

Abstract
Purpose
Many human breast tumors become resistant to endocrine therapies and recur due to estrogen receptor (ERα) mutations that convey constitutive activity and a more aggressive phenotype. Here, we examined the effectiveness of a novel adamantyl antiestrogen, K-07, in suppressing the growth of breast cancer metastases containing the two most frequent ER-activating mutations, Y537S and D538G, and in extending survival in a preclinical metastatic cancer model.
Methods
MCF-7 breast cancer cells expressing luciferase and Y537S or D538G ER were injected into NOD-SCID-gamma female mice, and animals were treated orally with the antiestrogen K-07 or control vehicle. Comparisons were also made with the antiestrogen Fulvestrant. The development of metastases was monitored by in vivo bioluminescence imaging with phenotypic characterization of the metastases in liver and lung by immunohistochemical and biochemical analyses.
Results
These breast cancer cells established metastases in liver and lung, and K-07 treatment reduced the metastatic burden. Mice treated with K-07 also survived much longer. By day 70, only 28% of vehicle-treated mice with mutant ER metastases were alive, whereas all K-07-treated D538G and Y537S mice were still alive. K-07 also markedly reduced the level of metastatic cell ER and the expression of ER-regulated genes.
Conclusion
The antiestrogen K-07 can reduce in vivo metastasis of breast cancers and extend host survival in this preclinical model driven by constitutively active mutant ERs, suggesting that this compound may be suitable for further translational examination of its efficacy in suppression of metastasis in breast cancers containing constitutively active mutant ERs.
Keywords: antiestrogen, estrogen receptor, metastasis, breast cancer
INTRODUCTION
Breast cancer mortality is almost always associated with metastatic spread of the cancer. Although estrogen receptor (ER)-positive breast cancers are considered to be a subtype that is quite often successfully treated with antiestrogens (selective ER modulators, SERMs) or aromatase inhibitors, it is now recognized that some of these breast cancers can recur at later times and that over one-third of these metastatic breast cancers now harbor ER mutations that render the ER proteins constitutively active in the absence of hormone [1-4]. In fact, deaths due to metastatic ER-positive breast cancers account for the largest subgroup of breast cancer mortality [5]. The most frequent of these mutations are single nucleotide changes that result in single amino acid changes (Y537S and D538G) in these ER-positive metastatic breast cancers [6-12]. Of note, these mutations are in the critical activation function 2 (AF2) region of the receptor and enable coactivator binding and stimulation of cell proliferation and ER signaling in the absence of hormone, with the result that tumors are resistant to aromatase inhibitors (AIs) and require increased concentrations of antiestrogens for effective suppression of growth and ER pathway activities [12-16]. In addition, these mutations are associated with more aggressive disease and shorter overall patient survival [17-20].
Because chemotherapies and radiation treatments aimed at reducing recurrence and metastatic spread are often associated with toxicity and deleterious side effects [21], it would be desirable to utilize targeted therapies with more limited adverse effects. We showed previously that novel SERM compounds developed by us work through ER and effectively suppress the growth of breast cancers driven by these mutant ERs in orthotopic xenograft tumor models in NOD-SCID-gamma (NSG) mice [16, 22]. However, because compounds that reduce the growth of primary tumors may not necessarily have antimetastatic activity and vice versa [23-25], the aim of this study was to specifically examine the ability of our lead SERM compound, K-07, to suppress the metastatic growth of mutant ER-containing breast cancer cells and to extend host animal survival. We have also focused on defining the effects of K-07 on the phenotypic and biochemical properties of the metastatic lesions. Notably, we find that K-07 suppresses the expression of ER and proliferation markers, reduces the metastatic burden of breast cancers containing these mutant ERs, and extends host survival. Hence, this compound may be suitable for further translational examination of its efficacy in suppressing metastasis in breast cancers containing constitutively active mutant ERs.
MATERIAL AND METHODS
Cell cultures and reagents
Preparation of the compound K-07 has been described [22]. MCF7 cells containing mutant Y537S- or D538G-ERα determined by DNA sequencing and digital drop PCR analyses as detailed previously [26, 27], were generated by adenovirus-associated viral infection. Cells were transfected with firefly luciferase/GFP using RediFect Red-FLuc-GFP Lentiviral Particles (Perkin Elmer CLS960003) as described by the manufacturer, and sorted for GFP fluorescence by flow cytometry with the fluorescence-activated cell sorter BDFACS AriaII. Cells were cultured in DMEM with 5% premium grade fetal bovine serum (VWR Life Science Seradigm, cat# 97068-085) and 1% penicillin/streptomycin. Cells were used in experiments within the first five passages after thawing. All cells were tested for mycoplasma using Real-Time PCR Mycoplasma Detection Kit (Agilent Technologies).
In vivo breast cancer metastasis studies
In preparation for injection into animals, Y537S and D538G cells were harvested with trypsin-EDTA, resuspended in medium containing 2 mM EDTA, passed through a cell strainer, adjusted in concentration to 5x106 cells/ml, and stored on ice until injection. 0.5x106 cells per mouse were then introduced by intracardiac [28] or tail vein injection as described [29].
Intact female NOD-SCID-gamma (NSG) mice (8 weeks of age), obtained from Jackson Labs, were divided into randomized groups with equal numbers receiving Y537S and D538G cell inoculations. Animals began receiving compound treatment 2 days post tumor cell inoculation. Treatments included control vehicle or K-07 by oral gavage (sterile 9/0.5/0.5/90 parts of PEG400/Tween80/Povidone/0.5% carboxymethylcellulose in sterile water) with 80 mg/kg delivered in 200 μl per treatment, 6 days/week for 30 days and then 40 mg/kg for the remainder of the study. In some studies comparing the effects of Fulvestrant (from Sigma Aldrich), Fulvestrant was administered by s.c. injection (80mg/kg) six times per week since it is not active orally.
IVIS bioluminescence imaging
The extent of metastasis was followed biweekly using an IVIS Spectrum CT imaging system. Animals were injected with luciferin (150 mg/kg), luciferase activity was measured, and bioluminescence as total flux (photon counts/sec) was quantified using Living Image software (PerkinElmer) as previously described [30].
RNA isolation and real-time PCR
Liver and lung tissues were pulverized, and total RNA was isolated using TRIzol (Invitrogen) and reverse transcribed using MMTV reverse transcriptase (New England BioLabs). Real-time PCR was performed using SYBR Green PCR Master Mix (Quanta) on a BioRad CFX384 PCR detection system. Expression of estrogen responsive genes is the mean ±SD from all animals in each group.
Immunohistochemistry (IHC)
Hematoxylin and eosin staining or IHC was performed as previously described [16, 31] with minor modifications. Tissues were fixed in 10% buffered formalin phosphate for 24 h at room temperature, transferred to 70% ethanol, and embedded in paraffin. Tissues were blocked in Rodent Block M (Biocare Medical, Pacheco, CA) as well as 3% H2O2. Immunostaining was performed using antibodies to firefly luciferase (Abcam 181640), ERα (Estrogen Receptor Clone 6F11 Leica NCL-L-ER-6F11, dilution 1:100, 1 h, room temperature), or Ki67 (Dako Code M7240. dilution 1:100 1 h, room temperature). Following primary antibody treatment, samples were treated with secondary antibody (MM HRP-Polymer, Biocare Medical, Pacheco, CA) for 30 min at room temperature. Chromagen treatment consisted of 3,3’-Diaminobenzidine (5 min, room temperature), followed by Harris hematoxylin counterstain (1 min). Quantitation of staining was done using AxioVision (Zeiss Microscopy) image analysis software.
RESULTS
Antiestrogen K-07 reduces the metastatic load of D538G and Y537S containing breast cancer cells and extends animal host survival
Because Y537S and D538G are the two most commonly found constitutively active ER mutations in recurrent, metastatic ER-positive breast cancers, we used MCF-7 cells containing each of these mutant ERs, and we also introduced luciferase into these cells so that we could monitor the location and progression of these cells as metastases over time. The luciferase/GFP-containing cells were sorted by flow cytometry so that only cells containing luciferase were used in these studies.
Figure 1a shows the location of these single amino acid changes in the mutant ER proteins Y537S and D538G, and also the structure of the adamantyl antiestrogen K-07 used in these studies. Using a model of metastatic breast cancer where cancer cells are introduced by tail vein and allowed to establish prior to intervention, we found that administration of K-07 by oral gavage reduced the metastatic load of ER mutant D538G-Luc breast cancer cells (Fig. 1b) and K-07 treatment also prolonged survival of the host NSG mice injected with these cells (Fig. 1c). Notably, all host animals receiving K-07 survived over the 90 days monitored, whereas only 28% of vehicle-treated animals were alive at 65 days after cell inoculation (Fig. 1c).
Fig. 1.

Mutant ER-containing breast cancer cells and antiestrogen K-07 used in metastasis and host survival studies. (a) Schematic of the human Estrogen Receptor alpha (ER) protein showing the location of the two most common amino acid alterations in mutant, constitutively active ER proteins present in recurrent, metastatic breast cancers and studied in this manuscript (top left). Chemical structure of the adamantyl antiestrogen K-07 utilized in these studies (top right). (b) Metastatic load and (c) animal survival were followed over time. Metastatic load was assessed by bioluminescence after tail vein injection of NOD-SCID-gamma (NSG) female mice with MCF-7 breast cancer cells expressing luciferase and D538G ER. Animals received 0.5 x 106 cells per mouse and were treated 6-times per week with vehicle or K-07 (80 mg/kg orally for 30 days and then 40 mg/kg) and metastatic burden was monitored over time by bioluminescence (IVIS) imaging after luciferin injection, and animal survival was followed to 90 days.
Likewise, following tail vein injection of ER mutant Y537S-Luc breast cancer cells into NSG mice, K-07 reduced the metastatic load of these cells, as seen in the reduced total photon flux monitored by IVIS bioluminescence imaging over time (Fig. 2a), and by comparisons of the metastatic burden in individual animals in K-07-treated versus vehicle-treated groups at day 57 (Fig. 2b). K-07 treatment also prolonged the survival of host mice receiving Y537S mutant ER cells (Fig. 2c).
Fig. 2.

Metastasis and host animal survival after tail vein injection of NOD-SCID-gamma (NSG) female mice with MCF-7 breast cancer cells expressing luciferase and mutant Y537S ER (0.5 x 106 cells per mouse). Animals were treated 6-times per week with vehicle or K-07 (80 mg/kg orally for 30 days and then 40 mg/kg). (a) Metastatic load was monitored over time and (b) at day 57 by bioluminescence (IVIS) imaging after luciferin injection, and (c) animal survival was followed to 100 days.
Metastasis of ER mutant D538G-Luc breast cancer cells was reduced with K-07 treatment even following intracardiac injection of these cells, which we found resulted in more rapid establishment of metastatic lesions compared to tail vein injection (Fig. 3a). This is seen by comparison of total photon flux with time after cell injection between Fig. 3a (intracardiac breast cancer cell injection) and Fig. 2b (intravenous tail vein cell injection), which show similar metastatic burden, monitored by IVIS total flux, at day 25 and day 57, respectively. Survival of mice was also improved significantly with K-07 treatment after intracardiac cell injection (Fig. 3b). However, comparison of Fig. 3b with Fig. 1c shows that host animal survival was prolonged less effectively after intracardiac cell injection than following tail vein injection where metastasis spread developed more slowly over time.
Fig. 3.

Metastasis progression and host animal survival after intracardiac injection of NOD-SCID-gamma (NSG) female mice with MCF-7 breast cancer cells expressing luciferase and mutant D538G ER. After injection of 0.5 x 106 cells, animals were treated 6-times per week with vehicle or K-07 (80 mg/kg orally for 30 days and then 40 mg/kg). (a) Metastatic load was monitored at 25 days by bioluminescence (IVIS) imaging after luciferin injection, and (b) animal survival was followed to 70 days.
Evaluation of the biochemical and phenotypic properties of the breast cancer metastases
In a repeat experiment that showed similar development of metastatic burden with D538G and Y537S mutant ER-containing cells over time, we also compared the efficacy of K-07 and the standard-of-care antiestrogen Fulvestrant in reducing D538G-Luc-MCF7 metastatic load following tail vein injection (Supplementary Fig. S1). K-07 and Vehicle were administered orally six times weekly, as done in the previous study groups, and Fulvestrant was administered by s.c. injection six times per week since it is not active orally. We found that K-07 and Fulvestrant were equally effective in greatly reducing the metastatic load in the treated animals, as monitored by IVIS imaging.
Tumors were harvested at day 42 for IHC and analysis of ER and ER target gene expression, and for assessment of proliferation monitored by expression of markers PCNA and Ki67. The representative panels in Fig. 4 show that metastases were readily observable by immunohistochemistry using an antibody against luciferase, indicating positive staining in the human breast cancer cells within the host mouse liver. Metastases were also seen in the host lungs. IHC for ER and Ki67 in metastases indicated markedly reduced ER levels (to less than 10% of vehicle control) and also greatly reduced Ki67 with K-07 treatment (Fig. 5a-5h and Table 1). Also, analyses of the expression of the ER target gene GREB1 and the proliferation associated gene PCNA in metastases revealed that their RNA expression levels were also reduced in lung and liver metastases of mice receiving the antiestrogen K-07 or Fulvestrant compared with vehicle-treated animals (Fig. 6).
Fig. 4.

Immunohistochemical (IHC) detection of luciferase in liver metastases. Animals inoculated with D538G-Luc or Y537S-Luc cells were treated with vehicle and liver tissue was harvested at 6 weeks. Tissue sections were stained with antibodies to luciferase to visualize the human breast cancer cells in the host animal liver. Scale bar is 100 μm.
Fig. 5.

IHC for ER (panels a, b, e, f) or for Ki67 (panels c, d, g, h) in the livers of mice receiving D538G or Y537S cells, followed by treatment with vehicle or K-07 for 42 days prior to tissue harvest. Scale bar is 25 μm
Table 1.
Quantitation of ER and Ki67 in liver metastases of mice receiving mutant D538G ER or mutant Y537S ER breast cancer cells.a
| D538G | Y537S | |||||
|---|---|---|---|---|---|---|
| ER | Veh | K-07 | % K-07/Veh (p-value) |
Veh | K-07 | % K-07/Veh (p-value) |
| Stained nuclei/field (%) | 42.52 ± 7.11 | 4.83 ± 1.70 | 11% (** 0.0021) |
52.03 ± 5.18 | 1.03 ± 0.29 | 2% (*** 0.0006) |
| Stain intensity/nucleus | 2.75 ± 0.21 | 2.30 ± 0.07 | 86% ns |
2.60 ± 0.10 | 1.83 ± 0.07 | 70% (** 0.0027) |
| Total staining/field | 395.98 ± 93.65 | 38.22 ± 14.60 | 10% (** 0.0092) |
399.23 ± 52.04 | 4.99 ± 1.74 | 1% (** 0.0016) |
| Ki67 | Veh | K-07 | % K-07/Veh (p-value) |
Veh | K-07 | % K-07/Veh (p-value) |
| Stained nuclei/field (%) | 13.73 ± 2.46 | 1.60 ± 0.42 | 12% (** 0.009) |
28.36 ± 4.80 | 8.76 ± 2.10 | 31% (** 0.0057) |
| Stain intensity/nucleus | 3.69 ± 0.28 | 3.53 ± 0.49 | 96% ns |
2.73 ± 0.39 | 3.18 ± 0.52 | 116% ns |
| Total staining/field | 158.54 ± 27.19 | 21.12 ± 4.36 | 13% (** 0.0081) |
183.46 ± 25.20 | 72.67 ± 14.21 | 40% (** 0.0050) |
Treatment with Vehicle or K-07 was six times per week for 42 days prior to liver metastasis collection and analysis. Detection of ER (top) and Ki67 (bottom) was by IHC and quantitation by AxioVision Image Analysis software. ns= not significant.
Fig. 6.
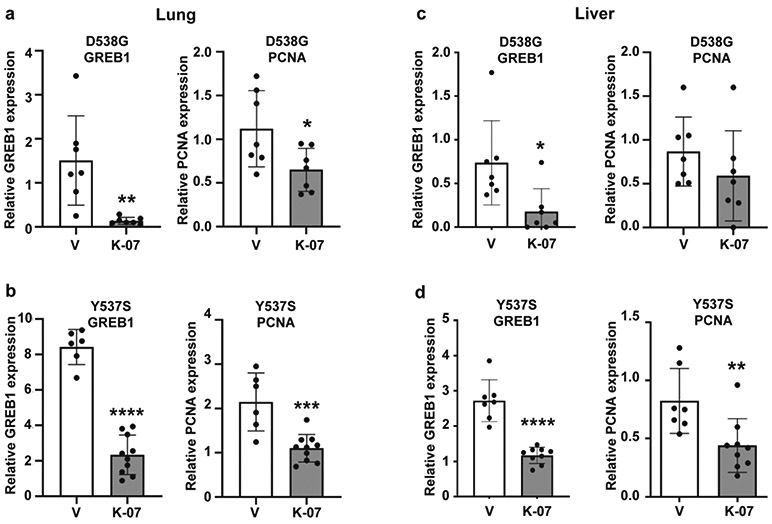
Expression of the ER target gene GREB1 and the proliferation associated gene PCNA in lung metastases (a, b) or liver metastases (c, d) of host mice receiving D538G or Y537S mutant ER-containing breast cancer cells. Animals received control vehicle or K-07 orally (80 mg/kg) 6 days per week or Fulvestrant s.c. (80 mg/kg) 6 days per week, and lung and liver tissues were harvested at day 42 of treatment. RNA was prepared, and RT-PCR with human specific primers was conducted. Values are from lungs or livers of different mice in each treatment group and are mean ± SD. *, p<0.05; **, p<0.01; ***, p<0.001, ****, p<0.0001 by Student t-test for Y537S data and by Dunnett’s multiple comparisons test for D538G data.
DISCUSSION
Cancer metastasis is a leading cause of morbidity and mortality in patients with breast cancer. Our studies reveal that the orally active SERM K-07 suppressed the overall metastatic burden resulting from constitutively active mutant ER-containing breast cancer cells, and increased host animal survival. In the experimental metastasis system we have used here, whereby tumor cells are injected into the bloodstream, we have circumvented the first steps in the metastatic process [25]; however, this model does require the survival of the cancer cells in the circulation and their metastatic colonization, the latter known to involve interactions between tumor cells and cells in the microenvironment of the distant site [25, 32]. This well-established experimental model “provides good quantification in a reasonable time frame” [33] and is useful for drug development and for assessing potential inhibitor drug efficacy, especially in the context of established metastatic lesions.
It is noteworthy that these mutant ER-containing breast cancer cells form metastatic lesions in the host liver and lung in the absence of any added exogenous estradiol, which was needed for the establishment of xenograft tumors with wild type ER [16]. Hence, these mutations not only support estrogen-independent growth, but they also promote a metastatic phenotype seen in their distinctive cistromes and transcription networks [1].
The ability of breast cancer cells with these mutant ERs to engender metastases very much mimics what is observed in breast cancer patients where cancer recurrence and metastasis, often after treatment with aromatase inhibitors, is associated with acquisition of these ER mutations that foster a more aggressive cancer biology and are associated with reduced patient survival [3, 17, 18, 20]. Hence, it is of note that host animal survival was extended with K-07 treatment after introduction of breast cancer cells containing either of the two most common ER mutations, D538G and Y537S, even when metastatic spread was very rapid after intracardiac inoculation of cells.
Although we and others have shown that breast cancer cells expressing these mutant ERs are more resistant to the antiproliferative effects of K-07 or other standard-of-care antiestrogens such as tamoxifen and fulvestrant [16, 22], our studies here reveal that K-07, which we showed previously to suppress the growth of xenograft tumors expressing these mutant ERs [16], was also effective in reducing metastatic burden and extending survival of animals with breast cancer cells containing these constitutively active ERs. In addition, in the D538G mutant ER cell cohort where we compared the efficacy of K-07 with the standard-of-care antiestrogen Fulvestrant, we found that both similarly greatly reduced metastases to liver and lung and suppressed the expression of ER target genes such as GREB1.
It was of interest that we found the metastatic load from the D538G mutant ER cells to be more readily suppressed by K-07 than was the metastatic load from the Y537S mutant ER cells. This is consistent with transcriptome analyses revealing that the D538G mutation is distinct from the Y537S mutation and that these mutations induce different transcriptional programs in which the Y537S ER induces many unique genes and shows an aggressive phenotype more difficult to inhibit with standard-of-care antiestrogens tamoxifen or Fulvestrant [1]. Interestingly, we also reported K-07 to more fully suppress in vitro cell proliferation, and in vivo xenograft growth of D538G tumors compared with Y537S tumors [16].
Analysis of gene expression regulation in the metastases indicated that K-07 was targeting ER and suppressing the expression of ER itself and of ER target genes. Of note, the level of ER RNA and protein expression, monitored by qRT-PCR and by IHC, and the mRNAs of well-known ER target genes in liver and lung metastases were markedly reduced by K-07 treatment. These findings suggest that decreased expression of the constitutively active ERs could be an important aspect in the ability of K-07 to reduce the metastatic burden of mutant ER-containing cancer cells. K-07 also binds to these ERs and changes the conformation of the ER ligand binding domain which is known to perturb interactions with key coactivators [34], that may also contribute to its inhibitory efficacy [2].
Agents with anti-metastatic efficacy may also be effective in reducing primary tumor size, or may be different from those effective in reducing primary tumor size [23-25]. For example, some factors, such as the type I insulin-like growth factor receptor protein, the metastasis suppressor Nm23, and the transcriptional repressor EZH2 regulate cancer metastasis independently of primary tumor growth [23, 24, 35]. By contrast, K-07 reduced the growth of primary xenograft breast tumors driven by Y537S and D538G mutant ERs [16] and also effectively reduced the metastatic burden of breast cancer cells containing these mutant ERs. This might indicate the potential clinical utility of this compound in treating metastatic breast cancer lesions with either of the most common ER mutations.
In this study, we examined metastatic spread and response to antiestrogen in two MCF-7 mutant ER cell lines. We worked previously with MCF-7 and T47D cells containing these two most common mutant ERs in human ER-positive breast cancers and compared K-07 in these two cell backgrounds. Results showed K-07 to be equally effective in cell culture experiments in suppressing cell proliferation [16]. We also found K-07 and Fulvestrant to be equally good antitumor agents in blocking MCF-7 D538G and Y537S mutant ER xenograft growth. However, there are major difficulties in using T47D ER-containing cells for in vivo tumor and metastasis studies. As opposed to MCF-7 cells, T47D cells form only very small, slow growing tumors [36] and both MCF-7 and T47D cells do not metastasize from xenografts in the mammary gland. A mammary intraductal injection (MIND) model might prove useful for future studies. In this model, MCF-7 cells generate slow growing tumors that expand predominantly within the mammary duct and after a latency of ca. six months, show some metastatic cells in liver and/or lung [37]. This mammary intraductal model might also be worthwhile in the study of ER-positive patient derived xenografts [38], some of which might metastasize from the ductal tumors over time, especially if they contain constitutively active mutant ERs.
Chemotherapies to reduce metastatic spread and risk of recurrence are often associated with adverse side effects. Because K-07 is a compound targeted to the ER, it would be expected to have fewer deleterious off-target effects. K-07 is also orally active, making it potentially suitable for clinical use. Our findings here, documenting reduced metastatic burden and extended host survival in this preclinical model, suggest that K-07 may be suitable for further translational examination of its efficacy in suppression of metastasis in breast cancers driven by constitutively active mutant ERs.
Supplementary Material
Supplementary Fig. S1 Progression of mutant ER-Luc metastases over time in NSG mice inoculated via tail vein with (a) mutant D538G ER-Luc cells or (b) Y537S ER-Luc cells. Animals received treatment with vehicle or K-07 (80 mg/kg) orally 6 days per week, or with Fulvestrant s.c. (80 mg/kg) 6 days per week, and metastatic burden was monitored by IVIS imaging over time. Total Flux values are mean ± SD. ** p<0.01 by Student t-test
Acknowledgments
Funding This research was supported by grants from the Breast Cancer Research Foundation (BCRF-083 to BSK and BCRF-084 to JAK and BSK), IIDRP-16-006 to BHP, NIH/NCI grant 1R01CA220284 (to BSK, JAK, and KWN), Susan G. Komen SAC 170079 to BHP, and NIH grant R01CA234025 and DOD Breast Cancer Research Program grant BC171214 to ERN.
Abbreviations
- ER
estrogen receptor α
- IHC
immunohistochemistry
- NSG
NOD-SCID-gamma
- SERM
selective estrogen receptor modulator
Footnotes
Disclosure statement
JAK is a consultant and stockholder of Radius Health, Inc. BSK and JAK have ownership interest in Celcuity, Inc. Patent applications covering K-07 that include JAK, SHK, and BSK as inventors have been filed by the University of Illinois. BHP receives royalties from Horizon Discovery, LTD, was a scientific advisory board member for Loxo Oncology and had ownership interest in Loxo Oncology, was a paid consultant for Foundation Medicine, Inc., H3 Biomedicine, Casdin Capital, Roche, Eli Lilly, Astra Zeneca, is an unpaid consultant for Tempus, and is a paid consultant for Jackson Laboratories, Celcuity, Pathovax, with ownership interest in Celcuity, and has research contracts with Abbvie, Foundation Medicine Inc. and Pfizer. The other authors declare that they have no conflict of interest
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All animal experiments were performed in accordance with institutional protocols and approved by the University of Illinois Institutional Animal Care and Use Committee (PHS Assurance: D16-00075 (A3118-01)). This article does not contain any studies with human participants performed by any of the authors.
Informed consent
Not applicable.
Data Availability
All data generated or analyzed in this study are available from the corresponding author on reasonable request.
REFERENCES
- 1.Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. 2018;Allele-Specific Chromatin Recruitment and Therapeutic Vulnerabilities of ESR1 Activating Mutations. Cancer Cell. 33(2):173–186 e175. 10.1016/j.ccell.2018.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Katzenellenbogen JA, Mayne CG, Katzenellenbogen BS, Greene GL, Chandarlapaty S 2018;Structural underpinnings of oestrogen receptor mutations in endocrine therapy resistance. Nat Rev Cancer. 18(6):377–388. 10.1038/s41568-018-0001-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richman J, Dowsett M 2019;Beyond 5 years: enduring risk of recurrence in oestrogen receptor-positive breast cancer. Nat Rev Clin Oncol. 16(5):296–311. 10.1038/s41571-018-0145-5 [DOI] [PubMed] [Google Scholar]
- 4.Tryfonidis K, Zardavas D, Katzenellenbogen BS, Piccart M 2016;Endocrine treatment in breast cancer: Cure, resistance and beyond. Cancer Treat Rev. 50:68–81. 10.1016/j.ctrv.2016.08.008 [DOI] [PubMed] [Google Scholar]
- 5.Waks AG, Winer EP 2019;Breast Cancer Treatment: A Review. JAMA. 321(3):288–300. 10.1001/jama.2018.19323 [DOI] [PubMed] [Google Scholar]
- 6.Chu D, Paoletti C, Gersch C, VanDenBerg DA, Zabransky DJ, Cochran RL, et al. 2016;ESR1 Mutations in Circulating Plasma Tumor DNA from Metastatic Breast Cancer Patients. Clin Cancer Res. 22(4):993–999. 10.1158/1078-0432.CCR-15-0943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. 2016;Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife. 5 10.7554/eLife.12792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. 2014;Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 20(7):1757–1767. 10.1158/1078-0432.CCR-13-2332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lazennec G, Ediger TR, Petz LN, Nardulli AM, Katzenellenbogen BS 1997;Mechanistic aspects of estrogen receptor activation probed with constitutively active estrogen receptors: correlations with DNA and coregulator interactions and receptor conformational changes. Mol Endocrinol. 11(9):1375–1386. 10.1210/mend.11.9.9983 [DOI] [PubMed] [Google Scholar]
- 10.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. 2013;Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 4(6):1116–1130. 10.1016/j.celrep.2013.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. 2013;Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 45(12):1446–1451. 10.1038/ng.2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toy W, Weir H, Razavi P, Lawson M, Goeppert AU, Mazzola AM, et al. 2017;Activating ESR1 Mutations Differentially Affect the Efficacy of ER Antagonists. Cancer Discov. 7(3):277–287. 10.1158/2159-8290.CD-15-1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bardia A, Iafrate JA, Sundaresan T, Younger J, Nardi V 2016;Metastatic Breast Cancer With ESR1 Mutation: Clinical Management Considerations From the Molecular and Precision Medicine (MAP) Tumor Board at Massachusetts General Hospital. Oncologist. 21(9):1035–1040. 10.1634/theoncologist.2016-0240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fanning SW, Jeselsohn R, Dharmarajan V, Mayne CG, Karimi M, Buchwalter G, et al. 2018;The SERM/SERD bazedoxifene disrupts ESR1 helix 12 to overcome acquired hormone resistance in breast cancer cells. Elife. 7 10.7554/eLife.37161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miller CA, Gindin Y, Lu C, Griffith OL, Griffith M, Shen D, et al. 2016;Aromatase inhibition remodels the clonal architecture of estrogen-receptor-positive breast cancers. Nat Commun. 7:12498 10.1038/ncomms12498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao Y, Laws MJ, Guillen VS, Ziegler Y, Min J, Sharma A, et al. 2017;Structurally Novel Antiestrogens Elicit Differential Responses from Constitutively Active Mutant Estrogen Receptors in Breast Cancer Cells and Tumors. Cancer Res. 77(20):5602–5613. 10.1158/0008-5472.CAN-17-1265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chandarlapaty S, Chen D, He W, Sung P, Samoila A, You D, et al. 2016;Prevalence of ESR1 Mutations in Cell-Free DNA and Outcomes in Metastatic Breast Cancer: A Secondary Analysis of the BOLERO-2 Clinical Trial. JAMA Oncol. 2(10):1310–1315. 10.1001/jamaoncol.2016.1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fribbens C, Garcia Murillas I, Beaney M, Hrebien S, O'Leary B, Kilburn L, et al. 2018;Tracking evolution of aromatase inhibitor resistance with circulating tumour DNA analysis in metastatic breast cancer. Ann Oncol. 29(1):145–153. 10.1093/annonc/mdx483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lopez-Knowles E, Pearson A, Schuster G, Gellert P, Ribas R, Yeo B, et al. 2019;Molecular characterisation of aromatase inhibitor-resistant advanced breast cancer: the phenotypic effect of ESR1 mutations. Br J Cancer. 120(2):247–255. 10.1038/s41416-018-0345-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Spoerke JM, Gendreau S, Walter K, Qiu J, Wilson TR, Savage H, et al. 2016;Heterogeneity and clinical significance of ESR1 mutations in ER-positive metastatic breast cancer patients receiving fulvestrant. Nat Commun. 7:11579 10.1038/ncomms11579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ratajczak MZ, Jadczyk T, Schneider G, Kakar SS, Kucia M 2013;Induction of a tumor-metastasis-receptive microenvironment as an unwanted and underestimated side effect of treatment by chemotherapy or radiotherapy. J Ovarian Res. 6(1):95 10.1186/1757-2215-6-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Min J, Guillen VS, Sharma A, Zhao Y, Ziegler Y, Gong P, et al. 2017;Adamantyl Antiestrogens with Novel Side Chains Reveal a Spectrum of Activities in Suppressing Estrogen Receptor Mediated Activities in Breast Cancer Cells. J Med Chem. 60(14):6321–6336. 10.1021/acs.jmedchem.7b00585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marshall JC, Collins JW, Nakayama J, Horak CE, Liewehr DJ, Steinberg SM, et al. 2012;Effect of inhibition of the lysophosphatidic acid receptor 1 on metastasis and metastatic dormancy in breast cancer. J Natl Cancer Inst. 104(17):1306–1319. 10.1093/jnci/djs319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sachdev D, Zhang X, Matise I, Gaillard-Kelly M, Yee D 2010;The type I insulin-like growth factor receptor regulates cancer metastasis independently of primary tumor growth by promoting invasion and survival. Oncogene. 29(2):251–262. 10.1038/onc.2009.316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Steeg PS 2006;Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 12(8):895–904. 10.1038/nm1469 [DOI] [PubMed] [Google Scholar]
- 26.Bahreini A, Li Z, Wang P, Levine KM, Tasdemir N, Cao L, et al. 2017;Mutation site and context dependent effects of ESR1 mutation in genome-edited breast cancer cell models. Breast Cancer Res. 19(1):60 10.1186/s13058-017-0851-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang P, Bahreini A, Gyanchandani R, Lucas PC, Hartmaier RJ, Watters RJ, et al. 2016;Sensitive Detection of Mono- and Polyclonal ESR1 Mutations in Primary Tumors, Metastatic Lesions, and Cell-Free DNA of Breast Cancer Patients. Clin Cancer Res. 22(5):1130–1137. 10.1158/1078-0432.CCR-15-1534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Waning DL, Mohammad KS, Reiken S, Xie W, Andersson DC, John S, et al. 2015;Excess TGF-beta mediates muscle weakness associated with bone metastases in mice. Nat Med. 21(11):1262–1271. 10.1038/nm.3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baek AE, Yu YA, He S, Wardell SE, Chang CY, Kwon S, et al. 2017;The cholesterol metabolite 27 hydroxycholesterol facilitates breast cancer metastasis through its actions on immune cells. Nat Commun. 8(1):864 10.1038/s41467-017-00910-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nelson ER, Li S, Kennedy M, Payne S, Kilibarda K, Groth J, et al. 2016;Chemotherapy enriches for an invasive triple-negative breast tumor cell subpopulation expressing a precursor form of N-cadherin on the cell surface. Oncotarget. 7(51):84030–84042. 10.18632/oncotarget.12767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J, Sato M, Li Q, Lydon JP, Demayo FJ, Bagchi IC, et al. 2008;Peroxisome proliferator-activated receptor gamma is a target of progesterone regulation in the preovulatory follicles and controls ovulation in mice. Mol Cell Biol. 28(5):1770–1782. 10.1128/MCB.01556-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Janiszewska M, Tabassum DP, Castano Z, Cristea S, Yamamoto KN, Kingston NL, et al. 2019;Subclonal cooperation drives metastasis by modulating local and systemic immune microenvironments. Nat Cell Biol. 21(7):879–888. 10.1038/s41556-019-0346-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steeg PS 2016;Targeting metastasis. Nat Rev Cancer. 16(4):201–218. 10.1038/nrc.2016.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gates LA, Gu G, Chen Y, Rohira AD, Lei JT, Hamilton RA, et al. 2018;Proteomic profiling identifies key coactivators utilized by mutant ERalpha proteins as potential new therapeutic targets. Oncogene. 37(33):4581–4598. 10.1038/s41388-018-0284-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anwar T, Arellano-Garcia C, Ropa J, Chen YC, Kim HS, Yoon E, et al. 2018;p38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat Commun. 9(1):2801 10.1038/s41467-018-05078-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mohammed H, Russell IA, Stark R, Rueda OM, Hickey TE, Tarulli GA, et al. 2015;Progesterone receptor modulates ERalpha action in breast cancer. Nature. 523(7560):313–317. 10.1038/nature14583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sflomos G, Dormoy V, Metsalu T, Jeitziner R, Battista L, Scabia V, et al. 2016;A Preclinical Model for ERalpha-Positive Breast Cancer Points to the Epithelial Microenvironment as Determinant of Luminal Phenotype and Hormone Response. Cancer Cell. 29(3):407–422. 10.1016/j.ccell.2016.02.002 [DOI] [PubMed] [Google Scholar]
- 38.Ozdemir B, Sflomos G, Brisken C 2018;The challenges of modelling hormone-receptor positive breast cancer in mice. Endocrine-related cancer. 25(5):R319–R330. 10.1530/ERC-18-0063 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. S1 Progression of mutant ER-Luc metastases over time in NSG mice inoculated via tail vein with (a) mutant D538G ER-Luc cells or (b) Y537S ER-Luc cells. Animals received treatment with vehicle or K-07 (80 mg/kg) orally 6 days per week, or with Fulvestrant s.c. (80 mg/kg) 6 days per week, and metastatic burden was monitored by IVIS imaging over time. Total Flux values are mean ± SD. ** p<0.01 by Student t-test
Data Availability Statement
All data generated or analyzed in this study are available from the corresponding author on reasonable request.