

Abstract
To fight against the present pandemic scenario of COVID-19 outbreak, medication with drugs and vaccines is extremely essential other than ventilation support. In this paper, we present a list of ligands which are expected to have the highest binding affinity with the S-glycoprotein of 2019-nCoV and thus can be used to make the drug for the novel coronavirus. Here, we implemented an architecture using 1D convolutional networks to predict drug–target interaction (DTI) values. The network was trained on the KIBA (Kinase Inhibitor Bioactivity) dataset. With this network, we predicted the KIBA scores (which gives a measure of binding affinity) of a list of ligands against the S-glycoprotein of 2019-nCoV. Based on these KIBA scores, we are proposing a list of ligands (33 top ligands based on best interactions) which have a high binding affinity with the S-glycoprotein of 2019-nCoV and thus can be used for the formation of drugs.
Keywords: COVID-19, Ligand, S-glycoprotein, Binding affinity, KIBA, Drug–target interaction values, 1D CNN, Protein Sequence Composition, ECFP4
Introduction
A worldwide pandemic has been declared by the World Health Organization (WHO) following the global spread of COVID-19 (coronavirus disease 19). Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) has been traced down as the virus causing this global havoc [1]. Originated at Wuhan, China, in December 2019 [2], currently this newly found virus has caused numerous deaths all over the world including India as well [3]. A few clinical trials have been made with some commonly used drugs all over the world, like chloroquine, hydroxychloroquine, lopinavir, ritonavir and remdesivir [4]. But the experts have stated that experiments performed in laboratories do not always give conclusive results which can lead to the recommendation of this medicine for cure. Therefore in the present scenario, the medical community is yet to get a vaccine or drug or medication that can help fight the COVID-19 outbreak. In this paper, we propose a deep learning-based drug prediction model to control the outbreak of the COVID-19 pandemic. Here, an end-to-end deep learning-based framework will work on the protein–ligand binding of various chemical molecules and give us a prediction of the type of drug which may work on the protein part of RNA of SARS-CoV-2.
The world medical fraternity is yet to come across any prominent medicine or drug to fight the havoc caused by COVID-19, as per recent updates from WHO [6]. Thereby, our goal is to help medical fraternity in inventing medicines by suggesting them effective ligands. Various countries have gone into complete lockdown administering social distancing as the sole tool to prevent the citizens from getting affected. Health workers are provided with Personal Protective Equipment (PPE) for securing themselves while treating COVID-positive patients. Not only the devastating health effects, this pandemic impacted hugely on world economy too [7]. Many universities and laboratories across the world are conducting various experiments to find out a new and effective drug or compound which can help to structure a medicine that will help us combat COVID-19 [8–10]. But till date, the results have not been fruitful. There had been an unexpected enthusiasm among various individuals in administering hydroxychloroquine (HCQS) as cure of this disease [11]. But medical professionals are not ready to administer the same in mass quantity as the result is not proven to be helpful. Though when administered has been found to be safe in the USA, usage of remdesivir for the treatment of COVID-19-affected patients is yet to be considered a safe option until large trial is conducted with the same [12]. Recent news shows there has been a clinical trial in New York on COVID-19 patients with heartburn medicines [13]. But the authorities have kept the trial under wrap off now until they get a concrete result. Therefore to summarize the situation, there is no proven drug or medicine which can fight COVID-19 virus till now. Adoption of artificial intelligence (AI) techniques in medical platform can lead to a probable solution toward it [5]. In recent years, the new DL techniques [12] have been adopted in drug discovery and development, opening a new opportunity to computational decision making in pharmaceutical science. After studying the structures of proteins, active small molecules toward the protein targets can be discovered from the structure-based drug design methodologies. Therefore, this research is of utmost importance where the proposed new compound, if validated by biochemists as an effective solution, can help mankind survive this tough time. In this paper, a deep learning-based architecture has been used to implement a drug prediction model which may work on the protein part of RNA of SARS-COV-2. Our contribution here is to select the number of filters to get the best possible result and also the input dimensions to suit the proposed CNN models.
The rest of the work has been organized in few sections. Background section (Section 2) deals with discussion on some recent published works to prevent COVID-19. Section 3 describes the proposed methodology we have followed. Here, we have used neural networks and 1D CNN to predict protein–ligand interaction value. Protein sequence composition (PSC) and ProtVec are used for featuring protein sequence, and graph neural network and ECPF4 are used for featuring ligand. Comparing the performance of these models, we picked PSC and ECPF4 for featuring protein and ligands. Concatenation of these two models yields 9444-dimensional input vector where the output of the model will be a real number, signifying the binding tendency between the protein and the ligand. We have created one python dictionary with SMILES code as keys and model’s output corresponding to SMILES code as values. After sorting, we are getting the top 33 ligands which are supposed to have the highest binding capacity with the S-protein part of SARS-CoV-2. The results are commendable here, reported in Section 5. Section 6 draws the final conclusion and an open problem as future work.
Background
Megha [14] has presented an approach for molecular docking analysis of selected natural products from plants for inhibition of SARS-CoV-2 main protease. The paper primarily focuses on proposing a new and naturally found compound that can be included in our daily food habit which will help us fight the dangerous SARS-CoV-2 viruses. The results of the research show that many of these compounds portray binding abilities with the SARS-CoV-2 protease. The focus of the entire study lies on finding binding affinity of various ligands (27) with the COVID-19 6LU7 and 6Y2E proteases. Studying these binding affinity efforts has been made to draw a conclusion to whether a natural product-based solution can be developed which will help us to fight the pandemic. The data that were chosen to be worked on include COVID-19 3CLpro/Mpro (PDB ID: 6LU7) 1, 5 and free enzyme of the SARS-CoV-2 (2019-nCoV) main protease (PBD ID: 6Y2E) 3, the active site of protease obtained using Computed Atlas for Surface Topography of Proteins (CASTp), the 3D structure of selected ligand, anti-HIV drug, saquinavir (has been used as a positive control). The result of the study shows that high virulence and spread of COVID-19 are reduced by 15 out of 27 natural products that are active in binding the protease. These 15 natural products are present in our day to day cuisine and can help us with providing the first line of defense against COVID-19. Some of these products are found in curcumin and coriandrin, compounds found in apple peels (ursolic acid), in olive oil (oleanolic acid), in cucurbit vegetables (hederagenin), in red pepper (apigenin), in Glycyrrhiza glabra (glabridin), rosemary and thyme or mint family plants (sageone), to name a few.
Another approach was presented to show protein–ligand scoring with convolutional neural networks [15]. This research helps us to get a faster computational approach for drug discovery compared to the long procedure of trial and error carried out in a clinic and laboratory. Scoring functions are one of the primary parameters against which the structure-based drug design method is evaluated. Here, the scoring function of a convolutional neural network has been adapted which takes a 3D representation of protein–ligand interaction as input and gives an output that helps to differentiate between correct and incorrect binding poses. For pose prediction and virtual screening, two different datasets have been used. For pose prediction, CSAR-NRC has been used which contains around 466 ligand-bound cocrystals. Those targets have been excluded which shows a binding affinity of less than 5pK units. For virtual screening, Database of Useful Decoys: Enhanced is used which contains 102 targets and more than 20,000 active molecules and over a million decoy molecules. To give an acceptable input to the CNN, the 3D structural data were transformed into a grid. Each grid point holds information about the heavy atom residing at that point. Caffe deep learning framework has been used to train this model. For model evaluation, threefold cross-validation has been done for both pose prediction and virtual screening.
Another group of scientists introduced us to an approach of deep learning-based drug screening for novel coronavirus 2019-nCov [16]. To avoid the long traditional clinic-based drug discovery method and to rapidly come up with a solution to fight COVID-19, a deep learning-based alternate approach has been adapted in the paper. Firstly, the RNA of the virus is collected from the GISAID database and transformed into a protein sequence. Next, using homology modeling, a protein 3D structure is constructed. DFCNN, a deep learning-based method, is developed by the researchers which can perform quick virtual screening and identifies potential drugs for SARS-CoV-2 protease after performing a thorough drug screening process against 4 different chemical compound databases. Also, drug screening is performed against tripeptides. The DFCNN does not have the gradient vanishing problem, and the layers are fully connected in the neural network. The database used for DFCNN is from PDBbind. The primary advantage of DFCNN is that it does not involve docking run and the dataset can have non-binding decoys. Check for any kind of mutation in the virus has been done by matching the protein sequence of the virus when collected from 18 different patients. As of now, the virus is stable. The 4 different compound databases against which the performance of the DFCNN has been checked are the Chemdiv dataset [17], Targetmol-Approved_Drug_Library [18], Targetmol-Natural_Compound_Library [19] and Targetmol-Bioactive_Compound_Library [20]. Results show the compounds with DFCNN have a score as high as 0.997 (Targetmol-Natural compound library). The DFCNN system when checked against the tripeptides set showed a score of 0.995. This high value indicates that peptides are most likely to bind with the pocket of SARS-CoV-2 main protease. Study shows peptides formed by I, K, P amino acids have the highest probability of binding with the pockets.
Proposed Methodology
Overview
We introduce a methodology for finding out the probable candidate drug molecule, also known as a ligand which binds with the S-protein sequence of SARS-CoV-2. The hunt here is to find a ligand that can bind with the active site of the SARS-CoV-2 protein chain. Biochemically, a drug is called an effective drug if it has a stable binding state with the active site of the S-protein chain of a certain virus. There are two different approaches to drug discovery of which Target-Based Discovery is the dominant approach. This approach involves screening of compounds for specific activity against known targets. This is where machine learning and neural networks come in. Convolutional neural networks are excellent in finding spatial and temporal patterns in a dataset. A drug’s efficiency may be affected by the degree to which it binds. The drug–target interaction (DTI) refers to the effective binding capacity of the drug molecule (ligand) and the target molecule (protein) chain. The use of neural networks and artificial intelligence for drug prediction is a well-versed field of research. We used a model consisting of 1D CNNs to find the best ligand with the highest effective potential binding. The raw protein names or SMILES codes [21] of ligands (as given in the downloaded dataset) have not been used directly as inputs to our model. Rather, we have vectorized both the protein sequences and SMILES codes and generated vectors corresponding to each protein sequence and each SMILES code. The input to our model is a vector formed by the concatenation of the vector corresponding to a protein sequence, with the vector corresponding to a SMILES code. Vectorization plays a vital role in proper drug–target interaction value prediction and is discussed in details in later sections. The KIBA score corresponding to a particular protein–ligand pair is not a 0-1 value, but is a real–valued number.
Description of the Dataset
We used the Kinase Inhibitor Bioactivity (KIBA) dataset to train our network [22] architecture for drug–target interaction prediction. The KIBA dataset, on the other hand, originated from an approach called KIBA, in which Kinase Inhibitor Bioactivities from different sources such as Ki, Kd and IC50 were combined. KIBA scores were constructed to optimize the consistency between Ki, Kd and IC50 by utilizing the statistical information they contained. The KIBA dataset originally comprised 467 targets and 52 498 drugs. The KIBA dataset has the following descriptions: Protein-229, Compound-2111 and Interaction-118254. It was later filtered to contain only drugs and targets with at least 10 interactions yielding a total of 229 unique proteins and 2111 unique drugs. Table 1 summarizes the dataset in the forms that we used in our experiments along with the distribution graphs. The workflow is shown in Fig. 1. The detailed KIBA score, length of SMILES characters and length of protein sequence are shown in Fig. 2.
Table 1.
KIBA Dataset Description
| Protein | Compound | Interaction |
|---|---|---|
| 229 | 2111 | 118254 |
Fig. 1.

The block diagram of the proposed work
Fig. 2.

(a) KIBA scores, (b) length of SMILES (chars), (c) length of protein sequence (chars)
Vectorization of Protein Sequence and Ligand SMILES Code
Vectorization of Protein Sequence
The primary focus of the work in the processed dataset is protein names, ligand SMILES code and the KIBA score corresponding to this pair. Now, it is hard to convert protein names to vectors. So, first, we had to generate protein sequence corresponding to the protein names. The task has been accomplished using the propy [23] library of python. Next, we had to convert the protein sequences into vectors. There are multiple ways existing in the literature for representing proteins as feature vectors. We have used protein sequence composition (PSC) descriptors [24] for vectorizing the protein sequence in this research. Initially, we converted the protein names to their corresponding protein sequences using GetProteinSequence function of propy library and then generated the PSC descriptors for the protein sequences with the help of the GetProDes function of the same library of python. Thus, we received a 8420-dimensional feature vector for each protein sequence. PSC descriptors consist of amino acid composition (AAC), dipeptide composition (DC) and tripeptide composition (TC) [24]. AAC is the frequency of each amino acid in the protein sequence and needs 20 feature values for it. DC is the frequency of dipeptide, that is, every two amino acid combination. It is represented by 400 feature values. TC is the frequency of three amino acid combination and is represented by 8000 feature values. Thus, PSC descriptors convert a protein sequence into (20+400+8000) = 8420 feature vectors.
Vectorization of Ligand SMILES Code
We have the Simplified Molecular-Input Line-Entry System (SMILES) codes of the ligands. SMILES is a chemical notation to represent a chemical structure in the form of text. SMILES code gives us information about the atoms present, different kinds of bonds, branching, aromaticity, etc., of a molecule. Some architectures work with the ligand SMILES codes directly as input. But, we have focused on vectorizing the SMILES code of ligands and then using that, vector for our model has been purposes. The most commonly used methods for featurizing the SMILES codes are Extended Connectivity Fingerprints (ECFP) and Neural Graph Fingerprints. ECFPs are circular topological fingerprints used for molecular characterization and structure–activity modeling. Basically, a molecule is converted into a bit vector or count vector of a given length. Some of the main properties of ECFPs are: They represent molecular structure by circular atom neighborhood; they represent the presence of particular sub-structures; they can be rapidly calculated; and they are not predefined and can represent essentially infinite number of different molecular features (including stereo-chemical information). The 2 important parameters in ECFPs are diameter and length. Diameter specifies the maximum diameter of the circular neighborhoods considered for each atom and is generally kept at 4. ECFP4 means maximum diameter is set to 4; similarly, ECFP6 means diameter is set to 6. Diameter parameter controls the number and the maximum size of considered atom neighborhoods. Length specifies the length of bit-string representation, that is, the length of the bit vector which is generally 1024 [25].
Instead of molecular fingerprint vectors like ECFP, we can use a vector generated by a differentiable neural network which takes a graph as input. This is done by Neural Graph Fingerprints. That graph is a representation of a molecule with vertices representing individual atoms and edges representing bonds. The length of the generated vector can be fixed by user. Important information like the atoms present, bonds present, degree of bonds, atom features, etc., is collected from the SMILES codes with the help of the RDKit library of python [26], and this information is used to build a molecular graph, representing the molecule. Then, the algorithm followed in this work [27] is used to get a real-valued vector corresponding to a molecule. The advantages of neural fingerprints over fixed length fingerprints are better predictive performance than fixed fingerprints. Neural fingerprints can be optimized to encode only the relevant features, thus reducing computation, while fixed length fingerprints will need large vectors to encode all possible sub-structures. We tried out both ECFP4 and Neural Graph Fingerprints with the PSC descriptors of proteins. We found that ECFP4 performs better than Neural Graph Fingerprints. Hence, we decided to use ECFP4 in our paper.
Generating Concatenated Input Vector for our Model
Till now, we have vectorized each protein sequences into a 8420-dimensional vector using PSC descriptors. Also, we have vectorized each SMILES code into a 1024-dimensional vector using ECFP4. Then, we concatenated the PSC descriptor vector of each protein sequence with the ECFP4 bit vector of its corresponding ligand SMILES code, as is done in [28]. Thus, we have a concatenated vector of length (8420+1024) = 9444. This concatenated vector was used as input to our model.
The Architecture
Convolutional neural networks (CNNs) are mainly used for image classification problems where we use 2D CNNs. But the power of CNNs can also be used for one-dimensional sequences of data with the help of 1D CNNs. We have used 1D CNNs to build up our architecture. The very first area of interest was the input dimensions to the architecture. We implemented 1D CNNs using keras library in python [29]. Since our input vector is 9444-dimensional vector, we either have to input it as (9444, 1) or (1, 9444). We found that for any given architecture, the performance was much better when the input shape was (1, 9444). Hence, we used this input shape for our architecture. The input vector reshaped to (1, 9444) was first passed through a 1D convolutional layer with 64 filters. Convolution involves the multiplication and addition of the input vector with another vector, called the weight or filter, thus producing a feature map. Over those feature maps, activation functions are applied. We have used the ReLU activation function. The output was passed through another 1D convolutional layer with 64 filters. Then, BatchNormalization was done. BatchNormalization is used to stabilize the learning process and to reduce the number of training epochs. After that, we applied two more 1D convolutional layers each having 128 filters followed by BatchNormalization. Then, we flattened the output from BatchNormalization, which was a (1, 128)-dimensional vector to generate a 128-dimensional vector. This was connected to a dense layer having 100 neurons which was finally connected to a single neuron, the output neuron. We used the ReLU activation function in all the convolutional layers. The kernel size was kept to 1. The dense layer with 100 neurons also had activation function ReLU. The last output layer with 1 neuron had linear activation function since we are trying to predict regression values. The diagram of the model is shown in Fig. 3.
Fig. 3.

The proposed architecture for DTI model
Training
Training was done on the KIBA dataset. We only used the first 19,000 rows due to lack of adequate computational resources. We trained our model on Google Colab using its Tesla K80 GPU. The training set was split into validation set by a factor of 0.1. The batch size was 100, and we trained the model for 100 epochs. The model was compiled using Adam optimizer. The learning rate of the Adam optimizer was set to 0.001. The loss function used was root mean squared loss (RMSE) loss function which is a common loss function for regression-based problems. We used mean squared error (MSE) and mean average error (MAE) as metrics. We used model checkpoint of keras to save the best model only. We monitored the validation loss, and the model corresponding to minimum validation loss was saved. The best model (using model checkpoint) was saved corresponding to minimum validation loss of 0.83. The minimum recorded values of validation MSE and validation MAE were 0.70 and 0.63, respectively. Graphs were plotted to see the progress of MSE, MAE and loss against the epoch number. The graphs are shown in Fig. 4.
Fig. 4.
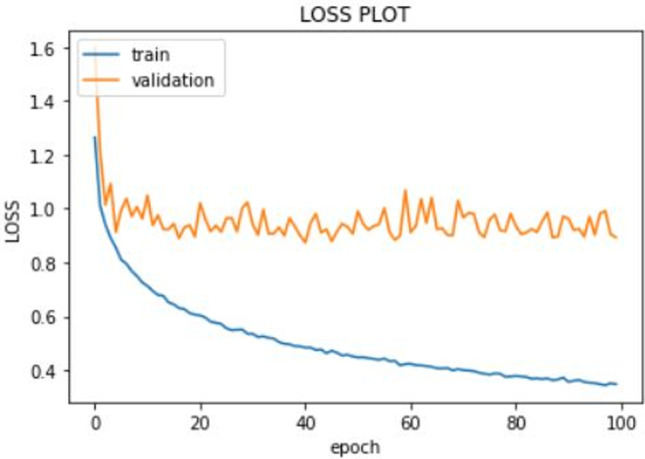
Training loss of RMSE vs the epoch number
The train loss is shown in blue line and the validation loss in orange line. The validation loss for the best model was 0.83. The plot of MSE vs the epoch number is shown in Fig. 5. The training MSE is marked by blue line and the validation MSE by orange line. The minimum recorded value of validation MSE was 0.70. Finally, the MAE vs the epoch number plot is shown in Fig. 6. The training MAE is blue, and the validation MAE is orange in color. The minimum recorded value of validation MAE was 0.63.
Fig. 5.
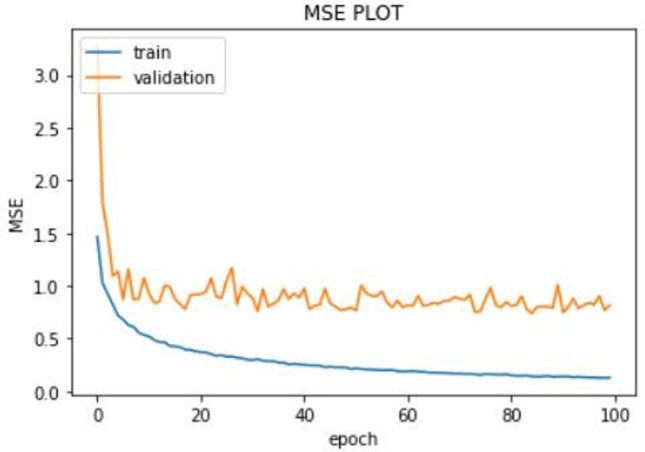
MSE vs the epoch number
Fig. 6.
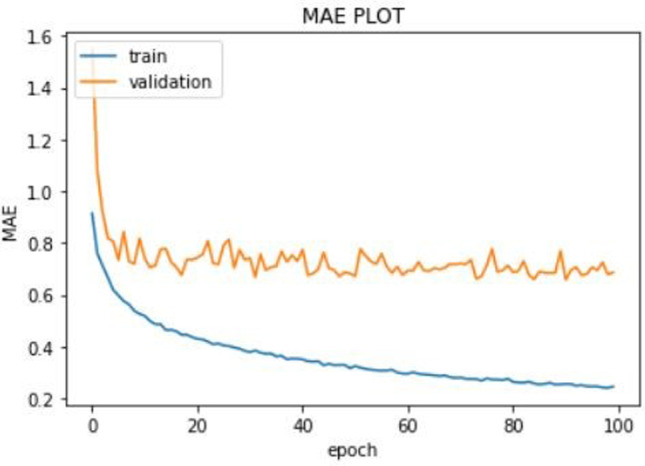
MAE vs the epoch number
Results and Discussion
After the completion of the training process, we have a model whose input is the vector formed by the concatenation of the ECFP4 descriptor of ligand SMILES code and the PSC descriptor of a protein sequence. The output of the model is the corresponding KIBA score for the protein–ligand pair which is a real-valued number. Hence, the task is a regression task. The KIBA score gives an indication of the binding affinity of the protein–ligand pair. Now, we use our model for predictive analysis. We want to predict those ligands which are supposed to have the highest binding affinity with the S-glycoprotein of SARS-CoV-2. Three monomeric subunits of spike (S) glycoprotein trimerized to form a functional spike protein interact with host ACE2 receptors and mediate host cell entry. Destabilization or inhibition of formation of functional spike glycoprotein may prevent the entry of virus inside the host cell. Hence, we need to predict ligands which can bind with the S-protein chains, thus stopping the tripeptide formation. So, our input to the model was the PSC descriptor of the S-protein sequence of SARS-CoV-2, concatenated with the ECFP4 descriptor of a ligand. A large database of ligands was essential to test their binding affinity with the S-protein. For that, we used the data available [30] from GitHub repository [31]. We have used the data available there and converted that into a CSV file containing the SMILES code of over 615,000 distinct ligands. So, we have the SARS-CoV-2 S-glycoprotein sequence on the one hand and the ligand SMILES code on the other hand. We iterated over the entire length of the ligand dataset of over 615,000 distinct ligands. At each iteration, the 9444-dimensional vector formed by concatenating the 8420-dimensional PSC descriptor of the S-protein with the 1024-dimensional ECFP4 vector of a ligand was given as input to the model and the predicted score was stored in a python dictionary. The keys of the dictionary were the SMILES code of the ligands, and the values of the dictionary were the predicted model scores corresponding to those ligands. Higher KIBA score indicates a lower binding affinity between a drug and the corresponding target (protein) and vice versa. Hence, we printed the dictionary in ascending order of the predicted KIBA scores and saved the top 33 ligands. These ligands have the lowest predicted KIBA scores and thus the highest possible binding affinity with the S-protein of SARS-CoV-2 among this set of about 615000 ligands. These 33 ligands with their SMILES codes are presented in Table 2. The binding affinity of the ligands with S-protein is expected to gradually decrease as we go down the table.
Table 2.
SMILES codes of the top 33 ligands in the descending order of their binding affinity with the S-glycoprotein of SARS-CoV-2
| SMILES code No | SMILES codes |
|---|---|
| 16 | NCCNC(=O)c1cccc(c1)c2cnc(Nc3cc(ccn3)N4CCC(F)(F)CC4)s2 |
| 17 | Cc1cn2c(cnc2c(Nc3ccc(C(=O)N4C[C@H]5CC[C@@H]4CN5)c(Cl) |
| c3)n1)c6cn[nH]c6 | |
| 19 | COCCCc1cc(Nc2nc(NCc3onc(C)c3)ncc2Br)n[nH]1 |
| 20 | Cc1cn2c(cnc2c(Nc3ccc(C(=O)N4CCCNCC4)c(Cl)c3)n1)c5cn[nH]c5 |
| 29 | Clc1cc(Nc2nc(cn3c(cnc23)c4cn[nH]c4)C5CC5)ccc1C(=O)N6C[C@H] |
| 7CC[C@@H]6CN7 | |
| 32 | CN1CC(CN(C)C1=O)c2ccc(NC(=O)c3nc(c[nH]3)CN) |
| c(c2)C4=CCCCC4 | |
| 36 | Cc1cn2c(cnc2c(Nc3ccc(C(=O)N4CCNCC4)c(Cl)c3)n1)c5cn[nH]c5 |
| 37 | Cc1cn2c(cnc2c(Nc3ccc(C(=O)N4CCNC5(CC5)C4)c(Cl)c3)n1) |
| c6cn[nH]c6 | |
| 39 | CN1CC(CN(C)C1=O)c2ccc(NC(=O)c3nc(c[nH]3)CN) |
| c(c2)C4=CCCCCC4 | |
| 40 | Cc1cc(CNc2ncc(Br)c(Nc3cc([nH]n3)C4CC4)n2)on1 |
| 44 | Cn1ncc(NC(=O)c2nc(sc2N)c3c(F)cccc3F)c1N4CCCN(CC4) |
| C5CNC5 | |
| 47 | CC(C)c1cn2c(cnc2c(Nc3ccc(C(=O)N4CCNCC4)c(Cl)c3)n1) |
| c5cn[nH]c5 | |
| 51 | Clc1cc(Nc2nc(cn3c(cnc23)c4cn[nH]c4)C5CC5)ccc1C(=O) |
| N6CCNCC6 | |
| 54 | Cc1cn2c(cnc2c(Nc3ccc(cc3F)C(=O)N4CCNCC4)n1)c5cn[nH]c5 |
| 55 | Cc1cn2c(cnc2c(Nc3ccc(C(=O)N4CCN(CCO)CC4)c(Cl)c3)n1) |
| c5cn[nH]c5 | |
| 56 | CN1CC(CN(C)C1=O)c2ccc(NC(=O)c3nc(c[nH]3)CN)c(c2) |
| C4=CCC(C)(C)CC4 | |
| 63 | C[C@H](Nc1nc(nc2c1cc(C(=O)NCCN(C)C)n2C)n3cnc4ccncc34) |
| c5ccccc5 | |
| 64 | Cc1cc2c(Nc3ccc4nc(N)sc4c3)c(cnc2cc1OCCCN5CCNCC5)CN |
| 67 | Cc1cn2c(cnc2c(Nc3ccc(C(=O)N4CCNCC45CC5)c(Cl)c3)n1)c6cn |
| [nH]c6 | |
| 68 | O=C(Nc1ccc(cc1C2=CCCCC2)C3CCN(CCCN)CC3)c4nc(c[nH]4) |
| CN | |
| 70 | CC1(C)CNCCN1C(=O)c2ccc(Nc3nc(cn4c(cnc34)c5cn[nH]c5) |
| C6CC6)cc2Cl | |
| 71 | CCN1CCC(C1)N=CC(C=N)c2ccn3c(cnc3c2)c4cccc |
| (NC(=O)NCC(F)(F)F)c4 | |
| 74 | COc1ccc(CCNC(=O)c2cc3C(=O)N4C=CC=C(C)C4=Nc3s2)cc1OC |
| 75 | CC(C)c1nc2c(Nc3ccc(C(=O)N4CCNCC4)c(Cl)c3)nc(C) |
| cn2c1c5cn[nH]c5 | |
| 79 | Clc1cc(Nc2nc(cn3c(cnc23)c4cn[nH]c4)C5CC5)ccc1C(=O) |
| N6CCNC7(CC7)C6 | |
| 88 | Clc1cc(Nc2nc(cn3c(cnc23)c4cn[nH]c4)C5CC5)ccc1C(=O) |
| N6CCNCC67CC7 | |
| 90 | Cc1cn2c(cnc2c(Nc3ccc(C(=O)N4CCNCC4)c(c3)C5CC5)n1)c6cn[nH]c6 |
| 91 | Cc1cn2c(cnc2c(Nc3ccc(cc3F)C(=O)N4CCNCC4(C)C)n1)c5cn[nH]c5 |
| 93 | Fc1cc(ccc1Nc2nc(cn3c(cnc23)c4cn[nH]c4)C5CC5)C(=O)N6CCNCC6 |
| 95 | Cc1cn2c(cnc2c(Nc3ccc(C(=O)N4CCNCC4(C)C)c(Cl)c3)n1)c5cn[nH]c5 |
| 96 | CC(C)(N)CC(=O)N1CCC(CC1)c2ccc(NC(=O)c3nc(c[nH]3)CN) |
| c(c2)C4=CCCCC4 | |
| 99 | CN1CC(CN(C)S1(=O)=O)c2ccc(NC(=O)c3nc(c[nH]3)CN) |
| c(c2)C4=CCCCC4 | |
| 100 | Fc1ccc(NC(=O)C2=C(CCC2)c3nc(Nc4cc([nH]n4)C5CC5) |
| c6cccn6n3)cn1 |
We present the 2D structures of first four of those ligands in Fig. 7. We used the chimera software [32] to visualize the PDB file of the S-protein. The PDB structure was downloaded from the site [33]. The PDB structure contained a trimeric complex formed by combining 3 S-protein monomeric units. These 3 monomeric units were represented as chain A, chain B, chain C in chimera. We removed chains B and C and also ligands, water molecules and ions, thus getting only chain A, that is, a single monomeric S-protein unit. The visualization image is shown in Fig. 8.
Fig. 7.

2D diagram corresponding to the 3D structure of the ligands, 16, 20, 54, 75 and 99, given in Table 2
Fig. 8.

Visualization of the PDB file representing chain A of S-glycoprotein of SARS-COV-2
At this stage, druggability test of all the predicted ligands is an essential task to validate the results. We calculated physicochemical property parameters of the ligands, such as partition coefficient (log p), molecular weight (MW), number of hydrogen bond donors (HBDs), hydrogen bond acceptors (HBAs) and rotatable bonds (Rot B) by following Lipinski’s rule of 5 [34] and compared the parameters with the rules. Lipinski’s rule of five is a thumb rule for druggability test of a determinate molecule. In the drug discovery setting, the rule of 5 predicts that poor absorption or permeation is more likely when there are more than 5 H-bond donors, 10 H-bond acceptors, the molecular weight is greater than 500, and the calculated Log P (CLog P) is greater than 5 [35]. We performed a check for each parameters using RDKit [26] and validated the proposed ligands which satisfy the rule of 5. Thirty-three proposed ligands have molecular weight less than 500, less than 5 H-bond donors, less than 10 H-bond acceptors, number of rotatable bonds less than 10 and CLog P less than 5. But, few ligands out of total 50 partially satisfy the rule of five, i.e., having less than 5 H-bond donors, less than 10 H-bond acceptors, number of rotatable bonds less than 10 but molecular weight and CLog P value are out of range. So, we have not considered those ligands as best performers in this work. The detailed report of first few predicted ligands is given in Table 3.
Table 3.
Druggability test of few predicted ligands taken from Table 2
| Serial no. | SMILES code | Molecular weight | No. of hydrogen bond donors | No. of hydrogen bond acceptors | No. of rotatable bonds | Partition coefficient | No of rules satisfied |
|---|---|---|---|---|---|---|---|
| 1 | 16 | 458.538 | 3 | 7 | 4 | 3.8727 | 5 |
| 2 | 17 | 462.945 | 3 | 7 | 2 | 3.4012 | 5 |
| 3 | 19 | 422.287 | 3 | 8 | 4 | 3.1933 | 5 |
| 4 | 20 | 450.934 | 3 | 7 | 2 | 3.26032 | 5 |
| 5 | 29 | 488.983 | 2 | 6 | 5 | 3.97 | 5 |
| 6 | 32 | 418.501 | 2 | 8 | 3 | 3.572 | 5 |
| 7 | 36 | 436.907 | 3 | 7 | 2 | 2.8702 | 5 |
| 8 | 37 | 462.945 | 3 | 7 | 2 | 3.4028 | 5 |
| 9 | 39 | 432.528 | 2 | 4 | 3 | 3.962 | 5 |
| 10 | 40 | 390.245 | 3 | 7 | 3 | 3.492 | 5 |
| 11 | 44 | 488.568 | 3 | 9 | 3 | 2.1402 | 5 |
| 12 | 47 | 464.961 | 3 | 7 | 2 | 3.6852 | 5 |
| 13 | 51 | 462.945 | 3 | 7 | 2 | 3.4392 | 5 |
| 14 | 54 | 420.452 | 3 | 7 | 2 | 2.3559 | 5 |
| 15 | 55 | 480.96 | 3 | 8 | 2 | 2.5749 | 5 |
| 16 | 56 | 446.555 | 2 | 4 | 3 | 4.20798 | 5 |
| 17 | 63 | 483.58 | 2 | 9 | 5 | 3.1667 | 5 |
| 18 | 64 | 473.606 | 3 | 9 | 5 | 3.4028 | 5 |
| 19 | 67 | 462.945 | 3 | 7 | 2 | 3.4028 | 5 |
| 20 | 68 | 428.54 | 2 | 5 | 3 | 4.584 | 5 |
| 21 | 70 | 490.99 | 3 | 7 | 2 | 4.2178 | 5 |
| 22 | 71 | 499.541 | 3 | 6 | 5 | 4.583 | 5 |
| 23 | 74 | 423.494 | 1 | 7 | 5 | 3.2073 | 5 |
| 24 | 75 | 478.988 | 3 | 7 | 2 | 3.993 | 5 |
| 25 | 79 | 488.983 | 3 | 7 | 2 | 3.971 | 5 |
| 26 | 88 | 488.983 | 3 | 7 | 2 | 3.9718 | 5 |
| 27 | 90 | 442.527 | 3 | 7 | 2 | 3.0942 | 5 |
| 28 | 91 | 462.945 | 3 | 7 | 2 | 2.92498 | 5 |
| 29 | 93 | 446.49 | 3 | 7 | 2 | 2.9249 | 5 |
| 30 | 95 | 464.961 | 3 | 7 | 2 | 3.6488 | 5 |
| 31 | 96 | 474.609 | 3 | 5 | 3 | 4.324 | 5 |
| 32 | 99 | 454.556 | 2 | 5 | 3 | 2.696 | 5 |
| 33 | 100 | 444.474 | 3 | 7 | 3 | 4.1837 | 5 |
Comparison Analysis
We compared our architecture to a few renowned architectures ever researched upon. The RMSE metric was chosen since it is a standard evaluation metric for regression problems, which is the case for our model. A state-of-the-art architecture was proposed in the PADME research paper [28] which adopts Molecular Graph Convolutions (MGC) which is more flexible than ECFP. This architecture achieved a RMSE of 0.79 and was one of the finest DTI architectures in action. Yet another revolutionary architecture was incorporated in the KronRLS-MKL [36] paper, where a multiple kernel learning algorithm was enforced to find the drug–target interaction which achieved a standard 0.6566 RMSE score. Yet another famous architecture is SimBoost [37] which achieved RMSE score of 0.4711. We present a table showing the summary of the compared architecture along with ours on the KIBA Dataset (Table 4). The comparison graph of the models for the KIBA Dataset is given in Fig. 9.
Table 4.
Comparison analysis
| Architectures | RMSE Values |
|---|---|
| PADME-ECFP | 0.7915 |
| KronRLS | 0.6566 |
| SimBoost | 0.4711 |
| Our Architecture | 0.83 |
Fig. 9.
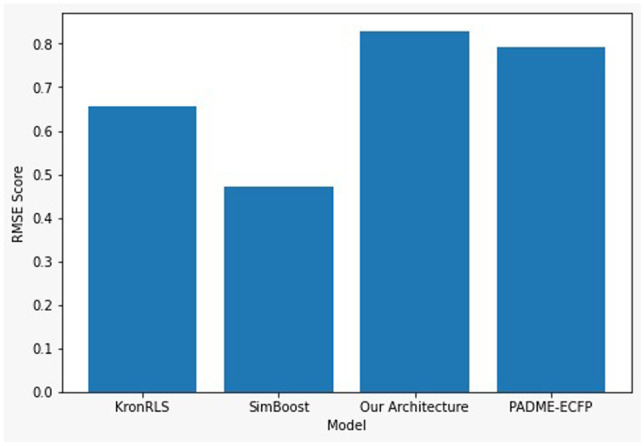
Comparative analysis based on RMSE score
Conclusion
According to various reports placed by the United Nations, at present we are facing such a health crisis that has not been witnessed on Earth in the last 75 years. The spread of COVID-19 is not only causing health crisis but affecting the society and economy of various nations giving them a tremendous blow. Due to its high virulence and ability to contaminate at a very fast rate, the virus is spreading in the community. The symptoms are that of the common cold, and therefore, many times a patient is not even able to realize that he/she has been affected. And by the time realization dawns, more people have been contaminated. This virus is proving to be fatal for old people with low immunity systems. To stop this chain, governments have administered lockdown situations where day-to-day life’s proceedings have come to a standstill. This has especially affected the poor and daily wage employees who earn their bread from everyday income. Homeless people, migrant workers or people stuck in places away from their homeland are facing even more trouble trying to connect to their families and provide them with necessities. Different projects have halted as people are not able to travel to their workplace and not everything can be resolved over the Internet. With the economy taking a backseat, job seekers are believed to face an even tougher situation in coming days. Situations are getting worse with every passing day, and only stopping the spread of this virus or finding a cure for the same can revive our societal condition. Therefore, this research is of utmost importance where the proposed new compound, if validated by biochemists as an effective solution, can help mankind survive this tough time. In this work, we have trained a machine learning model for the prediction of KIBA scores for a pair of protein–ligand. Using that model, we have identified the top 33 ligands which can be used to find a potential cure for SARS-CoV-2. We are very much thankful to Mr. Prasenjit Paria, SRF, CIFRI Lab, Barrackpore, India, for validation of our results with two standard software [38, 39] and other measures. In future, we will try to find the available druggable pockets of the S-glycoprotein of SARS-CoV-2 and also further validate our results with the help of docking.
Author Contributions
This work was carried out in close collaboration between all coauthors. The first author defined the research theme and contributed in early design of the system. Further, the idea was implemented and refined by other system developers. Corresponding author wrote the full paper. All authors have contributed in the work, seen and approved the final manuscript. We are very much thankful to Mr. Prasenjit Paria, SRF, CIFRI Lab, Barrackpore, India, for validation of our results with two standard software and other measures.
Compliance with Ethical Standards
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ethical Approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed Consent
Informed consent was obtained from all individual participants included in the study.
References
- 1.Shinde GR, Kalamkar AB, Mahalle PN, Dey N, Chaki J, Hassanien AE. Forecasting Models for Coronavirus Disease (COVID-19): A Survey of the State-of-the-Art. SN Computer Science. 2020;1(4):1–15. doi: 10.1007/s42979-020-00209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fong SJ, Li G, Dey N, Crespo RG, Herrera-Viedma E. Finding an accurate early forecasting model from small dataset: A case of 2019-ncov novel coronavirus outbreak. arXiv preprint 2020. arXiv:200310776
- 3.of India G. #IndiaFightsCorona COVID-19. MyGov.in; 2020. Available from: https://mygov.in/covid-19/.
- 4.Bamford C. Coronavirus treatments: what drugs might work against COVID-19? The Conversation; 2020. Available from: http://theconversation.com/coronavirus-treatments-what-drugs-might-work-against-covid-19-135352.
- 5.Allam Z, Jones DS. On the coronavirus (COVID-19) outbreak and the smart city network: universal data sharing standards coupled with artificial intelligence (AI) to benefit urban health monitoring and management. In: Healthcare. vol. 8. Multidisciplinary Digital Publishing Institute; 2020. p. 46. [DOI] [PMC free article] [PubMed]
- 6.WHO. Solidarity clinical trial for COVID-19 treatments; 2020. Library Catalog: www.who.int. Available from: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/global-research-on-novel-coronavirus-2019-ncov/solidarity-clinical-trial-for-covid-19-treatments.
- 7.Fong SJ, Li G, Dey N, Crespo RG, Herrera-Viedma E. Composite Monte Carlo decision making under high uncertainty of novel coronavirus epidemic using hybridized deep learning and fuzzy rule induction. Applied Soft Computing. 2020. p. 106282. [DOI] [PMC free article] [PubMed]
- 8.Chakrabortya HJ, Paria P, Gangopadhyay A, Ganguli S. Drug Repurposing against SARS-CoV-2 RDRP-a computational quest against CoVID-19. Research Square. 2020. p. 1–19.
- 9.Dai W, Zhang B, Jiang XM, Su H, Li J, Zhao Y, et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. 2020;368(6497):1331–1335. doi: 10.1126/science.abb4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhou Y, Hou Y, Shen J, Huang Y, Martin W, Cheng F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell discovery. 2020;6(1):1–18. doi: 10.1038/s41421-020-0153-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.FDA-CDER. FDA cautions against use of hydroxychloroquine or chloroquine for COVID-19 outside of the hospital setting or a clinical trial due to risk of heart rhythm problems. FDA; 2020. Available from: https://www.fda.gov/drugs/drug-safety-and-availability/fda-cautions-against-use-hydroxychloroquine-or-chloroquine-covid-19-outside-hospital-setting-or.
- 12.Jing Y, Bian Y, Hu Z, Wang L, Xie XQS. Deep learning for drug design: an artificial intelligence paradigm for drug discovery in the big data era. The AAPS journal. 2018;20(3):58. doi: 10.1208/s12248-018-0210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Borrell B, 2020, Pm. New York clinical trial quietly tests heartburn remedy against coronavirus. Science | AAAS; 2020. Available from: https://www.sciencemag.org/news/2020/04/new-york-clinical-trial-quietly-tests-heartburn-remedy-against-coronavirus.
- 14.Sampangi-Ramaiah MH, Vishwakarma R, Shaanker RU. Molecular docking analysis of selected natural products from plants for inhibition of SARS-CoV-2 main protease. Current Science. 2020;118(7):1087–1092. [Google Scholar]
- 15.Ragoza M, Hochuli J, Idrobo E, Sunseri J, Koes DR. Protein-ligand scoring with convolutional neural networks. Journal of chemical information and modeling. 2017;57(4):942–957. doi: 10.1021/acs.jcim.6b00740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Saravanan KM, Yang Y, Hossain MT. Deep learning based drug screening for novel coronavirus 2019-nCov. Interdisciplinary Sciences: Computational Life Sciences; 2020. p. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.https://www.targetmol.com/library-collection-2/Special-Bioactive-Compound-Libraries1https://www.chemdiv.com/ Accessed 28 Sep 2020.
- 18.https://www.targetmol.com/compound-library/Approved-Drugs-Library. Accessed 28 Sep 2020.
- 19.https://targetmol.com/library-collection-2/Natural-Compound-Library-for-HTS. Accessed 28 Sep 2020.
- 20.https://www.targetmol.com/library-collection-2/Special-Bioactive-Compound-Libraries. Accessed 28 Sep 2020.
- 21.Weininger D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. Journal of chemical information and computer sciences. 1988;28(1):31–36.
- 22.Tang J, Szwajda A, Shakyawar S, Xu T, Hintsanen P, Wennerberg K, et al. Making sense of large-scale kinase inhibitor bioactivity data sets: a comparative and integrative analysis. Journal of Chemical Information and Modeling. 2014;54(3):735–743. doi: 10.1021/ci400709d. [DOI] [PubMed] [Google Scholar]
- 23.Cao DS, Xu QS, Liang YZ. propy: a tool to generate various modes of Chou PseAAC. Bioinformatics. 2013;29(7):960–2. [DOI] [PubMed]
- 24.Lee I, Keum J, Nam H. DeepConv-DTI: Prediction of drug-target interactions via deep learning with convolution on protein sequences. PLoS computational biology. 2019;15(6):e1007129. doi: 10.1371/journal.pcbi.1007129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rogers D, Hahn M. Extended-connectivity fingerprints. Journal of chemical information and modeling. 2010;50(5):742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 26.Landrum G, Tosco P, Kelley B, sriniker, gedeck, Vianello R, et al.. rdkit/rdkit: 2020\_03\_4 (Q1 2020) Release. Zenodo; 2020. Available from: https://zenodo.org/record/3929204#.XxKQ9p5KjD4.
- 27.Duvenaud DK, Maclaurin D, Iparraguirre J, Bombarell R, Hirzel T, Aspuru-Guzik A, et al. Convolutional networks on graphs for learning molecular fingerprints. In: Advances in neural information processing systems; 2015. p. 2224–2232.
- 28.Feng Q, Dueva E, Cherkasov A, Ester M. Padme: A deep learning-based framework for drug-target interaction prediction. arXiv preprint 2018. arXiv:180709741 p. 1–29.
- 29.Keras. Keras: the Python deep learning API; 2020. Available from: https://keras.io/.
- 30.Öztürk H, Ozkirimli E, Özgür A. A novel methodology on distributed representations of proteins using their interacting ligands. Bioinformatics. 2018;34(13):i295–i303. doi: 10.1093/bioinformatics/bty287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.SMILES Vec Protein Representation;.
- 32.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera a visualization system for exploratory research and analysis. Journal of computational chemistry. 2004;25(13):1605–12. [DOI] [PubMed]
- 33.Chimera U. Download UCSF Chimera; 2019. Available from: https://www.cgl.ucsf.edu/chimera/download.html.
- 34.Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Advanced drug delivery reviews. 1997;23(1–3):3–25. doi: 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- 35.Benet LZ, Hosey CM, Ursu O, Oprea TI. BDDCS, the rule of 5 and drugability. Advanced drug delivery reviews. 2016;101:89–98. doi: 10.1016/j.addr.2016.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nascimento AC, Prudêncio RB, Costa IG. A multiple kernel learning algorithm for drug-target interaction prediction. BMC bioinformatics. 2016;17(1):46. doi: 10.1186/s12859-016-0890-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He T, Heidemeyer M, Ban F, Cherkasov A, Ester M. SimBoost: a read-across approach for predicting drug-target binding affinities using gradient boosting machines. Journal of cheminformatics. 2017;9(1):1–14. doi: 10.1186/s13321-017-0209-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Virtual Combinatorial Library of cheminfo.org. Available online: http://www.cheminfo.org/Chemistry/Cheminformatics/Virtual-combinatorial-library/index.html. Accessed 8 Dec 2020.
- 39.Williams AJ. Public chemical compound databases. Current Opinion in Drug Discovery and Development. 2008 May 1;11(3):393. Accessed 8 Dec 2020. [PubMed]