect on cell proliferation was observed when cells were treated with NLSP for 20 min only, and about 75% of proliferation rates were maintained when the infected cells were incubated with75 μM NLSP up to 12 hpi. Interestingly, the basal levels of cFOS were higher in cells treated with NLSP under either treatment condition at 0 hpi, and the protein remained in the cytoplasm. Consequently, these treatments delayed the replication of IBV and the induction of cFOS. The underlying mechanism is yet to be elucidated, but the data presented in this study do not support the involvement of the cytoplasmic function of cFOS in IBV replication.
MATERIALS AND METHODS
Antibodies, chemicals, and reagents.
Antibodies against β-actin (catalog number 4967), cFOS (catalog number 2250), ERK1/2 (catalog number 9194), and PARP (catalog number 9532) were purchased from Cell Signaling Technology. Goat anti-rabbit IgG(H+L) (Alexa Fluor 488) (catalog number ab150077) was purchased from Abcam. A TransDetect cell counting kit (CCK) was purchased from TransGen Biotech. Antisera against IBV S and N proteins were prepared in rabbits immunized with bacterially expressed fusion proteins as previously described (45, 46).
The AP-1 inhibitor T-5224 was purchased from Selleckchem and dissolved in DMSO for a 50 mM stock solution. The p38 inhibitor SB-203580 was purchased from Selleckchem and dissolved in DMSO for a 100 mM stock solution. The JNK inhibitor SP600125 was purchased from Selleckchem and dissolved in DMSO for a 50 mM stock solution. The peptide NLSP (amino acid sequence AAVALLPAVLLALLAPVQRKRQKLMP) was synthesized by Sangon (Shanghai, China). The synthetic peptides and inhibitors were aliquoted and stored at −80°C.
Eight-day-old SPF embryonated chicken eggs were obtained from the Laboratory Animal Center of South China Agricultural University and incubated at 37°C for 24 h, and dead embryos were discarded before infection experiments. Handling of the chicken embryos was performed by strictly following the experimental animal care guidelines and the experimental protocol approved by the Animal Welfare and Ethical Censor Committee at South China Agricultural University. Chicken embryos that did not die during the experiments were euthanized before execution.
Virus and cells.
The egg-adapted Beaudette strain of IBV (ATCC VR-22) was obtained from the American Type Culture Collection (ATCC) and adapted to Vero cells as previously described (47–49). PEDV virulent strain DR13 (GenBank accession no. JQ023162) was obtained from suckling pigs in 1999 and adapted for growth in Vero cells (50, 51). Vero and H1299 cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies, Carlsbad, CA, USA) supplemented with 6% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. All cells were grown in a 37°C incubator supplied with 5% CO2. In all the experiments, cells were washed twice with serum-free medium before being infected with IBV at a multiplicity of infection (MOI) of approximately 2 or incubated with an equal volume of UV-inactivated IBV in serum-free medium. After 2 h of absorption, cells were washed twice with serum-free medium and incubated at 37°C before being harvested.
To prepare the virus stock, monolayers of Vero cells were infected at an MOI of approximately 0.1 and cultured in DMEM at 37°C for 24 h. After three freeze-thaw cycles, total cell lysates were clarified by centrifugation at 1,500 × g at 4°C for 30 min. The supernatant was aliquoted and stored at −80°C as a virus stock.
UV inactivation of IBV was performed by exposing the virus stock to 120,000 mJ/cm2 of 254-nm shortwave UV radiation for 15 min with a CL-1000 cross-linker (UVP). To demonstrate that IBV had been inactivated, Vero cells were incubated with UV-IBV, and the cell lysates were analyzed by Western blotting to confirm that no viral proteins could be detected.
Unless stated otherwise, H1299 cells were first inoculated with IBV at an MOI of ∼2. After 2 to 4 h of virus adsorption, the unbound viruses were removed, and the cells were washed twice with serum-free medium. The inhibitors were diluted to final concentrations using serum-free medium and added to the cells. An additional well treated with the same amount of solvent was used as the solvent control.
Microarray hybridization and image analysis.
Microarray hybridization and image analysis were carried out as previously described (24, 52). Briefly, RNA was independently prepared from IBV-infected Vero cells harvested at 24 h postinfection and hybridized to a GeneChip human genome U133A array (Affymetrix, USA) according to the manufacturer’s instructions (Affymetrix). GeneChip arrays were scanned on an Affymetrix probe array scanner, and data were analyzed using Microarray Suite version 5.0 (MAS5.0) statistics software from Affymetrix.
Transcriptomic analysis.
Transcriptomic analysis was carried out by Biomarker Technologies Co., Ltd., Beijing, China. Briefly, RNA was independently prepared from IBV-infected H1299 cells harvested at 20 h postinfection and sequenced using the Illumina HiSeq sequencing technology platform to construct transcriptome libraries and obtain sequencing data.
IBV infection of chicken embryos.
A total of 500 PFU of IBV in 0.2 ml were injected into the allantoic cavities of 10-day-old embryonated SPF eggs. The infected eggs were incubated at 37°C for 60 h and harvested. Total RNAs were extracted and analyzed by RT-qPCR.
Virus titration by TCID50.
Supernatant samples were harvested from IBV-infected cells and clarified by centrifugation at 16,000 × g at 4°C for 5 min. Cell lysates were harvested by subjecting IBV-infected cells to three freeze-thaw cycles and clarified by centrifugation at 16,000 × g at 4°C for 5 min. Virus samples were kept at −80°C for less than 2 weeks before the titration experiment. The virus titer was determined by a 50% tissue culture infective dose (TCID50) assay. Briefly, virus samples were 10-fold serially diluted. Confluent monolayers of Vero cells seeded on 96-well plates were washed once with plain DMEM, and 100 μl of the diluted virus sample was added to each well, with 8 wells used for each dilution. Cells were incubated at 37°C for 3 to 5 days and examined with a phase-contrast microscope. Wells were determined as either positive (with cytopathic effect [CPE]) or negative (without CPE), and the TCID50 was calculated using the method of Reed and Muench (53). The virus titer was expressed in units of log TCID50 per milliliter. Each sample was titrated in duplicate or triplicate in each experiment.
Cell proliferation assay.
The TransDetect CCK is a cell proliferation and cytotoxicity detection kit based on water-soluble tetrazolium salt. In the presence of the electron-coupling reagent 1-methoxy phenazine methyl sulfate, tetrazolium can be reduced to soluble formazan by dehydrogenase in mitochondria. Compared with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetrazolium hydroxide (XTT), 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS), and water-soluble tetrazolium-1[2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulfophenyl)]-2H-tetrazolium monosodium salt (WST-1), this method has a high detection sensitivity and a wide linear range and is suitable for drug screening, cell proliferation assays, cytotoxicity assays, and tumor drug sensitivity assays.
H1299 cells (5 × 103) were seeded on 96-well plates, incubated at 37°C for 16 h, and then treated with different concentrations of nuclear localization signal peptide (NLSP) or other indicated inhibitors. At the indicated times posttreatment, 10 μl of the CCK solution was added to the plates, and the plates were incubated at 37°C for 2 h. The absorbance at 450 nm was detected by using an automatic microplate reader.
Immunofluorescence staining.
H1299 cells were seeded on 96-well plates and incubated at 37°C for 16 h. Cells were treated with 75 μM NLSP for 20 min and infected with IBV (MOI of ∼2) for 2 h. The inoculum was then changed to serum-free medium in the presence or absence of 75 μM NLSP for the indicated times. Cells were fixed with 4% paraformaldehyde for 20 min, permeabilized by treatment with 0.25% Triton X-100 in phosphate-buffered saline (PBS) for 10 min, washed three times with PBS, and blocked with PBS containing 1% bovine serum albumin (BSA) plus Tween 20 (0.1% [vol/vol]) for 2 h at room temperature. Cells were incubated with the primary anti-cFOS antibody at 4°C overnight and with secondary antibody [Alexa Fluor 488 goat anti-rabbit IgG(H+L)] for 2 h at room temperature, washed with PBS three times, nuclear stained with 4′,6-diamidino-2-phenylindole (DAPI), and washed with PBS. Images were obtained with a fluorescence microscope.
RNA interference.
The siRNA duplexes were purchased from Sangon Biotech (Shanghai, China). The sequences of the siRNA sense strands are as follows: , CCUGCUGAAGGAGAAGGAAdTdT for cFOS, GACCGGAUGUUAACCUUUAdTdT and GUUCGAGUAGCUAUCAAGAdTdT for ERK1/2, and GCUGACCCUGAAGUUCAUCdTdT for EGFP (negative control). Transfection of siRNA was performed using the TransIntro EL transfection reagent (TransGen Biotech) according to the manufacturer’s instructions. Briefly, H1299 cells were plated on a 12-well plate the day before transfection. For each well, 5 μl of 20 μM siRNA duplex and 2.5 μl of TransIntro EL were diluted with 100 μl of Opti-MEM (Gibco) and incubated for 20 min. H1299 cells were replenished with 900 μl Opti-MEM containing 5% FBS, and the transfection mixture was added to each well dropwise. Virus infection was performed at 36 to 48 hpi.
RNA extraction and RT-qPCR analysis.
Total RNA was extracted using the TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. Briefly, cells were lysed with 1 ml TRIzol per 10-cm2 effective growth area, and the lysates were vigorously mixed with a one-fifth volume of chloroform. The mixture was then centrifuged at 12,000 × g at 4°C for 15 min, and the aqueous phase was mixed with an equal volume of isopropanol. The RNA was precipitated by centrifugation at 12,000 × g at 4°C for 15 min, washed twice with 70% ethanol, and dissolved in 30 to 50 μl RNase-free water.
The total RNA was reverse transcribed using the FastKing gDNA Dispelling RT SuperMix kit (Tiangen) according to the manufacturer’s instructions. Briefly, 1 μg total RNA was mixed with 2 μl 5× FastKing-RT SuperMix [containing RT enzyme, an RNase inhibitor, random primers, an oligo(dT) primer, deoxynucleoside triphosphate (dNTP), and reaction buffer] in a 10-μl reaction mixture. Using a thermocycler, reverse transcription was performed at 42°C for 15 min, and the RT enzyme was then inactivated at 95°C for 3 min. The cDNA was then diluted 20-fold with RNase-free water for quantitative PCR (qPCR) analysis using the Talent qPCR PreMix SYBR green kit (Tiangen) according to the manufacturer’s instructions. Briefly, 8.4 μl diluted cDNA was mixed with 10 μl 2× qPCR PreMix, 0.4 μl 50× ROX, 0.6 μl 10 μM forward primer, and 0.6 μl 10 μM reverse primer for a 20-μl reaction mixture. qPCR analysis was performed using a QuantStudio 3 real-time PCR system (Applied Biosystems). The standard protocol included enzyme activation at 50°C for 3 min and an initial denaturation step at 95°C for 3 min, followed by 40 cycles of denaturation (95°C for 5 s) and annealing/extension (60°C for 30 s), with fluorescence acquisition at the end of each cycle. The results obtained were in the form of cycle threshold (CT) values. Using the ΔΔCT method, the relative abundance of a transcript was calculated using glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as an internal control and normalized to the respective control sample.
cDNAs were then subjected to qPCR using appropriate primers. The qPCR primers for IBV and PEDV used in this study are as follows: IBV gRNA primers 5′-GTTCTCGCATAAGGTCGGCTA-3′ and 5′-GCTCACTAAACACCACCAGAAC-3′, IBV sgRNA2 primers 5′-GCCTTGCGCTAGATTTTTAACTG-3′ and 5′-AGTGCACACAAAAGAGTCACTA-3′, and PEDV gRNA primers 5′-AGTAGCCATCGCAAGTGCTG-3′ and 5′-AACCGGAGGAAGGCTGTTTG-3′.
The primer pairs for human and Vero cells used for PCR were as follows: GAPDH primers 5′-CTGGGCTACACTGAGCACC-3′ and 5′-AAGTGGTCGTTGAGGGCAATG-3′, cFOS primers 5′-GGGGCAAGGTGGAACAGTTAT-3′ and 5′-CCGCTTGGAGTGTATCAGTCA-3′, FOSB primers 5′-GCTGCAAGATCCCCTACGAAG-3′ and 5′-ACGAAGAAGTGTACGAAGGGTT-3′, cJUN primers 5′-AACAGGTGGCACAGCTTAAAC-3′ and 5′-CAACTGCTGCGTTAGCATGAG-3′, JUNB primers 5′-ACAAACTCCTGAAACCGAGCC-3′ and 5′-CGAGCCCTGACCAGAAAAGTA-3′, JUND primers 5′-TCATCATCCAGTCCAACGGG-3′ and 5′-TTCTGCTTGTGTAAATCCTCCAG-3′, and IL-8 primers 5′-ATAAAGACATACTCCAAACCTTTCCAC-3′ and 5′-AAGCTTTACAATAATTTCTGTGTTGGC-3′.
The nucleotide sequences of qPCR primers for chicken were as follows: GAPDH primers 5′-GACCACTGTCCATGCCATCA-3′ and 5′-TTTCCCCACAGCCTTAGCAG-3′, cFOS primers 5′-AGCCTCACCTACTACCCGTC-3′ and 5′-GGTGCAGAAATCCTGCGAGT-3′, cJUN primers 5′-CCTCCCCTGTCCCCTATTGA-3′ and 5′-CCTTTTCCGGCATTTGGACG-3′, and JUND primers 5′-CCCCATCGATATGGACACGC-3′ and 5′-AGATGCGCTCCAGTTTCCTC-3′.
SDS-PAGE and Western blot analysis.
To obtain whole-cell lysates for protein analysis, cells were harvested at the indicated time points using cell scrapers (Corning) and collected by centrifugation at 16,000 × g for 1 min. The supernatant was discarded, and the cell pellet was lysed in radioimmunoprecipitation assay (RIPA) buffer (10 mM Tris-HCl [pH 8.0], 140 mM NaCl, 0.1% SDS, 1% Triton X-100, 0.1% sodium deoxycholate, 1 mM EDTA, and 0.5 mM EGTA). After clarification by centrifugation, the protein concentration of the cell lysate was determined. The cell lysate was then mixed with 5× Laemmli sample buffer (0.3125 M Tris-HCl [pH 6.8], 10% SDS, 50% glycerol, 25% β-mercaptoethanol, and 0.025% bromophenol blue) (54). The culture supernatant was clarified by brief centrifugation and mixed with 5× Laemmli sample buffer.
Protein samples were boiled at 90°C for 5 min and centrifuged at 16,000 × g for 5 min. Equal amounts of protein samples were loaded into each well and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using the Bio-Rad Mini-Protean Tetra cell system. The resolved proteins were then transferred to a 0.2-μm nitrocellulose membrane using the Bio-Rad Trans-Blot protein transfer system. To block off nonspecific binding sites, the membrane was incubated with 5% skim milk in Tris-buffered saline–Tween 20 (TBST) buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 0.1% Tween 20) at room temperature for 1 h. The membrane was then incubated with 1 μg/ml specific primary antibody dissolved in TBST with 3% (wt/vol) BSA at 4°C overnight. The membrane was washed three times with TBST and incubated with 1:10,000-diluted goat anti-rabbit or goat anti-mouse IgG secondary antibodies (Licor) at room temperature for 2 h. The membrane was washed three times with TBST, and fluorescence imaging was performed using the Azure c600 imager according to the manufacturer’s instructions. Densitometric measurement was performed using AzureSpot software. All experiments were repeated at least three times, with similar results, and one of the representative results is shown.
Statistical analysis.
One-way analysis of variance (ANOVA) was used to analyze significant differences between the indicated samples and the respective control samples. Significance levels are presented by the P value (ns, nonsignificant; *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
ACKNOWLEDGMENTS
This work was partially supported by National Natural Science Foundation of China grants 31972660 and 31900135, Natural Science Foundation of Guangdong Province grant 2018A030313472, and Guangdong Province Key Laboratory of Microbial Signals and Disease Control grants MSDC-2017-05 and MSDC-2017-06.
We declare no conflict of interest.
REFERENCES
- 1.Wu F, Zhao S, Yu B, Chen Y-M, Wang W, Song Z-G, Hu Y, Tao Z-W, Tian J-H, Pei Y-Y, Yuan M-L, Zhang Y-L, Dai F-H, Liu Y, Wang Q-M, Zheng J-J, Xu L, Holmes EC, Zhang Y-Z. 2020. A new coronavirus associated with human respiratory disease in China. Nature 579:265–269. doi: 10.1038/s41586-020-2008-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou P, Yang X-L, Wang X-G, Hu B, Zhang L, Zhang W, Si H-R, Zhu Y, Li B, Huang C-L, Chen H-D, Chen J, Luo Y, Guo H, Jiang R-D, Liu M-Q, Chen Y, Shen X-R, Wang X, Zheng X-S, Zhao K, Chen Q-J, Deng F, Liu L-L, Yan B, Zhan F-X, Wang Y-Y, Xiao G-F, Shi Z-L. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fung TS, Liu DX. 2019. Human coronavirus: host-pathogen interaction. Annu Rev Microbiol 73:529–557. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- 4.To J, Surya W, Fung TS, Li Y, Verdià-Bàguena C, Queralt-Martin M, Aguilella VM, Liu DX, Torres J. 2017. Channel-inactivating mutations and their revertant mutants in the envelope protein of infectious bronchitis virus. J Virol 91:e02158-16. doi: 10.1128/JVI.02158-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fung TS, Liu DX. 2014. Coronavirus infection, ER stress, apoptosis and innate immunity. Front Microbiol 5:296. doi: 10.3389/fmicb.2014.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liao Y, Fung TS, Huang M, Fang SG, Zhong Y, Liu DX. 2013. Upregulation of CHOP/GADD153 during coronavirus infectious bronchitis virus infection modulates apoptosis by restricting activation of the extracellular signal-regulated kinase pathway. J Virol 87:8124–8134. doi: 10.1128/JVI.00626-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cavanagh D. 2007. Coronavirus avian infectious bronchitis virus. Vet Res 38:281–297. doi: 10.1051/vetres:2006055. [DOI] [PubMed] [Google Scholar]
- 8.Wit JJ, Swart WAJM, Fabri THF. 2010. Efficacy of infectious bronchitis virus vaccinations in the field: association between the alpha-IBV IgM response, protection and vaccine application parameters. Avian Pathol 39:123–131. doi: 10.1080/03079451003604639. [DOI] [PubMed] [Google Scholar]
- 9.Pensaert MB, de Bouck P. 1978. A new coronavirus-like particle associated with diarrhea in swine. Arch Virol 58:243–247. doi: 10.1007/BF01317606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bertasio C, Giacomini E, Lazzaro M, Perulli S, Papetti A, Lavazza A, Lelli D, Alborali G, Boniotti MB. 2016. Porcine epidemic diarrhea virus shedding and antibody response in swine farms. A longitudinal study. Front Microbiol 7:2009. doi: 10.3389/fmicb.2016.02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou YL, Ederveen J, Egberink H, Pensaert M, Horzinek MC. 1988. Porcine epidemic diarrhea virus (CV 777) and feline infectious peritonitis virus (FIPV) are antigenically related. Arch Virol 102:63–71. doi: 10.1007/BF01315563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoshikawa T, Hill TE, Yoshikawa N, Popov VL, Galindo CL, Garner HR, Peters CJ, Tseng C-TK. 2010. Dynamic innate immune responses of human bronchial epithelial cells to severe acute respiratory syndrome-associated coronavirus infection. PLoS One 5:e8729. doi: 10.1371/journal.pone.0008729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He R, Leeson A, Andonov A, Li Y, Bastien N, Cao J, Osiowy C, Dobie F, Cutts T, Ballantine M, Li X. 2003. Activation of AP-1 signal transduction pathway by SARS coronavirus nucleocapsid protein. Biochem Biophys Res Commun 311:870–876. doi: 10.1016/j.bbrc.2003.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Varshney B, Lal SK. 2011. SARS-CoV accessory protein 3b induces AP-1 transcriptional activity through activation of JNK and ERK pathways. Biochemistry 50:5419–5425. doi: 10.1021/bi200303r. [DOI] [PubMed] [Google Scholar]
- 15.Curran T, Franza BR. 1988. Fos and Jun. The AP-1 connection. Cell 55:395–397. doi: 10.1016/0092-8674(88)90024-4. [DOI] [PubMed] [Google Scholar]
- 16.Madrigal P, Alasoo K. 2018. AP-1 takes centre stage in enhancer chromatin dynamics. Trends Cell Biol 28:509–511. doi: 10.1016/j.tcb.2018.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Angel P, Karin M. 1991. The role of Jun, Fos and the AP-1 complex in cell-proliferation and transformation. Biochim Biophys Acta 1072:129–157. doi: 10.1016/0304-419x(91)90011-9. [DOI] [PubMed] [Google Scholar]
- 18.Hess J, Angel P, Schorpp-Kistner M. 2004. AP-1 subunits: quarrel and harmony among siblings. J Cell Sci 117:5965–5973. doi: 10.1242/jcs.01589. [DOI] [PubMed] [Google Scholar]
- 19.Wang S, Xu X, Xu F, Meng Y, Sun C, Shi L, Zhao E. 2016. Combined expression of c-jun, c-fos, and p53 improves estimation of prognosis in oral squamous cell carcinoma. Cancer Invest 34:393–400. doi: 10.1080/07357907.2016.1217422. [DOI] [PubMed] [Google Scholar]
- 20.Lim SP, Garzino-Demo A. 2000. The human immunodeficiency virus type 1 Tat protein up-regulates the promoter activity of the beta-chemokine monocyte chemoattractant protein 1 in the human astrocytoma cell line U-87 MG: role of SP-1, AP-1, and NF-kappaB consensus sites. J Virol 74:1632–1640. doi: 10.1128/jvi.74.4.1632-1640.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bussolino DF, Guido ME, Gil GA, Borioli GA, Renner ML, Grabois VR, Conde CB, Caputto BL. 2001. c-Fos associates with the endoplasmic reticulum and activates phospholipid metabolism. FASEB J 15:556–558. doi: 10.1096/fj.00-0446fje. [DOI] [PubMed] [Google Scholar]
- 22.Cardozo Gizzi AM, Caputto BL. 2013. Mechanistic insights into the nongenomic regulation of phospholipid synthesizing enzymes. IUBMB Life 65:584–592. doi: 10.1002/iub.1173. [DOI] [PubMed] [Google Scholar]
- 23.Alfonso Pecchio AR, Cardozo Gizzi AM, Renner ML, Molina-Calavita M, Caputto BL. 2011. c-Fos activates and physically interacts with specific enzymes of the pathway of synthesis of polyphosphoinositides. Mol Biol Cell 22:4716–4725. doi: 10.1091/mbc.E11-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao Y, Wang X, Huang M, Tam JP, Liu DX. 2011. Regulation of the p38 mitogen-activated protein kinase and dual-specificity phosphatase 1 feedback loop modulates the induction of interleukin 6 and 8 in cells infected with coronavirus infectious bronchitis virus. Virology 420:106–116. doi: 10.1016/j.virol.2011.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu LH, Huang M, Fang SG, Liu DX. 2011. Coronavirus infection induces DNA replication stress partly through interaction of its nonstructural protein 13 with the p125 subunit of DNA polymerase δ. J Biol Chem 286:39546–39559. doi: 10.1074/jbc.M111.242206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li FQ, Tam JP, Liu DX. 2007. Cell cycle arrest and apoptosis induced by the coronavirus infectious bronchitis virus in the absence of p53. Virology 365:435–445. doi: 10.1016/j.virol.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang J, Fang S, Xiao H, Chen B, Tam JP, Liu DX. 2009. Interaction of the coronavirus infectious bronchitis virus membrane protein with beta-actin and its implication in virion assembly and budding. PLoS One 4:e4908. doi: 10.1371/journal.pone.0004908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fung TS, Liu DX. 2017. Activation of the c-Jun NH2-terminal kinase pathway by coronavirus infectious bronchitis virus promotes apoptosis independently of c-Jun. Cell Death Dis 8:3215. doi: 10.1038/s41419-017-0053-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fung TS, Liao Y, Liu DX. 2014. The endoplasmic reticulum stress sensor IRE1α protects cells from apoptosis induced by the coronavirus infectious bronchitis virus. J Virol 88:12752–12764. doi: 10.1128/JVI.02138-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mirzaei H, Khodadad N, Karami C, Pirmoradi R, Khanizadeh S. 2020. The AP-1 pathway; a key regulator of cellular transformation modulated by oncogenic viruses. Rev Med Virol 30:e2088. doi: 10.1002/rmv.2088. [DOI] [PubMed] [Google Scholar]
- 31.Fowler T, Sen R, Roy AL. 2011. Regulation of primary response genes. Mol Cell 44:348–360. doi: 10.1016/j.molcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.He Y, Fu W, Cao K, He Q, Ding X, Chen J, Zhu L, Chen T, Ding L, Yang Y, Zhu C, Yuan S, Li Z, Zhao C, Zhang X, Xu J. 2020. IFN-κ suppresses the replication of influenza A viruses through the IFNAR-MAPK-Fos-CHD6 axis. Sci Signal 13:eaaz3381. doi: 10.1126/scisignal.aaz3381. [DOI] [PubMed] [Google Scholar]
- 33.Annunziata CM, Hernandez L, Davis RE, Zingone A, Lamy L, Lam LT, Hurt EM, Shaffer AL, Kuehl WM, Staudt LM. 2011. A mechanistic rationale for MEK inhibitor therapy in myeloma based on blockade of MAF oncogene expression. Blood 117:2396–2404. doi: 10.1182/blood-2010-04-278788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Z-G, Jiang G, Tang J, Wang H, Feng G, Chen F, Tu Z, Liu G, Zhao Y, Peng M-J, He Z-W, Chen X-Y, Lindsay H, Xia Y-F, Li X-N. 2016. c-Fos over-expression promotes radioresistance and predicts poor prognosis in malignant glioma. Oncotarget 7:65946–65956. doi: 10.18632/oncotarget.11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tao T, Wang Y, Luo H, Yao L, Wang L, Wang J, Yan W, Zhang J, Wang H, Shi Y, Yin Y, Jiang T, Kang C, Liu N, You Y. 2013. Involvement of FOS-mediated miR-181b/miR-21 signalling in the progression of malignant gliomas. Eur J Cancer 49:3055–3063. doi: 10.1016/j.ejca.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 36.Fleischmann A, Jochum W, Eferl R, Witowsky J, Wagner EF. 2003. Rhabdomyosarcoma development in mice lacking Trp53 and Fos: tumor suppression by the Fos protooncogene. Cancer Cell 4:477–482. doi: 10.1016/s1535-6108(03)00280-0. [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, Zhang L, Yang H, Huang X, Otu H, Libermann TA, DeWolf WC, Khosravi-Far R, Olumi AF. 2007. c-Fos as a proapoptotic agent in TRAIL-induced apoptosis in prostate cancer cells. Cancer Res 67:9425–9434. doi: 10.1158/0008-5472.CAN-07-1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Min L, Ji Y, Bakiri L, Qiu Z, Cen J, Chen X, Chen L, Scheuch H, Zheng H, Qin L, Zatloukal K, Hui L, Wagner EF. 2012. Liver cancer initiation is controlled by AP-1 through SIRT6-dependent inhibition of survivin. Nat Cell Biol 14:1203–1211. doi: 10.1038/ncb2590. [DOI] [PubMed] [Google Scholar]
- 39.Zhong Y, Liao Y, Fang S, Tam JP, Liu DX. 2012. Up-regulation of Mcl-1 and Bak by coronavirus infection of human, avian and animal cells modulates apoptosis and viral replication. PLoS One 7:e30191. doi: 10.1371/journal.pone.0030191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu C, Xu HY, Liu DX. 2001. Induction of caspase-dependent apoptosis in cultured cells by the avian coronavirus infectious bronchitis virus. J Virol 75:6402–6409. doi: 10.1128/JVI.75.14.6402-6409.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bai L, Mao R, Wang J, Ding L, Jiang S, Gao C, Kang H, Chen X, Sun X, Xu J. 2015. ERK1/2 promoted proliferation and inhibited apoptosis of human cervical cancer cells and regulated the expression of c-Fos and c-Jun proteins. Med Oncol 32:57. doi: 10.1007/s12032-015-0490-5. [DOI] [PubMed] [Google Scholar]
- 42.Murphy LO, Smith S, Chen R-H, Fingar DC, Blenis J. 2002. Molecular interpretation of ERK signal duration by immediate early gene products. Nat Cell Biol 4:556–564. doi: 10.1038/ncb822. [DOI] [PubMed] [Google Scholar]
- 43.Kudo T, Uda S, Tsuchiya T, Wada T, Karasawa Y, Fujii M, Saito TH, Kuroda S. 2016. Laguerre filter analysis with partial least square regression reveals a priming effect of ERK and CREB on c-FOS induction. PLoS One 11:e0160548. doi: 10.1371/journal.pone.0160548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Portal MM, Ferrero GO, Caputto BL. 2007. N-terminal c-Fos tyrosine phosphorylation regulates c-Fos/ER association and c-Fos-dependent phospholipid synthesis activation. Oncogene 26:3551–3558. doi: 10.1038/sj.onc.1210137. [DOI] [PubMed] [Google Scholar]
- 45.Liu DX, Inglis SC. 1991. Association of the infectious bronchitis virus 3c protein with the virion envelope. Virology 185:911–917. doi: 10.1016/0042-6822(91)90572-s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li FQ, Xiao H, Tam JP, Liu DX. 2005. Sumoylation of the nucleocapsid protein of severe acute respiratory syndrome coronavirus. FEBS Lett 579:2387–2396. doi: 10.1016/j.febslet.2005.03.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang SG, Shen S, Tay FPL, Liu DX. 2005. Selection of and recombination between minor variants lead to the adaptation of an avian coronavirus to primate cells. Biochem Biophys Res Commun 336:417–423. doi: 10.1016/j.bbrc.2005.08.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim KP, Liu DX. 1998. Characterization of the two overlapping papain-like proteinase domains encoded in gene 1 of the coronavirus infectious bronchitis virus and determination of the C-terminal cleavage site of an 87-kDa protein. Virology 245:303–312. doi: 10.1006/viro.1998.9164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shen S, Law YC, Liu DX. 2004. A single amino acid mutation in the spike protein of coronavirus infectious bronchitis virus hampers its maturation and incorporation into virions at the nonpermissive temperature. Virology 326:288–298. doi: 10.1016/j.virol.2004.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Song DS, Yang JS, Oh JS, Han JH, Park BK. 2003. Differentiation of a Vero cell adapted porcine epidemic diarrhea virus from Korean field strains by restriction fragment length polymorphism analysis of ORF 3. Vaccine 21:1833–1842. doi: 10.1016/s0264-410x(03)00027-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park S-J, Kim H-K, Song D-S, An D-J, Park B-K. 2012. Complete genome sequences of a Korean virulent porcine epidemic diarrhea virus and its attenuated counterpart. J Virol 86:5964. doi: 10.1128/JVI.00557-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nasirudeen AMA, Liu DX. 2009. Gene expression profiling by microarray analysis reveals an important role for caspase-1 in dengue virus-induced p53-mediated apoptosis. J Med Virol 81:1069–1081. doi: 10.1002/jmv.21486. [DOI] [PubMed] [Google Scholar]
- 53.Yamada Y, Liu DX. 2009. Proteolytic activation of the spike protein at a novel RRRR/S motif is implicated in furin-dependent entry, syncytium formation, and infectivity of coronavirus infectious bronchitis virus in cultured cells. J Virol 83:8744–8758. doi: 10.1128/JVI.00613-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]