

Abstract
Vps13 is a highly conserved lipid transfer protein found at multiple interorganelle membrane contact sites where it mediates distinct processes. In yeast, recruitment of Vps13 to different contact sites occurs via various partner proteins. In humans, four VPS13 family members, A–D, are associated with different diseases. In particular, vps13A mutants result in the neurodegenerative disorder Chorea-Acanthocytosis (ChAc). ChAc phenotypes resemble those of McLeod Syndrome, caused by mutations in the XK gene, suggesting that XK could be a partner protein for VPS13A. XK does, in fact, exhibit hallmarks of a VPS13A partner: it forms a complex with VPS13A in human cells and, when overexpressed, relocalizes VPS13A from lipid droplets to subdomains of the endoplasmic reticulum. Introduction of two different ChAc disease-linked missense mutations into VPS13A prevents this XK-induced relocalization. These results suggest that dysregulation of a VPS13A-XK complex is the common basis for ChAc and McLeod Syndrome.
INTRODUCTION
Chorea-Acanthocytosis (ChAc) and McLeod Syndrome, adult-onset neurodegenerative disorders with very similar symptomology, are caused by mutations in the VPS13A and XK genes, respectively (Ho et al., 1994; Rampoldi et al., 2001; Walker et al., 2008; Roulis et al., 2018). Both diseases are characterized by progressive loss of neurons in the striatum and choreic movements, as well as the presence of acanthocytes in peripheral blood smears (Walker et al., 2008; Roulis et al., 2018). The similar phenotypes of the two syndromes suggest that the two genes are functionally linked; however, the molecular basis for either disease has remained elusive.
The XK gene encodes a polytopic integral membrane protein (Ho et al., 1994). In red blood cells, the XK protein is located in the plasma membrane in a complex with the Kell blood group antigen protein (Russo et al., 1998). There are multiple other XK-related (XKR) genes in humans. In mice, XKR8 encodes a lipid scramblase localized at the plasma membrane, which flips phosphatidylserine to the outer leaflet of the plasma membrane in response to apoptotic signals (Suzuki et al., 2013). Two other family members in mice, XKR4 and XKR9, can similarly act as phosphatidylserine scramblases at the plasma membrane (Suzuki et al., 2014). This activity of XKR4, -8, and -9 suggests that proteins in the XK family might generally function as phospholipid scramblases, though no such activity has been shown for XK.
In humans, VPS13A encodes one of four members of the highly conserved Vps13 protein family (Velayos-Baeza et al., 2004). While mutations in VPS13A result in ChAc, mutations in each of the other members of the gene family result in distinct disorders often associated with neurological symptoms, including Cohen Syndrome and autism (VPS13B), Parkinson’s disease (VPS13C), and cerebellar ataxia (VPS13D) (Kolehmainen et al., 2003; Ionita-Laza et al., 2014; Lesage et al., 2016; Gauthier et al., 2018; Seong et al., 2018). The connection of mutations in VPS13 family genes to these different diseases makes understanding the molecular function of this protein family of particular importance.
Studies in fungi have demonstrated that proteins in the Vps13 family act at interorganelle contact sites to mediate the transfer of lipids between different organellar membranes (Lang et al., 2015; Park et al., 2016; Kumar et al., 2018). In baker’s yeast, Saccharomyces cerevisiae, there is a single VPS13 gene. The yeast Vps13 protein is found at several different interorganellar contacts, including mitochondrial–endosome contacts, endoplasmic–reticulum (ER)–vacuolar contacts, and mitochondrial–vacuolar contacts, depending on the growth state of the cell (Lang et al., 2015; Park et al., 2016; John Peter et al., 2017). The deletion of yeast VPS13 is pleiotropic, resulting in missorting of vacuolar proteins in the secretory pathway, failure in sporulation, and defects in mitochondrial homeostasis (Bankaitis et al., 1986; Brickner and Fuller, 1997; Park and Neiman, 2012; Park et al., 2016). These pleiotropic effects probably reflect loss of Vps13 function at different contact sites (Park et al., 2016; John Peter et al., 2017).
The yeast system has been an informative way to assess the possible functions of the mammalian orthologs of VPS13 that are affected by mutations associated with various diseases. For example, introduction of missense mutations in VPS13A, associated with ChAc into the cognate amino acids in yeast VPS13, creates mutants specifically defective in mitochondrial homeostasis, as well as redistribution of Vps13 away from mitochondrial contact sites (Park et al., 2016). These observations indicate that mitochondrial localization correlates with a specific function of Vps13 and suggest that loss of a mitochondrial function of VPS13A may be the basis of ChAc.
Recruitment of yeast Vps13 to different contact sites is mediated by different partner proteins, termed adaptors (Park et al., 2013; John Peter et al., 2017; Bean et al., 2018). Adaptor proteins are localized on different organelles and share a specific motif, centered around the amino acid sequence ΦxxΦxPxPΦxΦ, where Φ is a hydrophobic amino acid (hereafter referred to as the P-x-P motif), through which they compete for binding to the same region of Vps13, termed the Vps13 adaptor binding (VAB) domain (Bean et al., 2018). For example, Vps13 localizes to endosomal membranes through interaction with the adaptor protein Ypt35 (Bean et al., 2018). Spo71 is a meiosis-specific adaptor protein that recruits Vps13 to the prospore membranes used to package the haploid products of meiosis into spores (Park et al., 2013). When ectopically expressed in vegetative cells, Spo71 competes with Ypt35 for binding to Vps13, resulting in relocalization of Vps13 away from endosomes to the plasma membrane (Park et al., 2013; Bean et al., 2018). Importantly, while loss of VPS13 is pleiotropic, deletion of individual adaptor proteins causes only a subset of the vps13∆ phenotypes—those associated with loss of Vps13 activity at a specific organellar contact site (Park et al., 2013; John Peter et al., 2017; Bean et al., 2018).
Similar to yeast Vps13, human VPS13A localizes to mitochondria–ER, ER–lipid droplet, and mitochondria–endosome contact sites in human cells (Kumar et al., 2018; Munoz-Braceras et al., 2019; Yeshaw et al., 2019). The yeast model of Vps13 function predicts that VPS13A should be recruited to these different sites by different adaptor proteins to mediate distinct functions. We propose that loss of VPS13A function at one of these contact sites is responsible for ChAc, in which case, loss of the adaptor protein at that contact site should result in a disease very similar to ChAc.
The similar symptomology of ChAc and McLeod Syndrome caused by vps13A and xk mutants, respectively, suggests that XK and VPS13A might function in the same cellular pathway. Indeed, our results show that the two proteins interact and that XK exhibits the hallmarks of a VPS13A adaptor protein, which recruits VPS13A to a specific site in human cells. However, mutations in neither a P-x-P motif in XK nor the VAB domain of VPS13A disrupt the XK-VPS13A interaction, suggesting that XK partners VPS13A in a manner distinct from the yeast adaptors. Nonetheless, these results reveal a specific molecular link between ChAc and McLeod Syndrome.
RESULTS
VPS13A and XK coprecipitate from HeLa cells
A key prediction of an adaptor protein is that it stably interacts with VPS13A in human cells. This idea was tested biochemically by looking for coimmunoprecipitation (co-IP) of VPA13A and XK from HeLa cells. Endogenous VPS13A was detected using a commercially available antibody (see Materials and Methods). A HAP1 human cell line containing a small internal deletion in the VPS13A gene was used to validate the α-VPS13A antibody (Figure 1A). To monitor the XK protein, the green fluorescent protein gene (GFP) was fused to XK and expressed under the cytomegalovirus promoter. In soluble cell lysates probed with α-GFP antibodies, a protein of the molecular weight predicted for GFP-XK was observed only in HeLa cells transfected with the fusion gene (Figure 1B). The protein level of VPS13A in soluble lysates was too low to be detected by immunoblot analysis, but VPS13A was visible when the protein was concentrated by immunoprecipitation with the VPS13A antibody (Figure 1C).
FIGURE 1:
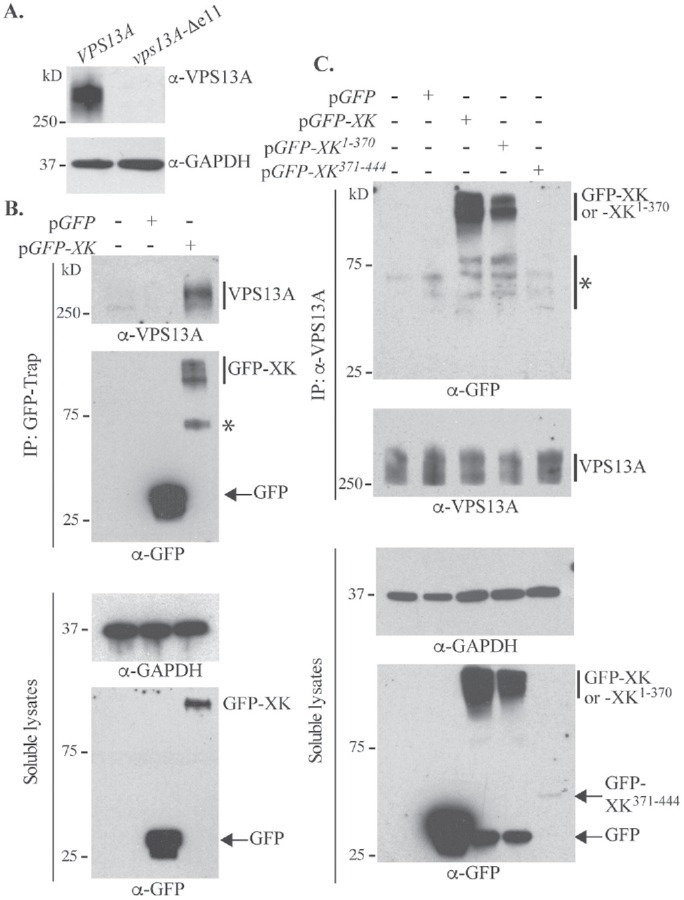
Coimmunoprecipitation of VPS13A and XK. (A) Lysates from human HAP1 cells containing either VPS13A or a 197 nucleotide deletion within exon 11 of VPS13A, vps13A-∆e11–– were probed with either α-VPS13A antibodies or α-GAPDH antibodies as a loading control. (B) Lysates from untransfected HeLa cells or cells transfected with plasmids expressing GFP (pEGFP-C1) or GFP-XK (pcDNA3.1(+)-N-eGFP-XK). (Top panels) GFP-Trap was used to precipitate GFP containing proteins and the precipitates were probed with either α-VPS13A or α-GFP antibodies. The asterisk indicates a likely GFP-XK degradation product. (Bottom panels) Soluble lysates before immunoprecipitation were examined by Western blot using α-GAPDH or α-GFP antibodies. (C) Lysates from untransfected HeLa cells or cells transfected with plasmids expressing GFP (pEGFP-C1), GFP-XK (pcDNA3.1(+)-N-eGFP-XK), GFP-XK1-370 (pJS135), or GFP-XK371-444 (pJS129). (Top panels) VPS13A immunoprecipitates using α-VPS13A probed with either α-GFP or α-VPS13A antibodies. The asterisk marks the position of likely GFP-XK degradation products and background bands. (Bottom panels) The soluble lysates before immunoprecipitation were examined by Western blot using α-GAPDH or α-GFP antibodies.
HeLa cells were transfected with plasmids expressing GFP alone or the GFP-XK fusion. Twenty-four hours after transfection, the GFP-trap reagent was added to soluble cell lysates to immunoprecipitate GFP proteins (Figure 1B). The immunoprecipitates were also probed with α-VPS13A antibodies, revealing that VPS13A coprecipitated specifically with GFP-XK (Figure 1B). Coimmunoprecipitation was also observed when the situation was reversed, that is, GFP-XK, but not GFP alone, was detected in immunoprecipitates generated using α-VPS13A antibodies (Figure 1C). Coimmunoprecipitation of VPS13A and XK was independently discovered by Urata et al. (2019). XK and VPS13A therefore form a complex in vivo.
Interaction between VPS13A and XK does not require the P-x-P motif
In yeast, organelle-specific partner proteins bind to Vps13 through a region of the protein carrying the P-x-P consensus sequence (Bean et al., 2018). The XK protein has 10 predicted transmembrane domains with both its N- and C-termini oriented into the cytoplasm (Ho et al., 1994; Suzuki et al., 2014). Within XK, there are two pairs of prolines properly spaced for P-x-P motifs (WCSGSPFPENI at amino acids 257–267 and SSKTSPEPGQF at amino acids 425–435). The first sequence is located in the fourth extracellular loop of the protein, while the second is found in the C-terminal cytoplasmic tail, making this the best candidate. To test whether XK binds to VPS13A through its C-terminal tail, GFP fusions containing either the last 74 amino acids of XK, including the putative P-x-P motif (GFP-XK371-444), or the XK lacking the C-terminal 74 amino acids (GFP-XK1-370) were analyzed.
Endogenous VPS13A protein was immunoprecipitated from Hela cells containing various GFP-XK fusions using the α-VPS13A antibodies. Similar amounts of VPS13A were precipitated from all the lysates (Figure 1C). GFP-XK1-370, but not GFP-XK371-444, bound to VPS13A (although the possibility that this negative result is due to the low amount of GFP-XK371-444 in the soluble lysates has not been ruled out). Therefore, the first 370 amino acids of XK are sufficient for VPS13A interaction and the potential P-x-P motif in the XK C-terminus is not required.
GFP-XK localizes throughout the endomembrane system
To determine whether VPS13A and XK form complexes at specific places within the cell, localization of the two proteins was examined by fluorescence microscopy. Background fluorescence was too great in HeLa cells for these experiments, so HEK293T cells were used. An N-terminal GFP fusion to human XK displayed a heterogeneous localization throughout the cytoplasm (Figure 2Ai). While localization was seen at the plasma membrane, extensive localization to intracellular membranes, including small foci, bright perinuclear fluorescence, and a reticular pattern throughout the cytosol was also visible. This was not a consequence of the position of the GFP tag, as a similar distribution was seen for a C-terminally tagged XK construct (Figure 2Aii). This is in contrast to a C-terminal mouse XK-GFP fusion that was previously reported to localize to the plasma membrane in HEK293T cells (Suzuki et al., 2014). The reason for this discrepancy is not clear. The GFP-XK1-370 construct had a similar distribution to the full-length proteins (Figure 2Aiii), but GFP-XK371-444, consistent with this protein lacking a transmembrane domain, was found diffusely throughout the cell (Figure 2Aiv).
FIGURE 2:
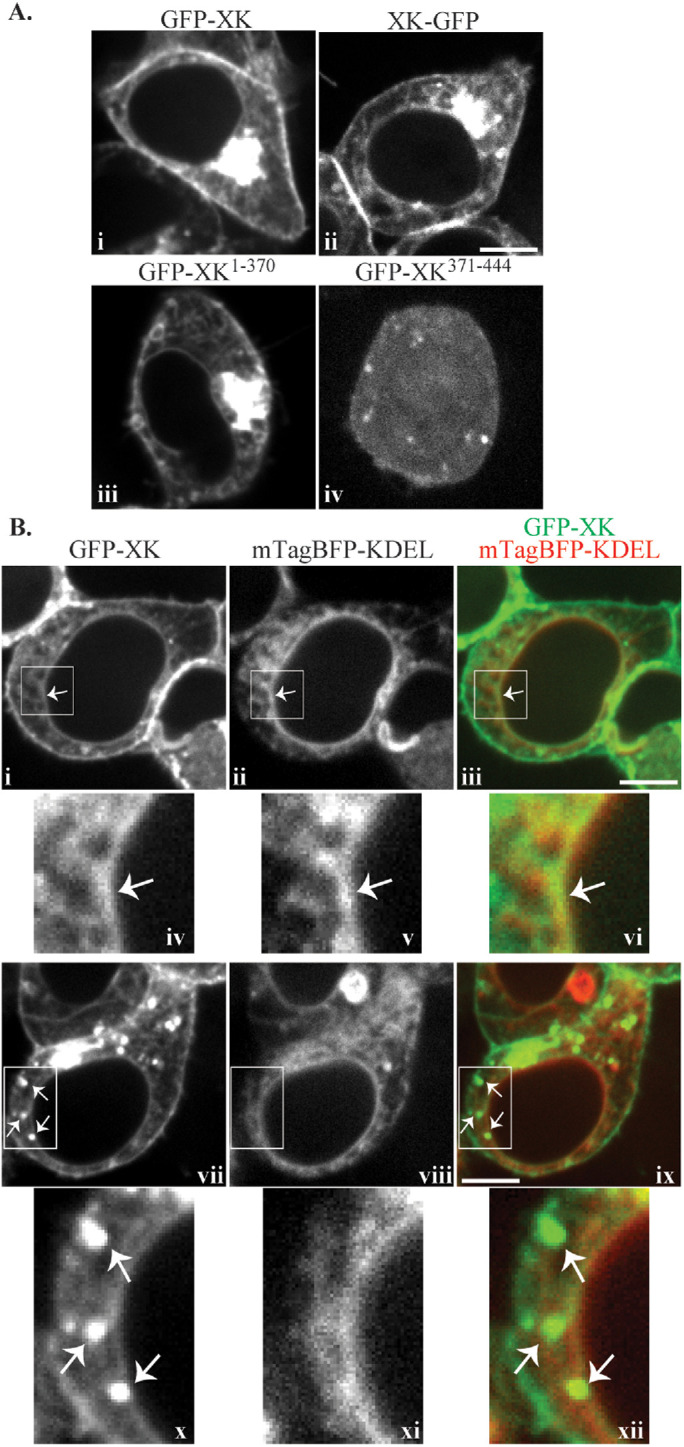
Localization of GFP-XK. (A) HEK293T cells were transfected with plasmids expressing GFP-XK (pcDNA3.1(+)-N-eGFP-XK), XK-GFP (pJS136), GFP-XK1-370 (pJS135), or GFP-XK371-444 (pJS129) and examined by fluorescence microscopy. (B) HEK293T cells were transfected with plasmids expressing GFP-XK (pcDNA3.1(+)-N-eGFP-XK) and the ER marker mTagBFP-KDEL (pEFIRES-mTagBFP-KDEL). Panels i–vi show a representative cell where GFP-XK displays a reticular pattern overlapping with the ER marker. Panels iv–vi are higher magnification views of the boxed areas in panels i–iii, respectively. The arrow indicates a region where the GFP-XK signal colocalizes with ER marker. Panels vii–xii show a representative cell where GFP-XK displays a punctate pattern. Panels x–xii are higher magnification views of the boxed areas in panels vii–ix, respectively. The arrows highlight examples of GFP-XK foci that lack corresponding foci of ER signal. Scale bars = 10 μm.
Several hundred cells expressing GFP-XK were examined and the extent of localization in each pattern varied between different cells. The reticular pattern resembles the ER and GFP-XK in this pattern extensively colocalized with the ER marker mTagBFP-KDEL (Salo et al., 2016) (Figure 2B, i–vi). In contrast, GFP-XK foci did not obviously correspond with the lumenal ER marker (Figure 2Bvii–xii), suggesting that these foci represent localization to some other compartment or are elaborations of ER that lack lumenal content. Thus, the GFP-XK protein is found in multiple compartments including the ER and the plasma membrane.
VPS13A was localized using the VPS13A^mCherry protein which contains an internal insertion of mCherry after amino acid residue 1371 of VPS13A (Kumar et al., 2018). As reported previously, two patterns of VPS13A^mCherry localization were seen: bright round puncta and short tubular structures (Figure 3). Consistent with previous reports (Kumar et al., 2018), costaining of cells with the lipid droplet dye 4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene (BODIPY 493/503) demonstrated that the bright puncta represent localization to the surface of lipid droplets (Figure 3A), while the tubular structures overlap with the ER marker mTagBFP-KDEL (Figure 3B).
FIGURE 3:

VPS13A^mCherry localizes to lipid droplets and the ER. (A) HEK293T cells transfected with a plasmid expressing VPS13A^mCherry (pVPS13A^mCherry) and stained with the lipid droplet marker BODIPY 493/503. (B) HEK293T cells transfected with plasmids expressing VPS13A^mCherry and ER marker mTagBFP-KDEL display a partial colocalization of VPS13A^mCherry and ER marker. Dashed lines indicate the cell outlines. Insets show higher magnification views of the boxed regions. Scale bars = 10 μm.
VPS13A and GFP-XK colocalize in human cells
Cells cotransfected with VPS13A^mCherry and GFP-XK exhibited colocalization of the two proteins to discrete puncta (Figure 4A, yellow arrows), extended linear elements (Figure 4B, pink arrows) or larger, ring-shaped structures (Figure 4B, white arrows). The XK fusions were more extensively distributed throughout cells than VPS13A^mCherry. Therefore, colocalization was quantified by determining the fraction of VPS13A^mCherry foci that also displayed GFP-XK. To control for possible effects of the GFP tag, colocalization XK-GFP was examined as well. Almost every VPS13A^mCherry focus in the cotransfected cells had an associated GFP-XK or XK-GFP focus (Figure 4C). In contrast, no correspondence between GFP and VPS13A^mCherry localization was observed, confirming that colocalization is due to the XK protein. Consistent with the co-IP results, GFP-XK1-370 colocalized with VPS13A^mCherry (though the colocalization was slightly reduced from full-length GFP-XK), but GFP-XK371-444 did not (Figure 4C). Similar results were seen when we quantified the colocalization of the different GFP fusions with VPS13A^mCherry at the extended linear elements or ring structures (Figure 4D).
FIGURE 4:

Colocalization of GFP-XK and VPS13A^mCherry in human cells. HEK293T cells were transfected with plasmids expressing VPS13A^mCherry and various versions of GFP-XK as indicated. (A) Representative cell showing colocalization of GFP-XK and VPS13A^mCherry in cytoplasmic foci (yellow arrows). Scale bar = 10 μm. (B) Representative cell showing GFP-XK and VPS13A^mCherry colocalization in ringlike structures (white arrows) and linear elements (pink arrows). (C) Quantification of VPS13A^mCherry colocalization in foci containing various GFP-XK proteins. Data are the average of at least three experiments with more than 25 foci scored in each experiment. Asterisks indicate p value < 0.00002 by unpaired t test. In graphs C–F, error bars represent one SD. (D) Quantification of VPS13A^mCherry/GFP-XK colocalization in rings and linear elements. Data are the average of three or more experiments with at least 20 cells scored in each experiment and at least 20 total rings or linear elements scored for the XK-GFP, GFP-XK, and GFP-XK1-370 constructs. *p < 0.0001 by one-way ANOVA with Dunnett’s test. (E) The number of VPS13A^mCherry foci per cell in cells coexpressing either GFP alone or the indicated GFP-XK fusions. Data are the average of at least three experiments with more than 25 foci scored for each construct in each experiment. At least 101 total foci scored for each construct. Horizontal lines indicate median values, boxes represent three quartiles, and whiskers indicate the range of values. *p < 0.0001 by unpaired t test. (F) The fraction of cells displaying rings or lines of VPS13A^mCherry fluorescence when coexpressing different GFP constructs. The fraction of cells displaying at least one ring or linear element is shown. Values are averages of three independent experiments with at least 19 cells scored per experiment. *p < 0.0005, unpaired t test
The number of bright VPS13A^mCherry foci visible was reduced in cells cotransfected with XK. Cells cotransfected with either GFP or GFP-XK371-444 displayed ∼5 foci per cell (Figure 4E). This number dropped to ∼1 focus per cell in cells cotransfected with any of the XK fusions capable of binding to VPS13A. Furthermore, extended linear elements and ring structures containing VPS13A^mCherry were only seen in the cells coexpressing GFP-XK, XK-GFP, or GFP-XK1-370, though significantly (p < 0.005, unpaired t test) more of these structures were seen with XK-GFP than the other fusions (Figure 4F). These results indicate that interaction with XK redistributes the VPS13A^mCherry protein either by concentrating it in fewer foci or in novel patterns.
GFP-XK relocalizes VPS13A from lipid droplets to subdomains on the ER
To compare the punctate localization of VPS13A^mCherry with and without GFP-XK coexpression, colocalization with the lipid droplets was quantified. In cells transfected with only VPS13A^mCherry, ∼99% of the bright VPS13A^mCherry puncta overlapped with the lipid droplet marker, consistent with localization on the lipid droplet surface (Figure 5). By contrast, in cells where GFP-XK was cotransfected with VPS13A^mCherry, only ∼5% of the VPS13A^mCherry puncta colocalized with lipid droplets. Thus, expression of GFP-XK releases VPS13A protein from lipid droplets.
FIGURE 5:

VPS13A^mCherry localization to lipid droplets is altered by GFP-XK expression. HEK293T cells were transfected with VPS13A^mCherry (pVPS13A^mCherry) without (left panels) or with (right panels) mTagBFP-XK (pJS143-E6) and stained with the lipid droplet dye BODIPY 493/503. Yellow arrows highlight examples of VPS13A^mCherry foci and pink arrows highlight examples of lipid droplets stained with BODIPY Green. Numbers below indicate the percentage of VPS13A^mCherry foci overlapped with the lipid droplet marker ± range. Data are the average of two independent experiments. In the absence of GFP-XK 255 foci were scored, while 96 foci were scored for cells containing GFP-XK. Scale bars = 10 μm. For illustration purposes, the cells shown have a greater than average number of VPS13A^mCherry foci (Figure 4).
GFP-XK is localized in multiple compartments (Figure 2), yet GFP-XK recruits VPS13A to discrete foci, not throughout the cell. To which compartment is VPS13A being directed? Staining GFP-XK and VPS13^mCherry cotransfected cells with the lipid droplet marker confirmed that the colocalizing foci are not at lipid droplets (Figure 6A). By contrast, when the ER marker mTagBFP-KDEL was cotransfected, 100% of the GFP-XK/VPS13A^mCherry foci overlapped with the blue fluorescence (average of two independent experiments with at least 20 foci scored in each experiment), indicating that these are specific regions of the ER (Figure 6B). Thus, induction of GFP-XK recruits VPS13A from lipid droplets to subdomains of the ER. This could result from release of VPS13A from the lipid droplet and recruitment to the ER, or alternatively, VPS13A could be located in an ER subdomain wrapped around lipid droplets (Kumar et al., 2018) and this entire subdomain released by GFP-XK expression
FIGURE 6:

VPS13A^mCherry and GFP-XK colocalize at the ER and mitochondria. HEK293T cells cotransfected with VPS13A^mCherry and GFP-XK or mTagBFP-XK were either stained with the lipid droplet marker BODIPY 493/503 or cotransfected with an additional plasmid expressing an ER marker (pEFIRES-mTagBFP-KDEL) or a mitochondrial marker (mito-BFP). (A) Representative cell showing the localization of a VPS13A^mCherry/mTagBFP-XK focus (yellow arrow) relative to a lipid droplet. (B) Representative cell showing the localization of a VPS13A^mCherry/GFP-XK focus (yellow arrow) relative to the ER. (C) Representative cell showing the localization of a VPS13A^mCherry/GFP-XK focus (yellow arrow) relative to the mitochondrial marker. Scale bars = 10 μm.
VPS13A localizes to ER–mitochondria contact sites (Kumar et al., 2018; Yeshaw et al., 2019). It is possible, therefore, that the specific regions of the ER to which XK recruits VPS13A are these contact sites. To investigate this possibility, a mitochondrial marker (mito-BFP; Friedman et al., 2011) was cotransfected along with GFP-XK and VPS13A^mCherry into HEK293T cells. For each GFP-XK/VPS13A^mCherry focus, fluorescence of the mitochondrial marker was examined and each focus was scored for overlap with the mitochondrial marker. Over half of the XK/VPS13A foci were found adjacent to or overlapping the mitochondrial signal (62%, average of three experiments with at least 50 foci scored per experiment; Figure 6C). This result indicates that a substantial fraction of the XK/VPS13A complexes in the ER could be at ER–mitochondrial contact sites, though nearly 40% are not associated with mitochondria. As a control, the association of VPS13A^mCherry foci with the mitochondrial marker was also examined in cells without GFP-XK overexpression (i.e., when the VPS13A^mCherry foci are at lipid droplets). Although an increase in association with mitochondria is seen with GFP-XK overexpression, the change was not statistically significant (p = 0.06; Student’s t test). Thus, it is not clear whether the XK/VPS13A structures represent mitochondrial contact sites.
VPS13A localization is disrupted by disease-associated mutations
In yeast, multiple Vps13 adaptors compete for binding to the VAB domain of the protein (Bean et al., 2018). One disease-associated VPS13A missense mutation, W2460R (Dobson-Stone et al., 2002), is found in the VAB domain, raising the possibility that it is the loss of interaction with an adaptor protein that is relevant to the disease state. Given that the P-x-P motif defined from yeast is not required for XK-VPS13A interaction, the question was whether the VPS13A VAB domain is necessary for XK to interact with VPS13A. The W2460R mutation was introduced into the VPS13A^mCherry construct and was cotransfected with a plasmid expressing GFP-XK into HEK293T cells. In Western blots of extracts probed with anti-VPS13A antibodies, the endogenous VPS13A protein was not visible in extracts of HEK293T cells, though both the wild-type and mutant VPS13A^mCherry proteins were observed (Figure 7A). These results demonstrate not only that the mutant is expressed as a full-length protein (albeit at a reduced level compared with the wild type) but also that VPS13A^mCherry is significantly overexpressed in the transfected cells. We found that our GFP-trap reagent cross-reacted with VPS13A^mCherry; therefore, to test for coprecipitation, anti-XK antibodies were used to pull down GFP-XK and the precipitates were probed with anti-VPS13A antibodies. Both the wild-type VPS13A^mCherry protein and VPS13AW2460R^mCherry coprecipitated with GFP-XK in these experiments (Figure 7A). The signal from VPS13AW2460R^mCherry was lower than with the wild-type protein, but above any background from the endogenous protein (Figure 7A, lane 3). The lower precipitation of VPS13AW2460R^mCherry may be due to the lower expression level of this construct. These results indicate that mutation of the VAB domain does not disrupt the physical interaction of XK with VPS13A.
FIGURE 7:
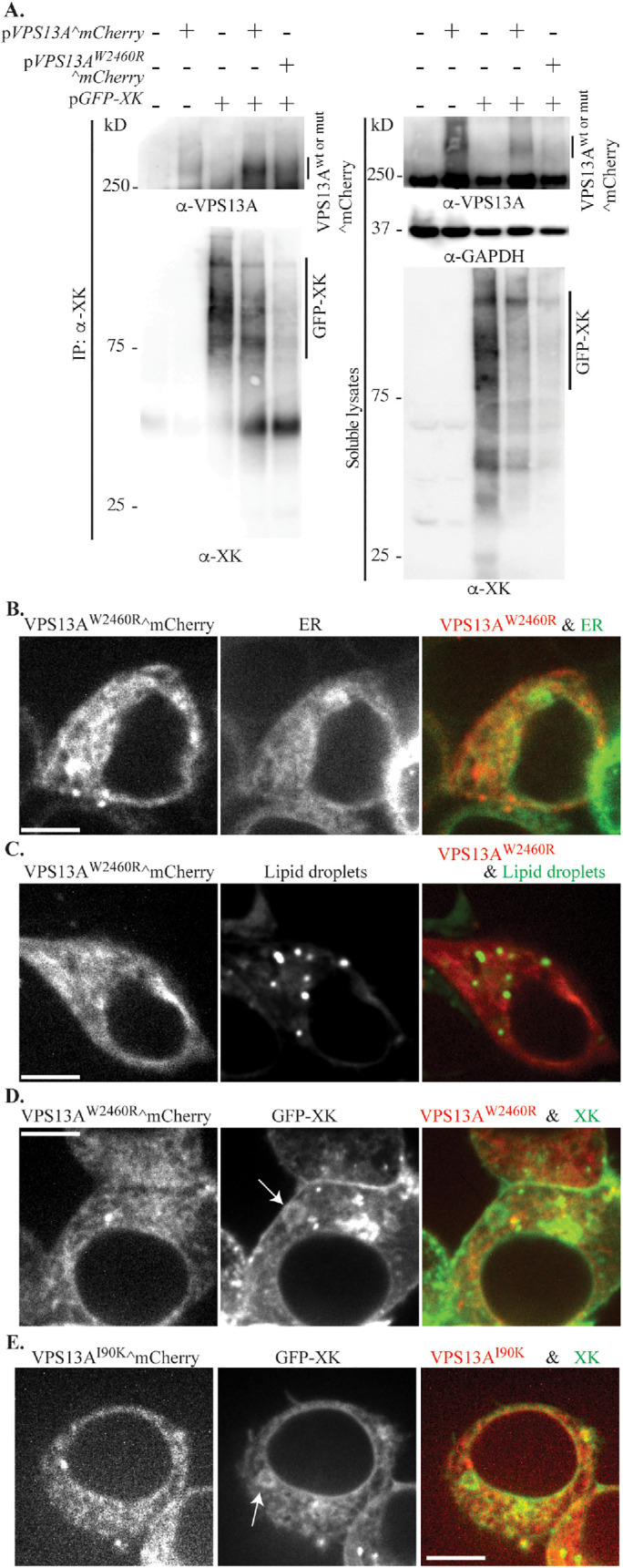
The disease mutation W2460R alters VPS13A^mCherry localization (A) Lysates of untransfected HEK293T cells, or cells transfected with single plasmids expressing VPS13A^mCherry (pVPS13A^mCherry) or GFP-XK (pcDNA3.1(+)-N-eGFP-XK), or two plasmids expressing wild-type VPS13A^mCherry or VPS13AW2460R^mCherry (pJS141-E4) with GFP-XK were analyzed by co-IP using polyclonal anti-XK antibodies and Western blot using anti-VPS13A antibodies, anti-XK antibodies, or anti-GAPDH antibodies as a loading control. (B) Localization of VPS13AW2460R^mCherry was examined in HEK293T cells cotransfected with plasmids expressing VPS13AW2460R^mCherry (pJS141-E4) and the ER marker mTagBFP-KDEL (pEFIRES-mTagBFP-KDEL). (C) VPS13AW2460R^mCherry localization was examined in HEK293T cells stained with the lipid droplet dye BODIPY 493/503. (D) Distribution of VPS13AW2460R^mCherry in cells cotransfected with plasmids expressing VPS13AW2460R^mCherry (pJS141-E4) and GFP-XK (pcDNA3.1(+)-N-eGFP-XK). (E) Distribution of VPS13AI90K^mCherry in cells cotransfected with VPS13AI90K^mCherry (pJS144-B2) and GFP-XK (pcDNA3.1(+)-N-eGFP-XK). Arrows in D and E indicate rings of GFP-XK in the ER. Scale bars = 10 μm.
The localization of VPS13AW2460R^mCherry was then examined by fluorescence microscopy. Similar to the wild-type protein, VPS13AW2460R^mCherry was found distributed in small patches tubules that partially overlapped with an ER marker (Figures 7B and 3B). However, in contrast to the wild-type protein, larger, bright foci were absent and no association of VPS13AW2460R^mCherry with the lipid droplet marker was seen (Figure 7C). Thus, the VAB domain is required for the association of VPS13A with the lipid droplets.
GFP-XK overexpression induces the colocalization of VPS13A^mCherry with GFP-XK in foci, elongated elements, and rings (Figure 4). Surprisingly, despite the coprecipiation of the two proteins (Figure 7A), when VPS13AW2460R^mCherry and GFP-XK were cotransfected, the distribution of VPS13AW2460R^mCherry did not appear to colocalize with GFP-XK (Figure 7D). To quantify this observation, the number of GFP-XK rings that display mCherry fluorescence in cells cotransfected with GFP-XK and either VPS13A^mCherry or VPS13AW2460R^mCherry was counted. While 87% of the GFP-XK rings displayed colocalization with the wild-type, only 3% colocalization was seen with VPS13AW2460R^mCherry (average of at least two experiments with at least 24 rings scored per experiment, p < 0.00015; Student’s t test).
Colocalization was also examined with VPS13A carrying a second disease-associated mutation, I90K, located in the conserved N-Chorein domain near the amino-terminus of VPS13A (Rampoldi et al., 2001). Similar to VPS13AW2460R^mCherry, VPS13AI90K^mCherry did not colocalize with overexpressed GFP-XK (Figure 7E). Thus, disease-associated mutations in two different domains of VPS13A disrupt colocalization with XK in vivo.
DISCUSSION
The neurogenerative disorders, ChAc and McLeod Syndrome, are caused by two different genes: VPS13A and XK, respectively. Because VPS13 family proteins are known to be recruited to different organelles through partner proteins, the phenotypic similarities between ChAc and McLeod Syndrome suggested that XK might be a VPS13A partner protein. This idea is strongly supported by the fact that the two proteins form a complex and colocalize within the cell (Urata et al., 2019; this work). Furthermore, XK is capable of relocalizing VPS13A from lipid droplets to ER subdomains, and two different disease-associated mutations in VPS13A disrupt this relocalization. We propose therefore that both ChAc and McLeod Syndrome result from disruption of XK-VPS13A complex formation.
In yeast, Vps13 partners, termed adaptors, bind to Vps13 through the conserved VAB domain. Indeed, a mutation in VPS13A that results in ChAc, VPS13A-W2460R resides in the adaptor binding domain and disrupts colocalization with XK. Despite the loss of colocalization, the VPS13AW2460R still coimmunoprecipitates with XK. While this is consistent with our finding that the P-x-P motif in XK is not required for interaction with VPS13A and suggests that XK does not bind to VPS13A through the VAB domain, it is puzzling that the W2460R mutation disrupts colocalization with XK but not co-IP of the two proteins. One possible explanation is that colocalization into specific ER subdomains requires an additional partner protein to stabilize the VPS13A-XK complex at these sites. In this model, the W2460R mutation disrupts the interaction with that additional partner protein. Given that W2460 lies in the VAB domain, that additional partner may bind VPS13A similarly to a canonical yeast adaptor protein. Moreover, because the W2460R mutation also disrupts the localization of the protein to lipid droplets, our results are consistent with the model that recruitment of VPS13A to lipid droplets is mediated through a yeast-like adaptor as well.
That the VAB domain mutation alters lipid droplet localization is somewhat surprising given that previous studies have identified C-terminal fragments of VPS13A that do not contain the VAB domain as sufficient to localize GFP to lipid droplets (Kumar et al., 2018; Yeshaw et al., 2019). In yeast, lipid-binding activities, with different lipid specificities, have been identified in several regions of the Vps13 protein, including the C-terminus (Rzepnikowska et al., 2017). The effect of the disease mutations on lipid droplet localization might result from an alteration in the folding of the C-terminus of the protein rather than a direct role for these domains in lipid droplet binding. Alternatively, it may be that, in the context of the intact protein, localization to the lipid droplet requires interactions through several domains of the protein, but that the C-terminal region is sufficient for lipid droplet localization when fused to GFP.
Our GFP-XK fusion localized broadly in different membrane compartments, yet VPS13A colocalized with XK only in small subdomains of the ER. This restricted localization suggests that additional factors might limit where XK and VPS13A interact. VPS13A contains an FFAT motif, through which it interacts with the ER-localized VAP-A protein (Kumar et al., 2018; Yeshaw et al., 2019). Mutation of the FFAT motif in VPS13A disrupts its colocalization with VAP-A and seems to broadly release VPS13A from the ER (Yeshaw et al., 2019). One possibility is that the combined action of VAP-A and XK is necessary to recruit VPS13A to specific subregions of the ER. It will be interesting in this regard to test whether overexpression of XK can still recruit the FFAT mutant of VPS13A to specific sites in the ER or, perhaps, just to the ER more broadly.
A high-throughput proteomics study in HEK293T cells identified XKR2, the closest paralog to XK as a VPS13A interacting protein (Huttlin et al., 2015). Human cells encode seven XK paralogs and four VPS13 family genes. It is possible that other XK paralogs function in conjunction with VPS13A at different locations or in different cell types or that these XK paralogs partner with other VPS13 family proteins.
Another question to be resolved is what is the XK/VPS13A subdomain of the ER? One obvious possibility was that these would represent ER–mitochondrial contact sites. Though we see some association of the mitochondria with these sites (Figure 6C), this association is not complete. One complicating factor is that both XK and VPS13A are overexpressed in our studies, which could cause abnormal localization. To resolve the question of where in the cell XK/VPS13A act, it will be important to determine where XK and VPS13A are localized at native expression levels.
Both McLeod Syndrome and ChAc are characterized by the appearance of acanthocytes in peripheral blood smears (Walker et al., 2008). In these cells, the XK protein is found in association with the Kell blood group antigen in the red blood cell plasma membrane (Russo et al., 1998). Although GFP-XK was seen at the plasma membrane in the HEK293T cells, this subpopulation of XK did not associate with VPS13A. It remains to be determined if VPS13A associates with XK to form a junction site at the red blood cell plasma membrane that is important for maintaining proper morphology of the cell or if, perhaps, the acanthocytosis phenotype is a consequence of loss of XK-VPS13A complexes at ER contact sites earlier in the differentiation of the red blood cells.
The results presented argue that dysfunction of an XK-VPS13A complex is the common basis for both McLeod Syndrome and ChAc. These proteins must function together to provide some activity essential for long-term survival of striatal neurons as well as normal red blood cell morphology. A critical outstanding issue is what is this function of the XK-VPS13A complex? Based on the behavior of the yeast ortholog, it is likely that VPS13A acts to transfer lipids between the ER and another organelle. The function of XK is unknown; however, some XKR proteins appear to act as plasma membrane localized lipid scramblases able to flip phosphatidylserine across the lipid bilayer in response to apoptotic signals (Suzuki et al., 2014). If XK also has scramblase activity, then an XK-VPS13A complex potentially links movement of lipids between leaflets of one membrane to transport of lipids between two membranes, providing an efficient way of moving a lipid from the luminal leaflet of one organellar membrane to the cytosolic leaflet of another, or vice versa. Given the localization of VPS13A to mitochondrial–ER contacts and the mitochondrial defects of VPS13A disease-associated mutations when expressed in yeast cells (Park et al., 2016; Kumar et al., 2018; Yeshaw et al., 2019), it is tempting to speculate that XK-VPS13A might act to transfer lipids between the inner leaflet of the ER and the outer leaflet of the mitochondrial outer membrane.
MATERIALS AND METHODS
Cell culture and transfections
HeLa (obtained from Sook-Young Sohn, Stony Brook University, Stony Brook, NY) and HEK293T cells (obtained from Kevin Czaplinski, Stony Brook University, Stony Brook, NY) were maintained at 37°C in humidified atmosphere at 5% of CO2 in DMEM supplemented with 1% penicillin and streptomycin and 10% fetal bovine serum (FBS). The HAP1 and HAP1-vps13A-∆e11 cell lines were purchased from Horizon Discovery. The vps13A-∆e11 allele carries a 197 base pair deletion in exon 11 resulting in a frameshift early in the coding region (https://www.horizondiscovery.com/human-vps13a-knockout-cell-line-197bp-deletion). HAP1 cells were incubated in Iscove’s Modified Dulbecco’s Medium supplemented with 1% penicillin and streptomycin and 10% FBS at 37°C in humidified atmosphere at 5% of CO2.
HeLa cells were transfected by using polyethyleneimine (PEI, 1 μg/μl). Briefly, when HeLa cells were 80% confluent in 35-mm glass bottom dishes (cat. P35G-1.5-14-C; MatTek Co.) or 10-cm tissue culture dishes (cat. 83.3902; SARSTEDT), 1.5 μg plasmid DNA plus 7.5 μg PEI or 10 μg plasmid DNA plus 40 μg PEI were mixed into 100 or 600 μl DMEM medium, respectively. After 10 min incubation at room temperature, this plasmid/PEI mixture in DMEM medium was added dropwise onto HeLa cells. Transfected cells were assayed by Western blot analysis 24 to 48 h after transfection. Transfection for HEK293T cells was done using Lipofectamine 2000 (cat. 11668-019; Thermo Fisher Scientific) following the manufacturer’s instructions. For live cell microscopy, HEK293T cells were grown in gelatin (cat. ES-006-B; Millipore)-coated 35-mm glass bottom culture dishes. Separately, 2 μg plasmid DNA and 4 μl Lipofectamine 2000 were added to 200-μl aliquots of OptiMEM (cat. 31985-062; Life Technologies). After 5 min incubation at room temperature, the Lipofectamine 2000 mixed with OptiMEM was added into the plasmid DNA plus OptiMEM and this mixture was incubated at room temperature for 20 min. As with HeLa cell transfection, the plasmid/Lipofectamine 2000 mixture in OptiMEM medium was then added dropwise onto HEK293T cells. For co-IP and Western blot analysis, HEK293T cells were grown in 10-cm culture dishes, and aliquots of 10 μg of plasmid DNA in 1 ml OptiMEM and 20 μl of Lipofectamine 2000 in 1 ml OptiMEM were used for transfection. The amount of two plasmids for cotransfection was 2.5 μg pGFP-XK and 7.5 μg pVSP13Awt or mutant^mCherry.
All cell lines tested free of mycoplasma contamination.
Plasmid construction
Plasmids and primers used in this study are listed in Tables 1 and 2, respectively. To construct plasmid JS135, expressing the C-terminally truncated XK protein, the region coding the first 370 amino acids of XK was first amplified from pcDNA3.1(+)-N-eGFP-XK (GenScript) using the PCR with oligos JSO587 and JSO588. pcDNA3.1(+)-N-eGFP-XK was digested with EcoRV and XbaI restriction enzymes to release the XK coding region and the vector backbone (∼6.2 kb) was purified by agarose gel electrophoresis using PureLink Quick Gel Extraction kit (cat. K2100-12; Invitrogen). The PCR fragment was then introduced into the linearized vector by Gibson assembly (cat. E2611L; New England BioLabs) according to the manufacturer’s instructions.
TABLE 1:
Plasmids used in this study.
| Name | Gene expressed | Source |
|---|---|---|
| JS113 | GFP-XK371-444 | This study |
| JS129 | GFP-XK371-444 | This study |
| JS135 | GFP-XK1-370 | This study |
| JS136 | XK-GFP | This study |
| JS141-E4 | VPS13AW2460R^mCherry | This study |
| JS141-E6 | VPS13AW2460R^mCherry | This study |
| JS143-E6 | mTagBFP-XK | This study |
| JS144-B2 | VPS13AI90K^mCherry | This study |
| pVPS13A^mCherry | VPS13A^mCherry | (Kumar et al., 2018) |
| pcDNA3.1(+)-N-eGFP-XK | GFP-XK | GenScript |
| mito-BFP | mitochondrial-BFP | (Friedman et al., 2011) |
| pEFIRES-mTagBFP-KDEL | ER localized BFP | (Salo et al., 2016) |
TABLE 2:
Primers used in this study.
| Name | Sequence |
|---|---|
| JSO558 | 5′-ACTAGTAACGGCCGCCAGTGTGCTGGAATTCATGAGTAAAGGAGAAGAACTTTT |
| JSO559 | 5′-TAGAATAGGGCCCTCTAGATGCATGCTCGAGTTAAGCAGAGCAGAGATCTTC |
| JSO587 | 5′-ACCGAGCTCGGATCCGAATTCTGCAGATATCTTATGAAATTCCCGGCCTC |
| JSO588 | 5′-CTGATCAGCGGGTTTAAACGGGCCCTCTAGATTAATAGAATACAAGCATGAAGAG |
| JSO600 | 5′-TGGTGGCGACCGGTGGATCCCGGGCCCGCGGAGCAGAGCAGAGATCTTCAG |
| JSO605 | 5′-CTTCACCATGGCTTTTTCTACATCTTCTCTTCAGCCTTCTAGAGCCC |
| JSO606 | 5′-TCCGGTGGGCTCTAGAAGGCTGAAGAGAAGATGTAGAAAAAGCCATGGTG |
| JSO607 | 5′-GTATGGCTGATTATGATCTAGAGTCGCGGCCGCTTCAGAGGCTCGGAGAAGGT |
| JSO623 | 5′-CGGCGGCATGGACGAGCTGTACAAGGGGCCCGCAACTGTGGTGACAGCTG |
| JSO637 | 5′-CTCCTTGATGATGGCCATGT |
| JSO638 | 5′-GAAGAAGGCACTTTAAGTAAATAAATTTCTTCCAATACGG |
| JSO639 | 5′-TTTATTTACTTAAAGTGCCTTCTTCTAGAATAAAATAT |
| JSO654 | 5′- AGACCCAAGCTGGCTAGCGTTTAAACTTAAGCTTGCCACCATGAGCGAGCTGATTAAGGAGAA |
| JSO655 | 5′-GCCGGGAATTTCATAAGATATCTGCAGAATTCGGATCCGAGCTCGGTACCATTAAGCTTGTGCCCCAGTT |
| CMV-F | 5′-CGCAAATGGGCGGTAGGCGTG |
To construct a plasmid for expression of a GFP fusion to the C-terminal 74 amino acids of XK in mammalian cells (pJS129), a GFP- XK371-444 fusion was first assembled in a yeast expression vector. The GFP gene was amplified by PCR to place SpeI and SmaI sites before the start and stop codons, and XK371-444 was amplified from a human cDNA library flanked with SmaI and XhoI sites. These two PCR products were incorporated into the SpeI/XhoI-digested yeast expression vector pRS425-TEF (Mumberg et al., 1995) by Gibson assembly, creating the plasmid, pJS113. To generate pJS129, pJS113 was used as a template to amplify GFP-XK371-444 with oligonucleotides JSO558 and JSO559, and the PCR product introduced into EcoRI and XhoI digested mammalian expression vector pcDNA3 (a gift from Sook-Young Sohn, Stony Brook University, Stony Brook, NY) by Gibson assembly. To construct the plasmid pJS136 for expression of C-terminally GFP tagged XK, the XK cDNA with EcoRI and SacII sites at 5′ and 3′ ends, respectively, was amplified from pcDNA3.1(+)-N-eGFP-XK using oligos JSO587 and JSO600. This PCR product and the expression plasmid pEGFP-N1 (Molecular Cloning Facility, Stony Brook University, Stony Brook, NY) were digested with EcoRI and SacII and ligated, resulting in C-terminally GFP-tagged XK. To construct a plasmid expressing N-terminally mTagBFP-tagged XK (pJS143-E6), mTagBFP cDNA was amplified from pEFIRES-mTagBFP-KDEL by using JSO654/JSO655 and pcDNA3.1(+)-N-eGFP-XK was linearized by HindIII/EcoRI double digestion in order to remove the fragment encoding. The mTagBFP cDNA was incorporated into the linear pcDNA3.1(+)-N-XK backbone by Gibson assembly. To generate the expression plasmids pJS141-E4 and pJS141-E6, which express pVSP13AW2460R^mCherry, two fragments were amplified using the pairs of oligos JSO623/JSO605 and JSO606/JSO607 with pVSP13A^mCherry as template. These PCR products encompass the 5′ and 3′ ends of the VPS13A^mCherry gene with the base change creating the W2460R mutation located at the overlap of the two fragments within the primer sequences. pVSP13A^mCherry was cut with ApaI and NotI to release the region of the VPS13A^mCherry gene 3′ of the mCherry coding region, and pVSP13AW2460R^mCherry was created using Gibson assembly to combine the two PCR products with the vector backbone. The expression plasmid pJS144-B2 expressing pVSP13AI90K^mCherry was constructed in a similar manner. To amplify sequences carrying the I90K mutation, the pairs of oligos used were CMV-F/JSO638 and JSO637/JSO639. pVPS13A^mCherry was cut with XhoI and SalI to release the region of the VPS13A^mCherry gene 5′ of the mCherry coding region and the expression vector was reassembled by Gibson assembly using the vector backbone and the two PCR products.
Immunoprecipitation and Western blot analysis
Transfected cells were washed with phosphate-buffered saline (PBS; cat. 14190-144; Life Technologies), harvested using a cell scraper into 1.5-ml microcentrifuge tubes, and stored at –80°C. For GFP-pull downs, cell pellets were resuspended in 200 μl lysis buffer (10 mM Tris-HCl, pH 7.5; 150 mM NaCl; 0.5 mM EDTA; 0.5% NP-40; protease inhibitor cocktail; cat. 04 693 159 001; Roche) and lysed by sonication with a microprobe (QSONICA Sonicators) at amplitude 40 for 10 s, twice per sample. Cell lysates were centrifuged at 11,000 × g for 5 min at 4°C and supernatants were collected. To precipitate GFP-tagged proteins, 25 µl GFP-trap agarose (cat. GTA020; Bulldog Bio) was added to the supernatants and incubated at 4°C for 1 h. GFP-trap agarose was precipitated by centrifugation at 2500 × g at 4°C for 2 min and washed twice with washing buffer (10 mM Tris/Cl, pH 7.5; 150 mM NaCl; 0.5 mM EDTA). Finally, 2× SDS sample buffer (100 mM Tris, pH 6.8; 4% SDS; 20% Glycerol; 0.2 mg/ml Bromophenol blue; 0.72 M β-Mercaptoethanol) was added and the beads were boiled at 100°C for 5 min.
For VPS13A or XK immunoprecipitations, cells were lysed with lysis buffer (50 mM HEPES, pH 7.4; 150 mM KCl; 1 mM EDTA; 0.5% NP-40; 1 mM DTT; protease inhibitor cocktail; cat. 04 693 159 001; Roche) and sonicated under the same conditions described above. Supernatants were incubated with 3 μl of anti-VPS13A antibody (0.4 mg/ml) (cat. HPA021652; Sigma) or 6 μl of anti-XK antibody (0.4 mg/ml) (cat. HPA019036; Sigma) at 4°C for 1.5 h; 25 μl of Dynabeads Protein A (cat. 81110110; Invitrogen) was added to the mixture of lysate and anti-VPS13A antibody or anti-XK antibody and incubated at 4°C for 1 h. After incubation the beads were washed twice with washing buffer (20 mM Tris/Cl, pH 7.5; 20 mM NaN3; 20 mM NaF) on magnet and boiled in sample buffer as described above.
Protein samples were run on precast SDS–PAGE gels (3–8% gradient) purchased from Invitrogen (cat. EA0375BOX). Transfer of the large molecular weight of VPS13A protein (350 kDa) to membranes was performed as described in Park et al. (2013). The primary antibodies used for Western blot analysis were anti-VPS13A at 1:500 dilution, anti-GFP (cat. 632381; Clontech) at 1:1000 dilution, and anti-GAPDH (cat. 60004; Proteintech) at 1:5000 dilution. For secondary antibodies, ECL anti-mouse IgG (cat. NA931V; GE Healthcare) and ECL anti-rabbit IgG (cat. NA934V; GE Healthcare) were used at 1:2500 and 1:5000 dilution, respectively.
Immunostaining and microscopy
To determine cellular localizations of VPS13A and/or XK, 70∼80% confluent HEK293T cells grown in gelatin-coated 35-mm glass bottom culture dishes were transfected with pVPS13A^mCherry (Kumar et al., 2018; Addgene plasmid #118758), pEGFP-C1, pcDNA3.1(+)-N-eGFP-XK, pEFIRES-mTagBFP-KDEL (Salo et al., 2016; Addgene plasmid # 87163), and/or mito-BFP (Friedman et al., 2011; Addgene plasmid # 49151) as described above. Twenty-four hours after transfection, cells were washed with Hank’s balanced salt solution (HBSS) (cat. 14025092; Life Technologies) buffer and incubated within FluoroBrite DMEM (cat. A18967-01; Life Technologies) supplemented with 10% FBS during live cell imaging. Live cells were observed using a wide-field Zeiss Observer.Z1 microscope with an attached Orca II ERG camera (Hamamatsu) or a confocal Zeiss Observer.Z1 microscope with a Yokogawa CSU-10 spinning disk, VersaLase 8 laser (VORTRAN), and attached PRIME 95B camera (Photometrics). ZEN 2012 Blue edition software or MetaMorph software was used to acquire and process images on the wide-field and confocal microscopes, respectively. To visualize lipid droplets in live cells, cells were washed once with PBS and HBSS buffer containing monodansylpentane at 0.1 mM (cat. SM1000a; Abgent) or 4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a-Diaza-s-Indacene (BODIPY 493/503) at 10 μM (cat. D3922; ThermoFisher) was added onto cells. Cells were incubated at 37°C in humidified atmosphere at 5% of CO2 for 15 to 30 min. Stained cells were washed twice with HBSS buffer, and 1 ml FluoroBrite DMEM media supplemented with 10% FBS was added before microscopy.
Supplementary Material
Acknowledgments
The authors thank Sook-Young Sohn and Kevin Czaplinski for reagents and advice for cell culture and Pietro de Camilli, Gia Voeltz, and Elina Ikonen for plasmids. We also thank David Matus and Rebecca Adikes for training and advice on confocal microscopy. We thank Rolf Sternglanz and Nancy Hollingsworth for comments on the manuscript and members of the Neiman, Futcher, and Leatherwood laboratories for helpful discussions. This work was supported by NIH Grant GM R01 072540 to A.M.N.
Abbreviations used:
- ChAc
Chorea-Acanthocytosis
- co-IP
coimmunoprecipitation
- ER
endoplasmic reticulum
- FBS
fetal bovine serum
- GFP
green fluorescent protein
- HBSS
Hank’s balanced salt solution
- PBS
phosphate-buffered saline
- PEI
polyethyleneimine
- VAB
Vps13 adaptor binding
- XKR
XK related.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www .molbiolcell.org/cgi/doi/10.1091/mbc.E19-08-0439-T) on August 26, 2020.
REFERENCES
- Bankaitis VA, Johnson LM, Emr SD (1986). Isolation of yeast mutants defective in protein targeting to the vacuole. Proc Nat Acad Sci USA , 9075–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BDM, Dziurdzik SK, Kolehmainen KL, Fowler CMS, Kwong WK, Grad LI, Davey M, Schluter C, Conibear E (2018). Competitive organelle-specific adaptors recruit Vps13 to membrane contact sites. J Cell Biol , 3593–3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickner JH, Fuller RS (1997). SOI1 encodes a novel, conserved protein that promotes TGN-endosomal cycling of Kex2p and other membrane proteins by modulating the function of two TGN localization signals. J Cell Biol , 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson-Stone C, Danek A, Rampoldi L, Hardie RJ, Chalmers RM, Wood NW, Bohlega S, Dotti MT, Federico A, Shizuka M, et al (2002). Mutational spectrum of the CHAC gene in patients with chorea-acanthocytosis. Eur J Hum Genet , 773–781. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Lackner LL, West M, DiBenedetto JR, Nunnari J, Voeltz GK (2011). ER tubules mark sites of mitochondrial division. Science , 358–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier J, Meijer IA, Lessel D, Mencacci NE, Krainc D, Hempel M, Tsiakas K, Prokisch H, Rossignol E, Helm MH, et al (2018). Recessive mutations in VPS13D cause childhood onset movement disorders. Ann Neurol , 1089–1095. [DOI] [PubMed] [Google Scholar]
- Ho MF, Chelly J, Carter N, Danek A, Crocker P, Monaco AP (1994). Isolation of the gene for mcleod syndrome that encodes a novel membrane-transport protein. Cell , 869–880. [DOI] [PubMed] [Google Scholar]
- Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, et al (2015). The BioPlex network: a systematic exploration of the human interactome. Cell , 425–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionita-Laza I, Capanu M, De Rubeis S, McCallum K, Buxbaum JD (2014). Identification of rare causal variants in sequence-based studies: methods and applications to VPS13B, a gene involved in Cohen syndrome and autism. PLos Genet , e1004729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John Peter AT, Herrmann B, Antunes D, Rapaport D, Dimmer KS, Kornmann B (2017). Vps13-Mcp1 interact at vacuole-mitochondria interfaces and bypass ER-mitochondria contact sites. J Cell Biol , 3219–3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, Traskelin AL, Perveen R, Kivitie-Kallio S, Norio R, Warburg M, et al (2003). Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet , 1359–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA, Wheeler H, Reinisch KM, De Camilli P (2018). VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol , 3625–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AB, John Peter AT, Walter P, Kornmann B (2015). ER-mitochondrial junctions can be bypassed by dominant mutations in the endosomal protein Vps13. J Cell Biol , 883–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun SM, Pujol C, Ciura S, et al (2016). Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent mitophagy. Am J Hum Genet , 500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumberg D, Muller R, Funk M (1995). Yeast vectors for the controlled expression of heterologous proteins in different genetic backgrounds. Gene , 119–122. [DOI] [PubMed] [Google Scholar]
- Munoz-Braceras S, Tornero-Ecija AR, Vincent O, Escalante R (2019). VPS13A is closely associated with mitochondria and is required for efficient lysosomal degradation. Dis Model Mech . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Neiman AM (2012). VPS13 regulates membrane morphogenesis during sporulation in Saccharomyces cerevisiae. J Cell Sci , 3004–3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Okumura Y, Tachikawa H, Neiman AM (2013). SPO71 encodes a developmental stage-specific partner for VPS13 in Saccharomcyes cerevisiae. Eukaryotic Cell , 1530–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Thorsness MK, Policastro R, McGoldrick L, Hollingsworth NM, Thorsness PE, Neiman AM (2016). Yeast Vps13 promotes mitochondrial function and is localized at membrane contact sites. Mol Biol Cell , 2435–2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampoldi L, Dobson-Stone C, Rubio JP, Danek A, Chalmers RM, Wood NW, Verellen C, Ferrer X, Malandrini A, Fabrizi GM, et al (2001). A conserved sorting-associated protein is mutant in chorea-acanthocytosis. Nat Genet , 119–120. [DOI] [PubMed] [Google Scholar]
- Roulis E, Hyland C, Flower R, Gassner C, Jung HH, Frey BM (2018). Molecular basis and clinical overview of mcleod syndrome compared with other neuroacanthocytosis syndromes a review. Jama Neurol , 1554–1562. [DOI] [PubMed] [Google Scholar]
- Russo D, Redman C, Lee S (1998). Association of XK and Kell blood group proteins. J Biol Chem , 13950–13956. [DOI] [PubMed] [Google Scholar]
- Rzepnikowska W, Flis K, Muñoz-Braceras S, Menezes R, Escalante R, Zoladek T (2017). Yeast and other lower eukaryotic organisms for studies of Vps13 proteins in health and disease. Traffic , 711–719. [DOI] [PubMed] [Google Scholar]
- Salo VT, Belevich I, Li SQ, Karhinen L, Vihinen H, Vigouroux C, Magre J, Thiele C, Holtta-Vuori M, Jokitalo E, Ikonen E (2016). Seipin regulates ER-lipid droplet contacts and cargo delivery. EMBO J , 2699–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong E, Insolera R, Dulovic M, Kamsteeg EJ, Trinh J, Bruggemann N, Sandford E, Li S, Ozel AB, Li JZ, et al (2018). Mutations in VPS13D lead to a new recessive ataxia with spasticity and mitochondrial defects. Ann Neurol , 1075–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Denning DP, Imanishi E, Horvitz HR, Nagata S (2013). Xk-related Protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science , 403–406. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Imanishi E, Nagata S (2014). Exposure of phosphatidylserine by Xk-related protein family members during apoptosis. J Biol Chem , 30257–30267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urata Y, Nakamura M, Sasaki N, Shiokawa N, Nishida Y, Arai K, Hiwatashi H, Yokoyama I, Narumi S, Terayama Y, et al (2019). Novel pathogenic XK mutations in McLeod syndrome and interaction between XK protein and chorein. Neurol. Genet , e328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velayos-Baeza A, Vettori A, Copley RR, Dobson-Stone C, Monaco AP (2004). Analysis of the human VPS13 gene family. Genomics , 536–549. [DOI] [PubMed] [Google Scholar]
- Walker RH, Saiki S, Danek A (eds) (2008). Neruoacanthocytosis Syndromes—A Current Overview, ed., Berlin Heidelberg: Springer-Verlag, 3–20. [Google Scholar]
- Yeshaw WM, van der Zwaag M, Pinto F, Lahaye LL, Faber AIE, Gomez-Sanchez R, Dolga AM, Poland C, Monaco AP, van IJzendoorn SCD, et al (2019). Human VPS13A is associated with multiple organelles and influences mitochondrial morphology and lipid droplet motility. Elife . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.