

Abstract
Gle1 is a nucleocytoplasmic shuttling protein with well-documented cytoplasmic roles as a modulator of ATP-dependent DEAD-box RNA helicases involved in messenger (m)RNA export, translation initiation and termination, and stress granule dynamics. Here, we identify a novel nuclear role for Gle1 during transcription termination. In HeLa cells treated with a peptide that disrupts Gle1 nucleocytoplasmic shuttling, we detected nuclear accumulation of specific mRNAs with elongated 3′-UTR (untranslated region). Enriched mRNAs were nascently transcribed and accumulated in the nucleus due to a change in transcription state and not due to altered nuclear export. Whereas Gle1 shuttling inhibition did not appear to perturb nuclear DDX19 functions, it did result in increased DDX1 nucleoplasmic localization and decreased DDX1 interactions with Gle1 and the pre-mRNA cleavage stimulation factor CstF-64. An increase in nuclear R-loop signal intensity was also observed with diminished Gle1 shuttling, as well as colocalization of Gle1 at R-loops. Taken together, these studies reveal a nuclear role for Gle1 in coordinating DDX1 function in transcription termination complexes.
INTRODUCTION
As nuclear messenger (m)RNA is synthesized, processed, and exported to the cytoplasm, a number of wide-ranging regulatory events impart indispensable plasticity for the range of gene expression required during eukaryotic cellular physiology. The generation of export-competent, protein-coding mRNAs begins with the transcription of nascent pre-mRNA by RNA polymerase II (RNA Pol II), wherein the 5′-end is then capped and the transcript is potentially edited and spliced (Ben-Yishay and Shav-Tal, 2019). Transcription is terminated at transcription termination sites (TTSs) with cleavage of the pre-mRNA, release from the RNA Pol II complex, and subsequent polyadenylation at the 3′-end. Throughout this pathway, the mRNA is assembled into a specific mRNA-protein (mRNP) complex that facilitates transport across the nuclear envelope through nuclear pore complexes (NPCs) (Natalizio and Wente, 2013). Through the actions of ATP-dependent DEAD-box helicases (yeast Dbps/human DDXs), the proteins bound to the mRNP are dynamically exchanged to direct each step in mRNP maturation and ultimately impart directionality to the nuclear export process (Xie and Ren, 2019). Coordination of each step in the mRNP maturation process requires a multitude of distinct factors that are the focus of intense investigation.
Transcription termination itself is a complex mechanism that requires the functions of three large protein complexes (CPSF, CFI, and CFII) (Porrua and Libri, 2015; Porrua et al., 2016). After the polyadenylation site (PAS) is transcribed, RNA Pol II elongation slows and stalls downstream at a G-rich sequence. The PAS is then recognized by the CPSF complex and designated as the TTS, initiating the process of 3′-end formation. Following this, the CFI and CFII complexes cleave the nascent pre-mRNA 18-30 nucleotides downstream of the PAS at U- or GU- rich sequences, and the 3′-end is subsequently polyadenylated. These processes are coupled by the function of Cleavage stimulation Factor-64 (CstF-64), a component of both the CFI and CFII complexes. Finally, the residual 5′ uncapped transcript is degraded by exonuclease Xrn2, enabling disengagement of RNA Pol II from genomic DNA (West et al., 2004; Richard and Manley, 2009; Proudfoot, 2011; Porrua and Libri, 2015).
Several RNA-dependent DEAD-box ATPases (DDX, DHX, Dbp) play a role in transcription termination. For example, DDX1 associates with CstF-64 to coordinate pre-mRNA cleavage (Bleoo et al., 2001). DHX9 and another RNA helicase, SETX, also have a required role in modulating transcription termination, by unwinding the 3′ DNA:RNA hybrids (R-loops) that slow Pol II (Skourti-Stathaki et al., 2011; Chakraborty et al., 2018). However, the mechanism by which RNA helicases coordinate with the transcription termination machinery remains unclear. During other steps in gene expression, activation and control of DDXs are dependent on specific cofactors. In the cytoplasm, eIF4G and PDCD4 regulate eIF4A (DDX2) during translation initiation (Chen et al., 2019). Likewise, our prior work shows that proper mRNA export through NPCs requires stimulation of yeast Dbp5/human DDX19B by Gle1 bound to inositol hexakisphosphate (IP6) for ATPase-dependent remodeling of the mRNP protein composition (reviewed in Folkmann et al., 2011; Adams et al., 2017). Human Gle1 also interacts with DDX3 during cellular stress to regulate translation and stress granule formation (Aditi et al., 2015, 2019). Importantly, other DDXs/Dbps act in the nucleus to resolve critical replication, mRNA transcription, and processing events, which may also be coordinated by cofactors. Dbp5 interacts with the TFIIH subunit of RNA Pol II (Estruch and Cole, 2003) and DDX19B is implicated in resolving DNA damage-induced R-loops, which can result in stalled transcription and replication fork catastrophes (Hodroj et al., 2017a,b). DDX1 and DDX5 also unwind R-loops (Li et al., 2008; Mersaoui et al., 2019) and are components of the R-loop interactome (Cristini et al., 2018; Wang et al., 2018), consistent with their interactions with transcriptional machinery (Bleoo et al., 2001; Mersaoui et al., 2019). Despite these many examples of nuclear roles for DDXs/Dbps, no cofactors have yet been described that stimulate or coordinate their actions.
To date, the established functions for Gle1 in regulating DDX/Dbp functions are in the cytoplasm or at the NPC cytoplasmic face (Adams et al., 2017; Aditi et al., 2019; and as reviewed in Folkmann et al., 2011). Although human Gle1 shuttles between the nucleus and cytoplasm, with both major isoforms Gle1A and Gle1B containing a 39 amino acid shuttling domain (SD) (Kendirgi et al., 2003), nuclear functions of Gle1 are not documented. By using a dominant cell-permeable peptide strategy in HeLa cells, we previously reported that inhibiting Gle1 nucleocytoplasmic shuttling results in nuclear accumulation of poly(A)+ RNA (Kendirgi et al., 2003). To determine the role of Gle1 in the nucleus and the mechanism by which Gle1 shuttling impacts nuclear poly(A)+ RNA localization, we conducted a series of molecular and cellular studies. Here we report a novel nuclear function of human Gle1 and find that it works in concordance with DDX1 to impact transcription termination and cleavage of pre-mRNA prior to polyadenylation.
RESULTS
Nucleocytoplasmic shuttling of human Gle1 is important for nuclear retention of specific mRNAs
We previously defined a 39 amino acid nucleocytoplasmic SD important for proper human Gle1 steady-state subcellular localization and normal nucleocytoplasmic distribution of poly(A)+ RNA. Specifically, a dominant-negative approach, wherein HeLa cells are incubated with a cell-permeable peptide generated by fusion of the SD peptide to the antennapedia peptide (herein called, Gle1-SD), reduces Gle1 shuttling and docking at the NPC and increases the relative levels of nuclear poly(A)+ RNA (Kendirgi et al., 2003). At the time, we concluded that Gle1 shuttling was necessary for its role at the NPC in stimulating efficient mRNA export. However, we have since discovered that Gle1 functions independent of its role in export at NPCs to also modulate DDX/Dbps involved in other stages of gene expression (Bolger et al., 2008; Aditi et al., 2015; Jao et al., 2016; Glass and Wente 2019). This led us to consider whether Gle1 might have a bona fide nuclear role that also requires its shuttling to allow proper poly(A)+ RNA distribution.
First, we sought to determine if the nuclear poly(A)+ RNA accumulation observed following Gle1-SD treatment represents the retention of bulk mRNA or of a specific class of mRNA. HeLa cells were treated with 5 μM Gle1-SD or control Gle1-scramble (Gle1-scr) peptide for 4 h at 37°C. Nuclear RNA was then isolated (Supplemental Figure S1A), and RNA-sequencing (RNA-seq) of respective poly(A)+ RNA libraries was performed. A marked increase in the levels of ribosomal RNA species was noted in the nuclear fraction of Gle1–SD-treated cells, a surprising observation given that previous analysis of Saccharomyces cerevisiae gle1 mutants did not uncover ribosomal RNA export defects (Neumann et al., 2016). Ribosomal RNA was depleted prior to generation of poly(A)+ RNA libraries for RNA-seq, precluding further analysis of the finding in this study. Differential expression analyses revealed that 3090 protein coding mRNAs significantly accumulated in the nucleus by at least twofold (Figure 1A and Supplemental Figure S1B). Although some variability in this cohort was observed between independent samples, we identified 70 unique protein coding mRNAs that consistently accumulated in the nucleus of Gle1–SD-treated cells by greater than fivefold (Supplemental Table S1). This set of nuclear-retained transcripts is referred to as Gle1-SD target mRNAs. Importantly, RNA-seq was also performed on nuclear RNA from HeLa cells treated with wheat germ agglutinin (WGA), which blocks NPC transport and mRNA export by binding to O-GlcNAc-ylated NPC proteins (Nups) (Dargemont 1992; Kehlenbach 2003; Li and Kohler 2014). No significant overlap was observed between the Gle1-SD target mRNAs and the transcripts accumulating in the nuclei of WGA-treated cells (Supplemental Figure S1C).
FIGURE 1:

Treatment of HeLa cells with Gle1-SD peptide leads to nuclear accumulation of specific mRNAs. (A) Heat map from RNA-seq data illustrating differences in nuclear RNA accumulated in HeLa cells when treated with 5 µM of Gle1-scr or Gle1-SD peptide. Heat map depicts z-transformed gene expression of all genes that showed a fold change of ≥2 and false discovery rate of ≤0.05. (B) Twelve gene targets from RNA-seq data were confirmed with RT-qPCR in CDS region. The qPCR reads were normalized to actin reads in the respective samples (ΔCT value). The difference between ΔCT values of treated and untreated samples (ΔΔCT) was used to calculate log2 fold change, plotted in the graph. The graph represents fold change values (mean ± SEM) from three biological replicates performed in triplicate. (C) Top panel depicts position of the primers designed to amplify CDS and 3′-UTR (defined in Supplemental Table S2). RT-qPCR analysis of fold change (mean ± SEM) in 3′-UTR of Fos from three biological replicates was performed in triplicate. Samples were normalized to actin (intron 1, In1) using ΔΔCT method. (D) Top panel depicts position of the primers designed to amplify CDS and 3′-UTR. RT-qPCR analysis of fold change (mean ± SEM) in 3′-UTR of FosB from three biological replicates was performed in triplicate. Samples were normalized as in C. ΔCT values were used to calculate statistical significance using one-tailed, paired t test (**p < 0.005, *p < 0.05).
To corroborate the RNA-seq differential expression analysis, we performed RT-qPCR for a subset of 12 Gle1-SD target mRNAs. Nuclear transcript levels were quantified in untreated, Gle1-scr, and Gle1-SD nuclear RNA. The qPCR reads from each primer set were first normalized to actin reads in their respective samples (ΔCT value). The difference between ΔCT values of treated and untreated samples (ΔΔCT) was used to calculate log2 fold change, which is plotted in the graph. For the 12 targets analyzed, similar transcript levels were detected in Gle1–scr-treated and -untreated samples. In contrast, significant nuclear enrichment was observed in Gle1–SD-treated cells (Figure 1B). Total RNA levels (Supplemental Figure S1D) of the 12 representative Gle1-SD target mRNAs were also determined by RT-qPCR. Although we observed an increase in total RNA levels for these target genes, nuclear enrichment of these target genes was much higher as seen by the level of fold change on the y-axis. An increase in nuclear:cytoplasmic ratio of these transcripts also validated nuclear enrichment (Supplemental Figure S1E). Fos and FosB transcript levels were most notably increased, and RT-qPCR of amplicons in the 3′-UTR (untranslated region) of Fos and FosB further confirmed this enrichment (Figure 1, C and D).
To further define characteristics of the enriched RNAs in Gle1–SD-treated cells, we analyzed the RNA-seq data to examine the abundance of reads across the Gle1-SD target transcripts 2 kb upstream and downstream of the TTS. This analysis revealed that transcripts were increased across the entire gene body but also distinctly at the TTS and 3′-UTR in the Gle1–SD-treated sample (Figure 2A). To directly measure elongation of Fos and FosB, HeLa cells were treated with Gle1-SD peptide and nuclear RNA was subjected to RT-qPCR for amplicons downstream of the TTS. Increased levels of Fos and FosB mRNA were detected in the region downstream of the cleavage site, confirming that the Fos and FosB transcripts were indeed elongated (Figure 2, B and C). Previous studies report that loss of termination factors can result in the elongation of transcripts beyond the TTS and usage of cryptic downstream polyadenylation signals (Yao et al., 2012; Vilborg et al., 2015; Proudfoot, 2016). The phenotype detected in our Gle1-SD RNA-seq data is consistent with such transcript lengthening, indicating a potential transcriptional termination defect. From these results, we concluded that disruption of nucleocytoplasmic shuttling of Gle1 results in nuclear accumulation of specific mRNA transcripts with extended 3′-UTRs.
FIGURE 2:

Gle1-SD peptide treatment indicates transcription termination defect in Gle1 target genes. (A) Gene coverage around TTS ± 2 kb of all upregulated genes from RNA-seq data is shown. Duplicate data sets are depicted individually. (B) Schematic depicts the position of primers D and E downstream of the 3′UTR and cleavage site (CS) of the Fos gene. RT-qPCR data represent fold change values (mean ± SEM) from at least three biological repeats in nuclear pre-mRNA levels on Gle1-scr or Gle1-SD treatment. (C) Schematic depicts the position of primers B and C downstream of the 3′UTR and cleavage site (CS) of the FosB. Shown is fold change (mean ± SEM) in nuclear levels of pre-mRNA detected using RT-qPCR of Gle1–scr- or Gle1–SD-treated cells from at least three biological repeats. Samples (B and C) were normalized to actin (In1) values and fold change was calculated using ΔΔCT method as in Figure 1. ΔCT values were used to calculate statistical significance using one-tailed, paired t test (*p < 0.05).
Nascently transcribed mRNAs show nuclear accumulation with Gle1-SD treatment
Both siRNA-mediated Gle1 depletion in human cell lines and temperature-sensitive gle1 mutants in S. cerevisiae result in a significant nuclear accumulation of bulk poly(A)+ RNA, reflecting the conserved, essential function of Gle1 in mRNA export (Murphy and Wente 1996; Folkmann et al., 2013). To further clarify if Gle1’s role in bulk mRNA export contributes to the nuclear accumulation of Gle1-SD target mRNAs, we tested whether the nuclear-retained transcripts were comprised of nascently transcribed RNA or pre-existing RNA sequestered in the nucleus. HeLa cells were treated with the transcription inhibitor actinomycin D for 30 min prior to Gle1-SD or Gle1-scr peptide treatment. Nuclear RNA was isolated and transcript levels were quantified by RT-qPCR. When transcription was inhibited, Gle1 target RNA transcript levels were not elevated in Gle1–SD-treated cells (Supplemental Figure S1F). This indicated that Gle1-SD treatment produces a change in transcription state and does not alter nuclear export.
To directly measure whether Gle1 shuttling specifically impacts transcription of the target RNAs, we employed a click chemistry approach to selectively isolate nascently transcribed RNA (Figure 3A). HeLa cells pretreated with Gle1-SD or Gle1-scr peptide for 1 h were incubated with ethynyl uridine (EU) in the presence of the peptide to specifically label newly synthesized RNA. The EU-tagged mRNAs were biotinylated by click chemistry, captured with streptavidin magnetic beads, and subjected to RT-qPCR. Increased levels of nascently transcribed target RNAs were detected following Gle1-SD treatment, as compared with Gle1-scr treatment (Figure 3B). Taken together, these data revealed that Gle1 has a novel function in the nucleus, potentially linked to transcription termination of specific nascent mRNAs.
FIGURE 3:

Gle1-SD peptide treatment leads to accumulation of nascent mRNAs. (A) Schematic diagram illustrates EU-based detection of nascent mRNA. (B) HeLa cells were labeled with EU in the presence of Gle1-scr or Gle1-SD peptide. Nascent nuclear RNAs were captured and subjected to RT-qPCR using primers in the CDS region. The graph depicts fold change relative to actin values in untreated samples (mean ± SEM) from three biological replicates performed in triplicate (as detailed in Figure 1). ΔCT values were used to calculate statistical significance using paired one-tailed Student t test (****p < 0.0001, ***p < 0.001, **p < 0.003).
Gle1’s role in transcription termination is independent of DDX19B
Given that Gle1 functions in nuclear export and translation by modulating DDXs, we next sought to determine if a DEAD box helicase is acted on by Gle1 during transcription termination. Human Gle1’s role in the activation of DDX19B at NPC for mRNA export is well documented (reviewed in Folkmann et al., 2011; Adams et al., 2017). DDX19B normally exhibits a pancellular distribution, localizing to the cytoplasm and nuclear rim and in the nucleus to a lesser extent (Zolotukhin et al., 2009; Hodroj et al., 2017a). However, a recent study reports that DDX19B is phosphorylated in response to DNA damage and translocates to the nucleus to resolve DNA–RNA hybrids (R-loops) (Hodroj et al., 2017a). This nuclear function is further bolstered by R-loop interactome studies that find DDX19B is one of the helicases associated with R-loops (Cristini et al., 2018; Wang et al., 2018). To investigate whether Gle1’s effects on transcription termination are mediated by DDX19B, we first examined the localization of DDX19B after Gle1-scr or Gle1-SD treatment by indirect immunofluorescence (IF) microscopy (Supplemental Figure S2, A and B). Quantification of the nuclear-to-cytoplasmic fluorescence intensity ratio demonstrated that no change in localization of DDX19B occurred with Gle1-scr or Gle1-SD treatment, whereas DDX19B redistributed to the nucleus as described (Hodroj et al., 2017a) following treatment with the DNA damage agent camptothecin (Supplemental Figure S2, C and D). Thus, impaired Gle1 shuttling did not result in altered levels of DDX19B in the nucleus.
We further reasoned that if Gle1 modulates the activity of DDX19B during the transcription termination process, then Gle1-SD target mRNAs should accumulate in the nucleus when a dominant-negative (DDX19R372G) or helicase-dead (DDX19E242Q) variant of DDX19B is overexpressed (Hodge et al., 2011; Noble et al., 2011). The GFP-DDX19WT, GFP-ddx19R372G, or GFP-ddx19E242Q plasmids were transfected into HeLa cells and GFP-positive cells were sorted by flow cytometry. Nuclear RNA was isolated and subjected to RT-qPCR for analysis of Gle1-SD target transcript levels. We found that none of the analyzed mRNAs showed nuclear accumulation with overproduction of either the dominant-negative or the helicase-dead ddx19, or with wild-type DDX19B (Supplemental Figure S2E). Taken together, we concluded that the observed effect of Gle1 on nuclear accumulation of target RNAs occurs independent of DDX19B.
Nuclear function of Gle1 is mediated through DDX1
In addition to DDX19B, the DEAD-box helicase DDX1 is independently implicated during transcription termination, as well as having a role in resolving R-loops (Bleoo et al., 2001; Cristini et al., 2018). DDX1 localizes primarily to the nucleus in widespread puncta throughout the nucleoplasm and to larger more distinct foci associated with the transcription termination cleavage factor, CstF-64 (Bleoo et al., 2001). To determine if Gle1-SD treatment alters the nuclear localization of DDX1, Gle1–SD- and Gle1–scr-treated cells were immunostained for DDX1 and the ratio of nuclear-to-cytoplasmic localization was quantified (Figure 4A). An increase in nucleoplasmic DDX1 was detected on Gle1-SD treatment, and its localization was notably altered (Figure 4B). In Gle1-scr cells, DDX1 primarily exhibited a punctate distribution throughout the nucleus, as previously reported. However, following Gle1-SD treatment, the DDX1 localization was markedly more diffuse. A histogram of the nuclear intensity variation revealed that there was a wider variation in the intensity of DDX1 peaks in Gle1–SD-treated cells, quantitatively affirming the reduction in DDX1 localization to nuclear foci (Figure 4C).
FIGURE 4:
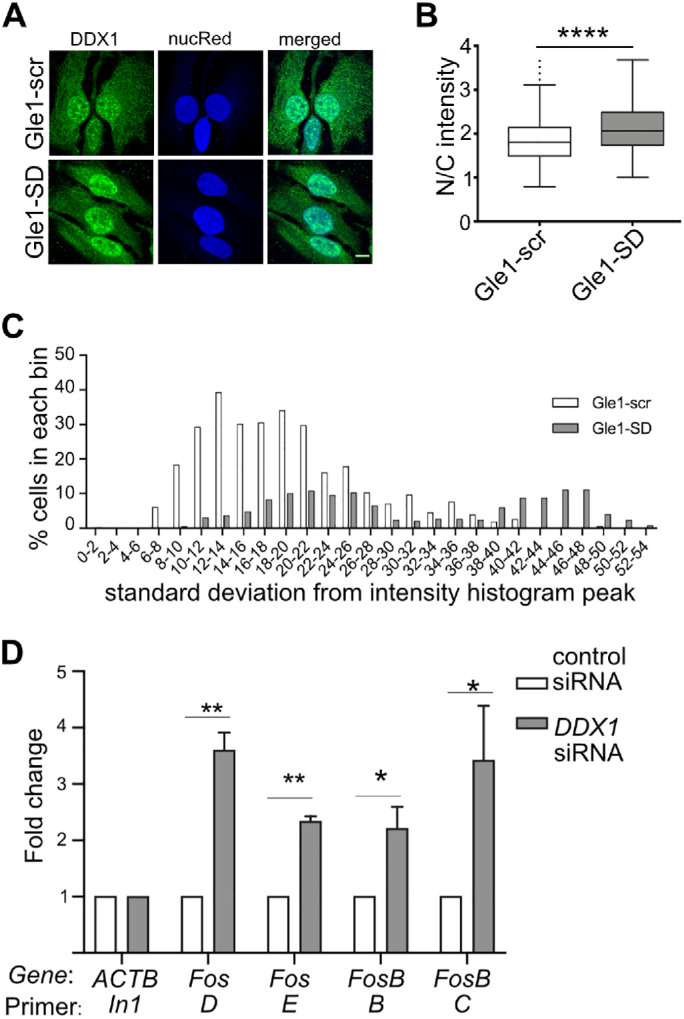
Gle1-SD effects are mediated through DDX1. (A) Gle1-scr or Gle1-SD peptide-treated HeLa cells were immunostained with DDX1 antibody. NucRed nuclear staining is pseudocolored as blue. Maximum intensity projected images are depicted and were used for subsequent quantification. Scale bar represents 10 μm. (B) Confocal images from three independent experiments were quantified using Image J and the nuclear/cytoplasmic ratio was plotted from over 290 cells. Unpaired, two-tailed t test was used to calculate statistical significance (****p < 0.0001). (C) Intensity Variation (SD of intensity histogram) analysis was done using Nikon Elements software over the nucleus region of interest from the above images. (D) HeLa cells were transfected with control or DDX1 siRNA for 72 h. RT-qPCR from nuclear RNA shows fold change (mean ± SEM) in nuclear pre-mRNA transcripts from three independent experiments. Samples were normalized to actin (In1) and fold change was calculated using ∆∆CT method (as detailed in Figure 1). ∆CT values were used to calculate statistical significance using one-tailed, paired t test (*p < 0.05 and **p < 0.005).
To examine whether DDX1 is involved in transcription termination of Gle1 target genes, we silenced DDX1 (Supplemental Figure S3A) and quantified the nuclear levels of uncleaved Fos and FosB transcripts. Importantly, significant nuclear accumulation of Fos and FosB amplicons beyond the cleavage site was observed in DDX1-depleted cells (Figure 4D). This analysis supported a model wherein Gle1 and DDX1 function together during transcription termination.
Gle1-SD disrupts pre-mRNA cleavage complex
To examine whether nuclear Gle1 localizes in close juxtaposition to DDX1, we utilized a proximity ligation assay (PLA), whereby an amplified fluorescent signal is produced only if Gle1 is within 40 nm of DDX1. Anti-Gle1 and anti-DDX1 antibodies were independently tested and shown to produce negligible background signal. Interestingly, together anti-Gle1 and anti-DDX1 exhibited a significant PLA signal above the background in untreated cells (Supplemental Figure S3B). Next, we tested if the PLA interaction between anti-Gle1 and anti-DDX1 changed with Gle1-SD treatment (Figure 5A). PLA signal was quantified from maximum intensity projected z-stacks as the number of PLA dots per nucleus, as well as nuclear fluorescence intensity (Integrated Density) (Figure 5, B and C). Whereas the interaction signal between anti-Gle1 and anti-DDX1 persisted in Gle1–scr-treated cells, the PLA signal decreased following Gle1-SD treatment. Taken together, these results indicated that Gle1 localizes in close proximity to nuclear DDX1 in a shuttling-dependent manner, and perturbing Gle1 shuttling alters DDX1 localization to nuclear foci.
FIGURE 5:

Gle1-SD disrupts colocalization between Gle1 and DDX1. (A) Shown are maximum intensity projected confocal images detecting proximity ligation between Gle1 and DDX1 in Gle1–scr- or Gle1–SD-treated samples. Scale bar represents 5 µm. (B) Number of PLA dots in the nucleus were quantified using ImageJ from over 200 cells per condition acquired over at least three independent experiments, and unpaired, two tailed t test was used to calculate statistical significance (***p < 0.0005). (C) Nuclear intensity of PLA dots was measured as Integrated Density using ImageJ from over 200 cells per condition acquired over three independent experiments, and unpaired, two tailed t test was used to calculate statistical significance (***p < 0.0005). (D) Proximity Ligation between DDX1 and CstF-64 was detected in Gle1–scr- or Gle1–SD-treated cells. Maximum intensity projected images are depicted. Scale bar represents 5 µm. (E) Number of PLA dots in the nucleus was measured using ImageJ from over 200 cells acquired over three independent experiments. (F) Nuclear intensity of PLA dots was measured as Integrated Density using ImageJ from over 200 cells from three independent experiments. Prism was used to calculate significance using unpaired, two-tailed t test (***p < 0.0005).
DDX1 is implicated in transcription termination by its colocalization to nuclear foci with CstF-64 (Bleoo et al., 2001), a component of the CSTF complex required for 3′-end cleavage and polyadenylation following transcription termination. Since impaired Gle1 shuttling altered the localization of DDX1 to nuclear foci, we further hypothesized that Gle1 plays a role in mediating interactions between DDX1 and CstF-64 at sites of active transcription termination. Using PLA, we confirmed that nuclear DDX1 and CstF-64 are colocalized in untreated cells (Supplemental Figure S3C) and in Gle1–scr-treated cells. Strikingly, Gle1-SD treatment resulted in a pronounced reduction in DDX1-CstF-64 PLA signal, based both on quantification of the number of nuclear PLA dots and on nuclear fluorescence intensity (Figure 5, D–F). In combination with our data demonstrating that Gle1-SD treatment results in accumulation of polyadenylated target RNAs with extended 3′-UTR, these results suggested that Gle1 impacts the localization and interactions of nuclear DDX1 during cleavage of nascent mRNA.
Abrogation of Gle1 shuttling increases the number of R-loops
R-loops act as a powerful inhibitor of RNA Pol II downstream of the TTS (Skourti-Stathaki et al., 2011; Ginno et al., 2013), and proper termination and cleavage depend on the removal of these R-loops. Indeed, increased R-loop persistence results in the accumulation of CstF-64 at cleavage sites (Katahira et al., 2019). We mined the R-loop database, which uses the quantitative R-loop forming sequence (RLFS) prediction model (QmRLFS; Supplemental Table S1), to identify Gle1-SD accumulated transcripts with a propensity for 3′-UTR R-loops (Wongsurawat et al., 2012; Jenjaroenpun et al., 2015). Strikingly, 74.3% (52/70) were predicted to form R-loops on the coding strand, of which 75% (39/52) were experimentally validated sites of R-loop formation (Figure 6A). Given that only ∼10% of genes across the human genome are predicted to contain R-loop prone sequences in the 3′-end (Ginno et al., 2013), these data reflected an enrichment of predicted 3′-UTR R-loops in the nuclear-retained mRNA of Gle1–SD-treated cells. This analysis indicated that proper Gle1 shuttling is linked to its role in the transcription termination of specific R–loop-forming mRNA transcripts.
FIGURE 6:

R-loops are increased following Gle1-SD treatment. (A) Pie chart depicts bioinformatic analyses of RNA-seq target genes with >5-fold change in nuclear RNA levels. (B) HeLa cells were treated with Gle1-scr or Gle1-SD peptide, immunostained with S9.6 antibody that recognizes RNA:DNA hybrids, and co-stained with nucleolin. Representative midplane images are shown, Scale bar represents 5 µm. (C) Box and whisker plot represents the nuclear intensity (Integrated Density) of S9.6 bound from midplane sections after subtracting nucleolin intensity, as determined from three independent experiments. (D) HeLa cells were transfected with mCherry or RNaseH1-NLS-mCherry for forty hours prior to Gle1-SD treatment and immunostained as described above. Scale bar represents 5 µm. (E) Nuclear S9.6 signal from midplane section was calculated by subtracting nucleolin as above from over 130 transfected cells from three independent experiments. P value was calculated in Prism using unpaired, two-tailed t test (***p < 0.0001).
Based on the correlation of accumulating Gle1-SD target transcripts with a high propensity to form R-loops, we speculated that disrupted Gle1 shuttling results in R-loop formation due to compromised pre-mRNA cleavage. To test this hypothesis, indirect IF microscopy was used to examine whether inhibiting Gle1 shuttling alters the occurrence of R-loops. HeLa cells treated with Gle1-SD or Gle1-scr peptide were immunostained with S9.6 antibody that specifically recognizes RNA–DNA hybrids. This antibody also commonly detects RNA–DNA structures in the cytoplasm, which are attributed to mitochondrial R-loops and Pol III-transcribed RNA–DNA hybrids (Koo et al., 2015; Vanoosthuyse 2018; Holt 2019). Since our RNA-seq analysis detected specific accumulation of R–loop-containing transcripts in the nucleus, we analyzed only the nuclear S9.6 signal to quantify R-loops for the purposes of this study. Co-staining for nucleolin was employed to allow subtraction of background signal from S9.6 binding to ribosomal R-loops in the nucleolus (Figure 6B). No change in nucleolar foci or fragmented nucleolin signal was detected between Gle1-scr and Gle1-SD cells. However, Gle1-SD treatment produced a statistically significant increase in S9.6 signal intensity in the nucleoplasm, supporting the bioinformatic data and linking altered Gle1 shuttling to changes in R–loop-containing mRNA (Figure 6C).
The S9.6 antibody used to identify RNA–DNA hybrids also recognizes dsRNA structure, albeit with a low affinity (Hartono et al., 2018). To further confirm that the increase in S9.6 signal intensity observed by IF microscopy was due to RNA–DNA hybrids and not due to RNA–RNA structures, we overexpressed mCherry-RNaseH1 (which degrades DNA–RNA hybrids but not dsRNA) and treated the cells with Gle1-SD peptide. Imaging was performed on mCherry-positive cells and scored for S9.6 signal intensity as described above (Figure 6D). Importantly, we observed a significant reduction in S9.6 signal intensity in cells expressing RNaseH1 (Figure 6E). These data verified our S9.6 staining as R-loops and further supported the conclusion that altered Gle1 shuttling increases R-loops.
To examine whether Gle1 is present near R-loops, we employed PLA. Anti-Gle1 and S9.6 antibodies were independently tested and produced negligible background signal (Supplemental Figure S4A). HeLa cells treated with Gle1-scr or Gle1-SD peptide were fixed, immunostained, and imaged to detect the proximity of anti-Gle1 and anti-S9.6 antibodies (Figure 7A). Nucleolin-localized anti-S9.6 signal was subtracted and PLA signals were quantified as described. A positive PLA signal was detected in untreated and Gle1–scr-treated samples, revealing that Gle1 localizes in close proximity to R-loops under wild-type conditions. Moreover, a small but noted increase in PLA signal was detected in the nuclei of cells treated with Gle1-SD peptide (Figure 7B). To test specificity of this assay, we silenced GLE1 using siRNA and employed PLA assay. PLA signal in the nucleus outside of the nucleolar region was calculated. Supplemental Figure S4B shows a significant reduction in PLA signal following GLE1 silencing. Thus, nuclear Gle1 was localized near R-loops under control conditions and remained there even when Gle1 shuttling was disrupted.
FIGURE 7:

Proximity of Gle1 to R-loops increases following Gle1-SD treatment. (A) Gle1–scr- or Gle1–SD-treated HeLa cells were subjected to PLA with Gle1 and S9.6 antibodies, counterstained with nucleolin antibody and imaged by confocal microscopy. NucRed is pseudocolored in blue. Scale bar represents 5 µm. (B) Nuclear PLA signal (in red) was quantified with nucleolin subtraction from midplane sections of over 225 cells from three independent experiments. Unpaired, two-tailed t test was used to calculate statistical significance (***p < 0.0005). (C) Model depicting a role for Gle1 in transcription termination.
Altered Gle1 shuttling does not induce DNA damage or predispose cells to DNA damage
In addition to playing a role in transcription termination, R-loop formation is associated with genomic instability and DNA damage (Santos-Pereira and Aguilera 2015; Crossley et al., 2019). Thus, if a disruption in Gle1 shuttling leads to DNA damage, an increase in unresolved R-loops would follow. To investigate this possibility, HeLa cells were immunostained for the well-established DNA damage marker γH2AX following Gle1-SD or Gle1-scr treatment. No significant difference in the nuclear intensity of γH2AX was observed after Gle1-scr or Gle1-SD peptide treatment (Supplemental Figure S5, A and B). As a positive control, HeLa cells were treated with hydroxyurea (HU) to induce DNA damage. HU treatment significantly increased the nuclear intensity of γH2AX in HeLa cells (Supplemental Figure S5C). To assess whether Gle1-SD treatment predisposes HeLa cells to increased DNA damage responses, cells were subjected to pretreatment with Gle1-scr or Gle1-SD peptide for 30 min followed by HU treatment in the presence of the peptide for 4 h. No increase in the nuclear intensity of γH2AX was observed with pretreatment of either peptide (Supplemental Figure S5, C and D). We concluded that the role for Gle1 in R-loop biology does not involve the DNA damage response. The absence of DNA damage following Gle1-SD treatment suggested that proper Gle1 shuttling is specifically linked to R-loops formed during normal transcription termination processes, wherein R-loops might be formed as a consequence of compromised pre-mRNA cleavage.
DISCUSSION
Although nucleocytoplasmic shuttling of human Gle1 has long been documented, we establish here a nuclear role for human Gle1, specifically in coordinating DDX1 and CstF-64 functions during transcription termination. Several pieces of evidence support this conclusion. First, inhibition of Gle1 nucleocytoplasmic shuttling leads to nuclear accumulation of specific nascent poly(A)+ RNAs and transcription termination defects in Gle1-SD target genes. Second, the abrogation of Gle1 shuttling decreases colocation between Gle1 and DDX1 and between DDX1 and the transcription termination factor CstF-64. This impairs pre-mRNA cleavage as evidenced by increased levels of transcripts downstream of the PAS and cleavage site. Third, under these conditions, the most highly accumulating transcripts are those that are prone to containing R-loops, and the overall detection of nucleoplasmic R-loops increases with Gle1-SD treatment. We propose that when Gle1 shuttling is disrupted, formation of the DDX1-CstF-64 complex is perturbed and pre-mRNA cleavage is not executed at the proper TTS. The consequential elongation of the 3′-UTR by RNA Pol II and downstream cleavage likely results in the associated increased R-loop formation (Figure 7C).
Under normal conditions, DDX1 is predominantly a nuclear protein localized to transcriptional foci at steady state, with nucleocytoplasmic shuttling ability (Figure 4; Bleoo et al., 2001). Our observed Gle1–SD-mediated displacement of DDX1 from transcriptional foci to diffuse nucleoplasmic localization is coincident with the reduced colocalization of Gle1 with DDX1 and DDX1 with CstF-64. Taken together, this indicates that nuclear localization of Gle1 is required for DDX1 to properly assemble or function with the transcription termination complex. In our prior studies, we show that the N-terminal domain of Gle1 facilitates self-association through a predicted coiled-coil domain (Folkmann et al., 2013). One possibility is that proper shuttling allows nuclear Gle1 self-association, creating some type of required scaffold for the activation and assembly of DDX1 with the transcription termination and pre-mRNA cleavage complexes.
An alternative mechanism by which Gle1 shuttling might impact transcription termination is through altering the nuclear transport, and thereby the stoichiometry, of proteins involved in transcription termination. From our prior studies, we know that Gle1-SD treatment does not impair the nuclear export of importin-β or nuclear import of its cargo (Kendirgi et al., 2003). However, a number of the components of the CPSF, CFI, and CFII complexes as well as several RNA helicases shuttle between the nucleus and cytoplasm, and inhibiting Gle1 shuttling might impact their shuttling through an unknown mechanism. For example, intracellular movement and localization of CstF-64 is mediated by its interaction with CstF-77 and is required for correct polyadenylation of mRNA transcripts (Ruepp et al., 2011). Thus, perturbed CstF-64 shuttling alone might generate the defects observed in transcription termination.
We also find a reduced interaction between DDX1 and CstF-64 on Gle1-SD treatment, which might reflect an altered stoichiometry of CstF-64 at TTSs. Silencing of CstF-64 affects a specific subset of transcripts for alternative polyadenylation processing (Yao et al., 2012). In the presence of CstF-64, proximal and weaker PAS sites are used for a specific subset of transcripts. Other studies reveal that CstF-64 is involved in changing the PAS from distal to proximal for IgM and NF-ATc mRNA transcripts (Takagaki et al., 1996; Takagaki and Manley 1997). Transcription termination can also be blocked during times of cellular stress (Vilborg et al., 2015, 2017) or viral infections (Bauer et al., 2018; Hennig et al., 2018), resulting in readthrough transcripts called “Downstream of Gene.” These transcripts extend beyond the 3′-UTR into intergenic regions. In these ways, Gle1 might participate with CstF-64 in altering cleavage site selection, leading to defective transcription termination. Further studies are needed to determine the full potential impact of Gle1 on all elements of 3′-UTRs.
Given Gle1 is known to regulate the activity of DDX/Dbps in the cytoplasm (Alcázar-Román et al., 2006; Weirich et al., 2006; Bolger et al., 2008; Folkmann et al., 2013; Aditi et al., 2015; Adams et al., 2017), it is possible that nuclear-localized Gle1 plays a direct role in facilitating the DDX1 RNA helicase activity required for resolving the R-loops during transcription termination. In two independent studies, DDX1 is found as a part of the R-loop interactome (Cristini et al., 2018; Wang et al., 2018). Although our PLA results indicate that Gle1 is colocalized with both R-loops and DDX1, Gle1 is not reported in the R-loop interactomes. This might be due to the Gle1-DDX1 PLA signal arising from transient interactions. Indeed, Gle1 association with other DEAD-box RNA helicases is quite transient (Weirich et al., 2006; Montpetit et al., 2011) and not easily detected in proteomic experiments (Hodge et al., 1999). In addition to DDX1, Gle1 might modulate other RNA helicases that are implicated in R-loop resolution at TTSs, such as SETX and DHX9. SETX resolves R-loops formed at G-rich transcriptional pause sites downstream of a PAS (Skourti-Stathaki et al., 2011) and we also see colocalization between Gle1 and SETX via PLA analysis (Supplemental Figure S3D). DHX9 also assists in transcription termination by preventing RNA Pol II read through downstream of PAS in response to stress (Cristini et al., 2018) or the absence of splicing factors (Chakraborty et al., 2018); however, we did not see a signal between anti-Gle1 and anti-DHX9 by PLA (Supplemental Figure S3D). Future studies will be needed to test for direct Gle1 modulation of DDX1 or other DDX/Dbp activities.
The precise mechanism by which the Gle1-SD disrupts shuttling has not been delineated. Presumably, the SD peptide competes with Gle1 protein interaction partners that are required for its transport or nuclear retention. Such interaction partners for the Gle1 39-residue SD domain are not known, and their identification will be the subject of our ongoing work to uncover important mechanistic contexts for Gle1’s function in transcription termination. Here, we have established principal connections that lay the foundation for this novel role for Gle1 in the nucleus. The routes by which Gle1 might act on transcription termination are many. Given its capacity to interact with DDX1 and R-loops and regulate DDX/Dbp activity through mRNA export and translation, the actions of Gle1 could bring about multiple effects on the stoichiometry, assembly, and activity of transcription termination complexes.
MATERIALS AND METHODS
Cell culture, treatments, transfections, and reagents
HeLa cells (CCL-2 from ATCC, passaged <12) were cultured in complete DMEM (Life Technologies, Thermo Fisher Scientific, Waltham, MA) media supplemented with 10% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), 1 mM HEPES solution (Life Technologies), and 10 mM sodium pyruvate (Life Technologies) at 37°C with 5% CO2. For Gle1 peptide treatment, 2.5 × 104 cells per well were plated on a 24-well tray, cultured overnight, and treated with 5 μM Gle1-scr or Gle1-SD peptide, 0.5 mg/ml WGA, 2.5 µg/ml actinomycin D, or 5 mM HU. Plasmids expressing GFP (pEGFP-C1, Takara Bio USA, Mountain View, CA), GFP-DDX19wt (pSW3254), GFP-DDX19R372G (pSW3477), GFP-DDX19E242Q (pSW3314), and mCherry-RNaseH1 (plasmid #60365, Addgene, Watertown, MA) were transfected using Fugene 6 according to manufacturer’s instructions.
Negative control siRNA (QIAGEN), GLE1 siRNA (QIAGEN; Aditi et al., 2015), and DDX1 siRNA pool (Dharmacon) were transfected in HeLa cells using HiPerfect according to the protocol published in Aditi et al. (2015). Cells were assayed after 72 h of transfection.
Antibodies used were: rabbit Gle1 (ASW48, 1:100 for IF and 1:1000 for IP), mouse S9.6 (Kerafast #ENH001, 1:100), rabbit nucleolin (Abcam #ab22758, 1:1000), phospho-γH2AX (EMD Millipore #05-636, 1:1000), rabbit DDX19 (Bethyl A300-547A, 1:100), mouse DDX1 (Novus Biologicals #NBP2-61745, 1:100 for IF), rabbit CstF-64 (Bethyl A301-092A, 1:100), Nucleolin-AF 488 (Abcam 364-5, 1:40), and NucRed (Molecular Probes, 2 drops/ml in phosphate-buffered saline [PBS]). Alexa Fluor secondary antibodies from Thermo Fisher Scientific were used at 1:500 dilution.
Peptide synthesis: Gle1-scr and Gle1-SD were custom synthesized by Genscript Biotech (Piscataway, NJ). Both sequences harbor antennapedia sequence at its amino terminus (RQIKIWFQNRRMKWKK).
Gle1-scr: VPSHLSTAFQDVSLYGVLPKSGQLKGLVSQFKENDIRGI
Gle1-SD: IFDKIHSLLSGKPVQSGGRSVSVTLNPQGLDFVQYKLAE
Nuclear and cytoplasmic RNA fractionation and RT-qPCR: Cytoplasmic and Nuclear RNA purification (Norgen Biotek, Thorold, ON, Canada) was performed according to manufacturer’s instructions. Briefly, cells were lysed directly in the wells with ice-cold lysis buffer J (containing β-mercaptoethanol) for 5 min on ice and spun at 13,000 rpm for 3 min. Supernatant (cytoplasmic fraction) and pellet (nuclear fraction) were resuspended in buffer SK and 100% ethanol. RNA was bound on the column, washed with wash solution A, and eluted in buffer E. DNase (Promega, Madison, WI) treatment was performed at 37°C for 30 min and the reaction was halted using 1× stop solution (Promega), 150 ng of RNA was used to generate cDNA using the high capacity cDNA reverse transcription kit (Thermo Fisher Scientific) according to manufacturer’s instructions, and 1.5 μl of cDNA was used for a 13-μl qPCR reaction using IQ Real time SYBR Green PCR Supermix (Bio-Rad Laboratories, Hercules, CA). Each reaction was done in triplicate and values were normalized to housekeeping gene as indicated based on ΔΔCT method. Statistical significance was calculated with a one-tailed, paired t test on ΔCT values using Prism software. Primers are listed in Supplemental Table S2.
RNA-seq library preparation and data analyses: Nuclear RNA from Gle1–scr- and Gle1–SD-treated HeLa cells was assessed for quality and enriched for mRNA, cDNA library was prepared, and polyA transcripts were sequenced by Vanderbilt Technologies for Advanced Genomics (VANTAGE) on Illumina HiSeq 2500. Data analyses was performed by VANTAGE Analysis and Research Design (VANGARD) using a standard pipeline. Reads were mapped to hg19 dataset using a HISAT2 scheme. Differential analyses between Gle1–scr- and Gle1–SD-treated samples were calculated using feature counts in SAMtools, and statistical analyses were performed using the edgeR Bioconducter software package. The false discovery rate was determined to be <0.05.
Click-IT nascent RNA capture: The Click-IT nascent RNA capture kit (#C-10365, Life Technologies, Thermo Fisher Scientific, Carlsbad, CA) was used according to manufacturer’s instructions. Briefly, HeLa cells were plated on a 24-well dish (2.5 × 104 cells per well) and treated as indicated; 0.2 mM of 5-EU was added to cells for 3 h, and nuclear RNA was isolated by fractionation from total RNA and treated with DNase as described. Nuclear RNA (550 ng) was biotinylated with 0.25 mM biotin-azide (Comp C) and precipitated with 12 ml of MyOne Streptavidin T1 magnetic beads (Comp H). Beads were washed and eluted and immediately used for cDNA synthesis using SuperScript VILO cDNA synthesis kit (#11754-050, Life Technologies) according to manufacturer’s instruction. qPCR was performed using SYBR Green Supermix as described above.
IF: Cells were processed for protein IF as previously described (Jao et al., 2016). Briefly, cells were fixed with 100% methanol at –20°C for 5 min, washed with PBS, and blocked overnight at 4°C in a humidified chamber with blocking buffer (2% sheep serum, 0.1% Triton X-100 and 10 mg/ml bovine serum albumin [BSA] in PBS). Primary antibodies were diluted in blocking buffer and incubated overnight at 4°C in a humidified chamber at dilutions indicated above. Cells were washed with PBS and immunostained with Alexa Flour antibodies for 1 h at room temperature (RT) in a humidified chamber. Cells were washed with PBS, incubated with NucRed for 45 min at RT, and mounted in Vectashield (Vector Laboratories, Burlingame, CA). Slides were imaged using a 63× 1.4 NA objective or 100× 1.47 NA objective on a Leica SP5 confocal microscope. Four color imaging in Figure 6 was performed using a 100X 1.42 N/A objective on an Olympus FV1000 confocal microscope. Postimaging processing was performed on maximum intensity projected z-stacks (excluding R-loop analyses, described below) using ImageJ software. GraphPad Prism 8 was used to remove outliers using recommended ROUT method (Q = 1%) and to graph data from at least three independent experiments. Statistical significance was determined using Prism software as indicated in figure legends.
R-loop IF (Wan et al., 2015) was performed with the following modifications. Following methanol fixation, cells were washed with 70% ethanol for 5 min on ice followed by PBS washes. Cells were blocked with 250 mg/ml BSA, 0.5% Triton X-100, and 2% normal goat serum in PBS. The same buffer was used to dilute primary and secondary antibodies. Cells were imaged as above and analyses were performed on a midplane section.
PLA and analyses: The Duolink PLA (Sigma-Aldrich, Saint Louis, MO) fluorescence protocol was followed with the changes described below. HeLa cells were plated on 8-well chamber slide (2 × 104 cell per chamber), treated as indicated, and blocked overnight at 4°C in a humidified chamber with the same blocking buffer as used for IF. Primary antibodies were diluted as described for IF and incubated overnight at 4°C in a humidified chamber. Chamber slides were washed in a Koplin chamber using buffer A. PLA probe incubations, ligation, amplification, and washes were performed according to manufacturer’s instruction. Counterstaining with Nucleolin-AF 488 (at 4°C overnight in a humidified chamber) and/or NucRed (for 45 min at RT in a humidified chamber) was performed after washes in buffer B, as directed by the manufacturer. Slides were mounted in Duolink mounting media and imaged on the Leica SP5 confocal microscope using 100× 1.47 NA objective lens. Z-stacks were acquired from top to bottom of the nucleus in 0.5-μm sections. Maximum intensity projected images were used to calculate the number of PLA dots per nucleus and Integrated Density (defined as product of area and mean gray value) in the nucleus. Images were processed and analyzed using ImageJ software.
Supplementary Material
Acknowledgments
We are grateful to the members of Wente Lab for discussions regarding this project, and particularly T. Renee Dawson for critical reading of the manuscript. We are thankful to Joshua Lin, Veronica Olaker, Angelica Zverovich, and Yiting Chen for technical assistance. This work was supported by the National Institutes of Health (NIH) Grant 5R37GM051219 (to S.R.W.). The authors acknowledge the expert technical support of VANTAGE with RNA sequencing and VANGARD with data analysis. VANTAGE and VANGARD are supported in part by the Vanderbilt Ingram Cancer Center (P30 CA68485) and the Vanderbilt Vision Center (P30 EY08126). VANTAGE is also supported by the CTSA Grant (5UL1 RR024975-03) and NIH/National Center for Research Resources (G20 RR030956). Microscopy was performed in part through the use of the Vanderbilt Cell Imaging Shared Resource, supported by the National Institutes of Health grants CA68485, DK20593, DK58404, DK59637 and EY08126.
Abbreviations used:
- BSA
bovine serum albumin
- CS
cleavage site
- DDX
DEAD-box protein
- EU
ethynyl uridine
- Gle1-scr
Gle1-scramble
- HU
hydroxyurea
- IF
immunofluorescence
- In1
intron 1
- IP 6
inositol hexakisphosphate
- m
messenger
- mRNP
mRNA-protein complex
- NPC
nuclear pore complex
- Nup
nucleoporin/NPC protein
- PAS
polyadenylation site
- PBS
phosphate-buffered saline
- PLA
proximity ligation assay
- RLFS
R-loop forming sequence
- RNA Pol II
RNA polymerase II
- RNA-seq
RNA-sequencing
- RT
room temperature
- SD
shuttling domain
- TTS
transcription termination site
- UTR
untranslated region
- VANGARD
Vanderbilt Technologies for Advanced Genomics Analysis and Research Design
- VANTAGE
Vanderbilt Technologies for Advanced Genomics
- WGA
wheat germ agglutinin.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-03-0215) on August 5, 2020.
REFERENCES
- Adams RL, Mason AC, Glass L, Aditi, Wente SR (2017). Nup42 and IP6 coordinate Gle1 stimulation of Dbp5/DDX19B for mRNA export in yeast and human cells. Traffic , 776–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aditi, Folkmann AW, Wente SR (2015). Cytoplasmic hGle1A regulates stress granules by modulation of translation. Mol Biol Cell , 1476–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aditi, Mason AC, Sharma M, Dawson TR, Wente SR (2019). MAPK- and glycogen synthase kinase 3-mediated phosphorylation regulates the DEAD-box protein modulator Gle1 for control of stress granule dynamics. J Biol Chem , 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alcázar-Román AR, Tran EJ, Guo S, Wente SR (2006). Inositol hexakisphosphate and Gle1 activate the DEAD-box protein Dbp5 for nuclear mRNA export. Nat Cell Biol , 711–716. [DOI] [PubMed] [Google Scholar]
- Bauer DLV, Tellier M, Martínez-Alonso M, Nojima T, Proudfoot NJ, Murphy S, Fodor E (2018). Influenza virus mounts a two-pronged attack on host RNA polymerase II transcription. Cell Rep , 2119–2129.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Yishay R, Shav-Tal Y (2019). The dynamic lifecycle of mRNA in the nucleus. Curr Opin Cell Biol , 69–75. [DOI] [PubMed] [Google Scholar]
- Bleoo S, Sun X, Hendzel MJ, Rowe JM, Packer M, Godbout R (2001). Association of human DEAD box protein DDX1 with a cleavage stimulation factor involved in 3-end processing of pre-mRNA. Mol Biol Cell , 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger TA, Folkmann AW, Tran EJ, Wente SR (2008). The mRNA export factor Gle1 and inositol hexakisphosphate regulate distinct stages of translation. Cell , 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty P, Huang JTJ, Hiom K (2018). DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat Commun , 4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Feng C, Fang Y, Zhou X, Xu L, Wang W, Kong X, Peppelenbosch PM, Pan Q, Yin Y (2019). The eukaryotic translation initiation factor 4F complex restricts rotavirus infection via regulating the expression of IRF1 and IRF7. Int J Mol Sci , 1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristini A, Groh M, Kristiansen MS, Gromak N (2018). RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep , 1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley MP, Bocek M, Cimprich KA (2019). R-Loops as cellular regulators and genomic threats. Mol Cell , 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargemont C (1992). Export of mRNA from microinjected nuclei of Xenopus laevis oocytes. J Cell Biol , 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estruch F, Cole CN (2003). An early function during transcription for the yeast mRNA export factor Dbp5p/Rat8p suggested by its genetic and physical interactions with transcription factor IIH components. Mol Biol Cell , 1664–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkmann AW, Collier SE, Zhan X, Aditi, Ohi MD, Wente SR (2013). Gle1 functions during mRNA export in an oligomeric complex that is altered in human disease. Cell , 582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folkmann AW, Noble KN, Cole CN, Wente SR (2011). Dbp5, Gle1-IP 6 and Nup159: A working model for mRNP export. Nucleus , 540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginno PA, Lim YW, Lott PL, Korf I, Chedin F (2013). GC skew at the 5’ and 3’ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res , 1590–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass L, Wente SR (2019). Gle1 mediates stress granule-dependent survival during chemotoxic stress. Adv Biol Regul , 156–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartono SR, Malapert A, Legros P, Bernard P, Chédin F, Vanoosthuyse V (2018). The Affinity of the S9.6 antibody for double-stranded RNAs impacts the accurate mapping of R-loops in fission yeast. J Mol Biol , 272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig T, Michalski M, Rutkowski AJ, Djakovic L, Whisnant AW, Friedl M-S, Jha BA, Baptista MAP, L’Hernault A, Erhard F, et al (2018). HSV-1-induced disruption of transcription termination resembles a cellular stress response but selectively increases chromatin accessibility downstream of genes. PLoS Pathog , e1006954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge CA, Colot HV, Stafford P, Cole CN (1999). Rat8p/Dbp5p is a shuttling transport factor that interacts with Rat7p/Nup159p and Gle1p and suppresses the mRNA export defect of xpo1-1 cells. EMBO J , 5778–5788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodge CA, Tran EJ, Noble KN, Alcazar-Roman AR, Ben-Yishay R, Scarcelli JJ, Folkmann AW, Shav-Tal Y, Wente SR, Cole CN (2011). The Dbp5 cycle at the nuclear pore complex during mRNA export I: dbp5 mutants with defects in RNA binding and ATP hydrolysis define key steps for Nup159 and Gle1. Genes Dev , 1052–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodroj D, Recolin B, Serhal K, Martinez S, Tsanov N, Abou Merhi R, Maiorano D (2017a). An ATR-dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J , 1182–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodroj D, Serhal K, Maiorano D (2017b). Ddx19 links mRNA nuclear export with progression of transcription and replication and suppresses genomic instability upon DNA damage in proliferating cells. Nucleus , 489–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt IJ (2019). The mitochondrial R-loop. Nucleic Acids Res , 5480–5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao L-E, Akef A, Wente SR (2016). A role for Gle1, a regulator of DEAD-box RNA helicases, at centrosomes and basal bodies. Mol Biol Cell , 120–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenjaroenpun P, Wongsurawat T, Yenamandra SP, Kuznetsov VA (2015). QmRLFS-finder: a model, web server and stand-alone tool for prediction and analysis of R-loop forming sequences. Nucleic Acids Res , W527–W534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katahira J, Ishikawa H, Tsujimura K, Kurono S, Hieda M (2019). Human THO coordinates transcription termination and subsequent transcript release from the HSP70 locus. Genes Cells , 272–283. [DOI] [PubMed] [Google Scholar]
- Kehlenbach RH (2003). In vitro analysis of nuclear mRNA export using molecular beacons for target detection. Nucleic Acids Res , e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendirgi F, Barry DM, Griffis ER, Powers MA, Wente SR (2003). An essential role for hGle1 nucleocytoplasmic shuttling in mRNA export. J Cell Biol , 1029–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo CX, Kobiyama K, Shen YJ, LeBert N, Ahmad S, Khatoo M, Aoshi T, Gasser S, Ishii KJ (2015). RNA polymerase III regulates cytosolic RNA:DNA hybrids and intracellular MicroRNA expression. J Biol Chem , 7463–7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Kohler JJ (2014). Glycosylation of the nuclear pore. Traffic , 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Monckton EA, Godbout R (2008). A role for DEAD Box 1 at DNA double-strand breaks. Mol Cell Biol , 6413–6425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mersaoui SY, Yu Z, Coulombe Y, Karam M, Busatto FF, Masson J-Y, Richard S (2019). Arginine methylation of the DDX5 helicase RGG/RG motif by PRMT5 regulates resolution of RNA:DNA hybrids. EMBO J e100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montpetit B, Thomsen ND, Helmke KJ, Seeliger MA, Berger JM, Weis K (2011). A conserved mechanism of DEAD-box ATPase activation by nucleoporins and InsP 6 in mRNA export. Nature , 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy R, Wente SR (1996). An RNA-export mediator with an essential nuclear export signal. Nature , 357–360. [DOI] [PubMed] [Google Scholar]
- Natalizio BJ, Wente SR (2013). Postage for the messenger: designating routes for nuclear mRNA export. Trends Cell Biol , 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann B, Wu H, Hackmann A, Krebber H (2016). Nuclear Export of Pre-Ribosomal Subunits Requires Dbp5, but Not as an RNA-Helicase as for mRNA Export. PLoS One , e0149571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble KN, Tran EJ, Alcázar-Román AR, Hodge CA, Cole CN, Wente SR (2011). The Dbp5 cycle at the nuclear pore complex during mRNA export II: nucleotide cycling and mRNP remodeling by Dbp5 are controlled by Nup159 and Gle1. Genes Dev , 1065–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porrua O, Boudvillain M, Libri D (2016). Transcription Termination: Variations on Common Themes. Trends Genet TIG , 508–522. [DOI] [PubMed] [Google Scholar]
- Porrua O, Libri D (2015). Transcription termination and the control of the transcriptome: why, where and how to stop. Nat Rev Mol Cell Biol , 190–202. [DOI] [PubMed] [Google Scholar]
- Proudfoot NJ (2011). Ending the message: poly(A) signals then and now. Genes Dev , 1770–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proudfoot NJ (2016). Transcriptional termination in mammals: Stopping the RNA polymerase II juggernaut. Science , aad9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard P, Manley JL (2009). Transcription termination by nuclear RNA polymerases. Genes Dev , 1247–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruepp M-D, Schweingruber C, Kleinschmidt N, Schümperli D (2011). Interactions of CstF-64, CstF-77, and symplekin: Implications on localisation and function ed. A.G. Matera. Mol Biol Cell , 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Pereira JM, Aguilera A (2015). R loops: new modulators of genome dynamics and function. Nat Rev Genet , 583–597. [DOI] [PubMed] [Google Scholar]
- Skourti-Stathaki K, Proudfoot NJ, Gromak N (2011). Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell , 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagaki Y, Manley JL (1997). RNA recognition by the human polyadenylation factor CstF. Mol Cell Biol , 3907–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagaki Y, Seipelt RL, Peterson ML, Manley JL (1996). The polyadenylation factor CstF-64 regulates alternative processing of IgM heavy chain pre-mRNA during B cell differentiation. Cell , 941–952. [DOI] [PubMed] [Google Scholar]
- Vanoosthuyse V (2018). Strengths and weaknesses of the current strategies to map and characterize R-loops. Non-Coding RNA , 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilborg A, Passarelli MC, Yario TA, Tycowski KT, Steitz JA (2015). Widespread inducible transcription downstream of human genes. Mol Cell , 449–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilborg A, Sabath N, Wiesel Y, Nathans J, Levy-Adam F, Yario TA, Steitz JA, Shalgi R (2017). Comparative analysis reveals genomic features of stress-induced transcriptional readthrough. Proc Natl Acad Sci , E8362–E8371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Y, Zheng X, Chen H, Guo Y, Jiang H, He X, Zhu X, Zheng Y (2015). Splicing function of mitotic regulators links R-loop–mediated DNA damage to tumor cell killing. J Cell Biol , 235–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IX, Grunseich C, Fox J, Burdick J, Zhu Z, Ravazian N, Hafner M, Cheung VG (2018). Human proteins that interact with RNA/DNA hybrids. Genome Res , 1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirich CS, Erzberger JP, Flick JS, Berger JM, Thorner J, Weis K (2006). Activation of the DExD/H-box protein Dbp5 by the nuclear-pore protein Gle1 and its coactivator InsP6 is required for mRNA export. Nat Cell Biol , 668–676. [DOI] [PubMed] [Google Scholar]
- West S, Gromak N, Proudfoot NJ (2004). Human 5’ → 3’ exonuclease Xrn2 promotes transcription termination at co-transcriptional cleavage sites. Nature , 522–525. [DOI] [PubMed] [Google Scholar]
- Wongsurawat T, Jenjaroenpun P, Kwoh CK, Kuznetsov V (2012). Quantitative model of R-loop forming structures reveals a novel level of RNA-DNA interactome complexity. Nucleic Acids Res , e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Y, Ren Y (2019). Mechanisms of nuclear mRNA export: A structural perspective. Traffic , 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, Biesinger J, Wan J, Weng L, Xing Y, Xie X, Shi Y (2012). Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc Natl Acad Sci , 18773–18778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolotukhin AS, Uranishi H, Lindtner S, Bear J, Pavlakis GN, Felber BK (2009). Nuclear export factor RBM15 facilitates the access of DBP5 to mRNA. Nucleic Acids Res , 7151–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.