

Abstract
Genetic sequencing has become a critical part of the diagnosis of certain forms of pancreatic beta cell dysfunction. Despite great advances in the speed and cost of DNA sequencing, determining the pathogenicity of variants remains a challenge, and requires sharing of sequence and phenotypic data between laboratories. We reviewed all diabetes and hyperinsulinism-associated molecular testing done at the Seattle Children’s Molecular Genetics Laboratory from 2009 to 2013. 331 probands were referred to us for molecular genetic sequencing for Neonatal Diabetes (NDM), Maturity-Onset Diabetes of the Young (MODY), or Congenital Hyperinsulinism (CHI) during this period. Reportable variants were identified in 115 (35%) patients with 91 variants in one of 6 genes: HNF1A, GCK, HNF4A, ABCC8, KCNJ11, or INS. In addition to identifying 23 novel variants, we identified unusual mechanisms of inheritance, including mosaic and digenic MODY presentations. Re-analysis of all reported variants using more recently available databases led to a change in variant interpretation from the original report in 30% of cases. These results represent a resource for molecular testing of monogenic forms of diabetes and hyperinsulinism, providing a mutation spectrum for these disorders in a large North American cohort. In addition, they highlight the importance of periodic review of molecular testing results.
Keywords: MODY, Hyperinsulinism, Neonatal diabetes, Mosaicism, Variant classification, Mutation spectrum
1. Introduction
Genetic sequencing is a critical part of the diagnosis of certain forms of pancreatic beta cell dysfunction, including Maturity Onset Diabetes of the Young (MODY), Neonatal Diabetes (NDM), and Congenital Hyperinsulinism (CHI). MODY is the most common form of monogenic diabetes, accounting for 2–5% of all patients with diabetes [1]. Initially defined by the presence of a positive family history, absence of insulin dependence, and early onset of symptoms (<25 years), the diagnosis of MODY is now defined by molecular genetic testing [2]. Mutations in 11 genes have now been described as causing MODY, although mutations in just three (HNF1A, GCK, HNF4A) account for over 70% of patients [3]. Only 5–20% of patients with MODY are correctly diagnosed [4]. Because patients with MODY are frequently young and non-obese, they are often misdiagnosed as having type 1 diabetes and treated with insulin [2,5]. Misdiagnosis with type 2 diabetes is possible as well [2,5]. Since the majority of MODY patients can be managed with oral hypoglycemic agents or diet alone, accurate molecular diagnosis of these patients is critical [2,6,7].
NDM is a disorder of pancreatic beta cell function characterized by hyperglycemia within the first 6 months of life. There are a variety of syndromic forms, which are categorized by the presence of other abnormalities, but most patients with NDM are non-syndromic and are classified as having either transient or permanent NDM. Transient NDM patients typically have hyperglycemia that resolves by 18 months;~70% of these patients have an imprinting abnormality at 6q24 [5]. The hyperglycemia in patients with permanent NDM typically does not resolve, and mutations in 4 different genes (KCNJ11, ABCC8, INS, GCK) account for ~70% of patients [8–11].
CHI is characterized by hypoglycemia with inappropriately elevated insulin. CHI can present in the neonatal period with severe hypoglycemia, although milder, childhood-onset presentations occur as well. Mutations in eight different genes, all of which function in the regulation of insulin secretion, have now been described as causing CHI, although mutations in just three (ABCC8, KCNJ11, GCK) account for ~50% of patients [12].
Here we report the results of diagnostic testing for pancreatic beta cell disorders at the Seattle Children’s Molecular Genetics Laboratory (SCHMGL) over a 4-year period. SCHMGL was the first laboratory in North America to offer such testing. Reportable variants were identified in 115 of 331 (35%) probands referred to us. A total of 91 variants were identified in 6 genes sequenced (HNF1A, GCK, HNF4A, ABCC8, KCNJ11, or INS), 23 of which are novel and not previously reported. We provide evidence for digenic and mosaic MODY presentations. Re-analysis of all variants using the latest available databases and literature altered variant interpretation in about 30% of cases. These results expand the spectrum of known variants associated with MODY, NDM, and CHI, and provide a resource for variant interpretation based on the largest North American cohort reported to date. In addition, they highlight the importance of periodic review of molecular testing results, in particular of variants of uncertain significance.
2. Research design and methods
2.1. Study design
The SCHMGL is a reference diagnostic laboratory that has been performing molecular diagnostic testing for MODY, NDM, and CHI since September 2009. Referrals come from physicians and other laboratories within North America. After approval by the Seattle Children’s Hospital Institutional Review Board, all molecular test results sent to SCHMGL for MODY, NDM, and CHI testing from 9/2009 to 10/2013 were reviewed. A total of 331 probands were referred for testing during this period. Reasons for referral included hyperinsulinism, neonatal diabetes or diabetes suspected to have a monogenic cause. Although efforts were always made to collect clinical indication for testing in some cases the indication was not given. Information that was reviewed included reason for referral, results of molecular genetic testing, and the interpretation (benign, variant of uncertain significance, or pathogenic) at the time the report was made. In addition, all variants were re-reviewed and classified according to a 5-tier system (see below).
2.2. Molecular genetic analysis
Genomic DNA was extracted from leukocytes using standard procedures. Coding exons were amplified by multiplex polymerase chain reaction followed by Sanger sequencing. The promoter regions of HNF1A, HNF4A, and INS were also included as known mutations have been reported in these areas. Primer sequences are available on request. Genes were sequenced in a serial manner, starting with the gene with the highest yield first and continuing to other genes if a pathogenic variant was not identified, according to consensus guidelines [2]. If a pathogenic variant was identified, additional genes were not sequenced. For MODY sequencing requests, the order of gene sequencing was HNF1A>GCK>HNF4A. For NDM sequencing requests, the order was KCNJ11>INS>ABCC8>GCK, and for CHI sequencing requests, the order was ABCC8>KCNJ11>GCK. Although HNF4A has been reported as a cause of CHI, this gene was not sequenced among our CHI patients. In a minority of patients, the ordering provider requested a single gene test. Many of these cases were from French Canada, where the incidence of GCK-related MODY is higher [13].
Low-level mosaicism for the GCK p.T209M was detected using restriction fragment length polymorphism analysis. This mutation(c.626C>T) introduces an NlaIII restriction site (5′-CATG-3′), producing a smaller 494 bp fragment in the mutant allele, versus the 539 bp fragment in the wild-type allele. PCR and restriction digests were performed with standard methods, and fragments were separated using an Agilent BioAnalyzer (Fig. 1).
Fig. 1.
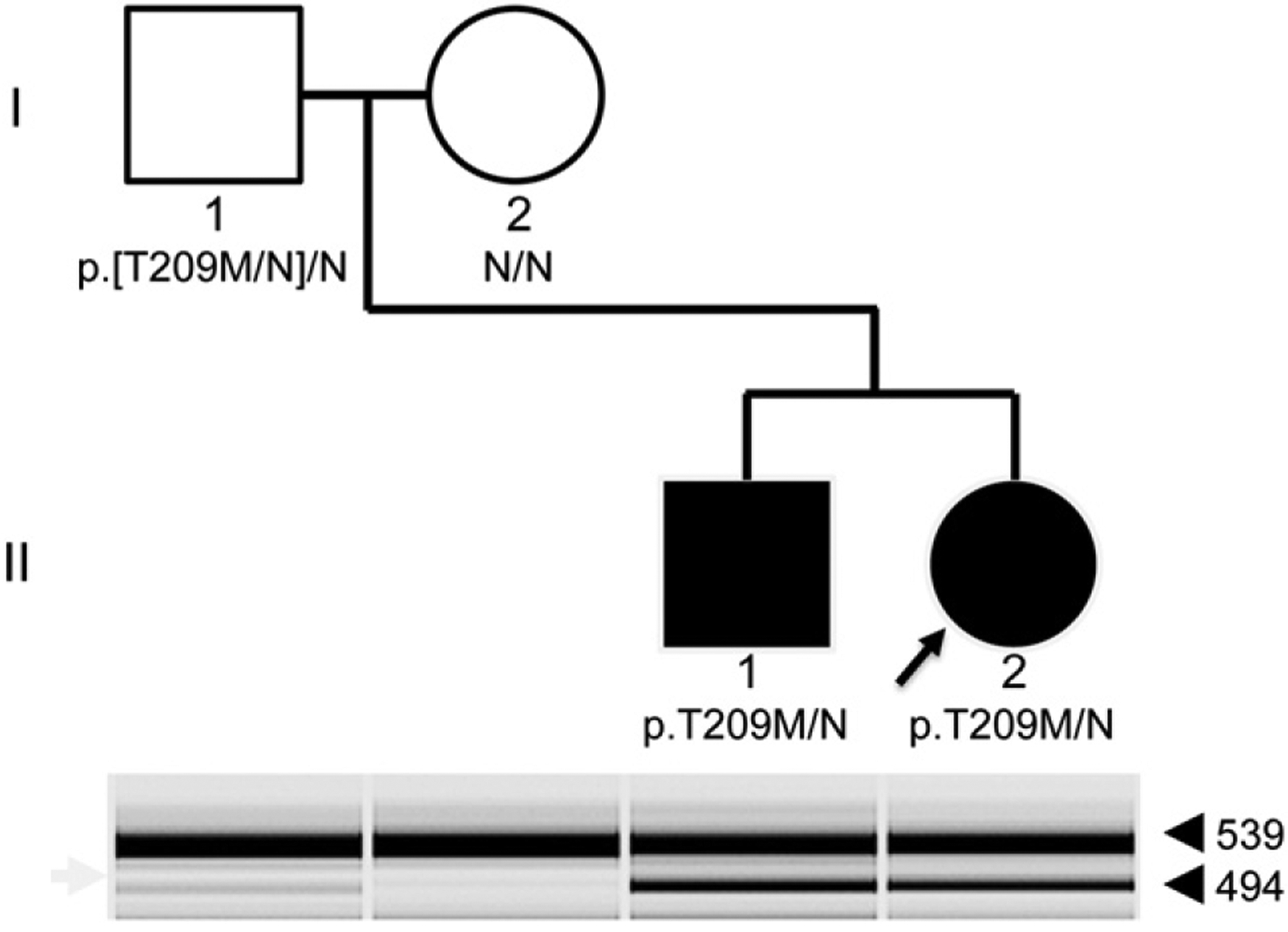
Recurrence of GCK-related MODY due to mosaicism. The pedigree of the family is shown. Genotypes based on Sanger sequencing shown below each individual, with N representing reference. Filled-in symbols represent individuals with MODY. Restriction-fragment based genotypes are shown in the gel image below the pedigree. The mutant allele is represented by the lower, 494 bp band (arrowhead), while the reference allele is represented by the upper, 539 bp band. The faint presence of the mutant band from blood-derived DNA from the father indicates evidence of mosaicism for this mutation.
Sequence variations were searched in PubMed (http://www.ncbi.nlm.nih.gov/pubmed) and in the following public databases. HGMD (www.hgmd.cf.ac.uk/ac/index.php); LOVD (www.grenada.lumc.nl/LOVD2/diabetes/); dbSNP (www.ncbi.nlm.nih.gov/projects/SNP/); ClinVar (www.ncbi.nlm.nih.gov/clinvar/); Exome Variant Server (www.evs.gs.washington.edu/EVS/), and GenBank accession numbers used are ABCC8 (NM_000352.3), KCNJ11 (NM_000525.3), INS (NM_000207.2), HNF1A (NM_000545.5), GCK (NM_000162.3), and HNF4A (NM_175914.4).
2.3. Variant interpretation
At the end of the review period, variants were re-classified into 5 tiers [14]: pathogenic, likely pathogenic (LP), variant of uncertain significance (VUS), likely benign (LB), or benign, using the following rules:
Variants with an allele frequency (AF) > 0.1% were classified as benign.
Synonymous variants that had not been previously reported that had no evidence of affecting splicing were classified as benign.
Missense and truncating variants with AF ≤ 0.1% that were previously reported in 1 or more unrelated individuals as being pathogenic were classified as pathogenic.
Novel (not previously reported in the literature or online databases) variants with AF ≤ 0.1% that are predicted to lead to nonsense mediated decay (NMD) were classified as pathogenic. NMD mutations are defined as any variant resulting in a stop-codon greater than 50 bases upstream from the 3′ end of the penultimate exon and include nonsense, frameshift, and canonical splice donor (GT) and acceptor (AG) variants that shift reading frame.
Novel missense variants with AF ≤ 0.1% that were demonstrated to be de novo were classified as pathogenic.
Novel missense variants with AF ≤ 0.1% involving an amino acid previously reported as pathogenic but to a different amino acid were classified as LP.
Novel missense variants with AF ≤ 0.1% that did not involve previously reported amino acid were classified as VUS. If additional information was available, this class of variants could be shifted up to LP or down to LB. For example, if family studies demonstrated segregation of the variant with a phenotype, the variant would be classified as LP. If there was evidence of a lack of segregation, or an additional variant was identified that had better evidence for pathogenicity, the variant would be classified as LB.
Novel intronic variants with AF ≤ 0.1% were evaluated using Human Splice Finder (www.umd.be/HSF/), an in silico splice prediction tool [16]. In no cases did HSF provide sufficient support for reporting out a variant as pathogenic. Previously reported intronic and promoter variants with AF ≤ 0.1% were evaluated on a case-by-case basis.
3. Results
3.1. Molecular genetic testing
A total of 331 probands were referred to the SCHMGL for molecular genetic sequencing of HNF1A, GCK, HNF4A, ABCC8, KCNJ11, or INS during the 4-year review period. For 184 probands (55%) the indication for diagnostic testing was some form of hyperglycemia; for 30 (9%) probands the indication was hypoglycemia/hyperinsulinism, and for 117 probands (35%) no clinical information was available. Ages of the patients at the time of testing ranged from birth to 67 years; 53% of the cohort was female.
Reportable variants, which include LB, VUS, LP, and pathogenic variants, were identified in 35% of the cohort (115/331 probands tested). These 115 patients had a total of 91 unique variants among the 6 genes sequenced (Tables 1 to 3). We identified 12 ABCC8 variants (5 of which are novel), 40 GCK variants (9 novel), 18 HNF1A variants (1 novel), 9 HNF4A variants (4 novel), 10 KCNJ11 variants (3 novel), and 2 INS variants (1 novel). 65 of the 91 variants were classified as pathogenic, 8 as likely pathogenic, 14 as VUS, and 4 as likely benign. The majority (68/91) of the variants were missense, but we also identified 6 nonsense variants, 9 frameshift variants, 2 splice variants, 2 promoter variants and one whole exon deletion variant. Only 13 of the 91 variants identified were seen greater than once within our cohort, and 23 variants have not previously been reported so are true private variants. 68 variants had been previously reported in the literature or in databases and so are recurrent variants. Of the 13 variants seen more than once within our cohort, only two were seen greater than 3 times: the previously described missense variant in GCK (c.676G>A, p.V226M, seen 10 times), which is a known founder mutation among French Canadians [13], and the previously described frameshift variant in HNF1A (c.872insC, p.G292Rfs*25, seen 8 times). In two patients, both with NDM, we were able to demonstrate that the variant occurred de novo: a previously reported [17] pathogenic missense variant in KCNJ11 (c.601C>T, p.R201C), and a previously reported [10] pathogenic missense variant in INS (c.265C>T, p.R89C).
Table 1.
List of ABCC8 (NM_000352.3), KCNJ11 (NM_000525.3), and INS (NM_000207.2) variants identified.
| Gene | Nucleotide | Protein | Pheno | dbSNP | Times seen | Interp. | LOVD | ClinVar | Reference |
|---|---|---|---|---|---|---|---|---|---|
| ABCC8 | c.1277A>G/c.3545G>A | p.N426S/p.R1182Q | NDM | –/rs193922400 | 1/2 | VUS/PATH | −/− | –/LP | This report/[9] |
| c.1433C>A | p.A478D | CHI | – | 1 | PATH | – | – | [41] | |
| c.1608T>G | p.F536L | NDM | – | 1 | LP | – | – | This report | |
| c.2422C>A | p.Q808K | “DM1”* | rs202189540 | 1 | LB | – | – | This report | |
| c.2506C>T/c.4138_4140delinsCA | p.R836*/p.T1380Qfs*80 | CHI | rs72559722/– | 1/1 | PATH/PATH | −/− | −/− | [42]/this report | |
| c.3976G>A | p.E1326K | CHI | rs200563930 | 1 | VUS | – | – | [8] | |
| ex 32 del | p.? | “Hypoglycemia”* | – | 1 | PATH | – | – | [15] | |
| c.4135C>A | p.R1379S | NDM | rs137852673 | 1 | PATH | – | PATH | ClinVar | |
| c.4307G>A | p.R1436Q | n.p. | rs387906407 | 2 | PATH | PATH | PATH | [43] | |
| c.4543A>G | p.T1515A | CHI | – | 1 | LP | – | – | This report | |
| KCNJ11 | c.137A>G | p.H46R | NDM | – | 1 | PATH | – | – | [44] |
| c.143A>T | p.N48I | NDM | – | 2 | PATH | – | – | [25] | |
| c.497G>A | p.C166Y | NDM | rs80356618 | 1 | PATH | – | PATH | [45] | |
| c.601C>T | p.R201C | NDM | rs80356625 | 1 | PATH | – | PATH | [17] | |
| c.602G>A | p.R201H | NDM | rs80356624 | 1 | PATH | VUS | – | [45] | |
| c.616C>T | p.R206C | CHI | – | 1 | LP | – | – | This report | |
| c.637G>A | p.A213T | CHI | – | 1 | PATH | – | – | [46] | |
| c.685G>A | p.E229K | NDM | – | 1 | PATH | – | – | [30] | |
| c.691G>C/c.970G>A | p.V231L/p.G324R | NDM | −/− | 1/1 | VUS/VUS | −/− | −/− | This report/this report | |
| INS | c.–331delC | p.? | NDM | – | 1 | VUS | – | – | This report |
| c.265C>T | p.R89C | NDM | rs80356669 | 1 | PATH | – | PATH | [10] |
Variants identified in ABCC8, KCNJ11, and INS are shown, along with phenotype, dbSNP number, clinical interpretation, and presence in two databases (LOVD and ClinVar). “n.p.” indicates no phenotypic information provided. In two individuals (indicated by *) the clinical information provided was insufficient to determine the diagnosis, but the available clinical information is shown. All identified variants are in the heterozygous state, and in three individuals compound heterozygosity was identified and is indicated.
Table 3.
List of HNF1A (NM_000545.5) and HNF4A (NM_175914.4) variants identified.
| Gene | Nucleotide | Protein | Pheno | dbSNP | Times seen | Interp. | LOVD | ClinVar | Ref |
|---|---|---|---|---|---|---|---|---|---|
| HNF1A | c.−160_−154dupTGGGGGT | p.? | MODY | – | 2 | PATH | Benign | – | [33] |
| c.155_156delinsCT | p.G52A | MODY | – | 1 | LB | Benign | – | [61] | |
| c.160C>T | p.R54* | MODY | – | 1 | PATH | PATH | – | [62] | |
| c.472A>G | p.K158E | MODY | – | 1 | PATH | LP | – | [26] | |
| c.476G>A/c.1537A>T | p.R159Q/p.T513S | MODY | −/− | 1/1 | PATH/VUS | PATH/− | −/− | [63]/[64] | |
| c.511C>T | p.R171* | MODY | – | 1 | PATH | PATH | – | [63] | |
| c.599G>A | p.R200Q | n.p. | – | 1 | PATH | PATH | – | [65] | |
| c.768delC | p.N257Tfs*85 | MODY | – | 1 | PATH | – | – | This report | |
| c.842T>C | p.L281P | n.p. | – | 1 | PATH | – | – | [64] | |
| c.872delC | p.P291Qfs*51 | n.p. | – | 2 | PATH | VUS | – | [63] | |
| c.872dupC | p.G292Rfs*25 | MODY | – | 8 | PATH | PATH | – | [66] | |
| c.1136C>A | p.P379H | MODY | rs371717826 | 1 | PATH | – | – | [67] | |
| c.1137delT | p.V380Sfs*4 | n.p. | – | 1 | PATH | – | – | [68] | |
| c.1405C>T | p.H469Y | MODY | rs201811844 | 1 | LB | Benign | – | [69] | |
| c.1456C>T | p.Q486* | MODY | – | 1 | PATH | – | – | [70] | |
| c.1522G>A | p.E508K | MODY | – | 1 | VUS | VUS | – | [64] | |
| c.1541A>G | p.H514R | n.p. | rs202039659 | 1 | VUS | Benign | – | [71] | |
| HNF4A | c.146A>G | p.H49R | MODY | – | 1 | VUS | – | – | This report |
| c.199C>T | p.R67W | MODY | – | 1 | PATH | PATH | – | [72] | |
| c.225–25T>A | p.? | “DM1”* | rs200071662 | 1 | LB | – | – | [73] | |
| c.281_282delinsC | p.R94Tfs*10 | MODY | rs193922471 | 2 | PATH | – | LP | ClinVar only | |
| c.318_331del | p.V108Gfs*11 | MODY | – | 1 | PATH | – | – | This report | |
| c.353G>A | p.R118Q | MODY | – | 1 | VUS | – | – | This report | |
| c.426+6G>A | p.? | “diabetes”* | rs182980547 | 1 | VUS | Benign | VUS | ClinVar & LOVD | |
| c.869GNA | p.R290H | n.p. | – | 1 | PATH | PATH | – | [74] | |
| c.878_881dup | p.Q294Hfs*15 | MODY | – | 1 | PATH | – | – | This report |
Variants identified in HNF1A and HNF4A are shown, along with phenotype, dbSNP number, clinical interpretation, and presence in two databases (LOVD and ClinVar). “n.p.” indicates no phenotypic information provided. In two individuals (indicated by *) the clinical information provided was insufficient to determine the diagnosis, but the available clinical information is shown. All identified variants are in the heterozygous state, and in one individual compound heterozygosity was identified and is indicated.
We examined two public databases in wide use, the locus-specific Leiden Open Variation Database (LOVD) and ClinVar, which is a more recently available genome-wide variant database. Over half (53/91) of the variants we identified were not present in either of these databases (Tables 1–3). In contrast, the Human Gene Mutation Database (HGMD) contained 59% (54/91) of the identified variants (data not shown).
3.2. Mosaic MODY presentation
We identified one family in our cohort with evidence of germline mosaicism for a previously reported [18] pathogenic GCK variant, c.626C>T (p.T209M). The proband presented with failure to thrive (FTT) in her first year of life, and diagnostic evaluation at 7 months of age identified consistent hyperglycemia in the 100–125 mg/dL range. In addition she was found to have GAD65 autoantibodies indicating that she may have both DM1 and GCK-MODY. Finger stick testing of the proband’s asymptomatic younger brother identified hyperglycemia, again in the 100–125 mg/dL range but he was autoantibody negative. Both siblings, but neither parent, possessed the p.T209M GCK pathogenic variant, and microsatellite testing confirmed family relationships (data not shown). We hypothesized that a post-zygotic, mosaic mutation that involved the germline was present in one of the parents, and designed a restriction digest based assay to detect low-level mosaicism from blood-derived DNA. The results of this testing, shown in Fig. 1, indicate low-level mosaicism present in the children’s father. His fasting serum glucose level was normal (69 mg/dL) at the age of 30 years.
3.3. Digenic MODY presentation
We identified one family in our cohort with individuals carrying known pathogenic variants in two MODY-related genes, HNF1A and GCK, suggesting the possibility of digenic inheritance (Fig. 2). The proband is a 3-year old female with hyperglycemia (fasting blood sugar is 110 mg/dL), a hemoglobin A1C of 5.8%, and negative autoantibodies. She is currently being managed by diet alone. Her mother, currently 32 years old, had diabetes since 18 years of age that had been well controlled on a sulfonylurea, but currently requires no medication. She did require insulin during each of her three pregnancies. The results of her oral glucose tolerance tests were not available. We identified what was initially classified as a VUS in the promoter of HNF1A (c.−160_−154dupTGGGGGT) in the proband, as well as a previously reported [19] pathogenic missense variant in GCK: c.781G>A (p.G261R). The proband’s mother and an older brother (II–1) were also found to carry both pathogenic variants, and the proband’s younger sister (II–3) carried only the HNF1A variant (Fig. 2). Neither of the proband’s siblings (ages 4 and 1) has shown signs of hyperglycemia to date, though neither has had fasting glucose or hemoglobin A1C levels measured. Subsequent to our initial clinical report, this HNF1A promoter variant was published as pathogenic based on its segregation with a MODY phenotype, in two unrelated families [20]. Intriguingly, one of the families reported in this paper also carried an additional GCK mutation (c.183C>A, p.Y61*).
Fig. 2.
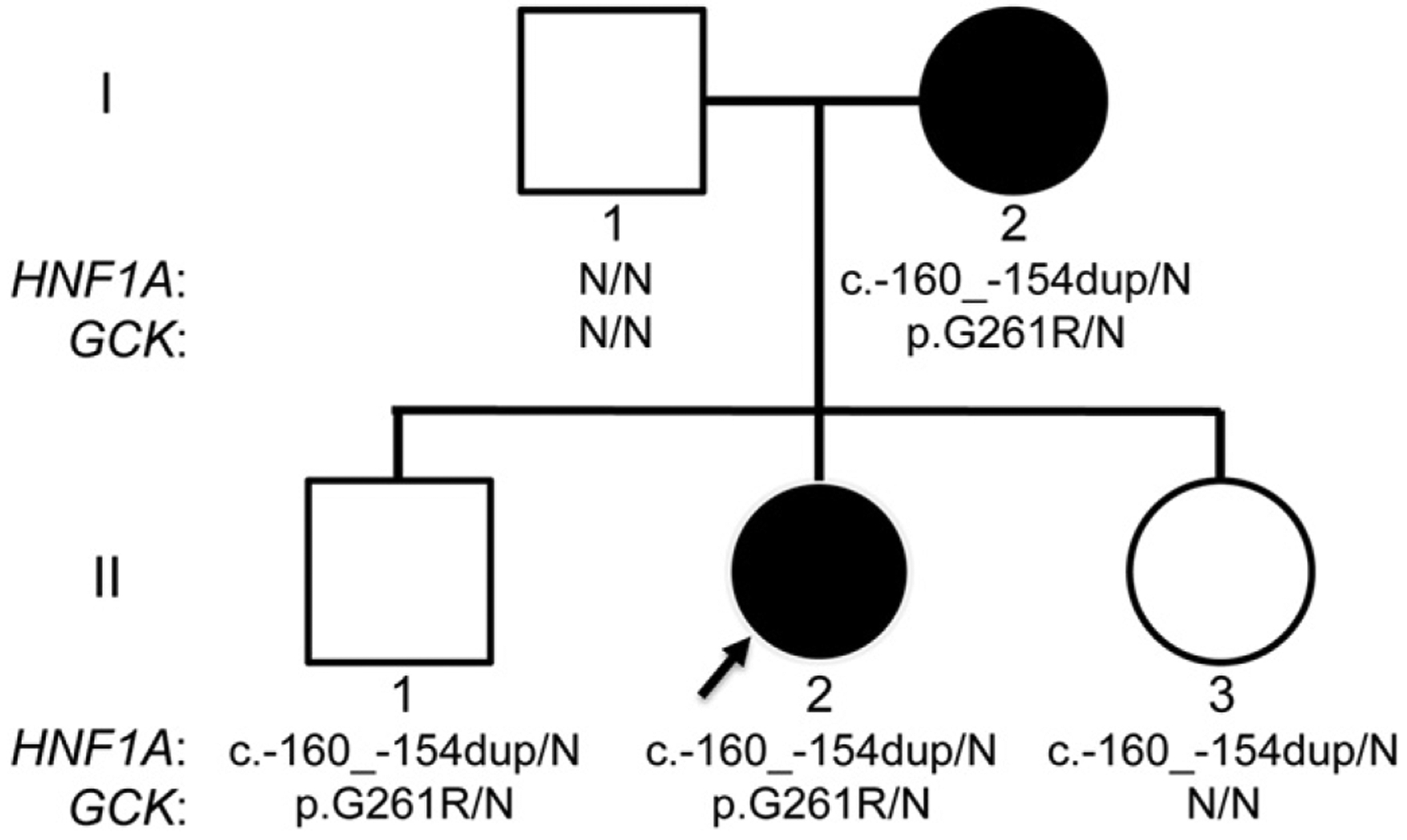
Coinheritance of HNF1A and GCK pathogenic variants. The pedigree of the family is shown. Filled-in symbols represent individuals with MODY. Results of Sanger sequencing are shown below each individual, with N representing reference.
3.4. Variant re-classification
The development of large, multicenter sequencing projects such as the NHLBI Exome sequencing project and the 1000 Genomes projects had a major impact on variant classification [21], primarily driven by a “downgrading” of variants previously believed to be pathogenic, but found to have allele frequencies (AFs) that are greater than the disease frequency. To determine how the interpretation of variants changed over the 4-year review period we re-classified all of them, using the 5-tier system described in the Research design and methods section. A total of 99 variants were identified as either VUS or pathogenic in the original reports, 30 of which underwent significant change in classification during the review period (Fig. 3), including 8 that were reclassified as benign and are no longer considered reportable. Of the 30 variants that underwent a classification change, 19 were “downgraded” from pathogenic to VUS (n = 7), VUS to likely benign (n = 4), or VUS to benign (n = 8). 11 were “upgraded” from VUS to pathogenic (n = 4) or likely pathogenic (n = 7). All variant re-classifications were the result of improved knowledge of the variant’s population frequency, or from additional literature published during the 4-year review period [22–25].
Fig. 3.
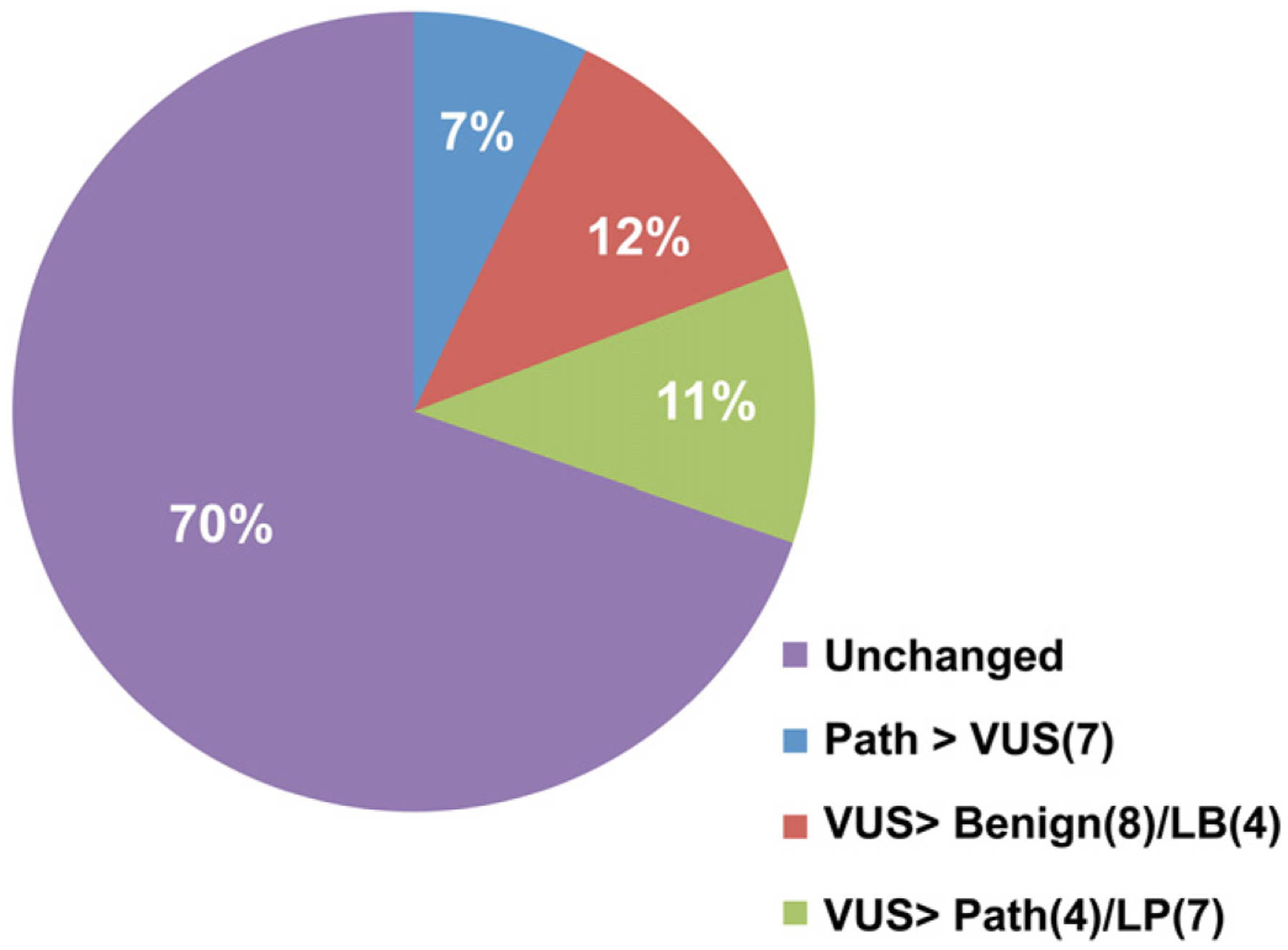
Variant re-classification. A total of 99 variants were originally reported out as either VUS or pathogenic. 30% (30/99 variants) changed classification during the 4-year review period. The types of classification changes are indicated in the legend, with number of variants within each class indicated in parentheses. “LB” is likely benign and “LP” is likely pathogenic.
4. Discussion
We present the results of molecular genetic testing for disorders of pancreatic beta cell function (MODY, NDM, and CHI) from 331 probands, the largest North American cohort of these patients reported to date. Reportable variants were identified in 35% of patients. In addition to providing a useful resource for variant classification, our results highlight some of the major hurdles facing molecular diagnostic testing, including challenges with variant interpretation, paucity and heterogeneity of variant databases, and the re-classification of variants with time.
4.1. Spectrum of mutations
A total of 91 variants were identified among 115 patients (Tables 1–3), 75% (68/91) of which have been previously reported. The most common mutation identified was c.676G>A (p.V226M) in GCK, found in 10 probands. This is a previously reported founder mutation known to be present in 0.057% of French Canadians [13], and reflects that a significant number of referrals are from Quebec. The second most common mutation was c.872insC (p.G292Rfs*25) in HNF1A, found in 8 probands. This previously reported recurrent mutation has been proposed to occur due to its presence within a highly mutable polyC tract [26]. Mechanisms for other recurrent variants are unknown at this time.
25% (23/91) of the variants were novel, despite a large number of publications related to these genes. Furthermore, only 14% (13/91) of the variants were seen in >1 patient within our cohort. This suggests that, although there is significant recurrent variation within these genes, novel variants remain unidentified. Interpretation of novel variants is, unsurprisingly, more difficult than interpretation of previously reported variants: 85% (58/68) of previously reported variants were unambiguously classified as pathogenic, versus only 30% (7/23) of novel variants. Our results highlight the importance of sharing variants between laboratories, as even over a 4-year period the majority (86%) of identified variants were seen in only one patient.
We received no phenotypic information for one-third of our cohort. The SCHMGL is a reference laboratory that receives referrals from providers and other diagnostic laboratories. Although attempts are always made to obtain phenotypic information, it is not always possible. Large research groups are able to collect more phenotypic data but most genetic testing is conducted by diagnostic laboratories that often receive only an ICD-9 code as the clinical indication. Referring clinicians need to be aware that phenotypic information is critical to variant classification.
Of the 91 variants, most (75%) were missense, though nonsense, frameshift, splice, and promoter variants were also identified. An exon 32 deletion of ABCC8 was identified in a 15 year-old patient with hypoglycemia. This variant has been previously reported in a patient with hyperinsulinism [15]. Novel frameshift and nonsense variants were identified within ABCC8, GCK, HNF1A, and HNF4A (see tables), and a novel promoter variant was identified in INS. Our results expand the spectrum of mutations associated with these genes, but there is insufficient phenotypic information for genotype-phenotype correlations.
In 65% of our cohort (216/331 probands) reportable variants were not identified. This is comparable to previous reports that did not identify mutations in 55–73% of probands referred for CHI or MODY testing [4,27]. There are several potential explanations for this. Given the absence of phenotypic information on many probands, it is possible that some do not meet clinical criteria to receive MODY, NDM, or CHI testing. The presence of undetected mutations within our genes is a possibility, as large deletions, duplications, and distant non-coding mutations would not be detected [8]. In addition there are additional genes that were not sequenced which are known to cause these phenotypes. The development of next-generation sequencing based panels that include a larger number of genes will improve the diagnostic yield of monogenic diabetes and hyperinsulinism testing.
4.2. Mosaic MODY presentation
An assumption underlying most genetic testing is that all cells in a multicellular organism have identical genomes; however, advances in DNA sequencing have made it increasingly apparent that post-zygotic, mosaic mutations are more common than is generally appreciated [28]. We identified a family with evidence of a post-zygotic, mosaic mutation in a previously reported pathogenic GCK mutation, c.626C>T (p.T209M), identified by Sanger sequencing in two siblings with a MODY phenotype but in neither parent. The same mutation was present at low-levels in blood-derived DNA from the children’s father (Fig. 1), who had no evidence of a MODY phenotype by random and fasting glucose levels. This suggests that GCK mutations may be required at a critical level within a critical tissue to manifest a MODY phenotype. Although “germline mosaicism” has frequently been used to describe this situation, this is misleading as it implies that the mutation is present only in the germline and not in somatic tissues. Although mosaicism has been previously described in families with HNF1B, INS, and KCNJ11 mutations [29–31], this is the first report of mosaicism in GCK-MODY. Post-zygotic mutations may represent an under-ascertained source of genetic variation contributing to phenotypic outcomes, although further research is required to test this hypothesis. Recognizing the possibility of “germline mosaicism” is critical for recurrence counseling, though the frequency of this genetic mechanism remains unknown.
4.3. Possible digenic MODY presentation
We identified a family with coinheritance of HNF1A c.−160_−154dupTGGGGGT and GCK c.781G>A (p.G261R). This is the 2nd report of a family with coinheritance of HNF1A and GCK pathogenic variants [20].
Evidence supporting the pathogenicity of the GCK p.G261R variant is very strong. Three unrelated families have previously been reported to possess this variant [19], and we have seen it in two additional unrelated MODY families in our cohort. Modeling of this variant using the yeast-derived glucokinase crystal structure suggests that this residue may be involved in glucose binding [19].
Several lines of evidence support the pathogenicity of the HNF1A c.−160_−154dupTGGGGGT variant. First, non-coding regulatory variants in the HNF1A promoter have been reported [32]. Second, this variant has been previously reported as pathogenic [20,33], as it was found it in three unrelated families with a MODY phenotype. In two of these families no other variant in a MODY related gene was reported; however, in one a nonsense variant in GCK (c.183C>A, p.Y61*) was identified, and the possibility that both the GCK and HNF1A variants could be contributing was asserted [20]. Third, this variant is in a highly conserved, DNAse sensitive region suggesting a role in regulating gene expression [34]. Complicating the analysis of this variant is the age-related penetrance of the MODY phenotype [35], which might explain the individuals (II–1 and II–3) who do not currently manifest evidence of hyperglycemia. Additionally the penetrance of MODY-related gene mutations may be lower than previously appreciated, as ~1.5% of euglycemic individuals from population-based cohorts were reported to possess previously reported pathogenic variants in MODY genes [36]. Coinheritance of multiple contributing loci may be more common than currently appreciated [37]. Given the shift from serial gene sequencing to more parallel, panel based approaches, it is likely that cases of possible coinheritance will be ascertained more frequently. Although more data is required to prove the pathogenicity of this HNF1A variant, our results reflect the difficulties in the interpretation of potential digenic inheritance.
4.4. Variant reclassification
Although the speed of DNA sequencing continues to accelerate, translating sequence data into clinically useful information depends on accurate variant classification. We found that over 30% of reported variants changed their classification during the 4-year study period (Fig. 3). Unsurprisingly, two-thirds of the reclassified variants (19/30) were downgraded in pathogenicity, from pathogenic to VUS (n = 7), VUS to LB (n = 4), or VUS to benign (n = 8). This is primarily driven by population-based sequencing projects, not available at the time of initial report, showing that the variants are too common in the general population to be pathogenic. However, a third (11/30) of reclassified variants were upgraded in pathogenicity. Most of these (7/11) were reclassified from VUS to LP due to publication of an individual with a variant involving the same codon. All four of the variants reclassified from VUS to pathogenic were due to publication of the same variant in an affected individual after the initial report [22,24–26].
The importance of variant reclassification remains underappreciated, particularly outside of the genetics community. Experienced clinicians may not realize that the interpretation of genetic testing can change, with implications for clinical management and counseling. The potential for confusion is even worse for patients. Our findings are not unique to the genes tested or to our diagnostic laboratory. For example, only 7.5% (18/239) of the variants reported by the Human Gene Mutation Database (HGMD) to be disease causing met rigorous criteria to remain classified as high-penetrance pathogenic variants [21]. Similarly, 66% of entries in a database of Lynch-syndrome associated variants changed classification after expert review [38]. Our results are consistent with these previous findings and highlight the importance of continued review of genetic test results, particularly when the result is a VUS. Issues directly related to variant reclassification, such as communicating reclassifications to the patient and ordering provider, have been addressed [39,40], though significant challenges remain.
A critical component of variant classification is the identification of unrelated individuals who share the variant and the phenotype. Because variants can be private or extremely rare, it is difficult or impossible for a single laboratory to ascertain all variation in a given gene. We found that, over a 4-year period, 25% of the variants we identified had never been previously reported, and 86% were seen in only one patient within our cohort. Several databases containing variant and phenotypic data have been created precisely to address this issue. In order for these databases to be useful they need to include as many variants as possible. We examined two databases: the Leiden Open Variation Database (LOVD), which is locus-specific, and the more recently developed ClinVar, which is an NIH-supported genome-wide database of variant and phenotypic data. Despite the fact that 68 of the 91 variants have been described in the literature, 58% (53/91) of the variants were not present in either database. There was no significant difference between the two, with 76 variants not present in ClinVar and 69 not present in LOVD. Although these databases are extremely useful, their utility remains limited by their incompleteness. Although concerns have been raised about the accuracy of variants listed within the Human Gene Mutation Database (HGMD) [21], it currently provides a more complete picture of the variants present within these genes, containing 60% (55/91) of the identified variants. A significant hurdle in the growth of these databases is the deposition of variants and phenotypic data by diagnostic and research laboratories. However, there can be little incentive for a laboratory to share their variants in this manner. In addition, diagnostic laboratories continue to be concerned about privacy related issues when it comes to sharing variant and phenotypic data. Efforts are being made to address these issues, which will hopefully lead to improvements in the ease and accuracy of variant interpretation for MODY, NDM, CHI, as well as other genetic diseases.
Table 2.
List of GCK (NM_000162.3) variants identified.
| Nucleotide | Protein | Pheno | dbSNP | Times seen | Interp. | LOVD | ClinVar | Reference |
|---|---|---|---|---|---|---|---|---|
| c.106C>T | p.R36W | MODY | – | 1 | PATH | PATH | – | [18] |
| c.130G>A | p.G44S | MODY | – | 1 | PATH | VUS | – | [23] |
| c.194C>T | p.T65I | NDM | – | 1 | PATH | – | – | [47] |
| c.203G>A/c.892A>G | p.G68D/p.M298V | MODY | rs373418736/− | 1/1 | VUS/PATH | −/− | −/− | [23]/[21] |
| c.241G>A | p.G81S | MODY | – | 1 | PATH | – | – | [23] |
| c.349G>C | p.G117R | MODY | – | 1 | VUS | – | – | This report |
| c.391T>C | p.S131P | MODY | rs104894010 | 1 | PATH | – | PATH | [48] |
| c.397T>C | p.F133L | MODY | – | 1 | LP | – | – | This report |
| c.431T>C/c.460G>C | p.L144P/p.V154L | MODY | −/− | 1/1 | VUS/PATH | −/− | −/− | This report/this report |
| c.533G>A | p.G178E | MODY | – | 1 | PATH | – | – | [49] |
| c.544G>A | p.V182M | MODY | – | 1 | PATH | PATH | – | [50] |
| c.556C>T | p.R186* | “Diabetes”* | rs104894006 | 1 | PATH | PATH | PATH | [51] |
| c.572G>A | p.R191Q | MODY | – | 1 | PATH | VUS | – | [52] |
| c.601G>C | p.A201P | MODY | 1 | LP | – | – | This report | |
| c.601G>T | p.A201S | MODY | – | 1 | PATH | – | – | [23] |
| c.608T>C | p.V203A | MODY | – | 2 | PATH | – | – | [50] |
| c.616A>C | p.T206P | MODY | – | 1 | PATH | – | – | [53] |
| c.626C>T | p.T209M | MODY | – | 1 | PATH | – | – | [18] |
| c.629T>C | p.M210T | MODY | rs80356654 | 1 | PATH | PATH | PATH | [54] |
| c.661G>A | p.E221K | MODY | rs193922317 | 1 | PATH | – | LP | [55] |
| c.667G>A | p.G223S | n.p. | – | 3 | PATH | – | – | [52] |
| c.676G>A | p.V226M | MODY | rs148311934 | 10 | PATH | PATH | PATH | [54] |
| c.683C>T | p.T228M | MODY | rs80356655 | 2 | PATH | – | PATH | [19] |
| c.766G>A | p.E256K | MODY | – | 1 | PATH | PATH | LP | [56] |
| c.781G>A | p.G261R | MODY | rs104894008 | 3 | PATH | – | PATH | [19] |
| c.787T>C | p.S263P | MODY | rs193922331 | 3 | PATH | – | LP | [57] |
| c.802G>A | p.E268K | MODY | 1 | PATH | – | – | [23] | |
| c.871A>T | p.K291* | n.p. | rs193922335 | 1 | PATH | – | PATH | ClinVar only |
| c.883G>T | p.G295C | MODY | – | 1 | LP | – | – | This report |
| c.891C>A | p.Y297* | n.p. | – | 1 | PATH | – | – | [58] |
| c.917T>C | p.L306P | MODY | rs193922337 | 1 | LP | – | LP | [23] |
| c.951delC | p.H317Qfs*36 | MODY | – | 1 | PATH | – | – | This report |
| c.971T>C | p.L324P | MODY | rs193922341 | 1 | PATH | PATH | LP | [59] |
| c.1007C>A | p.S336* | Diabetes | – | 1 | PATH | – | – | [23] |
| c.1113C>A | p.C371* | n.p. | – | 1 | PATH | – | – | This report |
| c.1142T>G | p.M381R | n.p. | rs193922266 | 1 | PATH | – | LP | [24] |
| c.1268T>C | p.F423S | “Fasting hyperglycemia”* | – | 1 | LP | – | – | This report |
| c.1364T>A | p.V455E | MODY | – | 1 | PATH | PATH | – | [60] |
Variants identified in GCK are shown, along with phenotype, dbSNP number, clinical interpretation, and presence in two databases (LOVD and ClinVar). “n.p.” indicates no phenotypic information provided. In two individuals (indicated by *) the clinical information provided was insufficient to determine the diagnosis, but the available clinical information is shown. All identified variants are in the heterozygous state, and in two individuals compound heterozygosity was identified and is indicated.
Acknowledgments
This work was funded by the NIH/NIGMS Medical Genetics Training Grant T32 GM007454 (J.T.B.). The authors would like to thank all of the providers, in particular Drs. David Cobb, who referred their patients to us and the patients and their families for participating. We also thank Drs. Catherine Pihoker and Lisa Gilliam at the University of Washington and Dr. Sian Ellard for their help in validating the clinical tests.
Abbreviations:
- MODY
Maturity Onset Diabetes of the Young
- CHI
Congenital Hyperinsulinism
- NDM
Neonatal Diabetes
- NMD
nonsense mediated decay
- VUS
variant of uncertain significance
Footnotes
Conflict of interest
There are no conflicts of interests.
References
- [1].Gat-Yablonski G, Shalitin S, Phillip M, Maturity onset diabetes of the young—review, Pediatr. Endocrinol. Rev 3 (Suppl. 3) (2006) 514–520. [PubMed] [Google Scholar]
- [2].Ellard S, Bellanne-Chantelot C, Hattersley AT, European Molecular Genetics Quality Network Mg: best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young, Diabetologia 51 (4) (2008) 546–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Steck AK, Winter WE, Review on monogenic diabetes, Curr. Opin. Endocrinol. Diabetes Obes 18 (4) (2011) 252–258. [DOI] [PubMed] [Google Scholar]
- [4].Shields BM, Hicks S, Shepherd MH, Colclough K, Hattersley AT, Ellard S, Maturity-onset diabetes of the young (MODY): how many cases are we missing? Diabetologia 53 (12) (2010) 2504–2508. [DOI] [PubMed] [Google Scholar]
- [5].Murphy R, Ellard S, Hattersley AT, Clinical implications of a molecular genetic classification of monogenic beta-cell diabetes, Nat. Clin. Pract. Endocrinol. Metab 4 (4) (2008) 200–213. [DOI] [PubMed] [Google Scholar]
- [6].Shepherd M, Shields B, Ellard S, Rubio-Cabezas O, Hattersley AT, A genetic diagnosis of HNF1A diabetes alters treatment and improves glycaemic control in the majority of insulin-treated patients, Diabet. Med 26 (4) (2009) 437–441. [DOI] [PubMed] [Google Scholar]
- [7].Pearson ER, Flechtner I, Njolstad PR, Malecki MT, Flanagan SE, Larkin B, Ashcroft FM, Klimes I, Codner E, Iotova V, et al. , Switching from insulin to oral sulfonylureas in patients with diabetes due to Kir6.2 mutations, N. Engl. J. Med 355 (5) (2006) 467–477. [DOI] [PubMed] [Google Scholar]
- [8].Ellard S, Flanagan SE, Girard CA, Patch AM, Harries LW, Parrish A, Edghill EL, Mackay DJ, Proks P, Shimomura K, et al. , Permanent neonatal diabetes caused by dominant, recessive, or compound heterozygous SUR1 mutations with opposite functional effects, Am. J. Hum. Genet 81 (2) (2007) 375–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Babenko AP, Polak M, Cave H, Busiah K, Czernichow P, Scharfmann R, Bryan J, Aguilar-Bryan L, Vaxillaire M, Froguel P, Activating mutations in the ABCC8 gene in neonatal diabetes mellitus, N. Engl. J. Med 355 (5) (2006) 456–466. [DOI] [PubMed] [Google Scholar]
- [10].Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, et al. , Insulin gene mutations as a cause of permanent neonatal diabetes, Proc. Natl. Acad. Sci. U. S. A 104 (38) (2007) 15040–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Polak M, Dechaume A, Cave H, Nimri R, Crosnier H, Sulmont V, de Kerdanet M, Scharfmann R, Lebenthal Y, Froguel P, et al. , Heterozygous missense mutations in the insulin gene are linked to permanent diabetes appearing in the neonatal period or in early infancy: a report from the French ND (Neonatal Diabetes) Study Group, Diabetes 57 (4) (2008) 1115–1119. [DOI] [PubMed] [Google Scholar]
- [12].Familial hyperinsulinism, http://www.ncbi.nlm.nih.gov/books/NBK1375/.
- [13].Henderson M, Levy E, Delvin E, Losekoot M, Lambert M, Prevalence and clinical phenotype of the p.Val226Met glucokinase gene mutation in French Canadians in Quebec, Canada, Mol. Genet. Metab 90 (1) (2007) 87–92. [DOI] [PubMed] [Google Scholar]
- [14].Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FB, Hoogerbrugge N, Spurdle AB, Tavtigian SV, et al. , Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results, Hum. Mutat 29 (11) (2008) 1282–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mohnike K, Wieland I, Barthlen W, Vogelgesang S, Empting S, Mohnike W, Meissner T, Zenker M, Clinical and genetic evaluation of patients with KATP channel mutations from the German registry for congenital hyperinsulinism, Horm. Res. Paediatr 81 (3) (2014) 156–168. [DOI] [PubMed] [Google Scholar]
- [16].Desmet FO, Hamroun D, Lalande M, Collod-Beroud G, Claustres M, Beroud C, Human Splicing Finder: an online bioinformatics tool to predict splicing signals, Nucleic Acids Res 37 (9) (2009) e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, et al. , Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes, N. Engl. J. Med 350 (18) (2004) 1838–1849. [DOI] [PubMed] [Google Scholar]
- [18].Hager J, Blanche H, Sun F, Vaxillaire NV, Poller W, Cohen D, Czernichow P, Velho G, Robert JJ, Cohen N, et al. , Six mutations in the glucokinase gene identified in MODY by using a nonradioactive sensitive screening technique, Diabetes 43 (5) (1994) 730–733. [DOI] [PubMed] [Google Scholar]
- [19].Stoffel M, Froguel P, Takeda J, Zouali H, Vionnet N, Nishi S, Weber IT, Harrison RW, Pilkis SJ, Lesage S, et al. , Human glucokinase gene: isolation, characterization, and identification of two missense mutations linked to early-onset non-insulin-dependent (type 2) diabetes mellitus, Proc. Natl. Acad. Sci. U. S. A 89 (16) (1992) 7698–7702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lopez-Garrido MP, Herranz-Antolin S, Alija-Merillas MJ, Giralt P, Escribano J, Co-inheritance of HNF1a and GCK mutations in a family with maturity-onset diabetes of the young (MODY): implications for genetic testing, Clin. Endocrinol 79 (3) (2013) 342–347. [DOI] [PubMed] [Google Scholar]
- [21].Dorschner MO, Amendola LM, Turner EH, Robertson PD, Shirts BH, Gallego CJ, Bennett RL, Jones KL, Tokita MJ, Bennett JT, et al. , Actionable, pathogenic incidental findings in 1,000 participants’ exomes, Am. J. Hum. Genet 93 (4) (2013) 631–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Capuano M, Garcia-Herrero CM, Tinto N, Carluccio C, Capobianco V, Coto I, Cola A, Iafusco D, Franzese A, Zagari A, et al. , Glucokinase (GCK) mutations and their characterization in MODY2 children of southern Italy, PLoS ONE 7 (6) (2012) e38906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanne-Chantelot C, Ellard S, Gloyn AL, Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia, Hum. Mutat 30 (11) (2009) 1512–1526. [DOI] [PubMed] [Google Scholar]
- [24].Yorifuji T, Fujimaru R, Hosokawa Y, Tamagawa N, Shiozaki M, Aizu K, Jinno K, Maruo Y, Nagasaka H, Tajima T, et al. , Comprehensive molecular analysis of Japanese patients with pediatric-onset MODY-type diabetes mellitus, Pediatr. Diabetes 13 (1) (2012) 26–32. [DOI] [PubMed] [Google Scholar]
- [25].Shahawy S, Chan NK, Ellard S, Young E, Shahawy H, Mace J, Peverini R, Chinnock R, Njolstad PR, Hattersley AT, et al. , A pathway to insulin independence in newborns and infants with diabetes, J. Perinatol 31 (8) (2011) 567–570. [DOI] [PubMed] [Google Scholar]
- [26].Colclough K, Bellanne-Chantelot C, Saint-Martin C, Flanagan SE, Ellard S, Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia, Hum. Mutat 34 (5) (2013) 669–685. [DOI] [PubMed] [Google Scholar]
- [27].Kapoor RR, Flanagan SE, Arya VB, Shield JP, Ellard S, Hussain K, Clinical and molecular characterisation of 300 patients with congenital hyperinsulinism, Eur. J. Endocrinol 168 (4) (2013) 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lupski JR, Gonzaga-Jauregui C, Yang Y, Bainbridge MN, Jhangiani S, Buhay CJ, Kovar CL, Wang M, Hawes AC, Reid JG, et al. , Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot–Marie–Tooth neuropathy, Genome Med 5 (6) (2013) 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Yorifuji T, Kurokawa K, Mamada M, Imai T, Kawai M, Nishi Y, Shishido S, Hasegawa Y, Nakahata T, Neonatal diabetes mellitus and neonatal polycystic, dysplastic kidneys: phenotypically discordant recurrence of a mutation in the hepatocyte nuclear factor-1beta gene due to germline mosaicism, J. Clin. Endocrinol. Metab 89 (6) (2004) 2905–2908. [DOI] [PubMed] [Google Scholar]
- [30].Edghill EL, Flanagan SE, Patch AM, Boustred C, Parrish A, Shields B, Shepherd MH, Hussain K, Kapoor RR, Malecki M, et al. , Insulin mutation screening in 1,044 patients with diabetes: mutations in the INS gene are a common cause of neonatal diabetes but a rare cause of diabetes diagnosed in childhood or adulthood, Diabetes 57 (4) (2008) 1034–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gloyn AL, Cummings EA, Edghill EL, Harries LW, Scott R, Costa T, Temple IK, Hattersley AT, Ellard S, Permanent neonatal diabetes due to paternal germline mosaicism for an activating mutation of the KCNJ11 Gene encoding the Kir6.2 subunit of the beta-cell potassium adenosine triphosphate channel, J. Clin. Endocrinol. Metab 89 (8) (2004) 3932–3935. [DOI] [PubMed] [Google Scholar]
- [32].Godart F, Bellanne-Chantelot C, Clauin S, Gragnoli C, Abderrahmani A, Blanche H, Boutin P, Chevre JC, Froguel P, Bailleul B, Identification of seven novel nucleotide variants in the hepatocyte nuclear factor-1alpha (TCF1) promoter region in MODY patients, Hum. Mutat 15 (2) (2000) 173–180. [DOI] [PubMed] [Google Scholar]
- [33].Awa WL, Thon A, Raile K, Grulich-Henn J, Meissner T, Schober E, Holl RW, Group DP-WS, Genetic and clinical characteristics of patients with HNF1A gene variations from the German–Austrian DPV database, Eur. J. Endocrinol 164 (4) (2011) 513–520. [DOI] [PubMed] [Google Scholar]
- [34].Consortium EP, Bernstein BE, Birney E, Dunham I, Green ED, Gunter C, Snyder M, An integrated encyclopedia of DNA elements in the human genome, Nature 489 (7414) (2012) 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Shepherd M, Ellis I, Ahmad AM, Todd PJ, Bowen-Jones D, Mannion G, Ellard S, Sparkes AC, Hattersley AT, Predictive genetic testing in maturity-onset diabetes of the young (MODY), Diabet. Med 18 (5) (2001) 417–421. [DOI] [PubMed] [Google Scholar]
- [36].Flannick J, Beer NL, Bick AG, Agarwala V, Molnes J, Gupta N, Burtt NP, Florez JC, Meigs JB, Taylor H, et al. , Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes, Nat. Genet 45 (11) (2013) 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Girirajan S, Rosenfeld JA, Coe BP, Parikh S, Friedman N, Goldstein A, Filipink RA, McConnell JS, Angle B, Meschino WS, et al. , Phenotypic heterogeneity of genomic disorders and rare copy-number variants, N. Engl. J. Med 367 (14) (2012) 1321–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, Al-Mulla F, Bapat B, Bernstein I, Capella G, den Dunnen JT, et al. , Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database, Nat. Genet 46 (2) (2014) 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Aronson SJ, Clark EH, Varugheese M, Baxter S, Babb LJ, Rehm HL, Communicating new knowledge on previously reported genetic variants, Genet. Med 14 (8) (2012) 713–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Wilcox AR, Neri PM, Volk LA, Newmark LP, Clark EH, Babb LJ, Varugheese M, Aronson SJ, Rehm HL, Bates DW, A novel clinician interface to improve clinician access to up-to-date genetic results, J. Am. Med. Inform. Assoc 21 (e1) (2014) e117–e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Macmullen CM, Zhou Q, Snider KE, Tewson PH, Becker SA, Aziz AR, Ganguly A, Shyng SL, Stanley CA, Diazoxide-unresponsive congenital hyperinsulinism in children with dominant mutations of the beta-cell sulfonylurea receptor SUR1, Diabetes 60 (6) (2011) 1797–1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Aguilar-Bryan L, Bryan J, Molecular biology of adenosine triphosphate-sensitive potassium channels, Endocr. Rev 20 (2) (1999) 101–135. [DOI] [PubMed] [Google Scholar]
- [43].Tanizawa Y, Matsuda K, Matsuo M, Ohta Y, Ochi N, Adachi M, Koga M, Mizuno S, Kajita M, Tanaka Y, et al. , Genetic analysis of Japanese patients with persistent hyperinsulinemic hypoglycemia of infancy: nucleotide-binding fold-2 mutation impairs cooperative binding of adenine nucleotides to sulfonylurea receptor 1, Diabetes 49 (1) (2000) 114–120. [DOI] [PubMed] [Google Scholar]
- [44].Moritani M, Yokota I, Tsubouchi K, Takaya R, Takemoto K, Minamitani K, Urakami T, Kawamura T, Kikuchi N, Itakura M, et al. , Identification of INS and KCNJ11 gene mutations in type 1B diabetes in Japanese children with onset of diabetes before 5 years of age, Pediatr. Diabetes 14 (2) (2013) 112–120. [DOI] [PubMed] [Google Scholar]
- [45].Flanagan SE, Edghill EL, Gloyn AL, Ellard S, Hattersley AT, Mutations in KCNJ11, which encodes Kir6.2, are a common cause of diabetes diagnosed in the first 6 months of life, with the phenotype determined by genotype, Diabetologia 49 (6) (2006) 1190–1197. [DOI] [PubMed] [Google Scholar]
- [46].Bellanne-Chantelot C, Saint-Martin C, Ribeiro MJ, Vaury C, Verkarre V, Arnoux JB, Valayannopoulos V, Gobrecht S, Sempoux C, Rahier J, et al. , ABCC8 and KCNJ11 molecular spectrum of 109 patients with diazoxide-unresponsive congenital hyperinsulinism, J. Med. Genet 47 (11) (2010) 752–759. [DOI] [PubMed] [Google Scholar]
- [47].Gloyn AL, Noordam K, Willemsen MA, Ellard S, Lam WW, Campbell IW, Midgley P, Shiota C, Buettger C, Magnuson MA, et al. , Insights into the biochemical and genetic basis of glucokinase activation from naturally occurring hypoglycemia mutations, Diabetes 52 (9) (2003) 2433–2440. [DOI] [PubMed] [Google Scholar]
- [48].Stoffel M, Bell KL, Blackburn CL, Powell KL, Seo TS, Takeda J, Vionnet N, Xiang KS, Gidh-Jain M, Pilkis SJ, et al. , Identification of glucokinase mutations in subjects with gestational diabetes mellitus, Diabetes 42 (6) (1993) 937–940. [DOI] [PubMed] [Google Scholar]
- [49].Johansen A, Ek J, Mortensen HB, Pedersen O, Hansen T, Half of clinically defined maturity-onset diabetes of the young patients in Denmark do not have mutations in HNF4A, GCK, and TCF1, J. Clin. Endocrinol. Metab 90 (8) (2005) 4607–4614. [DOI] [PubMed] [Google Scholar]
- [50].Froguel P, Zouali H, Vionnet N, Velho G, Vaxillaire M, Sun F, Lesage S, Stoffel M, Takeda J, Passa P, et al. , Familial hyperglycemia due to mutations in glucokinase. Definition of a subtype of diabetes mellitus, N. Engl. J. Med 328 (10) (1993) 697–702. [DOI] [PubMed] [Google Scholar]
- [51].Katagiri H, Asano T, Ishihara H, Inukai K, Anai M, Miyazaki J, Tsukuda K, Kikuchi M, Yazaki Y, Oka Y, Nonsense mutation of glucokinase gene in late-onset non-insulin-dependent diabetes mellitus, Lancet 340 (8831) (1992) 1316–1317. [DOI] [PubMed] [Google Scholar]
- [52].Massa O, Meschi F, Cuesta-Munoz A, Caumo A, Cerutti F, Toni S, Cherubini V, Guazzarotti L, Sulli N, Matschinsky FM, et al. , High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI, Diabetologia 44 (7) (2001) 898–905. [DOI] [PubMed] [Google Scholar]
- [53].Stern E, Strihan C, Potievsky O, Nimri R, Shalitin S, Cohen O, Shehadeh N, Weintrob N, Phillip M, Gat-Yablonski G, Four novel mutations, including the first gross deletion in TCF1, identified in HNF-4alpha, GCK and TCF1 in patients with MODY in Israel, J. Pediatr. Endocrinol. Metab 20 (8) (2007) 909–921. [DOI] [PubMed] [Google Scholar]
- [54].Velho G, Blanche H, Vaxillaire M, Bellanne-Chantelot C, Pardini VC, Timsit J, Passa P, Deschamps I, Robert JJ, Weber IT, et al. , Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families, Diabetologia 40 (2) (1997) 217–224. [DOI] [PubMed] [Google Scholar]
- [55].Guazzini B, Gaffi D, Mainieri D, Multari G, Cordera R, Bertolini S, Pozza G, Meschi F, Barbetti F, Three novel missense mutations in the glucokinase gene (G80S; E221K; G227C) in Italian subjects with maturity-onset diabetes of the young (MODY). Mutations in brief no. 162. Online, Hum. Mutat 12 (2) (1998) 136. [DOI] [PubMed] [Google Scholar]
- [56].Gidh-Jain M, Takeda J, Xu LZ, Lange AJ, Vionnet N, Stoffel M, Froguel P, Velho G, Sun F, Cohen D, et al. , Glucokinase mutations associated with non-insulin-dependent (type 2) diabetes mellitus have decreased enzymatic activity: implications for structure/function relationships, Proc. Natl. Acad. Sci. U. S. A 90 (5) (1993) 1932–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Cao H, Shorey S, Robinson J, Metzger DL, Stewart L, Cummings E, Hegele RA, GCK and HNF1A mutations in Canadian families with maturity onset diabetes of the young (MODY), Hum. Mutat 20 (6) (2002) 478–479. [DOI] [PubMed] [Google Scholar]
- [58].Toaima D, Nake A, Wendenburg J, Praedicow K, Rohayem J, Engel K, Galler A, Gahr M, Lee-Kirsch MA, Identification of novel GCK and HNF1A/TCF1 mutations and polymorphisms in German families with maturity-onset diabetes of the young (MODY), Hum. Mutat 25 (5) (2005) 503–504. [DOI] [PubMed] [Google Scholar]
- [59].McKinney JL, Cao H, Robinson JF, Metzger DL, Cummings E, Riddell DC, Sanderson SR, Pacaud D, Ho J, Hegele RA, Spectrum of HNF1A and GCK mutations in Canadian families with maturity-onset diabetes of the young (MODY), Clin. Invest. Med 27 (3) (2004) 135–141. [PubMed] [Google Scholar]
- [60].Gloyn AL, van de Bunt M, Stratton IM, Lonie L, Tucker L, Ellard S, Holman RR, Prevalence of GCK mutations in individuals screened for fasting hyperglycaemia, Diabetologia 52 (1) (2009) 172–174. [DOI] [PubMed] [Google Scholar]
- [61].Elbein SC, Teng K, Eddings K, Hargrove D, Scroggin E, Molecular scanning analysis of hepatocyte nuclear factor 1alpha (TCF1) gene in typical familial type 2 diabetes in African Americans, Metab. Clin. Exp 49 (2) (2000) 280–284. [DOI] [PubMed] [Google Scholar]
- [62].Lambert AP, Ellard S, Allen LI, Gallen IW, Gillespie KM, Bingley PJ, Hattersley AT, Identifying hepatic nuclear factor 1alpha mutations in children and young adults with a clinical diagnosis of type 1 diabetes, Diabetes Care 26 (2) (2003) 333–337. [DOI] [PubMed] [Google Scholar]
- [63].Vaxillaire M, Rouard M, Yamagata K, Oda N, Kaisaki PJ, Boriraj VV, Chevre JC, Boccio V, Cox RD, Lathrop GM, et al. , Identification of nine novel mutations in the hepatocyte nuclear factor 1 alpha gene associated with maturity-onset diabetes of the young (MODY3), Hum. Mol. Genet 6 (4) (1997) 583–586. [DOI] [PubMed] [Google Scholar]
- [64].Bellanne-Chantelot C, Carette C, Riveline JP, Valero R, Gautier JF, Larger E, Reznik Y, Ducluzeau PH, Sola A, Hartemann-Heurtier A, et al. , The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3, Diabetes 57 (2) (2008) 503–508. [DOI] [PubMed] [Google Scholar]
- [65].Hattersley AT, Maturity-onset diabetes of the young: clinical heterogeneity explained by genetic heterogeneity, Diabet. Med 15 (1) (1998) 15–24. [DOI] [PubMed] [Google Scholar]
- [66].Yamagata K, Oda N, Kaisaki PJ, Menzel S, Furuta H, Vaxillaire M, Southam L, Cox RD, Lathrop GM, Boriraj VV, et al. , Mutations in the hepatocyte nuclear factor-1alpha gene in maturity-onset diabetes of the young (MODY3), Nature 384 (6608) (1996) 455–458. [DOI] [PubMed] [Google Scholar]
- [67].Dominguez-Lopez A, Miliar-Garcia A, Segura-Kato YX, Riba L, Esparza-Lopez R, Ramirez-Jimenez S, Rodriguez-Torres M, Canizales-Quinteros S, Cabrera-Vasquez S, Fragoso-Ontiveros V, et al. , Mutations in MODY genes are not common cause of early-onset type 2 diabetes in Mexican families, JOP 6 (3) (2005) 238–245. [PubMed] [Google Scholar]
- [68].Hansen T, Eiberg H, Rouard M, Vaxillaire M, Moller AM, Rasmussen SK, Fridberg M, Urhammer SA, Holst JJ, Almind K, et al. , Novel MODY3 mutations in the hepatocyte nuclear factor-1alpha gene: evidence for a hyperexcitability of pancreatic beta-cells to intravenous secretagogues in a glucose-tolerant carrier of a P447L mutation, Diabetes 46 (4) (1997) 726–730. [DOI] [PubMed] [Google Scholar]
- [69].Hameed S, Ellard S, Woodhead HJ, Neville KA, Walker JL, Craig ME, Armstrong T, Yu L, Eisenbarth GS, Hattersley AT, et al. , Persistently autoantibody negative (PAN) type 1 diabetes mellitus in children, Pediatr. Diabetes 12 (3 Pt 1) (2011) 142–149. [DOI] [PubMed] [Google Scholar]
- [70].Ellard S, Hepatocyte nuclear factor 1 alpha (HNF-1 alpha) mutations in maturity-onset diabetes of the young, Hum. Mutat 16 (5) (2000) 377–385. [DOI] [PubMed] [Google Scholar]
- [71].Gragnoli C, Cockburn BN, Chiaramonte F, Gorini A, Marietti G, Marozzi G, Signorini AM, Early-onset Type II diabetes mellitus in Italian families due to mutations in the genes encoding hepatic nuclear factor 1 alpha and glucokinase, Diabetologia 44 (10) (2001) 1326–1329. [DOI] [PubMed] [Google Scholar]
- [72].Harries LW, Locke JM, Shields B, Hanley NA, Hanley KP, Steele A, Njolstad PR, Ellard S, Hattersley AT, The diabetic phenotype in HNF4A mutation carriers is moderated by the expression of HNF4A isoforms from the P1 promoter during fetal development, Diabetes 57 (6) (2008) 1745–1752. [DOI] [PubMed] [Google Scholar]
- [73].Kapoor RR, Locke J, Colclough K, Wales J, Conn JJ, Hattersley AT, Ellard S, Hussain K, Persistent hyperinsulinemic hypoglycemia and maturity-onset diabetes of the young due to heterozygous HNF4A mutations, Diabetes 57 (6) (2008) 1659–1663. [DOI] [PubMed] [Google Scholar]
- [74].Ellard S, Colclough K, Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha (HNF1A) and 4 alpha (HNF4A) in maturity-onset diabetes of the young, Hum. Mutat 27 (9) (2006) 854–869. [DOI] [PubMed] [Google Scholar]