

Abstract
Epithelia can eliminate apoptotic cells by apical extrusion. This is a complex morphogenetic event where expulsion of the apoptotic cell is accompanied by rearrangement of its immediate neighbors to form a rosette. A key mechanism for extrusion is constriction of an actomyosin network that neighbor cells form at their interface with the apoptotic cell. Here we report a complementary process of cytoskeletal relaxation that occurs when cortical contractility is down-regulated at the junctions between those neighbor cells themselves. This reflects a mechanosensitive Src family kinase (SFK) signaling pathway that is activated in neighbor cells when the apoptotic cell relaxes shortly after injury. Inhibiting SFK signaling blocks both the expulsion of apoptotic cells and the rosette formation among their neighbor cells. This reveals the complex pattern of spatially distinct contraction and relaxation that must be established in the neighboring epithelium for apoptotic cells to be extruded.
INTRODUCTION
Epithelia are mechanically integrated active materials (Saw et al., 2018). A key role is played by cell–cell adherens junctions (AJ), where E-cadherin (E-cad) adhesion receptors couple to the contractile actomyosin cytoskeleton. Coupling is achieved by molecular mechanisms that link the cadherin adhesion complex to actomyosin (Lecuit and Yap, 2015; Tang, 2018) and also signaling pathways that regulate actomyosin at junctions (Gomez et al., 2011; Charras and Yap, 2018). This yields a mechanical tension at AJ that supports epithelial barriers (Acharya et al., 2017; McRae et al., 2018) and cellular rearrangements during morphogenesis (Vasquez and Martin, 2016; Charras and Yap, 2018). Mechanotransduction pathways found at AJ (Dorland and Huveneers, 2017) also detect when junctional tension deviates from its resting state (Ratheesh et al., 2012; Priya et al., 2015; Acharya et al., 2018) to provide pathways for cell–cell communication. Thus, the mechanical regime of AJ plays an important role in epithelial biology (Harris et al., 2014).
However, morphogenetic transitions such as neighbor exchanges and intercalations often entail considerable changes in cell shape (Blankenship et al., 2006; Martin et al., 2009; Kuipers et al., 2014; Lubkov and Bar-Sagi, 2014; Monier et al., 2015; Curran et al., 2017; Teng et al., 2017). This raises the question of how such morphological plasticity can be achieved within the tensile network of epithelia. One possibility is that this is principally driven by local increases in contractility, such as occurs when cells generate enhanced actomyosin networks (Martin et al., 2009; Collinet et al., 2015; Curran et al., 2017). However, increased contractile stresses can disrupt epithelial integrity (Acharya et al., 2018). A number of mechanisms have been invoked to relieve local epithelial stresses, such as cell intercalation and oriented cell division (Wyatt et al., 2015; Firmino et al., 2016; Tetley and Mao, 2018), as well as changes in viscoelasticity (Clement et al., 2017; Iyer et al., 2019). However, these are responses to stress sustained over time scales that are often longer (hours–days) than the morphogenetic events themselves (minutes). Is it then possible that epithelia also possess early immediate mechanisms to limit tissue stress during morphogenetic transitions?
Here, we address this problem in the context of apoptotic cell extrusion, a striking form of epithelial homeostasis where fatally injured cells are physically expelled from the epithelium. Apoptotic extrusion is a complex morphogenetic process that encompasses both the apical expulsion of the apoptotic cell along with the rearrangement of its neighbors to form rosettes (Figure 1, a–c; Supplemental Movie S1). These two facets of the extrusion process are likely to be physiologically complementary. By eliminating apoptotic bodies, apical expulsion limits their potential inflammatory impact, while rosette formation may be necessary to preserve the epithelial barrier. However, loss of the apoptotic cell reduces cell volume (Saias et al., 2015) and apical cell area (Monier et al., 2015; Ambrosini et al., 2017), changes that are predicted to increase mechanical stress within the monolayer. We now identify mechanosensitive Src kinase signaling as an early response that is activated in the neighbors of apoptotic cells and causes AJ between those neighbor cells to relax. This junctional relaxation limits the mechanical stress that builds up in monolayers and is necessary for effective apoptotic extrusion.
FIGURE 1:

Apoptotic rosettes involve local changes in neighbor cell topology that are constrained by the mechanical regime of the epithelium. (a) Cartoon representation of the phenomenon of apoptotic cell extrusion. In gray is depicted the apoptotic cell, whereas in white are the surrounding cell neighbors. Distant, parallel, and orthogonal sets of junction are indicated as well as cell (area α and perimeter p) and model parameters (cell–cell adhesion J, junctional spring constant K, cell resting area α0, and energy E). (b) Still images of E-cad-GFP MCF-7 cell monolayers treated with etoposide showing an event of apoptotic cell extrusion and rosette formation. (c) Analysis of orthogonal junction and distant junction length as well as apoptotic cell area for the images shown in b (see also Supplemental Movie S1). Time = 0 was defined as the time when apoptotic cell area start to reduce (t = 20 min in images in b). Data are mean ± SEM for n = 8 junctions per group of junctions. (d) Frequency distribution of Δ number of junctions = number of junctions after extrusion-number of junctions before extrusion for cells that surrounds apoptotic cells. Data are the means ± SEM of five extrusion events (eight cells per extrusion event, n = 5, ****p < 0.0001; ***p < 0.001; two-way ANOVA Sidak’s multiple comparisons test). (e) 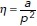 measured before and after extrusion for cells experiencing or not a reduction (–1) in their number of sides. Cell neighbors. Data are the mean of five extrusion events (*p < 0.05, two-way ANOVA Sidak’s multiple comparisons test. (f) Nonlinear regression analysis of junctional tension measurements on cells at different cell density. Results show
measured before and after extrusion for cells experiencing or not a reduction (–1) in their number of sides. Cell neighbors. Data are the mean of five extrusion events (*p < 0.05, two-way ANOVA Sidak’s multiple comparisons test. (f) Nonlinear regression analysis of junctional tension measurements on cells at different cell density. Results show 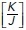 ratio values obtained as described in Section 5 of the Computational Supplement. (g) Time series sequence of the process of extrusion analyzed using a vertex model. (h and i) Analysis of monolayer energy (h) and junctional tension (i) on cell neighbors during extrusion in the model. Extrusion and rosette formation complete at time = 1. Calculations were performed for different values of
ratio values obtained as described in Section 5 of the Computational Supplement. (g) Time series sequence of the process of extrusion analyzed using a vertex model. (h and i) Analysis of monolayer energy (h) and junctional tension (i) on cell neighbors during extrusion in the model. Extrusion and rosette formation complete at time = 1. Calculations were performed for different values of 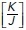 . (j) Theoretical predictions of the amount of junctional softening (reduction in
. (j) Theoretical predictions of the amount of junctional softening (reduction in 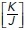 ratio) that is required for control monolayers (
ratio) that is required for control monolayers (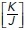 = 0.01401 ± 0.00314 μm–1 ) for topological cell changes to occur. Values are means ± SD of calculations using cell parameters (changes in area and η observed in four independent extrusion events, ****p < 0.0001; two-tailed paired t test). Scale bars, 20 µm.
= 0.01401 ± 0.00314 μm–1 ) for topological cell changes to occur. Values are means ± SD of calculations using cell parameters (changes in area and η observed in four independent extrusion events, ****p < 0.0001; two-tailed paired t test). Scale bars, 20 µm.
Movie S1.
E-cad-GFP MCF-7 cell monolayers treated with etoposide showing an event of apoptotic cell extrusion and rosette formation. Green: E-cadGFP, Magenta: Active Caspase 3/7. Scale bars, 20 μm.
RESULTS
Apoptotic rosettes involve local changes in neighbor cell topology
To evaluate how the shape of cells changed as they formed rosettes during apoptotic extrusion, we used MCF-7 cells in which endogenous E-cad was tagged with GFP at its C-terminus by CRISPR/Cas9 engineering (Liang et al., 2017). Treatment of confluent monolayers with etoposide induced sporadic apoptosis throughout the monolayers, leading to their apical extrusion (Figure 1, a–c; Supplemental Movie S1). Extrusion of the apoptotic cells was accompanied by significant junctional remodeling and reorganization of their neighbors to form rosettes, which typically consisted of 5–8 cells (Bement et al., 1993; Rosenblatt et al., 2001; Lubkov and Bar-Sagi, 2014; Michael et al., 2016).
To characterize this process, we first compared three sets of cell–cell junctions (Figure 1a): 1) “parallel” junctions, which are formed between the apoptotic cell and its immediate neighbors and mark the perimeter of the apoptotic cell. Parallel junctions progressively shrank as rosettes formed, as reflected in a progressive fall in the apoptotic cell area before being lost when the apoptotic cell was extruded (Figure 1c). 2) “Orthogonal” junctions: those that joined the immediate neighbor cells together and which linked orthogonally to the apoptotic cell perimeter. These typically elongated by ∼1.5-fold during rosette formation (Figure 1c; Supplemental Figure S1a). 3) “Distant” junctions that have no physical contact with the apoptotic cell (which for our experiments correspond to >2 cell diameters away from the apoptotic cell). Distant junctions did not change length during extrusion (Figure 1c; Supplemental Figure S1a).
Moreover, rosette formation was accompanied by a change in topology of the neighbor cells. Approximately 50% of nearest-neighbor cells (i.e., those in direct contact with the apoptotic cell) reduced their number of cell–cell contacts by one junction, whereas the number of cell–cell contacts was unaltered in cells that were >2 cell diameters away from the apoptotic cell (Figure 1d). We further quantitated this topological change using the polygonal type fingerprint, η = α/p2, where α is the polygon area and p2 is perimeter squared (see also Box 1 of the Computational Supplement). Nearest-neighbor cells that reduced their number of junctions as a result of extrusion displayed a significant reduction in η, which was unaltered in those cells that did not change junction number (Figure 1e). Together, these observations indicated that the morphological changes of apoptotic extrusion are local events, confined largely to the cells that come into direct contact with the apoptotic cell that will be expelled.
To test the generality of these morphological phenomenon in vivo, we used laser microirradiation to induce cell death (Michael et al., 2016) in the skin of one day postfertilization (1 dfp) zebrafish embryos (Supplemental Figure S1b; Supplemental Movie S2). Embryos expressed a fluorescently tagged membrane marker (Src-Bio-tk) to identify cell outlines. Time-lapse imaging confirmed that injured peridermal cells underwent apical extrusion, similar to that seen in tissue culture, associated with the formation of rosettes among their immediate neighbors (Supplemental Figure S1c; Supplemental Movie S2). Cells within these rosettes decreased their junction number and η ratio (Supplemental Figure S1, d and e), accompanied by elongation of the orthogonal junctions (Supplemental Figure S1f), exactly as we found in cultured epithelial cell monolayers. Therefore, topological transitions and junctional remodeling of apoptotic cell neighbors are common features of apoptotic rosettes.
Movie S2.
Apoptotic cell extrusion in the periderm of 1 dpf Zebrafish. Cell death was induced by laser mediated cell injury at time = 0 min as described in materials and methods and images were taken every 2 min.
Rosette formation is constrained by the mechanical regime of the epithelium
The topological changes that we observed have the potential to increase the mechanical stress in the monolayer (Charras and Yap, 2018), especially if already tensile AJ have to be stretched for cells to elongate and form rosettes. To more quantitatively consider how junctional elongation would affect the mechanical stress within a tensile network, we utilized a recently described vertex model (Coburn et al., 2016) that treats the epithelium as a surface of tiled polygons whose edges represent the apical AJ (described in detail in the Computational Supplement).
In this model, the energy of the system is governed by three whole-cell parameters: cell–cell adhesion energy (J), cell contractility (K), and target cell area (α0) and depends on three variables (cell area α, perimeter p, and monolayer density δ, as detailed in Figure 1a and Section 1 of the Computational Supplement). Energy then provides a useful descriptor that encompasses the ability of these polygons to adhere to one another, change their perimeter length, and alter their area as well as build up tension. Importantly, we could inform this model with experimentally measured parameters. Specifically, we directly measured monolayer density and derived the 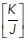 ratio by applying nonlinear regression analysis to our measurements of cell density and junctional tension (using Eq 26 in Sections 2 and 5 of the Computational Supplement and the data in Figure 1f). The
ratio by applying nonlinear regression analysis to our measurements of cell density and junctional tension (using Eq 26 in Sections 2 and 5 of the Computational Supplement and the data in Figure 1f). The 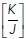 ratio can be intuitively understood as proxy for junctional tension, as its components—contractility and cell–cell adhesion—are key determinants of tension. This was consistent with the idea that junctional tension influences cell shape and patterning in epithelia (Loza et al., 2016; Mongera et al., 2018).
ratio can be intuitively understood as proxy for junctional tension, as its components—contractility and cell–cell adhesion—are key determinants of tension. This was consistent with the idea that junctional tension influences cell shape and patterning in epithelia (Loza et al., 2016; Mongera et al., 2018).
In validation of this, we experimentally confirmed two key predictions of the model (detailed in Sections 2 and 5 of the Computational Supplement): 1) junctional tension (measured as the initial recoil speed after E-cad-GFP junctions were cut with a laser) decreased with cell density (Figure 1f) as predicted by Eq. 26 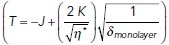 in Section 2 of the Computational Supplement. 2) The ratio of contractility to adhesion
in Section 2 of the Computational Supplement. 2) The ratio of contractility to adhesion 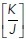 was decreased when contractility was reduced with the Rho kinase inhibitor, Y-27632 (Figure 1f). We therefore reasoned that we could use the model to calculate how the energy of the monolayer would change in response to the topological changes (changes in cell area/perimeter2 ratio) that are associated with rosette formation.
was decreased when contractility was reduced with the Rho kinase inhibitor, Y-27632 (Figure 1f). We therefore reasoned that we could use the model to calculate how the energy of the monolayer would change in response to the topological changes (changes in cell area/perimeter2 ratio) that are associated with rosette formation.
For this, we first considered the idealized state of a regular lattice of hexagonal cells that loses one cell and its associated cell–cell interactions by extrusion followed by the resolution of multicellular rosettes into tricellular junctions (Blankenship et al., 2006; Martin et al., 2009) (Figure 1g). We applied the set of equations in Box 6 of the Computational Supplement and informed the model with the ground-state 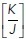 ratio derived from experimental data on control MCF-7 monolayers (Figure 1f). These calculations showed that the energy in the system rises progressively during the process of rosette formation, peaking at the moment when extrusion completes (t = 1, Figure 1, g–i) and later decreasing to a lower level as the rosettes resolved and the topology of the lattice was restored. Mechanical tension in the orthogonal junctions between neighbors was also predicted to increase as rosettes formed, peaking at the moment of extrusion (Figure 1i). The additional energy that needed to be injected into the system for rosettes to form could then be understood as representing an energy barrier for topological transitions, which would have to be overcome for extrusion to be accomplished.
ratio derived from experimental data on control MCF-7 monolayers (Figure 1f). These calculations showed that the energy in the system rises progressively during the process of rosette formation, peaking at the moment when extrusion completes (t = 1, Figure 1, g–i) and later decreasing to a lower level as the rosettes resolved and the topology of the lattice was restored. Mechanical tension in the orthogonal junctions between neighbors was also predicted to increase as rosettes formed, peaking at the moment of extrusion (Figure 1i). The additional energy that needed to be injected into the system for rosettes to form could then be understood as representing an energy barrier for topological transitions, which would have to be overcome for extrusion to be accomplished.
Interestingly, the calculated increase in monolayer energy was influenced by the pre-existing mechanical tension in the monolayer. We varied the 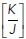 ratio that was present in the monolayer at ground state (i.e., in the hexagonal state) and calculated how this affects the change in energy associated with forming a rosette. The increase in energy and mechanical tension associated with rosette formation were both substantially enhanced when the pre-existing
ratio that was present in the monolayer at ground state (i.e., in the hexagonal state) and calculated how this affects the change in energy associated with forming a rosette. The increase in energy and mechanical tension associated with rosette formation were both substantially enhanced when the pre-existing 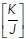 ratio of the epithelium was increased (Figure 1, h and i). Therefore, greater energy had to be injected into the monolayer for rosettes to form when monolayers were pretensioned than if they existed in more relaxed regimes. Intuitively, this might be understood to reflect the mechanical work that must be applied to stretch pretensioned junctions when cells have to elongate into a rosette.
ratio of the epithelium was increased (Figure 1, h and i). Therefore, greater energy had to be injected into the monolayer for rosettes to form when monolayers were pretensioned than if they existed in more relaxed regimes. Intuitively, this might be understood to reflect the mechanical work that must be applied to stretch pretensioned junctions when cells have to elongate into a rosette.
Then we informed the model with experimental data to predict the tensional regimes that might allow rosettes to form, that is, where the observed topological changes could occur with minimal energy cost. For this, we inputted the initial density of the monolayer (δ = 0.00162 ± 0.00023 μm–2) and the changes in cell topology that were observed to accompany extrusion (η* (before extrusion) = 0.0613 ± 0.0016, ηα (after extrusion) = 0.0521 ± 0.0030). Based on these morphological data, the model predicted that rosette formation would be favored for 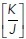 ratio values lower than a threshold
ratio values lower than a threshold 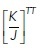 = 0.0049 ± 0.0003, as within this range the energy barrier for rosette formation is minimal (i.e., ∼0). Surprisingly, however, this predicted
= 0.0049 ± 0.0003, as within this range the energy barrier for rosette formation is minimal (i.e., ∼0). Surprisingly, however, this predicted 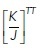 value was substantially lower than the ground-state
value was substantially lower than the ground-state 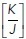 ratio derived from experimental measurements (0.01401 ± 0.00314 μm–1; Figure 1f). In other words, the model predicted that the already-existent mechanical state of the monolayers would tend to prohibit rosette formation, as substantial mechanical work would need to be performed to counteract the level of tensile stress that was already present. Yet, since rosettes did form in our experiments, this implied that epithelial cells had a mechanism to overcome this potential restriction.
ratio derived from experimental measurements (0.01401 ± 0.00314 μm–1; Figure 1f). In other words, the model predicted that the already-existent mechanical state of the monolayers would tend to prohibit rosette formation, as substantial mechanical work would need to be performed to counteract the level of tensile stress that was already present. Yet, since rosettes did form in our experiments, this implied that epithelial cells had a mechanism to overcome this potential restriction.
Extrusion is associated with AJ relaxation within the rosette
We considered two ways in which MCF-7 monolayers could overcome the challenge of tensile prestress to achieve apoptotic extrusion. First, constriction of the actomyosin cortex at the apoptotic:neighbor cell interface could do enough work to overcome the pretension in the monolayer. Alternatively, cells might relax to reduce the tensile prestress around the apoptotic cell. Indeed, the model predicted that MCF-7 monolayers could accommodate the topological changes of rosette formation if tension at their junctions were to decrease by ∼65% (Figure 1j; Section 6 of the Computational Supplement). To discriminate between these possibilities, we examined the orthogonal junctions in the nearest neighbors of the apoptotic cells. As noted earlier, these junctions elongate during rosette formation, a change that might reflect either increased contraction of the apoptotic:neighbor contractile interface or relaxation of contractility. In the first case, tension would increase in the orthogonal junctions, whereas the second case predicted a decrease in tension (Figure 2a).
FIGURE 2:

Junctional tension is reduced in the neighborhood of apoptotic cells. (a) Schematic representation of changes in junctional tension on orthogonal junctions in response to an increase in contractility of the apoptotic actomyosin purse string in the presence or absence of active softening on the orthogonal junction itself. (b) Example of the conditions used to measure junctional tension in the neighborhood of apoptotic cells. Apoptotic cells were identified by labeling with CellEvent Caspase-3/7 Green Detection Reagent, a dye that fluoresces green in the cytoplasm of apoptotic cells. (c) Junctional tension measurements (expressed as initial recoil) in untreated monolayers and 4 h etoposide-treated monolayers, where tension was measured on distant and orthogonal junctions. Data are mean ± SEM, n = 3 independent experiments, at least 15 junctions per experiment per condition. *p < 0.05, **p < 0.01. One-way ANOVA, Tukey’s multiple comparisons test; “theory” indicates the value of junctional tension for control/untreated monolayers to experience topological transitions without increasing monolayer mechanical stress (see Section 6 of the Computational Supplement). (d–f) Average staining of Vinculin (d), ratio α-18/α-catenin (e), and pMRLC (f) in the junctions between the neighbors of apoptotic cells (distant and orthogonal as defined in Figure 1a). Values corresponded to measurements on individual and independent extrusion events and are normalized by the average staining across all the junctions for an event (i.e., Orthogonal + Distant). Data are means ± SEM, n > 5 extrusion events, at least 5 junctions per extrusion event per condition. *p < 0.05, **p < 0.01; two-tailed paired t test. (g and h) Analysis of junctional MRLC content in response to laser-mediated cell injury. At time = 0 min, a cell in the center (red circle) was irradiated with a multiphoton laser as described in Materials and Methods. Images were taken at 30-s intervals and the area of the injured cell and the MRLC content in parallel (mean fluorescence intensity) and orthogonal (mean fluorescence intensity and integrated fluorescence intensity) junctions were measured. Still images of time sequence with rainbow pseudocolor (g) and quantitation (h) are shown. In h, data are mean ± SEM for n = 3 movies. *p < 0.05, **p < 0.001; two-way ANOVA Dunnet’s multiple comparisons test. Scale bars, 20 µm.
We therefore used recoil assays to characterize tension in the orthogonal junctions as they elongated. We performed our experiments when the apoptotic cells had constricted by ∼25%, compared with their immediate neighbors, but had not yet been extruded (Figure 2b). Strikingly, recoil (Supplemental Figure S2a) and the initial speed of recoil (Figure 2c) were significantly reduced in the orthogonal junctions compared with distant junctions. This implied that relaxation dominated at orthogonal junctions as they elongate, in contrast to the contractile behavior that increases tension at the apoptotic:neighbor cell interface (Michael et al., 2016). Of note, initial recoil velocity was reduced by ∼65%, which agreed exactly with the decrease in junctional contractility that the model predicted to be necessary for rosettes to form (compare Figures 2d and 1j).
Junctional relaxation was further corroborated by decreased staining for vinculin (Figure 2d; Supplemental Figure S2b) and for the α-18 epitope of α-catenin (normalized to total junctional α-catenin; Figure 2e; Supplemental Figure S2c), both effective proxies for molecular level tension at AJ (Yonemura et al., 2010; Acharya et al., 2017). Note that the decrease in these markers was largely confined to orthogonal junctions in the nearest-neighbor cells, again suggesting that junctional relaxation was a local event affecting the cells closest to the apoptotic event.
To test whether relaxation of the orthogonal junctions reflected changes in the contractile apparatus at the junctions, we first measured phosphorylated myosin regulatory light chain (pMRLC), a marker of nonmuscle myosin II (NMII) activity. This was reduced in orthogonal junctions compared with those located further away, suggesting that NMII might be down-regulated in these junctions (Figure 2f; Supplemental Figure S2d). We then performed time-lapse imaging of NMII (marked with MRLC-GFP) to characterize dynamic changes in the junctional contractile apparatus during rosette formation (Figure 2g). As previously described, MRLC-GFP in nearest-neighbor cells increases substantially in the contractile ring around the apoptotic cell (Figure 2, g and h) (Michael et al., 2016). However, in agreement with our analysis of endogenous pMRLC, we found that MRLC-GFP decreases in the orthogonal junctions within the rosette, something that was evident in the average fluorescence intensity of MRLC-GFP and also when we integrated MRLC-GFP intensity over the whole junction (Figure 2, g and h; Supplemental Movie S3). This occurred within 2 min of injury and was sustained for >10 min (Figure 2, g and h; Supplemental Movie S3), a point at which >50% of the extrusion process had been completed. This implied that actomyosin was down-regulated locally to mechanically relax orthogonal junctions during rosette formation, and this was especially evident at later time points when extrusion is being completed.
Movie S3.
Analysis of junctional MRLC content in response to laser mediated cell injury. A cell in the center was irradiated with a multiphoton laser as described in material and methods. Images were taken every 2 min.
Down-regulation of junctional tension is mediated by Src family kinase signaling
Then, we sought to identify a signaling pathway that was responsible for down-regulating tension in the orthogonal junctions. We focused on members of the Src family of protein tyrosine kinases (SFKs), which are activated during epithelial remodeling (Hunter et al., 2018; Shindo et al., 2008) and can down-regulate cadherin-mediated adhesion when overstimulated (McLachlan et al., 2007).
Immunostaining for an activation-specific phospho-epitope (pY419-SFK) revealed that active forms of SFK were selectively enriched in orthogonal junctions within apoptotic rosettes compared with more distant junctions in etoposide-treated cultures (Figure 3, a and b). This increase in Src activity in orthogonal junctions versus distant junctions was further confirmed in etoposide-treated MCF-7 cells stably expressing a plasma membrane-targeted Src-fluorescence resonance energy transfer (FRET)-based substrate sensor (Src-Bio-tK), a reporter that we have previously validated in our laboratory (Figure 3c) (Seong et al., 2009; Gomez et al., 2015). Orthogonal junctions showed a higher FRET index, indicating higher SFK activity than distant junctions (Figure 3c). Similarly, expression of the Src-Bio-tK sensor in 1 dpf zebrafish embryos showed that SFK signaling was increased at orthogonal junctions within rosettes in the periderm when apoptosis was induced by laser microirradiation (Figure 3d; Supplemental Figure S3a; Supplemental Movie S4). Movies of MCF-7 cells showed that SFK activity started to increase in the orthogonal junctions within 2 min after induction of injury and preceded junctional elongation by ∼2–4 min (Figure 3, e and f; Supplemental Movie S5). Furthermore, the increase in SFK activity correlated with an increase in the length of the orthogonal junctions (Figure 3, f and g). Together, these observations suggested that SFK signaling might be responsible for junctional elongation.
FIGURE 3:

Junctional relaxation in the neighborhood of apoptotic cells is mediated by SFK signaling. (a and b) Average staining of pY419 SFK and (c) Src Biosensor FRET activity (measured as FRET index = YFP/FRET ratio) on apoptotic neighbor cell junctions (distant and orthogonal). Apoptosis was stimulated by etoposide treatment and measurements were made on nonextruding apoptotic cells after 4 h etoposide treatment. Values corresponded to measurements on independent extrusion events and are normalized by the average FRET index across all the junctions for an event (i.e., orthogonal + distant). Data are mean ± SEM, n = 9 (b) and n > 7 (c) extrusion events per condition, at least 5 junctions per extrusion event per condition were analyzed. (b) ***p < 0.001, two-tailed paired t test and (c) *p < 0.05, one-way ANOVA with Tukey’s multiple comparisons test. (d) Analysis of junctional Src-Bio-tK FRET index after laser-mediated injury in the zebrafish periderm (see also Supplemental Movie S4). Average values of FRET index before and after injury (before rosette formation is complete) were measured. Values are mean ± SEM, n > 3 experiments (one embryo per experiment). *p < 0.05, two-tailed paired t test. (e) Analysis of junctional Src-Bio-tK FRET index and junctional length on orthogonal junctions as well as apoptotic cell area in MCF-7 cells in response to laser-mediated cell injury. At time = 0 min, cells in the center (red circle) were irradiated with a multiphoton laser as described in Materials and Methods. Then, images were taken evert 30 s and the area of the injured cell, FRET index, and junctional length in orthogonal junctions were measured. All values were normalized to the observed value observed immediately before injury (t = -2). Still images of time sequence with rainbow pseudocolor (e) and quantitation (f) are shown. Data are mean ± SEM for n = 4 movies (*p < 0.05, **p < 0.001, ***p < 0.0001, two-way ANOVA Dunnet’s multiple comparisons test). g) Plot of Src activity and junctional length for orthogonal junctions for all time points after injury in control (values derived from those in panel f and PP2-treated cell monolayers. Data are mean ± SEM for n = 3 movies. Also shown are the linear regression results for both conditions (p values correspond to statistical F test of whether the regression results were significant different from the null hypothesis of no correlation between both variables [i.e., slope = 0]). (h) Junctional tension measurements (expressed as initial recoil) in control siRNA, Src siRNA (Src KD), and Yes siRNA (Yes KD) Ecad-GFP MCF-7 cells. Initial recoil was measured on distant and orthogonal junctions around apoptotic cells which has not completely extruded (∼50% of monolayer cell area average). Monolayers were treated with Etoposide as described in Materials and Methods. Data are mean ± SEM for n = 8–10 extrusion events per condition. *p < 0.05; one-way ANOVA, Tukey’s multiple comparisons test. (i) Frequency of apoptotic cell extrusion in etoposide-treated E-cad-GFP MCF-7 monolayers that were treated with Control siRNA, Src siRNA (Src KD), and Yes siRNA (Yes KD). Ecad-GFP MCF-7 monolayers were treated for 4 h before fixation and apoptotic cells were identified using an anti-cleaved caspase 3 antibody. Data are mean ± SEM for n = 3 independent experiments. *p < 0.05; **p < 0.01; one-way ANOVA, Tukey’s multiple comparisons test. Scale bars, 20 µm.
Movie S4.
Analysis of junctional Src-Bio-tK FRET index (YFP/FRET) in zebrafish periderm in response to laser mediated cell injury. Cell death was induced by laser mediated cell injury at time = 0 min as described in materials and methods and images were taken every 2 min.
Movie S5.
Analysis of junctional Src-Bio-tK FRET index (YFP/FRET) in MCF-7 cells in response to laser mediated cell injury. Images were taken every 2 min.
To test this hypothesis, we treated MCF-7 cells with the broad-spectrum SFK inhibitor PP2. PP2 inhibited SFK activation around apoptotic cells and also blocked the elongation of orthogonal junctions (Figure 3g; Supplemental Movie S6). As expected, PP2 also prevented apoptotic cells from being extruded (Supplemental Movie S6). As an additional test for whether SFK signaling was necessary for orthogonal junctions to relax, we depleted MCF-7 cells of either c-Src or c-Yes (Supplemental Figure S3b, Supplemental Figure S5), which we earlier found to be the major contributors to SFK signaling at AJ (Gomez et al., 2015). c-Src or c-Yes RNAi did not affect the induction of apoptosis, but compromised SFK activation (Figure 3c) and prevented the relaxation of tension in orthogonal junctions compared with distant junctions located further away from the apoptotic cell (Figure 3h). Depleting these SFKs also compromised apical extrusion of apoptotic cells (Figure 3i). Overall, SFK signaling was necessary for orthogonal junctions to relax and lengthen and for apoptotic cells to be expelled. This supported the prediction of our model that effective extrusion and rosette formation required the mechanical relaxation of orthogonal junctions as well as the assembly of the contractile purse string.
Movie S6.
Analysis of junctional Src-Bio-tK FRET index (YFP/FRET) in MCF-7 cells pre-treated with the SFK inhibitor PP2 and subjected to laser mediated cell injury. Images were taken every 2 min (left, YFP fluorescence images, right, FRET index images). Green arrow points to the injured cell.
SFK signaling is activated by elastic relaxation of neighboring cells
Finally, we asked how SFK signaling becomes activated in the neighbor cells. We considered that relevant mechanisms should account for the observation that the SFK response was generally confined to cells in the immediate vicinity of the injured cells and occurred within ∼2 min after inducing cell injury (Figure 3, e and f). Here, we noted that the earliest detectable change in the neighbors was an initial recoil away from the injured cells. This was readily evident in movies as it occurred nearly instantaneously and consistently preceded SFK activation (Figure 3f) and junctional elongation (Supplemental Movies S3–S5). Similar recoil has been observed in earlier studies using different tissue systems and has been interpreted to reflect elastic recoil in the tissue (Abreu-Blanco et al., 2012; Antunes et al., 2013; Fernandez-Gonzalez and Zallen, 2013; Hunter et al., 2015; Coburn et al., 2018). Elastic recoil is also predicted to manifest as a decrease in junctional tension. Indeed, tension in orthogonal junctions between neighbors was reduced when recoil was measured immediately (<1 min) after injuring cells by laser microirradiation (Figure 4, a and b; Supplemental Movie S7). It should be noted that these measurements were made before orthogonal junctions began to elongate. This suggested that SFK signaling might have been activated in response to the mechanical stimulus of elastic recoil.
FIGURE 4:

SFK signaling is activated by elastic relaxation of neighboring cells. (a and b) Schematics of junctional tension measurements in the epithelium surrounding cells that were induced to cell death on ablation (ablated cells) (a). Initial recoil measurements were measured on cell junctions in either undamaged monolayers (Control) or in the neighborhood of ablated cells (after inducing cell damage). Data are mean ± SEM, for n = 20 initial recoil measurements per condition. *p < 0.05; unpaired t test. (c and d) pY419 SFK and E-cad-immunostained monolayers at different time points after mechanical relaxation of the underlying substrate (see Materials and Methods) and quantification of pY419 SFK at different time points Data are mean ± SEM, n = 100 junctions per condition; ***p < 0.001; ****p < 0.0001, one-way ANOVA, Tukey’s multiple comparisons test. Data representative of three independent experiments. (e and f) pY419 SFK junctional content in control or (e) blebbistatin (Blebb) or (f) Y-27632 (Y-27632)-treated monolayers. Data are mean ± SEM, n = 20 (e) and n = 8 (f) independent experiments; ****p < 0.0001, *p < 0.05, paired two-tailed t test. (g) Src activity (FRET index) in Src-Bio-tK MCF-7 cells treated (Y-27632) or not (Control) with Y-27632. Data are mean ± SEM, n = 4 independent experiments, at least 50 junctions per experiment were analyzed; ****p < 0.0001, paired two-tailed t test. Scale bars, 20 µm. (h) Model of SFK signaling activation in response to elastic relaxation of the tissue around apoptotic cells and how does it contribute to rosette formation and apoptotic cell extrusion.
Movie S7.
Measurements of junctional tension in the neighborhood of laser injured cells. Left, control experiments where junctional tension is analyzed in 2 distant junctions. Right, similar experiment but with the difference that a laser-induced cell injury was introduced within the monolayer before tension measurements.
However, injured cells also release chemical signals that could potentially activate SFK signaling (Hunter et al., 2018). To test if mechanical relaxation alone could activate junctional SFK signaling, confluent monolayers were grown on flexible substrata and tensioned by stretching the substrate (10% strain, 48 h) (Acharya et al., 2018). Tension on the substrate was then rapidly released, and the monolayer was fixed at different times and immunostained for pY419-SFK and E-cad to assess junctional SFK signaling. Junctional pY419-SFK began to increase 5 min after relaxation and peaked at 15 min (Figure 4, c and d), a time period that was comparable to that seen when SFK were activated in apoptotic rosettes (Figure 3, e and f). Note that SFK activation was transient, suggesting that its stimulation subsided as mechanical homeostasis was re-established.
Activation by mechanical relaxation of junctions in the absence of injury further predicted that junctional SFK signaling would be stimulated if we inhibited the actomyosin apparatus responsible for baseline junctional contractility. Consistent with this, junctional pY419-SFK levels were increased when we inhibited NMII directly with blebbistatin or when we blocked its upstream RhoA-ROCK pathway with Y-27632 (Figure 4, e and f; Supplemental Figure S4a). Y-27632 also reduced junctional SFK signaling assayed with the FRET biosensor (Figure 4g; Supplemental Figure S4b). Overall, these findings suggest that SFK signaling is activated when orthogonal junctions relax on elastic recoil of the tissue around acutely injured cells (Figure 4h).
DISCUSSION
Apoptotic extrusion involves complex changes in epithelial mechanics that are necessary both for the apical expulsion of the apoptotic cell and for its neighbors to rearrange into a rosette. To date, these mechanical changes have been best understood to reflect increased cortical contractility, both in the apoptotic cells and in their immediate neighbors. In apoptotic cells, contractility is up-regulated by the caspase-induced activation of procontractile kinases (such as ROCK and MRCK) (Coleman et al., 2001; Sebbagh et al., 2001; Gagliardi et al., 2018), whereas contractility in the neighbor cells is enhanced in response to RhoA signaling and actin regulation. The latter changes lead to the formation of an actomyosin ring at the apoptotic:neighbor interface whose contraction is thought to expel the apoptotic cell from the epithelium (Bement et al., 1993; Rosenblatt et al., 2001; Kuipers et al., 2014; Monier et al., 2015; Michael et al., 2016). Our experiments now show that neighbor cells also undergo a complementary relaxation of their orthogonal junctions that is necessary for rosettes to form and extrusion to be accomplished.
Junctional relaxation in the orthogonal junctions was revealed by reduced recoil following laser ablation and reduced immunostaining for proxies of tension across α-catenin: vinculin and α-18 mAb. Similar relaxation has been observed in Drosophila embryos when junctions elongate during cell rearrangements in the wing (Bardet et al., 2013) and when tissue boundaries elongate in the amnioserosa (Hara et al., 2016). Junctional relaxation was a local event, generally confined to the apoptotic rosette and not extending more than 1–2 cell diameters beyond the rosette. This is consistent with computational models (Coburn et al., 2016, 2018) and experimental analysis of traction forces (Ng et al., 2014) which suggest that the propagation of cellular forces is limited to a few cell diameters, similar to what we have observed in our present experiments. This indicated that extrusion is characterized by a complex mechanical landscape where localized contractility at the apoptotic:neighbor interface is complemented by relaxation at the orthogonal junctions formed between the immediate neighbor cells (Figure 4h).
Actomyosin at the junctional cortex is a major driver of junctional tension (Ratheesh et al., 2012). In contrast to the contractile ring, however, both the total levels of Myosin II and the levels of its activated pMLC decreased in the orthogonal junctions. This implied that orthogonal junctions may have relaxed because their contractile apparatus was being down-regulated. In our experiments, this appears to be mediated by SFK signaling. Thus, SFK activity increased at the orthogonal junctions, consistent with the recent demonstration that Src is activated in response to laser-mediated cell injury, as revealed using a newly designed Src biosensor (Wang et al., 2018). Importantly, blocking SFK with either inhibitors or kinase RNAi prevented junctions from relaxing and also interrupted their elongation. Therefore, the orthogonal junctions in rosettes elongate because they are being induced to actively relax and not just because they are stretched when contractile ring at the apoptotic:neighbor interface constricts.
Importantly, we also found that blocking SFK inhibited the two key features of the extrusion process: the ability of neighbor cells to rearrange into rosettes and to effectively expel apoptotic cells out of the epithelium. Active relaxation of junctions provides an attractive mechanism for neighbor cells to elongate, thereby sealing the rosette and the epithelium at sites of extrusion. But as well, although contractility at the apoptotic:neighbor interface has often been invoked to expel the apoptotic cell (Rosenblatt et al., 2001; Duszyc et al., 2017), our data imply that this must also be complemented by relaxation of the orthogonal junctions. One interpretation is that tension in its contiguous, orthogonal junctions would tend to oppose constriction of the contractile ring. This idea can also explain why increasing the pre-existing tension in an epithelium could antagonize extrusion as our calculations of energy change proposed (Figure 1h) and which we observed for oncogenic extrusion when tension in the epithelium was increased by depleting caveolae (Teo et al., 2020). Relaxation of the orthogonal junctions would reduce this opposing force, allowing the contractile ring to effectively expel the apoptotic cell at the same time that the epithelial barrier is preserved. SFK-induced junctional relaxation could also be considered to limit the amount of mechanical stress that the epithelium would have to bear during extrusion. Together these observations suggest that the relaxation of orthogonal junctions is critical for effective apoptotic extrusion. Thus, extrusion would require the complementary effects of contraction at the apoptotic:neighbor interface and relaxation of orthogonal junctions elsewhere in the immediate neighbor cells.
What stimulated SFK signaling at the orthogonal AJ? We propose that it was a mechanosensitive response triggered by the local release of tension on apoptotic injury. Thus, we observed an early immediate decrease in tension within the orthogonal junctions that occurred significantly earlier than the active relaxation associated with junction elongation (∼1 min vs. ∼ 10–30 min). This early immediate relaxation coincided with an abrupt recoil within the neighbor cells, a feature often seen in studies of epithelial injury (Abreu-Blanco et al., 2012; Antunes et al., 2013; Kuipers et al., 2014; Hunter et al., 2015). Together, these observations suggest that injured cells very quickly lose their ability to exert tension on their neighbors, perhaps because of reduced adhesive coupling (Teng et al., 2017), elasticity (Su et al., 2019), or transient swelling (Platonova et al., 2012) due to altered ion fluxes (Barros et al., 2001; Povea-Cabello et al., 2017). Irrespective of the underlying mechanism(s), this initial loss of tension would represent an early immediate change in the injured cell that is eventually superseded by hypercontractility as caspases activate procontractile kinases (Coleman et al., 2001; Sebbagh et al., 2001; Gagliardi et al., 2018).
Importantly, junctional SFK signaling could be stimulated by manipulating epithelial tension alone, without inducing cell injury. Thus, SFKs were activated when we passively reduced tension in monolayers grown on stretchable substrates. They were also stimulated when we inhibited the actomyosin apparatus that sustains the baseline tension of AJ. Therefore, although apoptotic cell injury will be associated with the release of many signals into the environment, these observations suggest that release of mechanical tension is a key factor. A number of mechanosensitive mechanisms have already been identified at AJ, ranging from tension-sensitive changes in the conformation of α-catenin (Buckley et al., 2014; Ishiyama et al., 2018) to activators of RhoA signaling (Acharya et al., 2018). However, these mechanisms respond to increases in junctional tension. Future work will be needed to identify how SFK are activated when junctional tension is reduced.
The mechanism(s) that allow SFK signaling to down-regulate tension also remain to be elucidated. SFKs can stimulate F-actin turnover (Ito et al., 2015), providing a potential mechanism to down-regulate actomyosin. It should also be noted these changes in junctional actomyosin may be complemented by changes in adhesion. Of note, recent evidence shows that the junctional apparatus is down-regulated in the vicinity of apoptotic cells to permit extrusion of the moribund cells to occur (Hunter et al., 2015). Also, SFK phosphorylate p120-catenin (Mariner et al., 2001), which can regulate epithelial viscoelasticity by modulating E-cad turnover (Iyer et al., 2019).
It is also interesting to note that SFK signaling contributes positively to support the junctional recruitment of Myosin IIB in steady-state monolayers (Gomez et al., 2015). Taken with our current observations, this suggests that the level of SFK signaling may critically influence whether it promotes or decreases contractility. Specifically, we would hypothesize that the baseline levels of SFK signaling found at steady-state junctions support their contractile apparatus, but the increased signaling that is triggered in response to apoptosis down-regulates contractility. Similarly, we earlier found that changing the level of SFK signaling also had a biphasic impact on cadherin adhesion (McLachlan et al., 2007), suggesting that sensitivity to signal strength is a fundamental feature of cellular regulation by SFKs.
In summary, we propose that mechanosensitive stimulation of SFK triggers a local active relaxation of AJ around the contractile interface during apoptotic extrusion. This reduces monolayer tension locally to permit effective extrusion and formation of an apoptotic rosette. Thus, effective apoptotic extrusion entails two spatially distinct biomechanical processes that are coordinated in the local neighborhood of the injured cell. The spatially limited nature of this response is noteworthy: local softening may confine the mechanical perturbations associated with extrusion so that the contractile stresses of extrusion are not propagated more extensively to challenge epithelial integrity (Acharya et al., 2018). Finally, our observations in zebrafish show that these heterogeneous mechanical landscapes are conserved in vivo, implying that the relaxation-sensitive process that we have identified may be relevant to morphogenetic events in other cell types and tissues. In particular, it would be interesting to test if local relaxation contributes to other morphogenetic events where cells elongate, such as intercalation during wound healing (Tetley et al., 2019) and neighbor exchange.
MATERIALS AND METHODS
Plasmids
pEGFP-MRLC1 (MRLC-GFP, Addgene, Plasmid #35680) was obtained from Addgene. pLL5.0-MRLC-GFP was obtained by cloning the MRLC gene into pll5.0 (Smutny et al., 2010). Src-Bio-tK-(ECFP-SH2-linker-substrate-YPet)-pcDNA3.1 was a generous gift from Yingxiao Wang (Seong et al., 2009).
Cell lines, siRNA transfection, and culture
MCF-7 from ATCC and grown in DMEM, supplemented with 10% fetal bovine serum (FBS), 1% l-glutamine, and 1% penicillin/streptomycin. MCF-7 Src-Bio-tk cell line was obtained by G418 selection (500 µg/ml) of MCF-7 transiently transfected with Src-Bio-tk-pcDNA3 and FACS sorted for CFP and YFP expression. MCF-7 MRLC-GFP was obtained by lentiviral infection of MCF-7 cells using lentiviral particles prepared with MRLC-GFP-pLL5.0 plasmid and protocols described in Priya et al. (2015). MCF-7 E-cad-GFP was obtained through CRISPR genome editing as described in Liang et al. (2017). All cell lines were maintained in low doses of Plasmocin (Invitrogen) and routinely tested for the presence of mycoplasma. For experiments, cells were seeded at 30–40% confluence 48 h before transfection with siRNA using RNAimax (Invitrogen, Cat #13778030) according to the manufacturer’s recommendations. For live-cell experiments, cells were cultured on 29-mm glass-bottom dishes (Shengyou Biotechnology) and imaged in clear Hank’s balanced salt solution supplemented with 5% FBS, 10 mM HEPES (pH 7.4), 5 mM CaCl2, glucose 4,5 g/l, and ProLong Live Antifade Reagent for live-cell imaging (Thermo Fisher Scientific, Cat #P36975).
Transfection and siRNA knockdown
MCF-7 cells were transfected with Src-Bio-tK using Lipofectamine 3000 (Invitrogen, Cat #L3000015) according to the manufacturer’s instructions for generation of stable cell lines. siRNA sequences were: Control (on-target plus nontargeting pool, UGGUUUACAUGUCGACUAA, UGGUUUACAUGUUGUGUGA, UGGUUUACAUGUUUUCUGA, UGGUUUACAUGUUUUCCUA); c-Src (on-target smart pool siRNA J-003175 DHARMACON; GGGAGAACCUCUAGGCACA, CCAAGGGCCUCAACGUAA, GCAGAGAACCCGAGAGGGA, GCAGUUGUAUGCUGUGGUU); c-Yes (on-target smart pool siRNA J-003184 DHARMACON, CAGAAGACCUUUCAUUUAA, GGAAAGUAUUUGAAGCUUC, GAUCUUCGGGCUGCUAAUA). For transfection of siRNA, we used the same double transfection protocol for siRNA as the one described in Gomez et al. (2015).
Antibodies
Primary antibodies used in this study were as follows: 1) phospho-Src family (Tyr416) antibody (Cell Signaling, Cat #2101); rabbit pAb against c-Yes (Cell Signaling, Cat #3201), mAb against c-Src (Santa Cruz, Cat #sc-8056); phospho-(Ser19)-MRLC ab (Cell Signaling, Cat #36755), Rat anti E-cad ab (ECCD-2, Cat#13-1900, Invitrogen), mouse anti-ZO-1 (Cat #33-9100, Invitrogen); rabbit pAb against GAPDH (R&D systems, Cat #2275-PC-100), rabbit pAb against α-Catenin (Invitrogen, Cat #71-1200); mouse mAb against vinculin (Invitrogen, #V-9131), rat pAb α-18 (a kind gift from Akira Nagafuchi, Nara Medical University, Japan), and rabbit mAb against cleaved caspase-3 (Cell Signaling, Cat #9664). F-actin was stained with Alexa Fluor 488-, 546-, 594-, 647-phalloidin (1:300 dilution; Invitrogen). CellEvent Caspase-3/7 Green Detection Reagent (Thermo Fisher Scientific, Cat #C10423) was used to identify apoptotic cells in live experiments. Secondary antibodies were species-specific antibodies conjugated with Alexa Fluor 488, 546, 594 or 647 (Invitrogen) for immunofluorescence, or with horseradish peroxidase (Bio-Rad Laboratories) for immunoblotting.
Drug treatments
MCF-7 cells were seeded at 30–40% confluency onto coverslips and allowed to recover to 90% confluency before drug treatment. Cells were treated for 1 h with Y-27632 (Merck, Cat #688000) or Blebbistatin (Merck, Cat #US1203390-5MG), at a final concentration of 25 and 100 μM, respectively.
Etoposide-induced apoptotic extrusion assay
Confluent cells were treated with 250 mM etoposide (Sigma) for 4 h (for live-cell imaging, tension measurements and immunofluorescence). DAPI, cell-event, or cleaved caspase-3 immuno-labeling was used to identify apoptotic cells. Cell extrusion was quantified after 5 h, when most of the WT apoptotic cells had fully extruded from the monolayer, with their nuclei—identified by DAPI staining—out of the plane of focus of the monolayer.
Immunofluorescence imaging
Cells were fixed in 4% paraformaldehyde (PFA) in cytoskeletal stabilization buffer (10 mM PIPES, pH 6.8, 100 mM KCl, 300 mM sucrose, 2 mM EGTA, 2 mM MgCl2) on ice for 30 min and subsequently treated with 50 mM NH4Cl for 10 min. Permeabilization was then performed with 0.2% Saponin/0.1% bovine serum albumin in Tris-buffered saline. Confocal images were taken using a Zeiss LSM-510 META inverted microscope and Zeiss LSM-710 FCS inverted microscope driven by ZEN software (ZEN 2009; Zeiss).
Quantitation of fluorescence at contacts
Quantitative analysis of fluorescence intensity at contacts in the periphery of apoptotic cells was performed using the “line” region of interest (ROI) tool function in ImageJ. A line of 3 μm in length along cell junctions was drawn on different types of cell–cell junctions (orthogonal junctions, parallel junctions, and distant junctions, at least five junctions per extrusion event). Mean values of fluorescence intensity for each line ROI were calculated and a global mean was calculated. Average values of junctional expression were then calculated for each junction type and normalized to the global media; >5 apoptotic regions were calculated per condition and data are representative of three or more experiments. Statistical analysis was then performed by using two-tailed t test or one-way ANOVA corrected for multiple comparisons as detailed in the figure legends.
Quantitative analysis of fluorescence intensity at contacts in cell monolayers (Figure 4, d–f) were performed in ImageJ using a line ROI of 10 μm length (averaged over 20 pixels) that was positioned orthogonal to, and centered on, randomly chosen contacts. Numerical values for the fluorescence intensity profile along this line were obtained using the Plot Profile feature of ImageJ and the baseline for each field of view was corrected by subtracting a constant value from each of the intensity profiles. Average profiles typically yielded peak-shaped curves with sides trending to zero, which were then fitted to a Gaussian function. Peak values for individual contacts were obtained by nonlinear regression using a custom-made MATLAB routine. A minimum of 50 contacts from three individual experiments were measured. Statistical analysis was then performed by using two-tailed t test or one-way ANOVA corrected for multiple comparisons as detailed in the figure legends.
Monolayer tension release experiments
MCF-7 monolayers were grown to 90% confluence on human fibronectin-coated 25-mm BioFlex culture plates (Acharya et al., 2018). Coating was performed at a density of 10 μg/cm2 at 37°C during 1 h from a solution of 1 mg/ml fibronectin in water. After monolayers were >90% confluence, cells were then subjected to static stretch (10% strain) using a Flex-cell Fx-5000TM Tension System (Flexcell International, Hillsborough, NC) for 48 h. Control wells were plugged at the bottom by rubber capping without application of any stretch. After 48 h, stretch was released and cells fixed with cold PFA 4% for 15 min. Then cells were permeabilized and immunolabeled as described above within the same wells. After labeling with secondary antibodies, cells were incubated in phosphate-buffered saline and imaged with a 60× water immersion objective (Zeiss) using an upright LSM 700 confocal microscope (Zeiss).
Src-Bio-tk FRET measurements
Cells were imaged live on a LSM 710 Zeiss confocal microscope equipped with a chamber incubator at 37°C. Images were acquired with a 40× (Figure 3, e and f) or 63× (Figures 3c and 4g) 1.4 NA oil Plan Apochromat immersion lens. A first scan was used to record simultaneously Donor and FRET channels using a 458-nm laser line, collecting the emission in the donor emission region (BP 470–500 nm) and acceptor emission region (BP 530–560 nm), respectively. A second scan was then used to acquire simultaneously cross-talk and Acceptor images using the 514-nm laser line for excitation and collecting the emission in the donor and acceptor emission regions. Scans were acquired sequentially line by line. Src activity, measured as a FRET index, was calculated for every image as the average (YFP [acceptor]/FRET) emission ratio for pixels located at cell–cell junctions (Seong et al., 2009; Gomez et al., 2015).
Laser ablation
Ablation experiments were performed on a LSM 510 meta Zeiss confocal microscope equipped with a 37°C heating stage as described previously (Liang et al., 2016). Images were acquired with a 63× objective, 1.4 NA oil Plan Apochromat immersion lens at 2× digital magnification. A total of six frames were acquired with an interval of ∼8 s per frame. The Ti Sapphire laser (Chameleon Ultra, Coherent Scientific, USA) was tuned to 790 nm for the ablation of cell–cell contacts labeled with either E-cad-GFP. A constant ROI, 3.8 × 0.6 μm, was positioned with the longer axis perpendicular to the cell–cell contact. Contacts were ablated with 30 iterations of the 790-nm laser at 24% transmission. The distance (d) between vertices that define the ablated contact was measured at every time (t). Distance values after ablation (d(t)) were subtracted from the initial contact length, d(0). The values of d(t) – d(0) were then calculated as a function of time (f(t)) and initial recoil values for each contact were obtained by nonlinear regression of the data to the following equation:
Finally, statistical analysis for control normalized average initial recoil between different groups was performed by ANOVA or t test as described in the corresponding figure legend.
Laser injury and extrusion
Laser-induced extrusion was performed on confluent cells stably expressing Src-Bio-tK or MRLC-GFP. To induce cell injury, we ablated a ROI at the center (nuclear plane) of cells following acquisition of the first frame. Ablation was carried out using 35% transmission of the 790-nm laser for 35 iterations. Image acquisition was performed every 2 min and GFP or FRET/YFP/CFP channels were imaged throughout the experiment.
Zebrafish husbandry and transgenesis
All animal work adhered to the guidelines of the animal ethics committee at the University of Queensland. To establish Tg(ubi: Src-Bio-tK), 1 nl of circular plasmid DNA was injected at 50 ng/μl into single cell stage embryos together with tol2 transposase mRNA (25 ng/μl). Embryos expressing alpha-crystaline:GFP in the eyes were raised to adulthood and screened for germline transmission to generate a stable transgenic line.
Gateway cloning
Src-Bio-tK (ECFP-SH2-linker-substrate-YPet) sequence was amplified from the Src-Bio-tK-pcDNA3.1 plasmid and cloned into the Gateway pME-vector (pDON-221) using Gateway technology (Hartley et al., 2000).
Primers used:
- pME-Src sensor-Forward (gateway homology arm = underlined, kozak sequence = italics):
5-GGGGACAAGTTTGTACAAAAAAGCAGGCTACCatggtgagcaagggcgag -3′
- pME-Src sensor-Reverse (gateway homology arm = underlined):
5′-GGGGACCACTTTGTACAAGAAAGCTGGGTAgtgtgatggatatctgcaga -3′
Subsequently, a Gateway LR reaction was performed combining a p5E-ubi, pME-Src-biosensor and p3E-polyA placing the final ubi:Src-biosensor sequence into pDestTol2pA2AC (containing the alpha-crystaline promoter driving GFP in the zebrafish lens).
Code availability
MATLAB Code for calculation of changes in monolayer energy and junctional tension during the process of cell extrusion is available in the supplemental material.
Supplementary Material
Acknowledgments
We thank our lab colleagues for their continuous support and fellowship and our many colleagues who generously provided reagents for this project. This work was supported by project grant funding from the National Health and Medical Research Council (NHMRC) Australia (1067405 and 1123816 to A.Y. and G.A.G.; 1037320 1163462 to A.Y.; 1140064 and 1150083 to RGP and 1099251 to T.E.H. and R.G.P.). A.Y. and R.G.P. are Research Fellows of the NHMRC (1136592 and 1156489, respectively). L.C. was funded under the Higher Education Authority of Ireland’s Programme for Research in Third Level Institutions (PRTLI) Cycle 5 Simulation Science and cofunded by the European Regional Development Fund (ERDF). G.A.G. is supported by an Australian Research Council Future Fellowship (FT160100366). R.G.P. is supported by the Australian Research Council (ARC) Centre of Excellence in Convergent Bio-Nano Science and Technology. R.W.M. was a recipient of a Queensland Cancer Council PhD scholarship. Optical microscopy was performed at the ACRF/IMB Cancer Biology Imaging Facility, established with the generous support of the Australian Cancer Research Foundation. G.A.G. and V.M.T. dedicate this manuscript to the memory of Jose L. Daniotti (CIQUIBIC-CONICET), who taught us how to look to the biology of cells.
Abbreviations used:
- AJ
adherens junctions
- E-cad
E-cadherin
- FBS
fetal bovine serum
- FRET
fluorescence resonance energy transfer
- MRLC
myosin regulatory light chain
- NMII
nonmuscle myosin II
- PFA
paraformaldehyde
- p-MRLC
phosphorylated myosin regulatory light chain
- SFK
Src family kinase.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-01-0084) on September 9, 2020.
REFERENCES
- Abreu-Blanco MT, Verboon JM, Liu R, Watts JJ, Parkhurst SM (2012). Drosophila embryos close epithelial wounds using a combination of cellular protrusions and an actomyosin purse string. J Cell Sci , 5984–5997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya BR, Nestor-Bergmann A, Liang X, Gupta S, Duszyc K, Gauquelin E, Gomez GA, Budnar S, Marcq P, Jensen OE, et al (2018). A mechanosensitive RhoA pathway that protects epithelia against acute tensile stress. Dev Cell , 439–452.e436. [DOI] [PubMed] [Google Scholar]
- Acharya BR, Wu SK, Lieu ZZ, Parton RG, Grill SW, Bershadsky AD, Gomez GA, Yap AS (2017). Mammalian diaphanous 1 mediates a pathway for E-cadherin to stabilize epithelial barriers through junctional contractility. Cell Rep , 2854–2867. [DOI] [PubMed] [Google Scholar]
- Ambrosini A, Gracia M, Proag A, Rayer M, Monier B, Suzanne M (2017). Apoptotic forces in tissue morphogenesis. Mech Dev , 33–42. [DOI] [PubMed] [Google Scholar]
- Antunes M, Pereira T, Cordeiro JV, Almeida L, Jacinto A (2013). Coordinated waves of actomyosin flow and apical cell constriction immediately after wounding. J Cell Biol , 365–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardet PL, Guirao B, Paoletti C, Serman F, Leopold V, Bosveld F, Goya Y, Mirouse V, Graner F, Bellaiche Y (2013). PTEN controls junction lengthening and stability during cell rearrangement in epithelial tissue. Dev Cell , 534–546. [DOI] [PubMed] [Google Scholar]
- Barros LF, Hermosilla T, Castro J (2001). Necrotic volume increase and the early physiology of necrosis. Comp Biochem Physiol A Mol Integr Physiol , 401–409. [DOI] [PubMed] [Google Scholar]
- Bement WM, Forscher P, Mooseker MS (1993). A novel cytoskeletal structure involved in purse string wound closure and cell polarity maintenance. J Cell Biol , 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blankenship JT, Backovic ST, Sanny JS, Weitz O, Zallen JA (2006). Multicellular rosette formation links planar cell polarity to tissue morphogenesis. Dev Cell , 459–470. [DOI] [PubMed] [Google Scholar]
- Buckley CD, Tan J, Anderson KL, Hanein D, Volkmann N, Weis WI, Nelson WJ, Dunn AR (2014). Cell adhesion. The minimal cadherin-catenin complex binds to actin filaments under force. Science , 1254211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras G, Yap AS (2018). Tensile forces and mechanotransduction at cell-cell junctions. Curr Biol , R445–R457. [DOI] [PubMed] [Google Scholar]
- Clement R, Dehapiot B, Collinet C, Lecuit T, Lenne PF (2017). Viscoelastic dissipation stabilizes cell shape changes during tissue morphogenesis. Curr Biol , 3132–3142.e3134. [DOI] [PubMed] [Google Scholar]
- Coburn L, Lopez H, Caldwell BJ, Moussa E, Yap C, Priya R, Noppe A, Roberts AP, Lobaskin V, Yap AS, et al (2016). Contact inhibition of locomotion and mechanical cross-talk between cell-cell and cell-substrate adhesion determine the pattern of junctional tension in epithelial cell aggregates. Mol Biol Cell , 3436–3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coburn L, Lopez H, Schouwenaar IM, Yap AS, Lobaskin V, Gomez GA (2018). Role of contact inhibition of locomotion and junctional mechanics in epithelial collective responses to injury. Phys Biol , 024001. [DOI] [PubMed] [Google Scholar]
- Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A, Olson MF (2001). Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol , 339–345. [DOI] [PubMed] [Google Scholar]
- Collinet C, Rauzi M, Lenne PF, Lecuit T (2015). Local and tissue-scale forces drive oriented junction growth during tissue extension. Nat Cell Biol , 1247–1258. [DOI] [PubMed] [Google Scholar]
- Curran S, Strandkvist C, Bathmann J, de Gennes M, Kabla A, Salbreux G, Baum B (2017). Myosin II controls junction fluctuations to guide epithelial tissue ordering. Dev Cell , 480–492.e486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorland YL, Huveneers S (2017). Cell-cell junctional mechanotransduction in endothelial remodeling. Cell Mol Life Sci , 279–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duszyc K, Gomez GA, Schroder K, Sweet MJ, Yap AS (2017). In life there is death: How epithelial tissue barriers are preserved despite the challenge of apoptosis. Tissue Barriers , e1345353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Gonzalez R, Zallen JA (2013). Wounded cells drive rapid epidermal repair in the early Drosophila embryo. Mol Biol Cell , 3227–3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firmino J, Rocancourt D, Saadaoui M, Moreau C, Gros J (2016). Cell division drives epithelial cell rearrangements during gastrulation in chick. Dev Cell , 249–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagliardi PA, Somale D, Puliafito A, Chiaverina G, di Blasio L, Oneto M, Bianchini P, Bussolino F, Primo L (2018). MRCKalpha is activated by caspase cleavage to assemble an apical actin ring for epithelial cell extrusion. J Cell Biol , 231–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez GA, McLachlan RW, Wu SK, Caldwell BJ, Moussa E, Verma S, Bastiani M, Priya R, Parton RG, Gaus K, et al (2015). An RPTPalpha/Src family kinase/Rap1 signaling module recruits myosin IIB to support contractile tension at apical E-cadherin junctions. Mol Biol Cell , 1249–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez GA, McLachlan RW, Yap AS (2011). Productive tension: force-sensing and homeostasis of cell-cell junctions. Trends Cell Biol , 499–505. [DOI] [PubMed] [Google Scholar]
- Hara Y, Shagirov M, Toyama Y (2016). Cell boundary elongation by non-autonomous contractility in cell oscillation. Curr Biol , 2388–2396. [DOI] [PubMed] [Google Scholar]
- Harris AR, Daeden A, Charras GT (2014). Formation of adherens junctions leads to the emergence of a tissue-level tension in epithelial monolayers. J Cell Sci , 2507–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley JL, Temple GF, Brasch MA (2000). DNA cloning using in vitro site-specific recombination. Genome Res , 1788–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter MV, Lee DM, Harris TJ, Fernandez-Gonzalez R (2015). Polarized E-cadherin endocytosis directs actomyosin remodeling during embryonic wound repair. J Cell Biol , 801–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter MV, Willoughby PM, Bruce AEE, Fernandez-Gonzalez R (2018). Oxidative stress orchestrates cell polarity to promote embryonic wound healing. Dev Cell , 377–387.e374. [DOI] [PubMed] [Google Scholar]
- Ishiyama N, Sarpal R, Wood MN, Barrick SK, Nishikawa T, Hayashi H, Kobb AB, Flozak AS, Yemelyanov A, Fernandez-Gonzalez R, et al (2018). Force-dependent allostery of the alpha-catenin actin-binding domain controls adherens junction dynamics and functions. Nat Commun , 5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito T, Taniguchi H, Fukagai K, Okamuro S, Kobayashi A (2015). Inhibitory mechanism of FAT4 gene expression in response to actin dynamics during Src-induced carcinogenesis. PLoS One , e0118336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer KV, Piscitello-Gomez R, Paijmans J, Julicher F, Eaton S (2019). Epithelial viscoelasticity is regulated by mechanosensitive E-cadherin turnover. Curr Biol , 578–591.e575. [DOI] [PubMed] [Google Scholar]
- Kuipers D, Mehonic A, Kajita M, Peter L, Fujita Y, Duke T, Charras G, Gale JE (2014). Epithelial repair is a two-stage process driven first by dying cells and then by their neighbours. J Cell Sci , 1229–1241. [DOI] [PubMed] [Google Scholar]
- Lecuit T, Yap AS (2015). E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat Cell Biol , 533–539. [DOI] [PubMed] [Google Scholar]
- Liang X, Budnar S, Gupta S, Verma S, Han SP, Hill MM, Daly RJ, Parton RG, Hamilton NA, Gomez GA, Yap AS (2017). Tyrosine dephosphorylated cortactin downregulates contractility at the epithelial zonula adherens through SRGAP1. Nat Commun , 790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang X, Michael M, Gomez GA (2016). Measurement of mechanical tension at cell-cell junctions using two-photon laser ablation. Bio Protoc , e2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loza AJ, Koride S, Schimizzi GV, Li B, Sun SX, Longmore GD (2016). Cell density and actomyosin contractility control the organization of migrating collectives within an epithelium. Mol Biol Cell , 3459–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lubkov V, Bar-Sagi D (2014). E-cadherin-mediated cell coupling is required for apoptotic cell extrusion. Curr Biol , 868–874. [DOI] [PubMed] [Google Scholar]
- Mariner DJ, Anastasiadis P, Keilhack H, Bohmer FD, Wang J, Reynolds AB (2001). Identification of Src phosphorylation sites in the catenin p120ctn. J Biol Chem , 28006–28013. [DOI] [PubMed] [Google Scholar]
- Martin AC, Kaschube M, Wieschaus EF (2009). Pulsed contractions of an actin-myosin network drive apical constriction. Nature , 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan RW, Kraemer A, Helwani FM, Kovacs EM, Yap AS (2007). E-cadherin adhesion activates c-Src signaling at cell-cell contacts. Mol Biol Cell , 3214–3223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae M, LaFratta LM, Nguyen BM, Paris JJ, Hauser KF, Conway DE (2018). Characterization of cell-cell junction changes associated with the formation of a strong endothelial barrier. Tissue Barriers , e1405774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael M, Meiring JCM, Acharya BR, Matthews DR, Verma S, Han SP, Hill MM, Parton RG, Gomez GA, Yap AS (2016). Coronin 1B reorganizes the architecture of f-actin networks for contractility at steady-state and apoptotic adherens junctions. Dev Cell , 58–71. [DOI] [PubMed] [Google Scholar]
- Mongera A, Rowghanian P, Gustafson HJ, Shelton E, Kealhofer DA, Carn EK, Serwane F, Lucio AA, Giammona J, Campas O (2018). A fluid-to-solid jamming transition underlies vertebrate body axis elongation. Nature , 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monier B, Gettings M, Gay G, Mangeat T, Schott S, Guarner A, Suzanne M (2015). Apico-basal forces exerted by apoptotic cells drive epithelium folding. Nature , 245–248. [DOI] [PubMed] [Google Scholar]
- Ng MR, Besser A, Brugge JS, Danuser G (2014). Mapping the dynamics of force transduction at cell-cell junctions of epithelial clusters. Elife , e03282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platonova A, Koltsova SV, Hamet P, Grygorczyk R, Orlov SN (2012). Swelling rather than shrinkage precedes apoptosis in serum-deprived vascular smooth muscle cells. Apoptosis , 429–438. [DOI] [PubMed] [Google Scholar]
- Povea-Cabello S, Oropesa-Avila M, de la Cruz-Ojeda P, Villanueva-Paz M, de la Mata M, Suarez-Rivero JM, Alvarez-Cordoba M, Villalon-Garcia I, Cotan D, Ybot-Gonzalez P, Sanchez-Alcazar JA (2017). Dynamic reorganization of the cytoskeleton during apoptosis: the two coffins hypothesis. Int J Mol Sci . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priya R, Gomez GA, Budnar S, Verma S, Cox HL, Hamilton NA, Yap AS (2015). Feedback regulation through myosin II confers robustness on RhoA signalling at E-cadherin junctions. Nat Cell Biol , 1282–1293. [DOI] [PubMed] [Google Scholar]
- Ratheesh A, Gomez GA, Priya R, Verma S, Kovacs EM, Jiang K, Brown NH, Akhmanova A, Stehbens SJ, Yap AS (2012). Centralspindlin and alpha-catenin regulate Rho signalling at the epithelial zonula adherens. Nat Cell Biol , 818–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt J, Raff MC, Cramer LP (2001). An epithelial cell destined for apoptosis signals its neighbors to extrude it by an actin- and myosin-dependent mechanism. Curr Biol , 1847–1857. [DOI] [PubMed] [Google Scholar]
- Saias L, Swoger J, D’Angelo A, Hayes P, Colombelli J, Sharpe J, Salbreux G, Solon J (2015). Decrease in cell volume generates contractile forces driving dorsal closure. Dev Cell , 611–621. [DOI] [PubMed] [Google Scholar]
- Saw TB, Xi W, Ladoux B, Lim CT (2018). Biological tissues as active nematic liquid crystals. Adv Mater , e1802579. [DOI] [PubMed] [Google Scholar]
- Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J, Breard J (2001). Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol , 346–352. [DOI] [PubMed] [Google Scholar]
- Seong J, Lu S, Ouyang M, Huang H, Zhang J, Frame MC, Wang Y (2009). Visualization of Src activity at different compartments of the plasma membrane by FRET imaging. Chem Biol , 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo M, Wada H, Kaido M, Tateno M, Aigaki T, Tsuda L, Hayashi S (2008). Dual function of Src in the maintenance of adherens junctions during tracheal epithelial morphogenesis. Development , 1355–1364. [DOI] [PubMed] [Google Scholar]
- Smutny M, Cox HL, Leerberg JM, Kovacs EM, Conti MA, Ferguson C, Hamilton NA, Parton RG, Adelstein RS, Yap AS (2010). Myosin II isoforms identify distinct functional modules that support integrity of the epithelial zonula adherens. Nat Cell Biol , 696–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X, Zhang L, Kang H, Zhang B, Bao G, Wang J (2019). Mechanical, nanomorphological and biological reconstruction of earlystage apoptosis in HeLa cells induced by cytochalasin B. Oncol Rep , 928–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang VW (2018). Cell-cell adhesion interface: orthogonal and parallel forces from contraction, protrusion, and retraction. F1000Res . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng X, Qin L, Le Borgne R, Toyama Y (2017). Remodeling of adhesion and modulation of mechanical tensile forces during apoptosis in Drosophila epithelium. Development , 95–105. [DOI] [PubMed] [Google Scholar]
- Teo JL, Gomez GA, Weeratunga S, Davies EM, Noordstra I, Budnar S, Katsuno-Kambe H, McGrath MJ, Verma S, Tomatis V, et al (2020). Caveolae control contractile tension for epithelia to eliminate tumor cells. Dev Cell , 75–91.e77. [DOI] [PubMed] [Google Scholar]
- Tetley RJ, Mao Y (2018). The same but different: cell intercalation as a driver of tissue deformation and fluidity. Philos Trans R Soc Lond B Biol Sci , 20170328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tetley RJ, Staddon MF, Heller D, Hoppe A, Banerjee S, Mao Y (2019). Tissue fluidity promotes epithelial wound healing. Nat Phys , 1195–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasquez CG, Martin AC (2016). Force transmission in epithelial tissues. Dev Dyn , 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Liang J, Shi LZ, Wang Y, Zhang P, Ouyang M, Preece D, Peng Q, Shao L, Fan J, et al (2018). Visualizing spatiotemporal dynamics of intercellular mechanotransmission upon wounding. ACS Photonics , 3565–3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt TP, Harris AR, Lam M, Cheng Q, Bellis J, Dimitracopoulos A, Kabla AJ, Charras GT, Baum B (2015). Emergence of homeostatic epithelial packing and stress dissipation through divisions oriented along the long cell axis. Proc Natl Acad Sci USA , 5726–5731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura S, Wada Y, Watanabe T, Nagafuchi A, Shibata M (2010). alpha-Catenin as a tension transducer that induces adherens junction development. Nat Cell Biol , 533–542. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.