

Key Points
SIRP Abs SIRP-1 and SIRP-2 induce macrophage phagocytosis of cancer cells.
They employ unique mechanisms to disrupt SIRPα/CD47 checkpoint and engage FcγRs.
SIRP-1 and SIRP-2 combine with targeted Abs including anti-CD47 mAb AO-176.
Visual Abstract
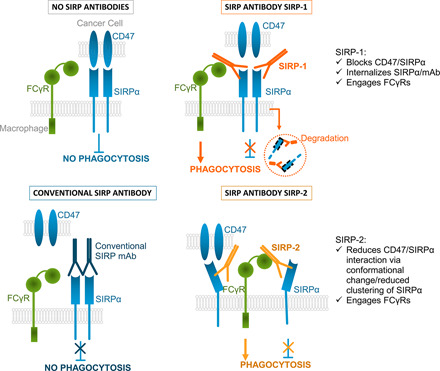
Abstract
The signal regulatory protein α (SIRPα)/CD47 axis has emerged as an important innate immune checkpoint that enables cancer cell escape from macrophage phagocytosis. SIRPα expression is limited to macrophages, dendritic cells, and neutrophils—cells enriched in the tumor microenvironment. In this study, we present novel anti-SIRP Abs, SIRP-1 and SIRP-2, as an approach to targeting the SIRPα/CD47 axis. Both SIRP-1 and SIRP-2 bind human macrophage SIRPα variants 1 and 2, the most common variants in the human population. SIRP-1 and SIRP-2 are differentiated among reported anti-SIRP Abs in that they induce phagocytosis of solid and hematologic tumor cell lines by human monocyte-derived macrophages as single agents. We demonstrate that SIRP-1 and SIRP-2 disrupt SIRPα/CD47 interaction by two distinct mechanisms: SIRP-1 directly blocks SIRPα/CD47 and induces internalization of SIRPα/Ab complexes that reduce macrophage SIRPα surface levels and SIRP-2 acts via disruption of higher-order SIRPα structures on macrophages. Both SIRP-1 and SIRP-2 engage FcγRII, which is required for single-agent phagocytic activity. Although SIRP-1 and SIRP-2 bind SIRPγ with varying affinity, they show no adverse effects on T cell proliferation. Finally, both Abs also enhance phagocytosis when combined with tumor-opsonizing Abs, including a highly differentiated anti-CD47 Ab, AO-176, currently being evaluated in phase 1 clinical trials, NCT03834948 and NCT04445701. SIRP-1 and SIRP-2 are novel, differentiated SIRP Abs that induce in vitro single-agent and combination phagocytosis and show no adverse effects on T cell functionality. These data support their future development, both as single agents and in combination with other anticancer drugs.
Introduction
Anticancer therapies to enhance adaptive immunity, including Abs against the T cell checkpoints, programmed cell death 1 (PD-1), programmed death ligand 1 (PD-L1), and CTLA-4, have raised the prospect of long-term remission or even cure for patients with metastatic disease (1, 2). Despite this promise, a significant patient population either fails to respond to checkpoint blockade or eventually develops resistance and experiences disease progression (3–5). Poor tumor antigenicity, additional inhibitory checkpoints on the tumors and in the tumor microenvironment, a lack of tumor infiltrating T cells, and/or the presence of immunosuppressive cells have all been attributed to a lack of efficacy (5). These findings highlight the need for alternative or synergistic approaches to boost antitumor immunity.
Myeloid cells (macrophages, dendritic cells, monocyte-derived suppressor cells, and granulocytes) represent the most abundant immune cell type in many solid tumors and are often linked to poor prognosis (6, 7). The antitumor activity of both tissue-resident and monocyte-derived macrophages (moMΦs) is rapidly compromised by the tumor microenvironment, which reprograms them into cancer-supporting, immunosuppressive tumor-associated macrophages. Harnessing the power of macrophages toward consuming and killing tumor cells has emerged as a promising therapeutic strategy. The CD47/SIRPα interaction regulates macrophage and dendritic cell phagocytosis of target cells, sending an inhibitory “do-not-eat-me” signal to the phagocyte. The physiological function of CD47 on normal cells is to act as a marker of self to prevent their being phagocytosed in addition to blocking a subsequent autoimmune response (8, 9). Upregulation of CD47 by cancer cells to evade innate immune surveillance has been associated with poor prognosis in multiple hematopoietic and solid tumor types (10).
The CD47 receptor, SIRPα, is a member of the closely related SIRP family of paired receptors, which also includes SIRPβ1, and the decoy receptor SIRPγ. SIRPα, less widely expressed than CD47, is mainly found on myeloid hematopoietic cells, including macrophages, dendritic cells, and granulocytes, and on neurons and some tumor cells (6). Binding of CD47 to macrophage SIRPα initiates its recruitment to the phagocytic synapse (11), phosphorylation of its cytoplasmic ITIMs (12, 13), recruitment and binding of SHP-1 and SHP-2, Src homology domain–containing protein tyrosine phosphatases (12, 14), inhibition of nonmuscle myosin IIA, and ultimately phagocytic function (6, 15–17). The gene encoding human SIRPα is polymorphic. The two most prevalent variants, SIRPαV1 and SIRPαV2, contain amino acid changes within the extracellular domain that do not appear to affect binding to CD47 or phagocytic activity (6, 18). Although the related SIRPβ1 receptor does not interact with CD47, binding of CD47 to SIRPγ, expressed by human, but not rodent, T cells and NK cells, has been shown to mediate T cell adhesion to APCs, resulting in T cell activation and proliferation (6, 19). Thus, blocking the SIRPα and CD47 interaction while preserving SIRPγ binding to CD47 may be an optimal strategy for cancer immunotherapy.
Blocking macrophage and dendritic cell SIRPα interactions with tumor cell CD47 has emerged as a viable strategy in cancer therapy. Numerous CD47-targeting agents, including Abs and SIRPα fusion proteins, are in early clinical development and have shown promising efficacy (20–22). Single-agent activity in humans appears to require anti-CD47 agents with an active Fc region to engage macrophage Fc receptors and release the “do-not-eat-me” checkpoint (23). The expression of CD47 on normal cells, particularly RBCs and platelets, has led to adverse events like acute anemia and thrombocytopenia (20, 22, 24–28). To address these, combination treatments using non–effector function–enabled anti-CD47 agents with a tumor-opsonizing Ab (29, 30) or performing intratumoral administration of anti-CD47 agents (21) have been used. AO-176, a next-generation anti-CD47 Ab that preferentially binds tumor versus normal cells and negligibly binds RBCs (31) provides another approach to increase the therapeutic index.
Because of the absence of SIRPα on RBCs and its restricted expression to myeloid cells, targeting SIRPα provides another potential therapeutic option to induce phagocytosis of tumor cells on the CD47 axis. Preclinical data have indicated that anti-SIRPα Abs that block the SIRPα/CD47 interaction exhibit antitumor efficacy in combination with opsonizing Abs or as single agents via a combination of Ab-dependent cellular phagocytosis (ADCP) and CD47/SIRPα blockade if the tumors also express SIRPα (7, 32, 33). However, because most tumors do not express SIRPα, lack of opsonization required combination of SIRPα Abs with tumor-targeting Abs to provide an “eat-me” signal to macrophages via engagement of the macrophage Fc receptors. The FcγR family is the largest family of Fc receptors and comprise FcγRI (CD64), FcγRII (CD32), and FcγRIII (CD16), all of which are expressed by human macrophages. All activating FcγRs signal via the ITAM pathway to remodel cytoskeleton for phagocytic internalization, engulfment, and release of proinflammatory mediators such as cytokines and reactive oxygen species.
Several anti-SIRP Abs are in early clinical (BI 765063) or preclinical development (KWAR23, ADU-168, and others), but none of them induce phagocytosis as single agents (32, 34–36). Furthermore, some block the beneficial interaction of SIRPγ with CD47 (33).
In the current study, we describe unique anti-SIRPα Abs with potentially best-in-class properties that can induce phagocytosis of tumor cells by human macrophages as single agents, unlike other published anti-SIRP Abs. These agents also combine with tumor-targeted Abs, including an anti-CD47 Ab. Finally, these unique anti-SIRPα Abs also disrupt the interaction of CD47 with human macrophages via two novel and distinct mechanisms while preserving T cell functionality.
Materials and Methods
Cell culture
Human tumor lines (Jurkat T-ALL, RAJI B cell lymphoma, DLD-1 colorectal adenocarcinoma, RL95-2 endometrial carcinoma, and ES-2 ovarian carcinoma), and the monocyte lines U937 and THP-1 were purchased from American Type Culture Collection and cultured as recommended by the vendor. Human moMΦs were differentiated from CD14+ monocytes purchased from Astarte Biologics or selected using Pan Monocyte Isolation Kit (Miltenyi Biotec) from PBMCs (AllCells and Hemacare). Monocytes were seeded onto 96-well, flat-bottom plates at 3 × 104 cells per well and differentiated into moMΦs in vitro for 7 d in AIM-V medium (Life Technologies) supplemented with 10% FBS and 50 ng/ml M-CSF (BioLegend). Human dendritic cells were generated by incubating monocytes in AIM-V medium (Life Technologies) supplemented with 10% human AB serum (Valley Biomedical), 200 ng/ml GM-CSF (BioLegend), and 50 ng/ml IL-4 (BioLegend) for 6 d. For T cell activation, healthy donor CD3+ T cells (Astarte Biologics) were stimulated with 5 μg/ml plate-bound anti-CD3 (clone UCHT1; eBioscience) and soluble anti-CD28 (clone 28.8; eBioscience) in AIM-V medium supplemented with 10% human AB serum (Valley Biomedical) and 1% penicillin/streptomycin. All cultures were maintained at 37°C in a humidified atmosphere containing 5% CO2.
mAb generation
mAbs against human SIRPα were generated by immunizing wild-type mice with recombinant human SIRPα containing all three extracellular immunoglobin domains fused to GST. Following repetitive immunization, the spleen cells were fused with the nonsecreting myeloma P3 × 63Ag8.653 (American Type Culture Collection), and clones were screened for reactivity to human SIRPαV1. For sequencing of successful clones, RNA was isolated from hybridoma cells, and immunoglobin cDNA was synthesized according to established methods using a deoxythymidine oligonucleotide primer and reverse transcriptase (31). Mouse clones of SIRP-1, SIRP-2, 18D5 (33), and KWAR23 (32) on a murine IgG1 backbone were expressed by Evitria (Schlieren, Switzerland) and purified in-house using MabSelect SuRe resin (GE Healthcare). Murine IgG1 control was purchased from BioLegend. Rituximab, avelumab, and cetuximab were sourced from Myoderm.
Genotyping of human macrophage donors
Genomic DNA was isolated from donor PBMCs using the PureLink Genomic DNA Mini Kit (Thermo Fisher Scientific). PCR amplification of the SIRPα target region (exon 3) and Sanger sequencing were carried out by Genewiz (Piscataway, NJ) as previously described (35). The following primer pairs were used for amplification: forward 5′-TGTCTGGAATACCAGGCTCCCTT-3′ and reverse 5′-TACCACCACACCTGATCATTGCTC-3′. Five different Sanger sequencing reactions were performed. The primers used for each of the five reactions were the following: 5′-GGCTCCCTTTCCGGAACTTCACACAG-3′; 5′-GTGTGAAGTTCCGGAAAGGGAGCCCCGAT-3′; 5′-GCTCCAGACTTAAACTCCACGTCATCGG-3′; 5′-CCTGCTCCAGACTTAAACTCCACGTCAG-3′; and 5′-GTGTGAAGTTCCGGAAAGGGAGCCCT-3′. Alignment of sequencing data was done using Sequencher DNA sequence analysis software (v5.2.4; Gene Codes Corporation, Ann Arbor, MI). The data were validated using U937 and THP-1 cell lines, which are homozygous for SIRPαV1 and SIRPαV2, respectively.
Binding to human or cynomolgus monkey SIRPα by ELISA
Fc-tagged human SIRPαV1 or polyhistidine-tagged cynomolgus monkey SIRPα (ACROBiosystems) was adsorbed to high-binding microtiter plates at 2 μg/ml in PBS, blocked with 1% casein in PBS containing 0.5% Tween 20 (PBST), and incubated with serially diluted SIRP Abs. The samples were then incubated with HRP-labeled donkey anti-mouse secondary Ab (Jackson ImmunoResearch Laboratories). Peroxidase substrate 3,3′,5,5′-tetramethylbenzidine (Thermo Fisher Scientific) color development was stopped using 1N H2SO4 and the absorbance at 450 nm determined using Synergy [1H] plate reader (BioTek). The apparent binding affinities were calculated using a nonlinear one-site fit model (Prism v8; GraphPad, San Diego, CA).
Cell-based binding of SIRP Abs
To assess the binding of Abs to cell-expressed SIRPα, U937 or THP-1 cell lines or moMΦs were used. For binding to cell-expressed SIRPγ, Jurkat T-ALL cell line or naive or activated human CD3+ T cells were used. The cells were incubated for 1 h at 37°C, 5% CO2 with serial dilutions of SIRP Abs in binding buffer containing 1 mM EDTA (Sigma-Aldrich), and 1% FBS (Biowest) in PBS without Ca2+ and Mg2+ (Corning). For Jurkat, DLD-1, RAJI, HEC1-A, RL95-2, and ES-2 tumor cell lines, 10 μg/ml of Abs was used. The cells were then stained with donkey anti-mouse IgG FITC-linked secondary Ab (Jackson ImmunoResearch Laboratories). moMΦs were additionally stained with anti-CD14 conjugated to Alexa Fluor 647 (AF647; BioLegend). T cells were additionally stained with anti-CD3 conjugated to 3-carboxy-6,8-difluoro-7-hydroxycoumarin (Pacific Blue; BioLegend). The cells were analyzed by flow cytometry (Attune NxT; Life Technologies) using FlowJo Software (v10 for Windows; Becton Dickinson). For moMΦ and T cells, the binding was assessed as the median FITC fluorescence intensity, subtracted from cells stained with secondary Ab alone. The apparent binding affinities were calculated using a nonlinear four parameter variable slope fit model (Prism v8; GraphPad).
Blocking CD47 binding to SIRPα by ELISA
Polyhistidine-tagged human CD47 (ACROBiosystems) was adsorbed to high-binding microtiter plates at a concentration of 2 μg/ml in PBS overnight at 4°C and blocked with 1% casein in PBST for 1 h at room temperature. Serially diluted SIRP Abs containing 1 μg/ml human Fc-tagged SIRPα (ACROBiosystems) were added to the plates for 1 h at room temperature. HRP-labeled donkey anti-human Ab (The Jackson Laboratory), diluted 1:20,000 in PBST, was added to the plate and incubated for 1 h at room temperature. Following addition of 3,3′,5,5′-tetramethylbenzidine, color development was stopped with 1 N H2SO4, and absorbance was measured at 450 nm. The apparent binding affinities were calculated using a nonlinear four parameter variable slope fit model (Prism v8; GraphPad).
Blocking the CD47/SIRPα interaction on macrophages
Fc receptors on moMΦ, THP-1, or U937 cells were blocked with human Fc receptor blocking solution (BioLegend) for 20 min at room temperature. The cells were then incubated for 1 h at 37°C, 5% CO2 with 10 μg/ml of SIRP mAbs in buffer containing 1 mM EDTA (Sigma-Aldrich), and 1% FBS (Biowest) in PBS (Corning). Subsequently, murine IgG2a Fc-tagged human CD47 (ACROBiosystems) was added at 20 μg/ml, and the cells were incubated as previously. Following staining with donkey anti-mouse IgG2a Ab–AF647 conjugate (BioLegend), the samples were analyzed by flow cytometry. Background murine IgG2a staining in the absence of Fc-tagged CD47 was subtracted from median AF647 fluorescence intensity. Blocking was assessed as the reduction in background-corrected median fluorescence intensity of AF647 in the presence of SIRP mAbs compared with murine IgG1 (clone MOPC-21; BioLegend) control. Ordinary one-way ANOVA was performed to compare SIRP mAb treatments to control using Dunnett multiple comparisons test (Prism v8; GraphPad).
Induction of phagocytosis
Human moMΦs, differentiated as described above, were cultured in AIM-V medium for 2 h. Human cancer cells or autologous PBMCs were labeled with 1 μM CFSE (Sigma-Aldrich) or with 10 μg/ml pHrodo Red (Incucyte pHrodo Red Cell Labeling Kit for Phagocytosis, Sartorius) and added to the macrophage cultures in 96-well plates at 8 × 104 cells per well in AIM-V medium without supplements. Anti-SIRP mAbs were added at varying concentrations immediately and incubated at 37°C for 3 h. For combination with tumor-opsonizing Abs, SIRP mAbs alone, tumor-targeting mAb alone, or a combination were added at varying concentrations as above. For combination with Fc blocking reagents, 10 μg/ml anti-CD16 (clone 3G8; Invitrogen), anti-CD32 (clone AT10; Invitrogen), anti-CD64 (clone 10.1; Invitrogen), or mIgG1 isotype control were added together or each separately immediately prior to adding SIRP Abs. Following phagocytosis assay, the nonadherent, nonphagocytosed cells were removed, and the remaining cells were washed in PBS. Cells were detached using Accutase (STEMCELL Technologies), collected into V-bottom plates, and incubated in 100 ng of allophycocyanin-labeled CD14 Abs (BD Biosciences) for 30 min, then analyzed by flow cytometry (Attune NxT; Life Technologies). Phagocytosis was calculated as the percentage of CFSE+ cells within the CD14+ population.
SIRP mAb internalization
SIRP Abs were labeled using pHrodo Green Microscale Protein Labeling Kit (Invitrogen), per manufacturer’s instructions. Labeling efficiency was comparable between all the Abs. The Abs were diluted into macrophage growth medium and warmed to 37°C prior to incubating 10 μg/ml mAbs with macrophages at 37°C for a maximum of 1 h. After incubation, the cells were washed in ice-cold PBS and analyzed by flow cytometry on Attune NxT (Life Technologies). Internalization was quantified as the percentage of pHrodo Green+ live single cells.
Flow cytometry of SIRPα/SIRPγ/CD47 surface expression
Following differentiation, human moMΦs were incubated in AIM-V medium without supplements for 2 h in V-bottom, 96-well plates. The cells were incubated with 10 μg/ml SIRP Abs for 2 h on ice or for 2, 4, 6, or 24 h at 37°C, 5% CO2. The cells were then washed in flow cytometry buffer (2% FBS in PBS) and stained for 30 min with 10 μg/ml fluorescently conjugated SIRP Abs in the presence of human TruStain FcX block (BioLegend). For detection of cell surface SIRPα on SIRP-1–, 18D5-, and KWAR23-treated macrophages, the noncompeting, AF647-labeled SIRP-2 was used, generated using AF647 Ab Labeling Kit (Life Technologies). In the case of SIRP-2 treatment, the noncompeting, PE-conjugated commercial clone SE5A5 (BioLegend) was used. The samples were analyzed by flow cytometry on Attune NxT (Life Technologies) using FlowJo Software. Fluorescence levels were normalized to median fluorescence intensity of mIgG1-treated moMΦ, stained with the respective fluorescent SIRP Abs on ice.
For tumor cell line surface expression, commercial Abs against SIRPα (clone SE5A5-PE; BioLegend), SIRPγ (clone LSB2.20-PE; BioLegend), or CD47 (clone B6H12-PE; Santa Cruz Biotechnology) in the presence of human Fc block (BioLegend) were incubated for 30 min on ice with tumor cell lines in flow cytometry buffer. Flow cytometry was performed as above.
Fluorescence resonance energy transfer analyzed by flow cytometry
PE and allophycocyanin-labeled Abs against SIRPα were used according to fluorescence resonance energy transfer analyzed by flow cytometry (FCET) methodology (37). An energy transfer (FCET) between an Ab–PE-conjugated donor and an Ab–allophycocyanin-conjugated acceptor indicate that the two molecules are in close proximity. Human moMΦs were incubated in AIM-V medium without supplements for 2 h in V-bottom, 96-well plates, then incubated with 10 μg/ml SIRP-1, SIRP-2, or mIgG1 control for 2 h at 37°C, 5% CO2. Equimolar concentration of SIRP Ab clone SE5A5-PE and SE5A5–allophycocyanin (BioLegend) were combined (for 10 μg/ml final) and added to the cell suspension after washing. For single-fluorophore staining, cells were labeled with SE5A5-PE or SE5A5–allophycocyanin and equimolar quantities of unlabeled SE5A5. The cells were stained for 30 min on ice, washed, and analyzed on Attune NxT flow cytometer (Life Technologies) using FlowJo Software. Fluorescence resonance energy transfer (FRET) efficiency was calculated using three-wavelength correction method (37), assuming 1:1 protein-to-dye ratio (information provided by BioLegend). Ordinary one-way ANOVA was performed to compare SIRP mAb treatments to mIgG1 control using Dunnett multiple comparisons test (Prism v8; GraphPad).
Allogeneic dendritic cell/T cell assay
Dendritic cells were plated onto a 96-well plate at 1 × 104 cells per well. CellTrace Violet (Life Technologies) dye–labeled allogeneic healthy donor CD3+ T cells from four different donors (Astarte Biologics) were added at a 1:5 dendritic cell/T cell ratio. SIRP Abs were added at the saturating concentration of 10 μg/ml immediately and the cells incubated at 37°C, 5% CO2, for 6–7 d. Cells were collected, incubated with PerCP-Cy5.5 fluorescent dye–labeled CD3 Ab (BioLegend) and analyzed by flow cytometry (Attune; Life Technologies). T cell proliferation was measured by the dilution of the CellTrace Violet dye within the CD3+ cell population.
Results
Generation and characterization of pan-allele SIRPα Abs
Murine hybridoma-generated Abs against human SIRPα were screened for binding to a recombinant human SIRPα ectodomain fusion protein using solid-phase ELISA (Fig. 1A). The affinities of SIRP-1 and SIRP-2 Abs were in the picomolar range (Kd of 300 ± 140 pM and 290 ± 250 pM, respectively) and were comparable to other SIRPα Abs in development, namely KWAR23 (32) (Kd of 200 ± 50 pM) and 18D5 (33, 36) (Kd of 130 ± 90 pM). SIRP-1 and SIRP-2 bound to cynomolgus monkey SIRPα with similar affinities (Kd of 310 and 210 pM, respectively) (Supplemental Fig. 1A). Because KWAR23 and 18D5 block the SIRPα/CD47 interaction (32, 33), we performed studies to assess whether SIRP-1 and SIRP-2 bind to distinct or overlapping regions on SIRPα. Competitive ELISA assays suggest that SIRP-2 does not significantly compete with SIRP-1, KWAR23, or 18D5 for SIRPα binding (Supplemental Fig. 1B). The binding of SIRP-1 only slightly impeded the binding of KWAR23 and SIRP-2 but competed with the binding of 18D5 to recombinant human SIRPα (Supplemental Fig. 1B). In contrast to SIRP-1, KWAR23 completely inhibited the binding of 18D5 (Supplemental Fig. 1B).
FIGURE 1.
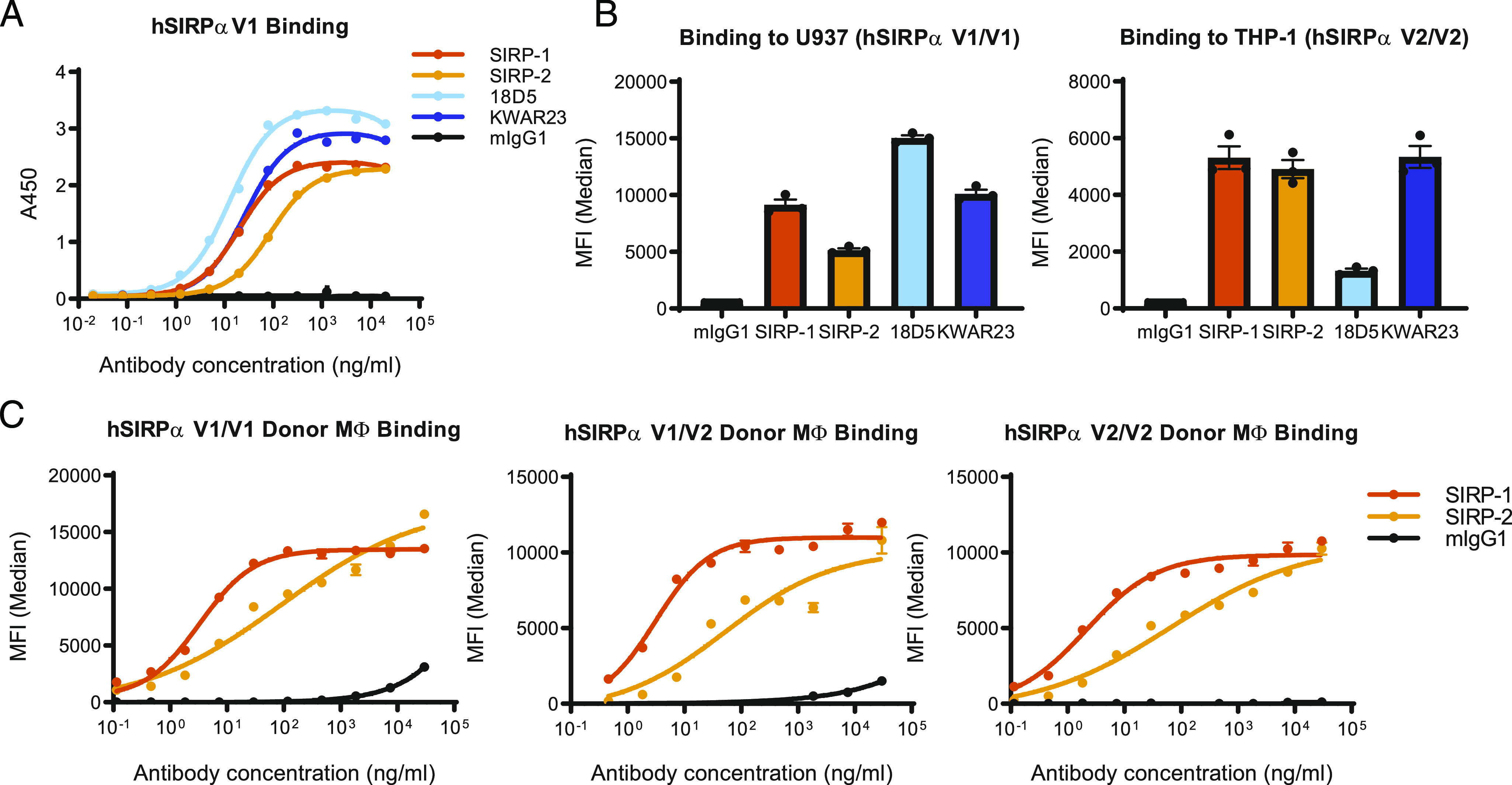
SIRP-1 and SIRP-2 bind to recombinant and cell-expressed human SIRPα. (A) SIRP-1 and SIRP-2 Abs were screened by solid-phase ELISA for binding to human SIRPαV1 (hSIRPαV1) and compared with competitor benchmarks KWAR23 and 18D5. (B) At 10 μg/ml SIRP-1, SIRP-2 and KWAR23 bound to cell-expressed SIRPαV1 and SIRPαV2 on promonocytic cell lines U937 and THP-1, respectively, whereas benchmark 18D5 only bound SIRPαV1. (C) SIRP-1 and SIRP-2 bound to human moMΦs from most-common allelic groups. All panels show mean ± SEM; a representative minimum n = 2 is shown.
Although up to 10 SIRPα haplotypes were initially reported (38, 39), three allelic groups (homozygous V1/V1, heterozygous V1/V2, and homozygous V2/V2) account for virtually all ethnic groups that have been genotyped (18, 35). A pan-allele–specific Ab against SIRPα would most broadly target the SIRPα/CD47 checkpoint in diverse populations, as heterozygotes require blocking of both alleles of SIRPα to enhance macrophage-mediated phagocytosis (35, 40). In cell-based binding assays, SIRP-1 and SIRP-2 bound to both human monocytic cell lines U937 (SIRPαV1/V1) and THP-1 (SIRPαV2/V2) (41), unlike 18D5, which only bound to U937, as previously described (Fig. 1B, Supplemental Fig. 1C) (42). We further confirmed that SIRP-1 and SIRP-2 both bound to human moMΦs expressing endogenous SIRPα of all three allelic groups (Fig. 1C, Supplemental Fig. 1D, 1E).
SIRP-1 and SIRP-2 induce macrophage-dependent phagocytosis of human cancer cell lines as single agents
To further characterize the SIRP-1 and SIRP-2 Abs, we assessed their ability to induce phagocytosis of a set of hematological and solid tumor lines in vitro. Human moMΦs expressing endogenous SIRPα were incubated with CFSE-labeled cancer cell lines expressing CD47 in the presence of Abs against SIRPα. SIRP-1 and SIRP-2 induced phagocytosis of the Jurkat human T-ALL cell line in the absence of a tumor-opsonizing Ab, in contrast to KWAR23 and 18D5 (Fig. 2A, Supplemental Fig. 2A, 2B), as measured by the appearance of CFSE labeling in the moMΦ population. To confirm that the assay measured true phagocytosis rather than conjugate formation between macrophages and tumor cells, we validated our findings in a second phagocytosis assay using Jurkat cells labeled with pHrodo Red, a fluorescent dye that increases fluorescence only in the acidic environment of the phagosome. The percentage of phagocytosis obtained by the two methods was comparable (Fig. 2A, 2B, Supplemental Fig. 2), confirming that SIRP-1 and SIRP-2 induced tumor cell engulfment by moMΦs. SIRP-1– and SIRP-2–induced phagocytosis was not limited to Jurkat cells but was also seen in the other tested solid tumor cell lines: RL95-2 (endometrial carcinoma) and DLD-1 (colorectal adenocarcinoma) (Fig. 2C). The ability of SIRP-1 and SIRP-2 to induce single-agent phagocytosis of cancer cell lines was observed in donors from all three major allelic groups of SIRPα (Fig. 2D, 2E). To assess the effect of SIRP-1 and SIRP-2 on the phagocytosis of normal noncancerous cells, human moMΦs were incubated either with normal autologous PBMCs or Jurkat T-ALL cells in the presence of SIRP Abs. SIRP-1 and SIRP-2 potently induced phagocytosis of Jurkat T-ALL cells, but not of PBMCs (Fig. 2F). Thus, we demonstrate SIRP Abs with single-agent phagocytosis activity in both hematologic and solid tumor cell lines.
FIGURE 2.

SIRP-1 and SIRP-2 Abs induce single-agent phagocytosis of a range of cancer cell lines. (A–C) SIRP-1 and SIRP-2 induced human moMΦs to phagocytose a panel of hematopoietic and solid tumor cell lines. CFSE-labeled (A, C, and D–F) or pHrodo Red–labeled (B) target cells were added to moMΦs and phagocytosis measured as the percentage of moMΦs that had engulfed target cells (CFSE+ or pHrodo+) of the total macrophage population. Single-agent phagocytosis of Jurkat T-ALL cells with SIRP-1 (D) and SIRP-2 used at 10 μg/ml (E) is seen in human moMΦs of all three most common allelic groups of SIRPα. The crosses in (A)–(D) indicate mIgG1 control at 10 μg/ml. (F) SIRP-1 and SIRP-2 did not induce moMΦ phagocytosis of autologous PBMCs in contrast to Jurkat T-ALL cells. All panels show mean ± SEM; a representative minimum n = 2 is shown.
SIRP-1 and SIRP-2 induce single-agent, macrophage-dependent phagocytosis by two distinct mechanisms
We next sought to further understand the ability of SIRP-1 and SIRP-2 to confer single-agent phagocytosis activity. Even though SIRP-1 and SIRP-2 are of the relatively inert murine IgG1 isotype (43), one possible mechanism could be through induction of ADCP. This mechanism has been described in syngeneic mouse models in which murine SIRPα Abs with an active Fc were able to reduce the growth of RENCA and B16BL6 cell lines, both of which express SIRPα (7). In contrast, in the syngeneic CT26 model and a Raji xenograft model, neither of which express SIRPα, the same Abs against SIRPα reduced tumor growth only when combined with tumor-opsonizing Abs (7). In our panel of cell lines, Jurkat cells expressed SIRPγ, but not SIRPα, whereas DLD-1 and RL95-2 cell lines expressed neither (Fig. 3A). As expected, all expressed CD47 (Fig. 3A). Only SIRP-2 bound to Jurkat cells, and neither SIRP-1 nor SIRP-2 bound DLD-1 or RL95-2 cells (Fig. 3B). However, both SIRP-1 and SIRP-2 were able to induce single-agent phagocytosis of all the cancer cell lines, suggesting that the efficacy of these Abs is independent of target expression on cancer cells and that single-agent activity is likely not mediated by ADCP.
FIGURE 3.

SIRP-1 and SIRP-2 binding to tumor cell lines correlates to their expression of SIRPα/SIRPγ. (A) Expression of CD47, SIRPα/β, and SIRPγ by cancer cell lines used in phagocytosis assays. Representative histograms from one of two technical replicates from one of two independent experiments are shown. (B) The binding of SIRP-1 and SIRP-2 at 10 μg/ml correlates well with SIRPα/β or SIRPγ expression seen by commercial anti-SIRP Abs. Data are expressed as mean ± SEM, representative of n = 2.
To further investigate the mechanism of SIRP-1 and SIRP-2 activity, we conducted experiments to elucidate the ability of these Abs to block interaction of SIRPα to its ligand, CD47. In ELISA-based blocking assays, we found that SIRP-1, but not SIRP-2, blocked the interaction of recombinant CD47 with SIRPα with almost identical potency as KWAR23 and 18D5 (Fig. 4A). We next tested the ability of SIRP-1 and SIRP-2 to block the binding of human CD47 to SIRPα on U937 and THP-1 cell lines. Consistent with the blocking data from solid-phase ELISA assays, SIRP-1 reduced the binding of recombinant CD47 to U937 and THP-1 cells (Fig. 4B, Supplemental Fig. 3A). Surprisingly, SIRP-2 also reduced the binding of recombinant CD47 to both these cell lines. Similarly, both SIRP-1 and SIRP-2 reduced CD47 binding to human moMΦs derived from SIRPα homozygous V1/V1, V2/V2, or heterozygous V1/V2 donors (Fig. 4C). Thus, SIRP-1 and SIRP-2 appear to be pan-allele–specific SIRPα Abs that disrupt the interaction of macrophage SIRPα with CD47. These data also demonstrate that blocking the CD47/SIRPα interaction through binding to SIRPα is not sufficient for phagocytosis induction because both KWAR23 and 18D5 do not exhibit single-agent phagocytosis activity (Fig. 2A, Supplemental Fig. 3B).
FIGURE 4.
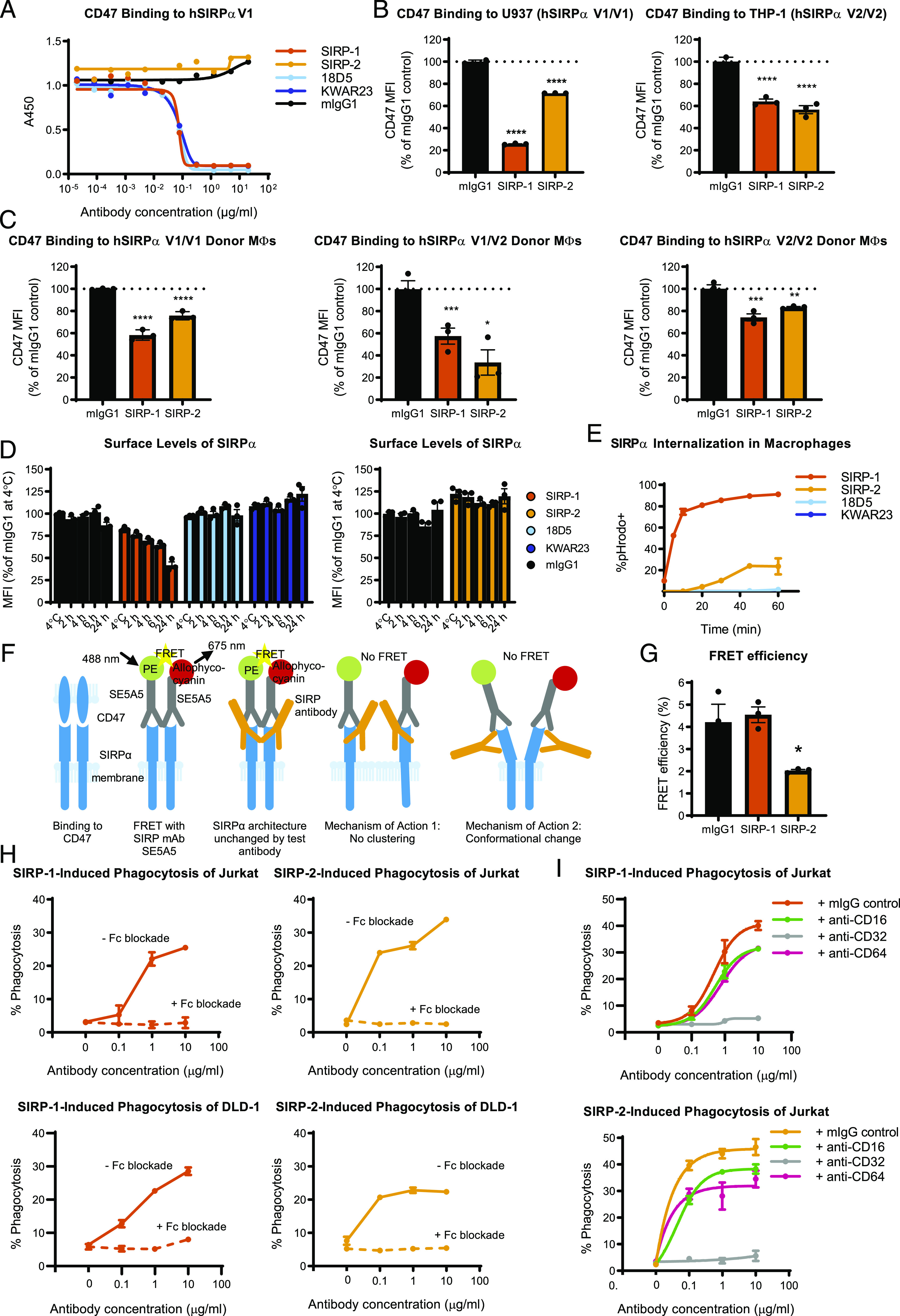
SIRP-1 and SIRP-2 act via distinct mechanism of action: internalization and conformational change/declustering of SIRPα. (A) Blocking soluble CD47 binding to human SIRPαV1 (hSIRPαV1) was assessed by ELISA. Reduction of 20 μg/ml soluble CD47 binding to cell-expressed SIRPαV1 and SIRPαV2 by 10 μg/ml SIRP Abs was assessed using (B) promonocytic cell lines U937 and THP-1 and (C) moMΦs from various donor genotypes. (D) SIRP-1, but not mIgG1 control, 18D5, KWAR23, or SIRP-2 at 10 μg/ml reduced surface SIRPα levels in moMΦs in a time-dependent manner. The cells were incubated with Abs for 2 h at 4°C or 2, 4, 6, or 24 h at 37°C. (E) A total of 10 μg/ml SIRP-1 and, to a lesser extent, SIRP-2 caused Ab internalization in a time-dependent manner when measured by the uptake of pHrodo dye–Ab conjugates. (F) SIRPα binds CD47 most efficiently when clustered. When SIRPα Ab clone SE5A5, labeled with PE or allophycocyanin in equimolar concentrations, is added to cells, FRET is observed if SIRPα molecules and Abs bound to them are in close proximity. When a noncompeting anti-SIRP Ab is added that does not alter SIRPα architecture, no reduction in FRET is seen. No FRET is observed when a noncompeting anti-SIRP Ab that disables SIRPα from clustering is added. No FRET also occurs if binding of a noncompeting SIRPα mAb induces a conformation change in SIRPα that increases the distance between SE5A5 epitopes. (G) Treatment of moMΦs with 10 μg/ml SIRP-2, but not SIRP-1, for 2 h under the same conditions as in phagocytosis assays reduced the FRET efficiency between anti-SIRP Abs SE5A5-PE and SE5A5–allophycocyanin, indicating SIRPα conformational change/receptor declustering upon SIRP-2 treatment. (H) Addition of 10 μg/ml blocking Abs against human CD16, CD32, and CD64 inhibits SIRP-1– and SIRP-2–induced phagocytosis of Jurkat and DLD-1 cells by human moMΦs. (I) Function-blocking Abs against CD32 at 10 μg/ml, but not CD16 or CD64, inhibit SIRP-1– and SIRP-2–mediated Jurkat phagocytosis of moMΦs. All panels show mean ± SEM; a representative minimum n = 2 is shown. Statistical differences compared with the mIgG1 control are indicated. *p < 0.05, **p < 0.01, ***p < 0.01, ****p < 0.0001.
We next tested whether the single-agent phagocytosis activity of SIRP-1 and SIRP-2 could be due to changes in the surface availability of macrophage SIRPα. Indeed, preincubation of macrophages at 37°C with SIRP-1 resulted in a time-dependent reduction of surface level of SIRPα compared with a 2-h incubation at 4°C (Fig. 4D, Supplemental Fig. 3C). No such reduction was observed when macrophages were incubated with mIgG1, 18D5, KWAR23, or SIRP-2 (Fig. 4D, Supplemental Fig. 3D, 3E). To test whether SIRP-1 mediated the internalization of endogenous SIRPα-Ab complexes, we used Abs labeled with the pH-sensitive fluorescent probe pHrodo. SIRP-1, and to a much lesser extent SIRP-2, but not KWAR23 or 18D5, caused rapid and sustained increase in pHrodo fluorescence within 30 min of macrophage treatment, suggesting rapid internalization of the SIRPα Ab-target complex (Fig. 4E, Supplemental Fig. 3F). This rapid time course is consistent with the assay length for measuring induction of phagocytosis by SIRP-1 in macrophages. Therefore, SIRP-1 acts via a dual mechanism of both blocking the interaction of SIRPα with CD47 and reducing cell surface levels of SIRPα by mediating its internalization.
Because SIRP-2 did not block the interaction of recombinant SIRPα/CD47 by ELISA, did not strongly promote SIRPα internalization by macrophages, and did not reduce the cell surface levels of SIRPα, we hypothesized that SIRP-2 might alter the avidity of cellular SIRPα for CD47. SIRPα has been shown to exist in clusters that are necessary for effective cell surface interaction with CD47 and inhibitory SIRPα signaling of the optimal “do-not-eat-me” signal (44, 45). In a separate study, SIRPα was shown to form cis-dimers (46). To investigate whether our SIRP Abs could interfere with the dimerization/clustering of SIRPα, required for efficient CD47 interaction and the “do-not-eat-me” signal, we performed an FRET applied in flow cytometry (FCET) (Fig. 4F) according to the method of Batard et al. (37). The authors calculated that for a PE/allophycocyanin donor/acceptor pair, the maximal theoretical FRET efficiency was 10% and that anything below 1% should be considered nondimerized/nonclustered. FRET efficiency values between 4 and 6% were observed for CD29/CD49, a known heterodimer pair. Therefore, we first incubated moMΦs with SIRP Abs for 2 h and counterstained them with commercial, noncompeting SIRP mAb clone SE5A5, labeled with PE, allophycocyanin, or both. Consistent with dimerization/clustering, we observed that FRET efficiency between SE5A5-PE and SE5A5–allophycocyanin in the presence of mIgG1 control was 4% (Fig. 4G). This was significantly reduced ∼2-fold in the presence of SIRP-2, suggesting that SIRP-2 may alter the avidity of cellular SIRPα for CD47 by impacting clustering on the cell surface. In contrast, no reduction in FRET efficiency was observed with SIRP-1, indicating that this mechanism of action is not shared by the two Abs and is unique to SIRP-2 (Fig. 4G).
In addition to blocking the CD47 “do-not-eat-me” signal, it is possible that SIRP-1 and SIRP2 could induce phagocytosis through an “eat-me” signal. We tested whether a prophagocytic signal from the macrophage Fc receptors is involved in mediating the single-agent phagocytosis activity of SIRP-1 and SIRP-2. A mixture of function-blocking Abs against CD16 (FcγRIII), CD32 (FcγRII), and CD64 (FcγRI) added immediately prior to incubating cancer cells with moMΦs completely inhibited both SIRP-1– and SIRP-2–mediated phagocytosis of Jurkat and DLD-1 cells (Fig. 4H). When assessed individually, functional blockade by mAbs against CD64 or CD16 had no impact on the ability of SIRP-1 and SIRP-2 to induce phagocytosis of Jurkat cells (Fig. 4I). Addition of function-blocking CD32 Abs, however, completely abrogated the single-agent phagocytosis activity of SIRP-1 and SIRP-2 (Fig. 4I). This suggests that single-agent phagocytic activity of SIRP-1 and SIRP-2 is dependent on CD32 (FcγRII) function. In addition to macrophage FcγR stimulation, other “eat-me” signals expressed on cancer cell surface such as phosphatidylserine, calreticulin, and heat shock proteins could also contribute “eat-me” signals toward phagocytosis of cancer cells by SIRP-1 and SIRP-2.
SIRP-1 and SIRP-2 preserve T cell functionality
The interaction of SIRPγ, expressed by both activated and naive T cells, with CD47 on APCs, has been shown to be required for maximal T cell responses (19). SIRP-1 and 18D5 did not bind the SIRPγ-expressing Jurkat T-ALL cell line, whereas SIRP-2 and KWAR23 did (Fig. 5A). We confirmed this effect in human donor-derived naive CD3+ T cells as well as T cells from the same donor activated with anti-CD3/anti-CD28 (Fig. 5B, Supplemental Fig. 4A). Likewise, SIRP-1 and 18D5 did not bind human T cells, whereas SIRP-2 bound them to a similar extent as an SIRPγ-specific Ab (clone LSB2.20) (Fig. 5B). The binding of KWAR23 to both naive and activated T cells was even higher (Fig. 5B).
FIGURE 5.
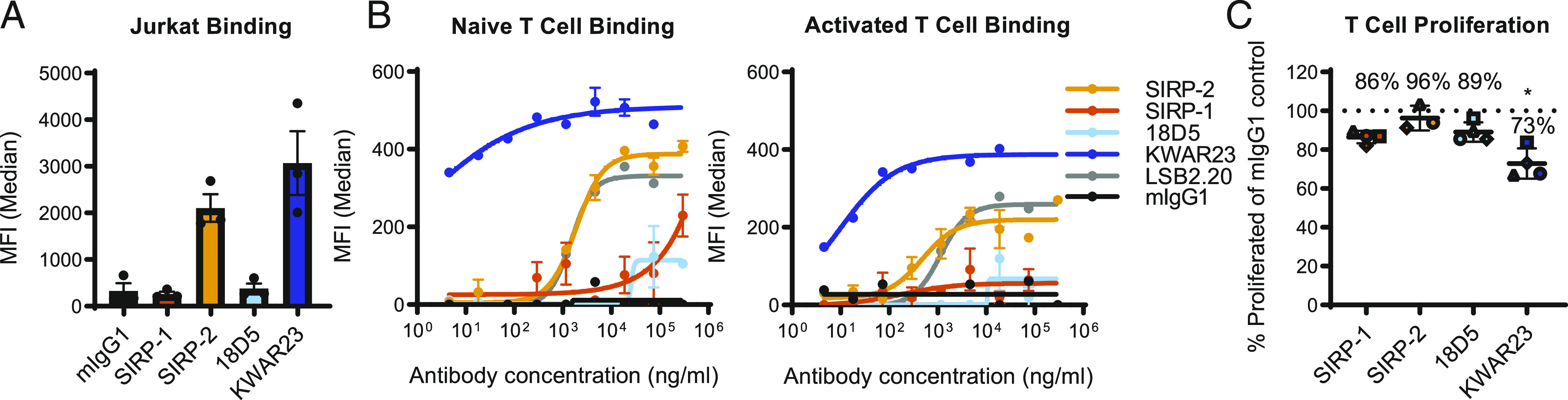
SIRP-1 and SIRP-2 do not inhibit T cell proliferation. (A) Binding of 10 μg/ml Abs to cell-expressed SIRPγ was assayed using T-ALL cell line Jurkat and compared with benchmarks 18D5 and KWAR23. (B) Binding to naive or 72-h-activated human T cells was determined by cell-based binding assays and compared with SIRPγ-specific Ab clone LSB2.20. (C) A total of 10 μg/ml SIRP-1 or SIRP-2 did not inhibit T cell proliferation in an allogeneic T cell/dendritic cell assay over the course of 6–7 d. Each data point represents mean from a separate donor (shown in different shapes), constituting an average of six technical replicates within a minimum of two independent replicates. The asterisks (*) indicate statistical differences compared with the mIgG1 control group. Unless otherwise stated, all panels show mean ± SEM; a representative minimum n = 2 is shown. *p < 0.05.
Because SIRP-2 bound human T cells and SIRP-1 did not, we evaluated the effects of SIRP-1 and SIRP-2 on T cell proliferation upon activation with allogeneic donor monocyte-derived dendritic cells. No impairment of T cell proliferation was observed upon stimulation in the presence of SIRP-1 or SIRP-2 (Fig. 5C, Supplemental Fig. 4B, 4C), suggesting that although SIRP-2 may bind T cells, it does not inhibit proliferation, unlike KWAR23, which reduced T cell proliferation (Fig. 5C, Supplemental Fig. 4C) in an allogeneic T cell/dendritic cell assay (33).
SIRP-1 and SIRP-2 potentiate ADCP of cancer cell lines in combination with tumor-targeted Abs
Many therapeutic tumor-targeting Abs use their Fc domain to exert multiple anticancer effects. Abs containing an IgG1 Fc are capable of Ab-dependent cellular cytotoxicity and ADCP in addition to complement fixation and variable domain-specific effects. We tested the ability of SIRP-1 and SIRP-2 to potentiate cancer cell phagocytosis in combination with IgG1 therapeutic tumor-opsonizing Abs. SIRP-1 and SIRP-2 induced macrophage phagocytosis of the Raji Burkitt lymphoma, ES-2 ovarian clear cell carcinoma, and DLD-1 colorectal adenocarcinoma cell lines in a dose-dependent manner as a single agent (Fig. 6A, 6B) in an in vitro coculture assay. The addition of tumor-opsonizing Abs against a range of targets: PD-L1 (avelumab), CD20 (rituximab), and epidermal growth factor receptor (cetuximab), combined with SIRP-1 or SIRP-2 to potentiate phagocytosis of a panel of hematologic and solid tumor cell lines (Fig. 6A, 6B) with varying levels of SIRPα/γ expression (Fig. 3, Supplemental Fig. 4D, 4E). We also observed increased phagocytosis with an anti-CD47 Ab, AO-176 (Fig. 6C), which could act via both tumor-opsonizing and additional CD47-blocking effects (31). Therefore, SIRP-1 and SIRP-2 have the potential to be used as single agents or in combination with tumor-opsonizing or CD47-blocking agents that are currently approved or being investigated for clinical use.
FIGURE 6.
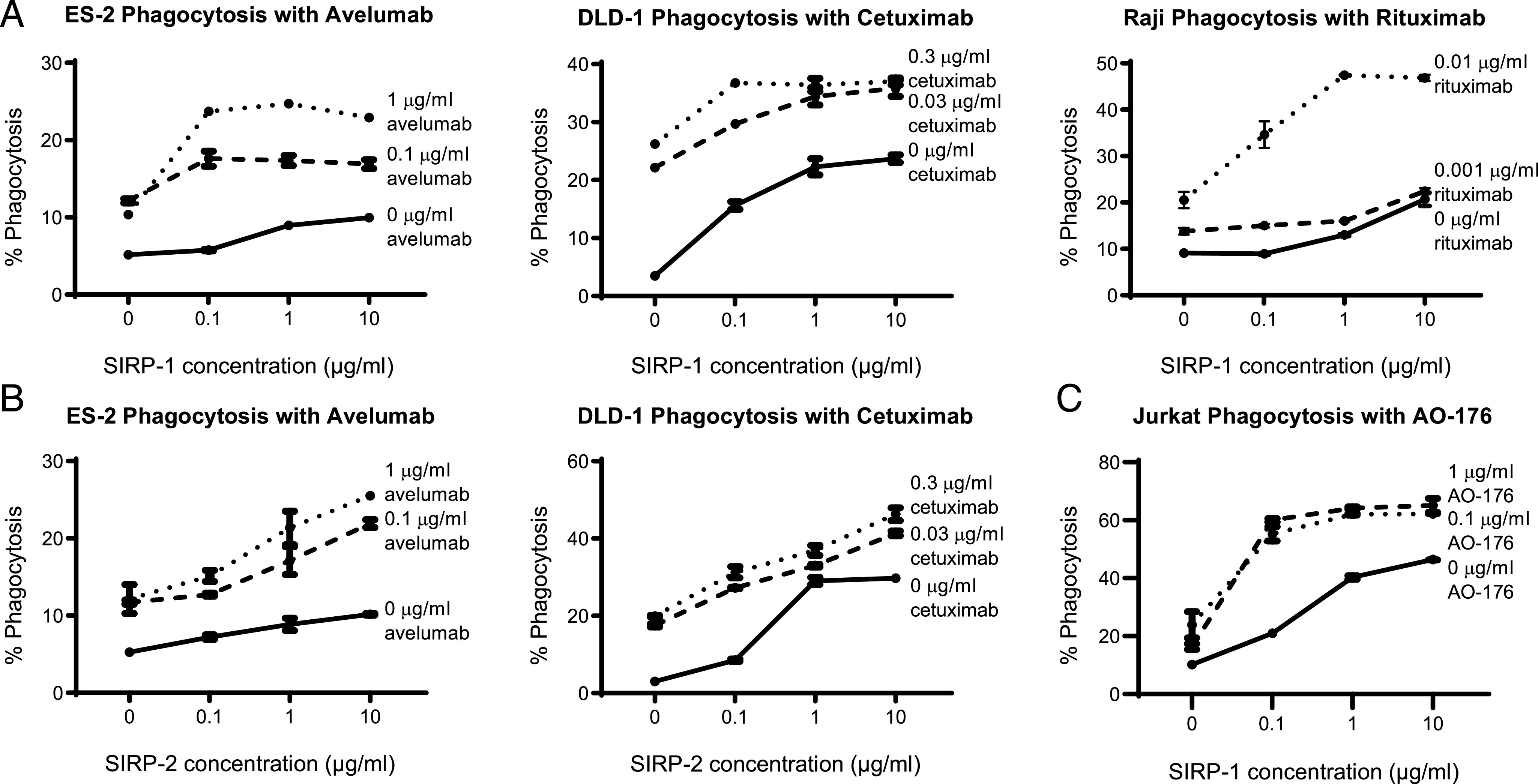
SIRP-1 and SIRP-2 combine with tumor-opsonizing Abs to potentiate phagocytosis of cancer cell lines. (A) SIRP-1 and (B) SIRP-2 combine with anti–PD-1 mAb avelumab, anti–epidermal growth factor receptor mAb cetuximab, and anti-CD20 mAb rituximab to potentiate human moMΦ phagocytosis of ES-2, DLD-1, and Raji cell lines, respectively. (C) SIRP-1 also combines with anti-CD47 mAb AO-176 to potentiate phagocytosis of Jurkat cells. All panels show mean ± SEM; a representative minimum n = 2 is shown.
Discussion
In this paper, we present two pan-allele–specific anti-human SIRPα Abs with unique properties that impede the interaction of macrophage SIRPα with CD47. Unlike other anti-human SIRPα agents, both anti-SIRPα Abs described in this study induce the phagocytosis of tumor cells by macrophages without the need of a secondary tumor-opsonizing agent (15, 32, 35), thus introducing a paradigm shift in the targeting of the SIRP/CD47 axis in cancer. The Abs described in this study bind to SIRPα on macrophages from all three common allelic groups, SIRPαV1/V1, V1/V2, and V2/V2, and induce phagocytosis by two novel mechanisms that are independent of ADCP and do not require binding to SIRPα or SIRPγ on tumor cells. Through one mechanism, SIRP-1 directly blocks SIRPα/CD47 interaction and induces the internalization of SIRPα on the surface of the macrophages, thereby reducing the availability of SIRPα to bind CD47 on the tumor cell and subsequently inhibiting phagocytosis. Through a second mechanism, SIRP-2 can alter SIRPα architecture/higher-order organization on the macrophage to impede the interaction with CD47, thereby blocking the “do-not-eat-me” signal. The Abs described in this study demonstrate novel mechanisms of action in SIRPα-mediated phagocytosis.
Under most, but not all, conditions, simply blocking the SIRP/CD47 axis may not be sufficient to induce phagocytosis by macrophages. A second, activating signal might be needed (17). We demonstrated that this is the case for these novel Abs: CD32 (FcγRII) was required for SIRP-1– and SIRP-2–mediated phagocytosis. Given the varying affinities of different FcγRs for specific IgG subtypes, it is possible that certain FcγR reliance is Ab isotype dependent. Even though SIRP-1 and SIRP-2 employed distinctly different molecular mechanisms to disrupt the SIRPα/CD47 interaction, they both also seemed to facilitate a cis-engagement of FcγRII. These multifunctional characteristics of SIRP-1 and SIRP-2 represent next-generation SIRP Abs, capable of simultaneously disrupting the SIRPα/CD47 interaction and engaging FcγRs to promote phagocytosis in macrophages.
SIRP-1 induced rapid internalization of SIRPα/Ab complexes, resulting in reduced surface levels of SIRPα. Additionally, it also directly inhibited the binding of CD47 to SIRPα. Extensive inhibition of macrophage SIRPα might not only lead to temporal unblocking of the “do-not-eat-me” signal but also durable changes in macrophage phenotype, signaled via downstream targets such as SHP-1 (47) and is subject to future investigation. Therapeutic Abs, such as trastuzumab, that partially act through target internalization have been described (48). To our knowledge, SIRP-1 is the first example of an Ab that leads to an inhibitory checkpoint internalization on the immune cell.
In the case of SIRP-2, we elucidated a second novel mechanism of action, demonstrating an altered architecture of SIRPα that reduces its ability to interact with CD47. SIRPα exists as both a homodimer and is clustered, with receptor clustering shown to be important for a high-affinity interaction with CD47 (44–46). It is unknown exactly what structural changes are induced by SIRP-2 binding that lead to dispersion, but both disruption of SIRPα homodimers or SIRPα clusters could play a role. SIRP-2 is differentiated from other non–CD47-blocking anti-SIRPα Abs by its ability to potentiate phagocytosis as a single agent and in combination with therapeutic opsonizing Abs, whereas other SIRPα Abs are only able to promote phagocytosis by combining with tumor-targeting opsonizing Abs (15, 35). Human epidermal growth factor receptor 2 (HER2) Abs provide the best-known examples in which altering dimerization or higher-order organization of receptors leads to inhibition of signaling (49).
The mechanistic changes that lead to disruption of SIRPα/CD47 by both SIRP-1 and SIRP-2 could potentially reduce the trans-interaction between macrophage SIRPα and tumor CD47 as well as a cis-interaction of SIRPα and CD47, which has been shown to be as important for blocking macrophage phagocytosis (50).
A further observation from our work was the enhanced phagocytic activity of these novel SIRP Abs with tumor-targeting Abs rituximab, cetuximab, and avelumab, demonstrating that enhanced phagocytosis results from the combination of SIRPα/CD47 blockade and tumor cell opsonization. Additionally, combination of these novel SIRP Abs with AO-176, a highly differentiated Ab targeting CD47, potently potentiated phagocytic activity. To our knowledge, the latter observation that dual targeting of both CD47 and SIRPα enhances phagocytosis is novel and offers additional therapeutic options such as combining both monotherapies or using a CD47xSIRPα-bispecific Ab.
In summary, SIRP-1 and SIRP-2 present a new class in the repertoire of agents targeting the CD47 axis. These agents capitalize on the limited expression of SIRPα to avoid an Ag sink and normal cell binding that most anti-CD47 agents show while demonstrating single-agent phagocytosis activity unlike any anti-SIRP Abs described so far. Disrupting SIRP/CD47 signaling on macrophages, either by internalization or disrupting SIRPα architecture together with engaging FcγRs, primes the macrophage for maximized phagocytic potential. It should be noted that the ability of certain tumor cell types to undergo phagocytosis following SIRPα/CD47 disruption may depend on the amount and type of prophagocytic signals on the tumor cell surface (such as phosphatidylserine, calreticulin, heat shock proteins, and others). SIRP-1 and SIRP-2 therefore have a strong potential for clinical combination strategies involving not only tumor-opsonizing but potentially other targeted therapies as well as immunomodulatory agents involving adaptive or innate immunity, including CD47 targeting agents.
Supplementary Material
Acknowledgments
We thank the hardworking and thoughtful members of Arch Oncology for significantly contributing to this paper (although not appearing).
This work was supported by Arch Oncology, Inc.
The online version of this article contains supplemental material.
- ADCP
- Ab-dependent cellular phagocytosis
- AF647
- Alexa Fluor 647
- FCET
- fluorescence resonance energy transfer analyzed by flow cytometry
- FRET
- fluorescence resonance energy transfer
- moMΦ
- monocyte-derived macrophage
- PBST
- PBS containing 0.5% Tween 20.
Disclosures
All authors are employees of Arch Oncology, Inc., and have stocks and/or stock options in Arch Oncology, Inc. G.A., B.J.C., R.J.P., and R.R.H. have filed a patent application that describes the invention of the anti-SIRPα and Abs SIRP-1 and SIRP-2. B.J.C. and R.J.P. have received and filed further patent applications that describe the invention of the anti-CD47 Ab AO-176. The other authors have no financial conflicts of interest.
References
- 1.Hodi, F. S., O’Day S. J., McDermott D. F., Weber R. W., Sosman J. A., Haanen J. B., Gonzalez R., Robert C., Schadendorf D., Hassel J. C., et al. 2010. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 363: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McDermott, D. F., Drake C. G., Sznol M., Choueiri T. K., Powderly J. D., Smith D. C., Brahmer J. R., Carvajal R. D., Hammers H. J., Puzanov I., et al. 2015. Survival, durable response, and long-term safety in patients with previously treated advanced renal cell carcinoma receiving nivolumab. J. Clin. Oncol. 33: 2013–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pitt, J. M., Vétizou M., Daillère R., Roberti M. P., Yamazaki T., Routy B., Lepage P., Boneca I. G., Chamaillard M., Kroemer G., Zitvogel L. 2016. Resistance mechanisms to immune-checkpoint blockade in cancer: tumor-intrinsic and -extrinsic factors. Immunity 44: 1255–1269. [DOI] [PubMed] [Google Scholar]
- 4.Restifo, N. P., Smyth M. J., Snyder A. 2016. Acquired resistance to immunotherapy and future challenges. Nat. Rev. Cancer 16: 121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma, P., Hu-Lieskovan S., Wargo J. A., Ribas A. 2017. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 168: 707–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barclay, A. N., Van den Berg T. K. 2014. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu. Rev. Immunol. 32: 25–50. [DOI] [PubMed] [Google Scholar]
- 7.Yanagita, T., Murata Y., Tanaka D., Motegi S. I., Arai E., Daniwijaya E. W., Hazama D., Washio K., Saito Y., Kotani T., et al. 2017. Anti-SIRPα antibodies as a potential new tool for cancer immunotherapy. JCI Insight 2: e89140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oldenborg, P. A. 2004. Role of CD47 in erythroid cells and in autoimmunity. Leuk. Lymphoma 45: 1319–1327. [DOI] [PubMed] [Google Scholar]
- 9.Oldenborg, P.-A., Gresham H. D., Chen Y., Izui S., Lindberg F. P. 2002. Lethal autoimmune hemolytic anemia in CD47-deficient nonobese diabetic (NOD) mice. Blood 99: 3500–3504. [DOI] [PubMed] [Google Scholar]
- 10.Willingham, S. B., Volkmer J. P., Gentles A. J., Sahoo D., Dalerba P., Mitra S. S., Wang J., Contreras-Trujillo H., Martin R., Cohen J. D., et al. 2012. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 109: 6662–6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrissey, M. A., Kern N., Vale R. D. 2020. CD47 ligation repositions the inhibitory receptor SIRPA to suppress integrin activation and phagocytosis. Immunity 53: 290–302.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oldenborg, P. A., Gresham H. D., Lindberg F. P. 2001. CD47-signal regulatory protein alpha (SIRPalpha) regulates Fcgamma and complement receptor-mediated phagocytosis. J. Exp. Med. 193: 855–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okazawa, H., Motegi S., Ohyama N., Ohnishi H., Tomizawa T., Kaneko Y., Oldenborg P.-A., Ishikawa O., Matozaki T. 2005. Negative regulation of phagocytosis in macrophages by the CD47-SHPS-1 system. J. Immunol. 174: 2004–2011. [DOI] [PubMed] [Google Scholar]
- 14.Veillette, A., Thibaudeau E., Latour S. 1998. High expression of inhibitory receptor SHPS-1 and its association with protein-tyrosine phosphatase SHP-1 in macrophages. J. Biol. Chem. 273: 22719–22728. [DOI] [PubMed] [Google Scholar]
- 15.Murata, Y., Tanaka D., Hazama D., Yanagita T., Saito Y., Kotani T., Oldenborg P. A., Matozaki T. 2018. Anti-human SIRPα antibody is a new tool for cancer immunotherapy. Cancer Sci. 109: 1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsai, R. K., Discher D. E. 2008. Inhibition of “self” engulfment through deactivation of myosin-II at the phagocytic synapse between human cells. J. Cell Biol. 180: 989–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Veillette, A., Chen J. 2018. SIRPα-CD47 immune checkpoint blockade in anticancer therapy. Trends Immunol. 39: 173–184. [DOI] [PubMed] [Google Scholar]
- 18.Treffers, L. W., Zhao X. W., van der Heijden J., Nagelkerke S. Q., van Rees D. J., Gonzalez P., Geissler J., Verkuijlen P., van Houdt M., de Boer M., et al. 2018. Genetic variation of human neutrophil Fcγ receptors and SIRPα in antibody-dependent cellular cytotoxicity towards cancer cells. Eur. J. Immunol. 48: 344–354. [DOI] [PubMed] [Google Scholar]
- 19.Piccio, L., Vermi W., Boles K. S., Fuchs A., Strader C. A., Facchetti F., Cella M., Colonna M. 2005. Adhesion of human T cells to antigen-presenting cells through SIRPβ2-CD47 interaction costimulates T-cell proliferation. Blood 105: 2421–2427. [DOI] [PubMed] [Google Scholar]
- 20.Sikic, B. I., Lakhani N., Patnaik A., Shah S. A., Chandana S. R., Rasco D., Colevas A. D., O’Rourke T., Narayanan S., Papadopoulos K., et al. 2019. First-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 37: 946–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thompson, J. A., Akilov O., Querfeld C., Taylor M. H., Johnson L., Catalano T., Petrova P. S., Uger R. A., Irwin M., Sievers E. L. 2017. A phase 1 dose-escalation trial of intratumoral TTI-621, a novel immune checkpoint inhibitor targeting CD47, in subjects with relapsed or refractory percutaneously-accessible solid tumors and mycosis fungoides. J. Clin. Oncol. 35(15_suppl). DOI: 10.1200/JCO.2017.35.15_suppl.TPS3101. [Google Scholar]
- 22.Advani, R., Flinn I., Popplewell L., Forero A., Bartlett N. L., Ghosh N., Kline J., Roschewski M., LaCasce A., Collins G. P., et al. 2018. CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin’s lymphoma. N. Engl. J. Med. 379: 1711–1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jain, S., Van Scoyk A., Morgan E. A., Matthews A., Stevenson K., Newton G., Powers F., Autio A., Louissaint A., Pontini G., et al. 2019. Targeted inhibition of CD47-SIRPα requires Fc-FcγR interactions to maximize activity in T-cell lymphomas. Blood 134: 1430–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnson, L., Pillai R. K., King R. L., Ansell S. M., Chen R. W., Flinn I., Maris M. B., Irwin M., Sievers E. L., Petrova P. S., Uger R. A. 2017. Effects of TTI-621 (SIRPαFc) on CD47 and serum cytokines associated with phagocytosis in subjects with relapsed, refractory hematologic malignancies: pharmacodynamic findings from a first-in-human clinical trial. J. Clin. Oncol. 35(7_suppl): 112. [Google Scholar]
- 25.Petrova, P. S., Viller N. N., Wong M., Pang X., Lin G. H. Y., Dodge K., Chai V., Chen H., Lee V., House V., et al. 2017. TTI-621 (SIRPαFc): a CD47-blocking innate immune checkpoint inhibitor with broad antitumor activity and minimal erythrocyte binding. Clin. Cancer Res. 23: 1068–1079. [DOI] [PubMed] [Google Scholar]
- 26.Brierley, C. K., Staves J., Roberts C., Johnson H., Vyas P., Goodnough L. T., Murphy M. F. 2019. The effects of monoclonal anti-CD47 on RBCs, compatibility testing, and transfusion requirements in refractory acute myeloid leukemia. Transfusion 59: 2248–2254. [DOI] [PubMed] [Google Scholar]
- 27.Reyland, L., Dwight M., Bullock T., Latham T., Lord K., Wardle A., Palmer D., Eggington J., Callaghan T., Seals D., Kulkarni S. 2020. Two case reports involving therapeutic monoclonal anti-CD47 (Hu5F9-G4), it’s effect on compatibility testing and subsequent selection of components for transfusion. Transfus. Med. 30: 157–160. [DOI] [PubMed] [Google Scholar]
- 28.Velliquette, R. W., Aeschlimann J., Kirkegaard J., Shakarian G., Lomas-Francis C., Westhoff C. M. 2019. Monoclonal anti-CD47 interference in red cell and platelet testing. Transfusion 59: 730–737. [DOI] [PubMed] [Google Scholar]
- 29.Kauder, S. E., Kuo T. C., Harrabi O., Chen A., Sangalang E., Doyle L., Rocha S. S., Bollini S., Han B., Sim J., et al. 2018. ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a favorable safety profile. PLoS One 13: e0201832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin, G. H. Y., Viller N. N., Chabonneau M., Brinen L., Mutukura T., Dodge K., Helke S., Chai V., House V., Lee V., et al. 2018. Abstract 2709: TTI-622 (SIRPα-IgG4 Fc), a CD47-blocking innate immune checkpoint inhibitor, suppresses tumor growth and demonstrates enhanced efficacy in combination with antitumor antibodies in both hematologic and solid tumor models. Cancer Res. 78: 2709. [Google Scholar]
- 31.Puro, R. J., Bouchlaka M. N., Hiebsch R. R., Capoccia B. J., Donio M. J., Manning P. T., Frazier W. A., Karr R. W., Pereira D. S. 2020. Development of AO-176, a next-generation humanized anti-CD47 antibody with novel anticancer properties and negligible red blood cell binding. Mol. Cancer Ther. 19: 835–846. [DOI] [PubMed] [Google Scholar]
- 32.Ring, N. G., Herndler-Brandstetter D., Weiskopf K., Shan L., Volkmer J. P., George B. M., Lietzenmayer M., McKenna K. M., Naik T. J., McCarty A., et al. 2017. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA 114: E10578–E10585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poirier, N., Mary C., Hove B., Gauttier V., Thepenier V., Pengam S. 2017. New anti-SIRPa antibodies and their therapeutic applications. World Intellectual Property Organization, Geneva, Switzerland. [Google Scholar]
- 34.Voets, E., Paradé M., Lutje Hulsik D., Spijkers S., Janssen W., Rens J., Reinieren-Beeren I., van den Tillaart G., van Duijnhoven S., Driessen L., et al. 2019. Functional characterization of the selective pan-allele anti-SIRPα antibody ADU-1805 that blocks the SIRPα-CD47 innate immune checkpoint. J. Immunother. Cancer 7: 340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sim, J., Sockolosky J. T., Sangalang E., Izquierdo S., Pedersen D., Harriman W., Wibowo A. S., Carter J., Madan A., Doyle L., et al. 2019. Discovery of high affinity, pan-allelic, and pan-mammalian reactive antibodies against the myeloid checkpoint receptor SIRPα. MAbs 11: 1036–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Delord, J.-P., Kotecki N., Marabelle A., Vinceneux A., Korakis I., Jungels C., Champiat S., Huhn R. D., Poirier N., Costantini D., et al. 2019. A phase 1 study evaluating BI 765063, a first in class selective myeloid sirpa inhibitor, as stand-alone and in combination with BI 754091, a programmed death-1 (PD-1) inhibitor, in patients with advanced solid tumours. Blood 134(Suppl. 1): 1040. [Google Scholar]
- 37.Batard, P., Szollosi J., Luescher I., Cerottini J.-C., MacDonald R., Romero P. 2002. Use of phycoerythrin and allophycocyanin for fluorescence resonance energy transfer analyzed by flow cytometry: advantages and limitations. Cytometry 48: 97–105. [DOI] [PubMed] [Google Scholar]
- 38.Takenaka, K., Prasolava T. K., Wang J. C. Y., Mortin-Toth S. M., Khalouei S., Gan O. I., Dick J. E., Danska J. S. 2007. Polymorphism in Sirpa modulates engraftment of human hematopoietic stem cells. Nat. Immunol. 8: 1313–1323. [DOI] [PubMed] [Google Scholar]
- 39.Brooke, G., Holbrook J. D., Brown M. H., Barclay A. N. 2004. Human lymphocytes interact directly with CD47 through a novel member of the signal regulatory protein (SIRP) family. J. Immunol. 173: 2562–2570. [DOI] [PubMed] [Google Scholar]
- 40.Zhao, X. W., van Beek E. M., Schornagel K., Van der Maaden H., Van Houdt M., Otten M. A., Finetti P., Van Egmond M., Matozaki T., Kraal G., et al. 2011. CD47-signal regulatory protein-α (SIRPα) interactions form a barrier for antibody-mediated tumor cell destruction. Proc. Natl. Acad. Sci. USA 108: 18342–18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tsai, R. K., Rodriguez P. L., Discher D. E. 2010. Self inhibition of phagocytosis: the affinity of ‘marker of self’ CD47 for SIRPα dictates potency of inhibition but only at low expression levels. Blood Cells Mol. Dis. 45: 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.van Eenennaam, H., van Elsas A., Voets E., Vink P., Lutje Hulsik D. 2018. Anti-sirp alpha antibodies. United States Patent and Trademark Office, Alexandria, Virginia. [Google Scholar]
- 43.Lubeck, M. D., Steplewski Z., Baglia F., Klein M. H., Dorrington K. J., Koprowski H. 1985. The interaction of murine IgG subclass proteins with human monocyte Fc receptors. J. Immunol. 135: 1299–1304. [PubMed] [Google Scholar]
- 44.Ha, B., Lv Z., Bian Z., Zhang X., Mishra A., Liu Y. 2013. ‘Clustering’ SIRPα into the plasma membrane lipid microdomains is required for activated monocytes and macrophages to mediate effective cell surface interactions with CD47. PLoS One 8: e77615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lopes, F. B., Bálint Š., Valvo S., Felce J. H., Hessel E. M., Dustin M. L., Davis D. M. 2017. Membrane nanoclusters of FcγRI segregate from inhibitory SIRPα upon activation of human macrophages. J. Cell Biol. 216: 1123–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lee, W. Y., Weber D. A., Laur O., Stowell S. R., McCall I., Andargachew R., Cummings R. D., Parkos C. A. 2010. The role of cis dimerization of signal regulatory protein α (SIRPalpha) in binding to CD47. J. Biol. Chem. 285: 37953–37963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sharma, Y., Bashir S., Bhardwaj P., Ahmad A., Khan F. 2016. Protein tyrosine phosphatase SHP-1: resurgence as new drug target for human autoimmune disorders. Immunol. Res. 64: 804–819. [DOI] [PubMed] [Google Scholar]
- 48.Fehling-Kaschek, M., Peckys D. B., Kaschek D., Timmer J., Jonge N. 2019. Mathematical modeling of drug-induced receptor internalization in the HER2-positive SKBR3 breast cancer cell-line. Sci. Rep. 9: 12709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ghosh, R., Narasanna A., Wang S. E., Liu S., Chakrabarty A., Balko J. M., González-Angulo A. M., Mills G. B., Penuel E., Winslow J., et al. 2011. Trastuzumab has preferential activity against breast cancers driven by HER2 homodimers. Cancer Res. 71: 1871–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hayes, B. H., Tsai R. K., Dooling L. J., Kadu S., Lee J. Y., Pantano D., Rodriguez P. L., Subramanian S., Shin J.-W., Discher D. E. 2020. Macrophages show higher levels of engulfment after disruption of cis interactions between CD47 and the checkpoint receptor SIRPα. J. Cell Sci. DOI: 10.1242/jcs.237800. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.