

Abstract
Higher fracture risk in type 2 diabetes (T2D) is attributed to disease-specific deficits in micro-structural and material properties of bone, although the primary cause is not yet established. The TallyHO (TH) mouse is a polygenic model of early-onset T2D and obesity analogous to adolescent-onset T2D in humans. Due to incomplete penetrance of the phenotype, ~25% of male TH mice never develop hyperglycemia, providing a strain-matched, non-diabetic control. Utilizing this model of T2D, we examined the impact of glucose-lowering therapy with canagliflozin (CANA) on diabetic bone. Male TH mice with or without hyperglycemia (High BG, Low BG) were monitored from ~8 to 20 weeks of age, and compared to age-matched, male, TH mice treated with CANA from ~8 to 20 weeks of age. At 20 weeks, untreated TH mice with high BG [High BG: 687±106 mg/dL] exhibited lower body mass, decrements in cortical bone of the femur (decreased cross-sectional area and thickness; increased porosity) and in trabecular bone of the femur metaphysis and L6 vertebra (decreased bone volume fraction, thickness, and tissue mineral density), as well as decrements in cortical and vertebral bone strength (decreased yield force and ultimate force) when compared to untreated TH mice with low BG [Low BG: 290±98 mg/dL; p<0.0001]. CANA treatment was metabolically advantageous, normalizing body mass, BG and HbA1c to values comparable to the Low BG group. With drug-induced glycemic improvement, cortical area and thickness were significantly higher in the CANA than in the High BG group, but deficits in strength persisted with lower yield force and yield stress (partially independent of bone geometry) in the CANA group. Additionally, CANA only partially prevented the T2D-related loss in trabecular bone volume fraction. Taken together, these findings suggest that the ability of CANA to lower glucose and normalized glycemic control ameliorates diabetic bone disease but not fully.
Keywords: type 2 diabetes, animal models, bone microarchitecture, bone μCT, fracture, canagliflozin, phosphate wasting
1. Introduction
Humans with T2D have an increased risk for bone fracture (1, 2), a risk that is neither explained by individual bone mineral density (BMD) measurements (3) nor by an increased propensity to fall (4). Instead, both cortical and trabecular bone quality are impaired in T2D, more so in those with poorer glycemic control (5) and longer duration of disease (2). At present, little is known about the onset and natural history of skeletal changes in youth diagnosed early with T2D, or of the impact of T2D during the adolescent years which are critical to acquisition of peak bone mass (6). However, coincident with the global epidemic of obesity among the pediatric population (7), the prevalence of T2D in children and adolescents is also increasing (8), with the incidence of T2D estimated to increase at a rate of ~7% per year (9), disproportionately impacting ethnic minority groups (7). Unlike adults with T2D, youth-onset T2D also displays a more aggressive phenotype (7, 8), characterized by: 1) a more rapid decline in ß-cell function (10, 11); 2) a more rapid deterioration in glycemic control (11), requiring multi-drug therapy; and 3) an earlier onset and more rapid progression of both microvascular and macrovascular complications of all types (12, 13). With both an earlier onset and greater severity of disease, pediatric patients with T2D face the potential for lifelong exposure to antidiabetic medications. Therefore, understanding the impact of early-onset hyperglycemia and relevant pharmacotherapy on the skeletal health in youth-onset T2D is critically important.
TallyHO/JngJ (TH) mice represent a polygenic model of early-onset T2D and obesity (14) consistent, maturationally, with adolescent-onset T2D in humans. In male TH mice, rapid weight gain is apparent by 4 weeks of age. Body weight increases progressively, and by ~10-14 weeks of age, insulin resistance progressing to frank hyperglycemia [blood glucose (BG) >300 mg/dL] is demonstrable; hyperinsulinemia and hyperlipidemia are also present (15). When compared with a genetically similar, non-diabetic, control strain (SWR/J mice) (16), adult TH male mice also exhibit lower bone formation markers (17, 18), lower trabecular bone volume (17, 18), lower bone toughness (18), and reduced fracture resistance (18), suggesting that skeletal acquisition and bone integrity are impaired in this model of T2D. Such skeletal comparisons, however, are confounded by a >30% increase in body size and a >100% increase in percent body fat in the obese TH mice, compared with the non-obese SWR/J strain (17). Moreover, the observed reduction in bone toughness and fracture resistance of TH mice, compared with SWR/J mice, does not relate to duration of diabetes (18), questioning the role of hyperglycemia in these strain differences that may be due, in part, to differences in the genetic makeup of SWR/J and TH mice.
Interestingly, the penetrance of the polygenic TH phenotype is incomplete, with ~20-25% of TH male mice maintaining normal glucose levels in our hands, despite advancing age and excess weight gain. (Female TH mice also remain euglycemic throughout life.) By selectively identifying and studying a cohort of non-hyperglycemic, yet comparably obese male TH mice, the skeletal effects of juvenile-onset diabetes in TH mice can be assessed independently of the co-existent, early-onset obesity and accomplished using littermates (i.e., same genetic background). Moreover, the TH model is amenable to testing the efficacy of therapies to lower blood glucose (19). One such FDA-approved therapy, canagliflozin (CANA), is a selective sodium-dependent glucose co-transporter 2 inhibitor (SGLT2I), a class of anti-hyperglycemic medications known to effectively lower serum glucose in an insulin-independent manner by inhibiting renal glucose reabsorption and promoting enhanced urinary glucose excretion. Despite the effectiveness of SGLT2Is in controlling glycemia and in reducing the risk of cardiovascular disease (20), CANA has been associated with a higher-than-expected fracture risk among T2D patients with certain comorbidities (21), though recent reports have indicated that this association was a chance observation (22, 23). Since hypercalciuria accompanies excessive glucose excretion, SGLT2Is may also decrease the mineral content in the extracellular matrix of bone, thereby reducing bone strength in T2D. Therefore, we sought to evaluate the hypothesis that SGLT2I treatment of hyperglycemic TH mice would improve their glycemic control, but would not fully rectify the diabetes-related skeletal deficits.
2. Research Design and Methods
2.1. Animals and experimental design
Six-week old male mice, TallyHO/JngJ (JAX #5314) were obtained from Jackson Laboratories (Bar Harbor, ME). All mice were maintained in a 14-hour light (lights on at 0600 hours): 10-hour dark cycle (lights off at 2000 hours), and provided ad libitum access to food (Teklad global 18% protein rodent diet; 6.2% fat, 44.2% carbohydrate; Envigo Corp., Indianapolis, IN) and water throughout all studies, unless otherwise specified below. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Kentucky (#2015-2293). Additionally, all methods were carried out in accordance with relevant guidelines and regulations.
Following documentation of hyperglycemia, juvenile diabetic TallyHO/JngJ male mice (hereafter designated High BG; average pre-treatment blood glucose (BG) = 496±145 mg/dL; 8-10 weeks of age) along with age-matched non-converting, normal glucose TallyHO/JngJ male mice (designated Low BG; average pre-treatment BG = 245±51 mg/dL) were thereafter monitored from study start (8-10 weeks of age) until 20 weeks of age. Concurrently, a mixed population of TH male mice (also 8-10 weeks of age) were treated with canagliflozin for a duration of 10 weeks by consuming Teklad chow compounded with CANA at 100 ppm (designated CANA; average pre-treatment BG = 449 ± 179 mg/dL, in which 12 of 52 mice had pretreatment BG ≤250 mg/dL and 40 of 52 mice had pretreatment BG > 250 mg/dL); to also reach a comparable 20 weeks of age. CANA dosing (at 100 ppm) was calculated to approximate 20 mg/kg/day of drug exposure; this would be comparable to the human adult canagliflozin therapeutic dose of 100 mg/day (equaling 1.7 mg/kg for a 60 kg reference adult body weight), according to recommendations for interspecies dose extrapolation (24).
At study conclusion, any mouse initially assigned to the Low BG group, but later identified as “a converter” with documented hyperglycemia [by BG>400 mg/dL at study end; n=4] was removed from further analysis. To achieve sufficient cohort size in the Low BG group, this experimental design was replicated three times and data from all three experiments were pooled for analysis (Sample size for the three study replicates: High BG, n=7, n=16, or n=20 mice, respectively; Low BG, n=5/7/4 mice per study; CANA, n= 12/24/16 mice per study). The final cohort size for analysis (Table 1, Supplemental Table S1) included High BG, n=43 mice; Low BG, n=16 mice; CANA, n= 52 mice.
Table 1:
Effect of treatment (CANA) or high blood glucose (BG) on selected outcome measurements (mean ± SD) from the evaluations of femur mid-shafts (cortical bone) and distal femur metaphysis (trabecular bone). CANA-treatment normalizes a bone parameter if the CANA vs. Low BG comparison is not significant (NS). p<0.05 (p<0.1).
| Parameter | Units | [A] | [B] | [C] | ANOVA | Adjusted p-value | ||
|---|---|---|---|---|---|---|---|---|
| CANA | High BG |
Low BG |
p-value | CANA effect |
CANA normalization |
T2D effect |
||
| Cortical Bone | (n = 52) | (n = 43) | (n = 14-16) | A vs. B | A vs. C | B vs. C | ||
| Ma.V | --- | 1.17 ± 0.09 | 1.16 ± 0.10 | 1.10 ± 0.09 | (0.0526) | NS | 0.0462 | (0.0886) |
| Ct.Ar | mm2 | 0.79 ± 0.04 | 0.76 ± 0.6 | 0.81 ± 0.04 | 0.0005 | 0.0103 | NS | 0.0011 |
| Tt.Ar | mm2 | 1.43 ± 0.07 | 1.39 ± 0.07 | 1.41 ± 0.08 | (0.0742) | (0.0592) | NS | NS |
| Imin | mm4 | 0.090 ± 0.010 | 0.085 ± 0.009 | 0.092 ± 0.010 | 0.0076 | 0.0166 | NS | 0.0368 |
| Section modulus | mm3 | 0.157 ± 0.012 | 0.151 ± 0.013 | 0.158 ± 0.012 | 0.0297 | 0.0424 | NS | NS |
| Ct.Th | mm | 0.212 ± 0.010 | 0.205 ± 0.013 | 0.222 ± 0.009 | <0.0001 | 0.0105 | 0.0073a | <0.0001a |
| Ct.TMD | mgHA/cm3 | 1282 ± 36 | 1268 ± 35 | 1289 ± 41 | (0.0699)b | NS | NS | NS |
| Ct.Po | % | 2.79 ± 0.31 | 2.95 ± 0.33 | 2.65 ± 0.34 | 0.0030 | 0.0378 | NS | 0.0047 |
| Stiffness | N/mm | 129 ± 15 | 122 ± 17 | 138 ± 15 | 0.0020 | (0.070) | NS | 0.0019 |
| Yield force | N | 17.6 ± 2.0 | 16.3 ± 2.3 | 19.3 ± 1.5 | <0.0001 | 0.0091 | 0.0151 | <0.0001 |
| Ultimate force | N | 20.5 ± 1.6 | 19.3 ± 2.3 | 21.4 ± 1.8 | 0.0025c | 0.0332 | NS | 0.0048 |
| PYD | mm | 0.267 ± 0.152 | 0.250 ± 0.108 | 0.218 ± 0.121 | NSb | No effects | ||
| Work-to-fracture | kJ/m2 | 6.18 ± 1.86 | 5.66 ± 1.45 | 5.95 ± 2.09 | NSb | No effects | ||
| Yield stress | MPa | 228 ± 25 | 220 ± 24 | 249 ± 20 | 0.0004 | NS | 0.0087 | 0.0002 |
| Ultimate stress | MPa | 262 ± 15 | 257 ± 24 | 271 ± 16 | 0.0486c | NS | NS | 0.0430 |
| Modulus | GPa | 15.3 ± 1.7 | 15.4 ± 1.9 | 16.1 ± 1.5 | NS | No effects | ||
| Toughness | MJ/m3 | 2.96 ± 0.86 | 2.84 ± 0.75 | 2.76 ± 0.94 | NSb | No effects | ||
| PY toughness | MJ/m3 | 1.99 ± 0.85 | 1.98 ± 0.75 | 1.73 ± 0.92 | NSb | No effects | ||
| Trabecular Bone | ||||||||
| Tb.BV/TV | % | 5.9 ± 1.6 | 4.6 ± 1.2 | 7.6 ± 1.8 | <0.0001b | 0.0006 | 0.0142 | <0.0001 |
| Conn.D | mm−3 | 72.8 ± 26.6 | 76.5 ± 23.6 | 80.5 ± 22.9 | NS | No effects | ||
| SMI | 0:plates; 3:rods | 2.76 ± 0.26 | 2.83 ± 0.20 | 2.56 ± 0.24 | 0.0007 | NS | 0.0111 | 0.0004 |
| Tb.N | mm−1 | 3.29 ± 0.36 | 3.32 ± 0.29 | 3.41 ± 0.31 | NS | No effects | ||
| Tb.Th | mm | 0.042 ± 0.005 | 0.036 ± 0.004 | 0.046 ± 0.004 | <0.0001b | <0.0001 | NS | <0.0001 |
| Tb.Sp | mm | 0.309 ± 0.036 | 0.303 ± 0.028 | 0.297 ± 0.028 | NSb | No effects | ||
| Tb.TMD | mgHA/cm3 | 983.5 ± 40.8 | 959.0 ± 39.7 | 984.9 ± 50.3 | 0.0003b | 0.0003 | NS | (0.058) |
P-value from Dunn’s test because only Ct.Th of TH – Low BG did not pass normality, but the residuals did pass normality.
P-value from Kruskal-Wallis test because residuals did not pass normality
P-value from Kruskal-Wallis test because the SDs were significantly different among the 3 groups
2.1.1. Sample size estimation
Based on the mean and SD values of yield force for untreated T1D mice and T1D mice treated with CANA in our previous study (25), we used a standardized mean difference (Cohen’s d) of 1.0951 in the estimate of the sample size. With an allocation ratio of 4 (penetrance estimate of 4 times more High BG mice than Low BG mice), an α error probability of 0.05, and a power (1 – β error probability) of 0.9, 12 mice in the Low BG group and 46 mice in the High BG group would be required to detect a difference in yield force that was similar to the difference in our previous CANA study involving a mouse model of T1D (25). Because of the uncertainty in which mouse would maintain High BG at the time of allocation to treatment (Rx) or no Rx during the replicate studies, our final sample size was similar but not exact. Nonetheless, by having a total sample size of 109-111 mice, we were powered (0.90) to detect an effect size f of 0.3457 (Cohen’s d = 0.6912) among 3 groups with 1 covariate at α=0.05. These power calculations were done using G*Power (v3.1.9.3, Heinrich Heine University Düsseldorf) (26, 27). The Cohen’s d for BV/TV in the previous T1D-CANA study was greater than the Cohen’s d to power the present study.
2.2. Assessment of physiology and phenotype
For all experiments, weekly (experimental replicate 1), or biweekly (experimental replicates 2, 3) animal assessments included measurement of individual body weight, food and water consumption, and random blood glucose concentration (so as to document glycemic status). Blood glucose (BG) was measured from tail vein sampling using a glucometer (AlphaTRAK 2 Veterinary Blood Glucose Monitoring System; Zoetis, Parsippany, NJ). During the 9-10th week of treatment (mouse age ~19 weeks) mice from all 3 cohorts were transferred to individual metabolic cages for urine collection. Urine was collected over defined intervals (range 6-18 hours for experimental replicates 1 and 2; 24-hour collection for replicate 3); for each experiment, total urine volume and collection times were recorded. Using an aliquot of each collection, urine calcium and phosphate concentrations were measured using calcium and phosphate colorimetric assays (Sigma-Aldrich Corporation, St. Louis, MO; #MAK022 and #MAK308, respectively) and urine creatinine was measured using an alkaline picrate chemical assay (Exocell, Inc., Philadelphia, PA; The Creatinine Companion, #1012). Calcium and phosphate concentrations were normalized to urine creatinine concentration, and reported as a urine calcium/creatinine ratio (CA/Cr) or urine phosphate/creatinine ratio (PHOS/Cr). In the third experimental replicate, a 24-hour urine collection was used to quantify total daily urine excretion of CA, PHOS, or glucose, reported as μg/g/day for calcium or phosphate and as mg/g/day for glucose, corrected to individual mouse body mass (g). Urine glucose for this parameter was quantified using the Mouse Crystal Chem glucose enzymatic assay (Elk Grove Village, IL; #81692).
At study end, mice were euthanized by isoflurane overdose followed by decapitation, and trunk blood was collected directly into 1.5 ml Eppendorf tubes on ice. Physiological assessments (diabetes parameters and bone biomarkers) and ex vivo analyses of bone phenotype [high-resolution micro-computed tomography (μCT) and biomechanical testing] were completed on all mice, as described in subsequent sections.
2.3. Serum Assays
At study end, fasting blood glucose (4-5 hour fast with free access to water) was measured and terminal Hemoglobin A1c [HbA1c; a long-term estimate of daily averaged blood glucose; (28)] was quantified using a mouse HbA1c whole blood assay (Crystal Chem; Downers Grove, IL, #80310). Fasting serum insulin levels were also quantified at study end, using an ultrasensitive mouse insulin ELISA (Crystal Chem: #90080). As a marker of bone formation, procollagen type 1 N-terminal propeptide (P1NP) was measured in serum at sacrifice, using the Rat/Mouse P1NP Enzyme immunoassay (Immunodiagnostics Systems, Inc., Fountain Hills, AZ; #AC-33F1). As a marker of bone resorption, C-terminal telopeptides of type I collagen (CTX) were measured in serum using the RatLAPs ELISA (Immunodiagnostics Systems, Inc., Fountain Hills, AZ; #AC-06F1).
2.4. Assessment of Skeletal Microarchitecture
At study end, left femurs and lumbar vertebrae were harvested, frozen in phosphate buffered saline (PBS) and stored at −20°C until analysis. Femur length (neck to condyle groove) was measured using calipers. For bone microarchitecture analyses, the mid-shaft and distal metaphysis regions of the femurs or centrum of the L6 vertebral bodies (VB) were scanned with a micro-computed tomography (μCT) scanner (Scanco Medical μCT50, Brϋttisellen, Switzerland): isotropic voxel size of 6.0 μm (12.0 μm for VB), peak x-ray energy setting of 70 kVp/0.114 mA (55 kVp/0.200 mA for VB), integration time of 300 ms (600 ms for VB), and a sampling rate of 1162 samples (500 for VB) per 1000 projections during a full rotation of the tube holder. Each scan was calibrated to the manufacturer provided QC standard [a hydroxyapatite (HA) phantom with peak density of 1200 mgHA/cm3] to account for any beam hardening artefacts, as previously described (29). The regions of interest (ROI) included the trabecular bone of the metaphysis (3.72 mm segment above the growth plate of the distal femur) and between the VB endplates (1.40-2.25 mm segment of the VB) as well as the cortical bone of the diaphysis (1.86 mm segment centered at the mid-point of the femur).
Each ROI was analyzed after the scan reconstruction using a custom auto-contour program in Scanco Image Processing (IPL) Software v6.6 to segment out the bone against the background. The entire image stack (310 axial slices) of the femur mid-shaft was contoured to capture the cortical bone properties, while for the image stack of the distal femur (350 slices after excluding 50 slices proximal to primary spongiosa) and L6VB (115-185 slices after excluding endplates) only the trabecular bone was included. Using our previously published methods for contouring these regions (18, 29) and then applying a Gaussian noise filter (sigma of 0.8 with support of 2 for cortical bone and sigma=0.2, support=1 for trabecular bone) and a global threshold (429.4 mgHA/cm3 and 900.5 mgHA/cm3 for trabecular and cortical bone, respectively), each ROI was evaluated using standard Scanco evaluations scripts IPL program applied to determine the architectural and structural properties of trabecular and cortical bone. Also, the femur mid-shafts dual contour (encompassing only the intra-cortical region) was used to estimate the porosity by subtracting its cortical bone volume fraction from unity.
2.5. Assessment of Bone Biomechanical Properties
After μCT analysis, each hydrated left femur mid-shaft was loaded to failure at 3 mm/min in three-point bending with a span of 8 mm, following our previously described procedures (30, 31). The stiffness (slope of the linear portion of the curve), yield force (0.2% offset method), and ultimate force (maximum or peak load endured by the bone before fracture) were determined from the resulting force vs. displacement curve. These structural-dependent properties were converted to estimates of material properties (modulus, yield stress, and ultimate stress, respectively) using the flexural equations from Euler-Bernoulli beam theory and the geometrical properties of each femur mid-shaft from the μCT evaluations, namely the moment of inertia for the bending orientation and the distance between the neutral axis and the periosteal surface in the direction of loading (29). Because the flexural equations are less valid after the bone tissue yields, we estimated the toughness and post-yield toughness as 3 × Wf / Ct.Ar / Span where Wf is the work-to-fracture (area under the entire force vs. displacement curve) or the post-yield work-to-fracture (area under the force vs. displacement curve between the yield point and failure point) and Ct.Ar is the bone cross-sectional area of the femur mid-shaft (32). Each hydrated L6 VB was loaded at 3.0 mm/min in compression to determine the ultimate compressive force as the measurement of whole-bone strength (18).
2.6. Statistical Analysis
Outcomes of interest were measured as pre-treatment (pre-treatment body mass, blood glucose), near end-of-experiment (urine CA/Cr, CA/day; urine PHOS/Cr, PHOS/day; urine glucose/day) or at the time of death for each mouse (post-treatment blood glucose and all other parameters). For these quantitative variables, group-level means and standard deviations (SD) were calculated as well as group-level medians and first and third quartiles when warranted. The approximate normality of data was checked prior to data analyses using the Anderson-Darling test. For normally distributed variables with homoscedasticity across groups, analysis of variance (ANOVA) was employed. If the group means were significantly different, the 3 pairwise comparisons of groups were subjected to Tukey’s multiple comparison test. For variables not normally distributed or heteroscedastic, Kruskal-Wallis non-parametric testing was employed, followed by Dunn’s multiple comparison test. Statistically significant group differences were defined as p≤0.05 unless otherwise stated in table or figure legends. A Spearman’s rank correlation coefficient was also calculated between variables of interest, given that most correlations were either heteroscedastic or not normally distributed. In a sub-group analysis comparing daily mineral excretion to daily glucose excretion for untreated and treated mice, the residuals from the ordinary least squares regression either did not pass the Anderson-Darling normality test or the runs test for homoscedasticity. Therefore, the data was bootstrapped with 1000 replicates to determine the p-values for the slope of the regression line(s) and the interaction between glucose excretion and group (untreated or CANA-treated). To determine whether group-level differences were independent of final body mass and whether the linear relationship between yield force (or BV/TV) depended on treatment, we used analysis of covariance, bootstrapping with 1000 replicates when the residuals did not pass the Anderson-Darling test or were heteroscedastic. Analyses of the group differences, Spearman correlations, and linear regressions were completed using GraphPad Prism 8.1.1 statistical software (San Diego, CA). To bootstrap the data for estimating the p-values of the regression coefficients, we used Stata 11.0 data analysis (College Station, TX).
3. Results
3.1. Physiology:
3.1.1. A comparison of mice with hyperglycemia vs. normoglycemia confirms differences in body mass and metabolic phenotype, by 20 weeks of age.
At the start of the experimental period, pre-treatment measurements of body mass for the 3 groups were similar (Fig. 1A; CANA: 33.3 ± 2.6 gm; High BG: 33.5 ± 2.5 gm; Low BG: 31.9 ± 3.3 gm; ANOVA p=0.1304). Additionally, at study start, mice in the High BG and CANA groups had similarly high pre-treatment blood glucose levels (Fig. 1C), compared with the designated Low BG group. Over the next 10 weeks, untreated hyperglycemic TH mice (High BG) demonstrated a failure to gain weight (Fig. 1A compared to Fig. 1B and Fig. S1 in supplemental materials); and, as shown in Figure 1, at study end, in addition to the ~15% lower body mass, High BG mice maintained a significantly higher blood glucose (Fig. 1D and Table S1 in supplemental materials), along with an elevated HbA1c (Fig. 1F) and a lower serum insulin concentration (Fig. 1E), compared with the non-diabetic cohort (Low BG). By comparison, following 10 weeks of treatment with canagliflozin (CANA), body mass (Fig. 1B), post-treatment BG (Fig. 1D) and HbA1c values (Fig. 1F) normalized, and serum insulin levels improved (Fig. 1E), compared with the High BG group (Table S1). Final femur lengths, however, were not significantly different among the 3 groups (CANA: 14.94 ± 0.33 mm; High BG: 15.01 ± 0.20 mm; Low BG: 14.87 ± 0.17 mm; Kruskal-Wallis test p=0.0624) indicating comparable linear bone growth across the groups by 20 weeks of age.
Figure 1: Differences in selected physiological measurements at pre-treatment or post-treatment.
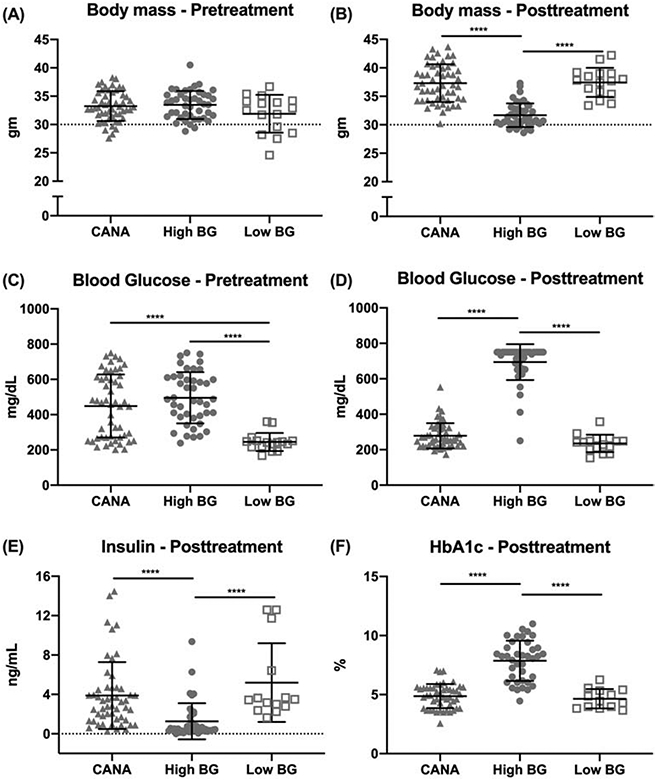
Pre-treatment body mass was similar among the 3 groups (A); 10-12-weeks later, untreated mice with high blood glucose (BG) weighed less than CANA-treated mice or mice with low blood glucose (B). Average pre-treatment BG was significantly elevated in both the CANA-treated and High BG groups, compared with the designated Low BG group (C); 10-12-weeks later, BG was similar between CANA-treated mice and Low BG mice (D). [Fig. 1D shows 1 outlier for the High BG cohort, with a post-treatment BG of 250 mg/dL. For this mouse, BG at the time of group assignment was 493 mg/dL with subsequent measurements of 505, 363, 399, and 368 mg/dL throughout the study. Because the average BG for study duration was 394 mg/dL, this mouse was retained in the High BG group despite an inconsistent final BG reading.] Accompanying the reduction in BG, circulating insulin (E) and HbA1c (F), a marker of glycemic control, was higher and lower, respectively, for CANA-treated than untreated mice with high BG. Group mean ± SD is shown. **** p<0.0001.
Individual values for HbA1c and post-treatment BG were correlated (Table S3; Spearman r = 0.689, p<0.0001), demonstrating consistency across these two measures of glycemic control. Additionally, individual values for final body mass were inversely correlated with both HbA1c (r = −0.676, p<0.0001) and post-treatment BG (r = −0.734, p<0.0001), supporting the expectation that chronic, untreated hyperglycemia, where present, contributed to the poor weight gain across time.
3.1.2. Hypercalciuria, hyperphosphaturia, and reduced bone formation are evident in diabetic mice; these abnormalities persist in CANA-treated mice despite a drug-related glycemic correction.
At study end, High BG mice had significantly higher urine calcium (CA) and urine phosphate (PHOS) excretion, compared with the Low BG group (Table S1; Fig. 2A and 2C, respectively). However, unlike the effects of CANA on glucose control, urinary mineral loss did not normalize with CANA. Instead, daily calcium excretion (urine CA/day) was highly correlated with daily glucose excretion (urine glucose/day), regardless of whether glucosuria was the result of untreated diabetes or of SGLT2I exposure (Fig. 2B). Daily phosphate excretion was also highly correlated with daily glucose excretion, although CANA treatment affected the relationship (Fig. 2D) such that there was less urine PHOS/day for a given urine glucose/day in CANA-treated mice, relative to untreated mice with hyperglycemia.
Figure 2: Differences in urine and serum bone biomarkers.
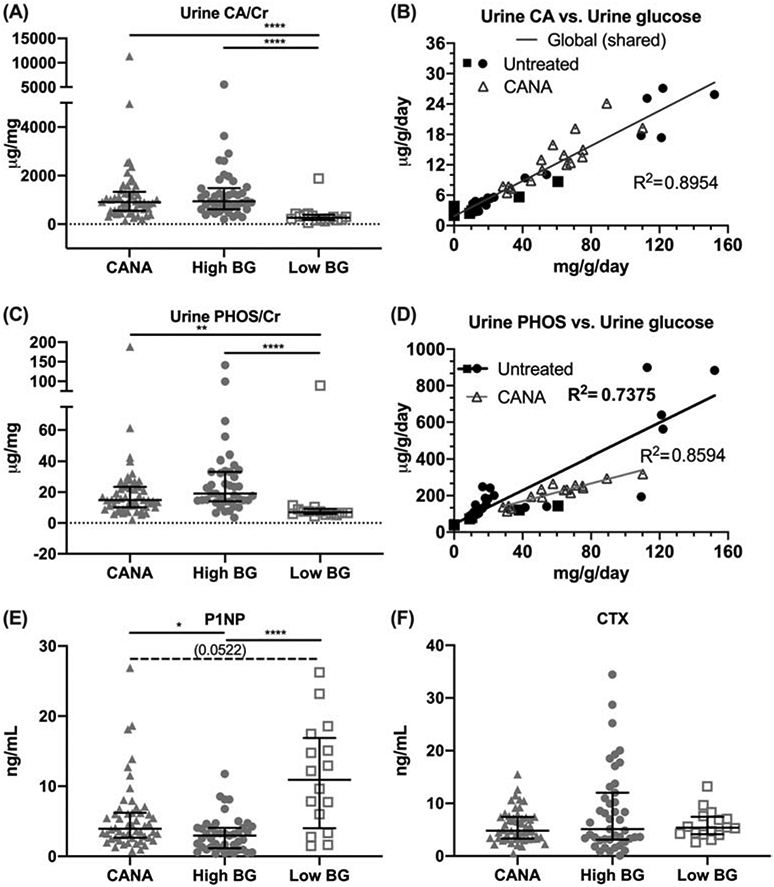
Urinary excretion of calcium (CA) normalized to creatinine (Cr), measured at study end, was elevated in CANA-treated mice and untreated High BG mice, compared to Low BG mice (A). Examined in a subset of mice, the daily excretion of CA was directly proportional to daily excretion of glucose (B) with one linear curve being required to explain the relationship for both untreated and treated mice (i.e., the interaction term was not significant, p=0.1960, and therefore removed). Urinary excretion of phosphorous (PHOS/Cr) was also higher in the CANA-treated mice and untreated High BG mice, compared with Low BG mice (C). In the same subset of mice, the daily excretion of PHOS was directly proportional to daily excretion of glucose (D), but the linear relationship was significantly different between untreated and treated mice (i.e., the interaction term was significant, p<0.0005). The serum marker of bone formation (P1NP) was reduced in the High BG group (E). There were no differences in the serum marker of bone resorption (CTX) between groups (F). Group median with the intra quartile range is shown. **** p<0.0001; ** p<0.01; *p≤0.05; and dashed line when adjusted p-value was greater than 0.05 but less than 0.1. In the linear regression graphs (B, D), closed symbols indicate untreated mice and the open triangle indicate CANA-treated mice.
Compared to the Low BG group, the mean for P1NP (i.e., the N-terminal propeptide) a marker of bone formation, was lower for both the CANA group and the untreated High BG group. (Table S1; Fig. 2E). However, CANA improved bone formation as P1NP was significantly higher in the CANA group compared with the High BG group. Consistent with these between group differences for bone formation, P1NP was also inversely correlated with HbA1c (Table S3, r= −0.452, p<0.0001) and with post-treatment BG (r= −0.473, p<0.0001), supporting the finding that bone formation is reduced in diabetic mice.
Mean values for CTX (i.e., the C-terminal telopeptide), a marker of bone resorption, were not significantly different between the groups (Table S1, Fig. 2F).
3.2. Skeletal Phenotype
3.2.1. Micro-computed tomography (μCT) analysis of the femur diaphysis (cortical bone) demonstrates deficits in cortical bone with diabetes, which are only partially corrected with CANA treatment.
When comparing structural properties of the diaphysis between the High BG vs. Low BG groups (Table 1), hyperglycemic mice exhibited a significant reduction in cortical area (Ct.Ar) and cortical thickness (Ct.Th), and in moment of inertia about the minimum axes (Imin), along with an increase in our estimate of cortical porosity (Ct.Po), as evidence of a diabetic bone phenotype in the TH model.
Mice treated with CANA showed a normalization of Ct.Ar and Imin after 10 weeks of treatment (Table 1). However, the lower Ct.Th and higher medullary volume (Ma.V) seen in High BG mice persisted in CANA-treated mice. Since mice with High BG did not gain weight while CANA treatment improved weight gain (Fig. 1B), the effect of T2D and the effect of CANA on these structural properties of bone could be due to differences in weight gain or lack thereof. When final body mass (BM) was included as a covariate in the analysis (Table S2), only Ct.Ar and Ct.Th depended on group (CANA, High BG, or Low BG). In effect, Ct.Ar and Ct.Th remained lower for CANA-treated mice than for untreated High BG mice, when accounting for the positive contribution of BM to these bone properties (Table S2). In contrast, the variance in Imin and Ct.Po was explained by BM, without any significant contribution from group (Table S2).
3.2.2. Micro-computed tomography (μCT) analysis of the femur metaphysis (trabecular bone) demonstrates deficits in trabecular bone with diabetes, which persist despite CANA treatment.
Hyperglycemic TH mice also exhibited significant deficits in trabecular bone (Table 1 and Fig. 3A, 3B), as a component of the diabetic bone phenotype. Specifically, reductions in trabecular bone volume fraction (Tb.BV/TV) and trabecular thickness (Tb.Th), along with an increase in structural model index (SMI) indicating a more rod-like characteristic, were apparent when comparing High BG and Low BG mice (Table 1 and Fig. 3C).
Figure 3: Differences in trabecular bone architecture and mineral density.

Representative micro-computed tomography evaluations of distal femur metaphysis are shown for the 3 groups (A) along with 3D renderings of the compartment with blue indicating low trabecular thickness and red indicating high trabecular thickness (B). There was less trabecular bone volume fraction (BV/TV) in the High BG group compared to the Low BG group and CANA group; CANA-treatment partially rescued BV/TV (C). Tissue mineral density of the trabecular bone (Tb.TMD) was also less in the High BG group compared to the other groups with no difference between CANA-treated and Low BG mice (D). Group median with the intra quartile range is shown. **** p<0.0001; ***p<0.001; *p<0.05; and dashed line when adjusted p-value was greater than 0.05 but less than 0.1.
While a trend toward improvement in trabecular parameters was seen in the CANA group (Table 1; significant differences noted between CANA and High BG), at study end Tb.BV/TV and SMI did not normalize with CANA treatment, but remained significantly different from the Low BG group, despite the period of improved metabolic control with CANA treatment. When adjusting for final body mass, group still significantly explained some of the variance in Tb.BV/TV, SMI, and Tb.Th (Table S2), but CANA treatment only affected Tb.Th (higher compared to High BG). In the multivariable regressions, mice with Low BG had significantly higher Tb.BV/TV and Tb.Th as well as lower SMI (Table S2) compared to mice with High BG (i.e., persistent T2D).
3.2.3. Micro-computed tomography (μCT) analysis of the L6 vertebra (trabecular bone) also demonstrates deficits in trabecular bone with diabetes, which persist despite CANA treatment.
Similar to the trabecular bone of the femur, deficits in trabecular bone of the L6 vertebra were noted with diabetes, including reductions in Tb.BV/TV, Tb.Th, and Tb.TMD along with an increase in SMI, when comparing High BG and Low BG mice (Table 2). And again, a trend toward improvement in these trabecular parameters was noted in the CANA group, but CANA treatment did not normalize the L6 VB trabecular bone (Table 2).
Table 2:
Effect of treatment (CANA) or high blood glucose (BG) on selected outcome measurements (mean ± SD) from the analysis of the L6 vertebral body (trabecular bone with a thin cortical shell). CANA-treatment normalizes a bone parameter if the CANA vs. Low BG comparison is not significant (NS). p<0.05 (p<0.1).
| Parameter | Units | [A] | [B] | [C] | ANOVA | Adjusted p-value | ||
|---|---|---|---|---|---|---|---|---|
| CANA | High BG |
Low BG |
p-value | CANA effect |
CANA normalization |
T2D effect |
||
| Centrum | (n = 52) | (n = 43) | (n = 14-16) | A vs. B | A vs. C | B vs. C | ||
| Tb.BV/TV | % | 25.4 ± 3.0 | 22.9 ± 3.5 | 28.8 ± 2.3 | <0.0001 | 0.0049a | 0.0031a | <0.0001 |
| Conn.D | mm−3 | 213.3 ± 36.8 | 232.6 ± 21.8 | 210.1 ± 24.0 | 0.0038b | 0.0136 | NS | 0.0201 |
| SMI | 0:plates; 3:rods | 0.80 ± 0.28 | 1.06 ± 0.31 | 0.49 ± 0.18 | <0.0001 | <0.0001 | 0.0002 | <0.0001 |
| Tb.N | mm−1 | 4.74 ± 0.33 | 4.81 ± 0.32 | 4.84 ± 0.24 | NSb | No effects | ||
| Tb.Th | mm | 0.056 ± 0.004 | 0.052 ± 0.005 | 0.061 ± 0.003 | <0.0001c | 0.0006 | 0.0024 | <0.0001 |
| Tb.Sp | mm | 0.209 ± 0.017 | 0.205 ± 0.015 | 0.205 ± 0.013 | NSb | No effects | ||
| Tb.TMD | mgHA/cm3 | 876.0 ± 17.0 | 862.7 ± 22.3 | 891.5 ± 10.2 | <0.0001c | 0.0167 | 0.0055 | <0.0001 |
| Ax.Led | mm | 1.80 ± 0.17 | 1.76 ± 0.15 | 1.85 ± 0.26 | NSa | No effects | ||
| Whole Bone | ||||||||
| Yield force | N | 21.8 ± 6.6 | 15.5 ± 4.5 | 27.9 ± 7.3 | <0.0001c | <0.0001 | (0.0594) | <0.0001 |
| Ultimate force | N | 26.0 ± 7.1 | 19.5 ± 5.4 | 32.1 ± 6.4 | <0.0001 | <0.0001 | 0.0011 | <0.0001 |
P-value from Dunn’s test because only BV/TV of CANA did not pass normality, but the residuals did pass normality.
P-value from Kruskal-Wallis test because residuals did not pass normality.
P-value from Kruskal-Wallis test because the SDs were significantly different among the 3 groups.
Axial length (Ax.Le) is the distance between endplates as determined by μCT evaluations.
3.2.4. Biomechanical testing demonstrates deficits in bone strength with diabetes.
Biomechanical testing of the femur demonstrated a reduction in both bone stiffness and bone strength in High BG mice compared with Low BG mice (Table 1), as indicated by significant reductions in stiffness (Table 1), yield force (Fig. 4A), ultimate force (Table 1), yield stress (Fig. 4B), and ultimate stress (Table 1) with T2D. In contrast, overall resistance to fracture (work-to-fracture, toughness, and post-yield toughness) was not significantly different between High BG and Low BG mice (Table 1). Biomechanical testing of the L6 VB also demonstrated reductions in bone strength (yield force, ultimate force) with T2D (Table 2).
Figure 4: Differences in bending strength.
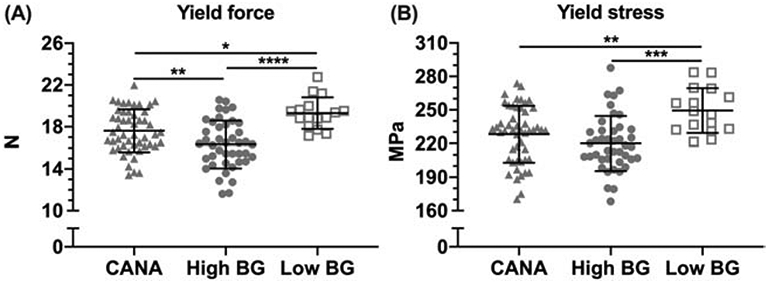
The force at yielding (proportional limit when permanent damage occurs) was highest in the Low BG group and lowest in the High BG group (A). The stress at yielding (partially independent of bone structure) was also highest for the Low BG group, compared with the other two groups. (B). ****p<0.0001; ***p<0.001; ** p<0.01; *p≤0.05.
In CANA treated mice, despite metabolic improvement with drug therapy, neither of the material strength estimates (yield stress or ultimate stress) of the femur improved; however, some increase in yield force (Fig. 4A) and ultimate force (Table 1) was apparent when comparing the CANA-treated mice to untreated High BG mice. When accounting for differences in final body mass between High BG and CANA and between High BG and Low BG, group was still a significant factor affecting yield force and yield stress, but the coefficients for the CANA effect or the T2D effect were not strictly significant. The effect of hyperglycemia on the strength measurements was not independent of its negative effect on weight gain (Table S2).
Similar to the femur, some improvement in strength of the L6VB was noted when comparing the CANA-treated mice to untreated High BG mice, although drug therapy did not normalize these parameters (Table 2).
3.2.5. The relationships between yield force and several outcomes differ between non-treated TH mice and CANA-treated TH mice.
To identify potential reasons for the treatment effect on yield force, we performed linear regressions pooling High BG and Low BG mice as an untreated group (no CANA) and included CANA treatment (Rx) as a potential explanatory factor. As expected, yield force was positively associated with body mass, but Rx was a significant factor such that for a given BM, mice treated with CANA tended to have weaker femur mid-shafts in bending than untreated mice (Table 3 and Fig. S2). Yield force was negatively associated with blood glucose, but again Rx was a significant factor such that this measure of whole bone bending strength tended to be less in the treated than in non-treated mice for a given BG. With respect to HbA1c as a potential predictor, the interaction term was significant (Table 3) indicating that the negative association between glycemic control and yield force was only significant for the untreated mice (slope = −0.8 N/%) but much less so for treated mice (slope = −0.002 N/%). For all other potential predictors – negative associations for CTX, uPHOS/Cr, and Ct.Po and positive associations for P1NP, section modulus or Imin/cmin, Ct.Ar, and Ct.Th – Rx was not a significant factor suggesting that the CANA-related increase in P1NP, section modulus, Ct.Ar, and Ct.Th improved bone strength. Likewise, the inability of CANA to improve uPHOS/Cr and CTX had a negative effect on yield force. Lastly, CANA treatment reduced the strength of the already weak negative association between uCA/Cr and yield force (slope = −0.001 N/μg/mg for untreated mice and slope = −0.0002 N/μg/mg).
Table 3:
Coefficients and p-values from linear regression models in which yield force was the dependent variable.
| Model | Ref if not Rx |
p-value | Add to Ref if Rx a |
p-value | Slope | p-value | Slope if Rx b |
p-value | Adj-R2 |
|---|---|---|---|---|---|---|---|---|---|
| Rx + Body mass | +2.57 | 0.1388 | −1.27 | 0.0020 | +0.45 | <0.0001 | NI | 0.414 | |
| Rx + Glucose | +21.58 | <0.0001 | −1.61 | 0.0015 | −0.007 | <0.0001 | NI | 0.293 | |
| Rx + HbA1c | +23.3 | <0.0001 | −5.3 | 0.0030 | −0.8 | <0.0001 | −0.002 | 0.0130 | 0.257 |
| Rx + Insulin c | +16.94 | <0.0001 | zero | 0.6500 | +0.19 | 0.0069 | NI | 0.063 | |
| Rx + P1NP d | +16.09 | <0.0005 | zero | 0.246 | +0.24 | <0.0005 | NI | 0.309 | |
| Rx + CTX c | +18.12 | <0.0001 | zero | 0.5131 | −0.10 | 0.0128 | NI | 0.051 | |
| Rx + uCA/CR | +18.64 | <0.0001 | zero | 0.5005 | −0.001 | <0.0001 | −0.0002 | 0.0047 | 0.142 |
| Rx + uPhos/CR | +17.95 | <0.0001 | zero | 0.1287 | −0.023 | 0.0005 | NI | 0.123 | |
| Rx + Imin/cmin | +2.2 | 0.3633 | zero | 0.9103 | +99.7 | <0.0001 | NI | 0.272 | |
| Rx + Ct.Ar | −8.3 | 0.0005 | zero | 0.7698 | +33.3 | <0.0001 | NI | 0.544 | |
| Rx + Ct.Th | −13 | <0.0001 | zero | 0.6894 | +146 | <0.0001 | NI | 0.611 | |
| Rx + Ct.Po | +29.0 | <0.0001 | zero | 0.5960 | −4.1 | <0.0001 | NI | 0.338 |
If zero, the contribution of the coefficient for treatment (Rx) to the prediction of yield force is negligible (not significant). Ref is the intercept term.
The interaction term was not included (NI) if it was not significant. When included, the coefficient of the slope is different between untreated mice and the CANA-treated mice.
The regression model explaining the variance in yield force is not robust (p<0.025 instead of p≤0.0006 like the other models)
P-values were generated from bootstrapped regression with 1000 replicates because residuals were heteroscedastic.
3.2.6. The relationships between Tb.BV/TV and several outcomes differ between non-treated TH mice and CANA-treated TH mice.
Treatment with CANA also affected relationships between trabecular bone volume fraction in the distal femur metaphysis and several outcome measurements (Table 4). It reduced the slope of the linear relationship between BV/TV and final body mass and caused a negative offset of the relationship between BV/TV and BG as well as between BV/TV and HbA1c. Essentially, a negative association exists between glycemic control and trabecular bone, and so the CANA-related improvement in HbA1c and BG helps preserve BV/TV but not fully, as evident by the significant coefficient for Rx. There were positive associations with P1NP and insulin; and CANA had little effect on their linear relationship with BV/TV. The linear relationships between BV/TV and a resorption marker or mineral excretion were not significant or very weak (Table 4).
Table 4:
Coefficients and p-values from linear regression models in which trabecular bone volume fraction (BV/TV) was the dependent variable.
| Model | Ref if not Rx |
p-value | Add to Ref if Rx a |
p-value | Slope | p-value | Slope if Rx b |
p-value | Adj-R2 |
|---|---|---|---|---|---|---|---|---|---|
| Rx + Body mass d | −7.48 | 0.002 | +6.63 | 0.050 | +0.39 | <0.0005 | +0.18 | 0.036 | 0.335 |
| Rx + Glucose d | +9.18 | <0.0005 | −1.41 | 0.001 | −0.007 | <0.0005 | NI | 0.402 | |
| Rx + HbA1c d | +9.51 | <0.0005 | −3.00 | 0.028 | −0.58 | <0.0001 | −0.11 | 0.062 | 0.221 |
| Rx + Insulin dc | +4.97 | <0.0005 | zero | 0.381 | +0.17 | <0.0005 | NI | 0.093 | |
| Rx + P1NP d | +4.32 | <0.0005 | +0.47 | 0.094 | +0.20 | <0.0005 | NI | 0.365 | |
| Rx + CTX dc | +6.02 | <0.0005 | zero | 0.292 | −0.08 | <0.0005 | NI | 0.065 | |
| Rx + uCA/CR dc | +5.77 | <0.0005 | +0.63 | 0.059 | Zero | 0.148 | NI | 0.072 | |
| Rx + uPhos/CR dc | +5.77 | <0.0005 | +0.58 | 0.086 | zero | 0.119 | NI | 0.084 |
If zero, the contribution of the coefficient to the prediction of yield force is negligible (not significant).
The interaction between the categorical variable (Rx) and the continuous variable was not included (NI) if it was not significant. When included, the coefficient of the slope is different between untreated mice and the Rx-treated mice.
The regression model explaining the variance in yield force is weak (p<0.025 instead of p≤0.0006 like the other models)
P-values were generated from bootstrapped regression with 100 replicates because residuals were heteroscedastic.
3.2.7. Correlations between metabolic parameters and bone parameters reveal different relationships when examined across all untreated mice (Table 5) vs. all CANA-treated mice (Table 6).
Table 5.
Spearman r between metabolic properties vs. bone properties pooling data from untreated mice (High BG + Low BG groups).
N = 51 to 59. Bold font: r ≥ ∣0.6. n.s. = not significant.
| X property | Glucose | HbA1c | Insulin | P1NP | CTX | uCA/Cr | uPHOS/Cr | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Y property | r | p-Value | r | p-Value | r | p-Value | r | p-Value | r | p-Value | r | p-Value | r | p-Value |
| Ma.V | n.s. | 0.3434 | n.s. | 0.1865 | n.s. | 0.6010 | n.s. | 0.2538 | 0.276 | 0.0342 | n.s. | 0.1708 | 0.413 | 0.0019 |
| Ct.Ar | −0.453 | 0.0003 | −0.570 | < 0.0001 | 0.357 | 0.0069 | 0.544 | < 0.0001 | n.s. | 0.4325 | −0.431 | 0.0007 | −0.420 | 0.0016 |
| Tt.Ar | n.s. | 0.1233 | −0.312 | 0.0258 | n.s. | 0.1354 | 0.305 | 0.0189 | n.s. | 0.9692 | n.s. | 0.1684 | n.s. | 0.5498 |
| Imin | −0.329 | 0.0108 | −0.433 | 0.0015 | 0.385 | 0.0034 | 0.457 | 0.0003 | n.s. | 0.7817 | −0.299 | 0.0227 | −0.306 | 0.0244 |
| Imin/cmin | −0.278 | 0.0329 | −0.420 | 0.0022 | 0.343 | 0.0096 | 0.462 | 0.0002 | n.s. | 0.8298 | −0.297 | 0.0238 | n.s. | 0.0596 |
| Ct.Th | −0.536 | < 0.0001 | −0.609 | < 0.0001 | 0.356 | 0.0071 | 0.593 | < 0.0001 | −0.293 | 0.0243 | −0.511 | < 0.0001 | −0.620 | < 0.0001 |
| Ct.TMD | n.s. | 0.4809 | n.s. | 0.9163 | n.s. | 0.1724 | n.s. | 0.2816 | −0.334 | 0.0098 | n.s. | 0.9129 | n.s. | 0.1231 |
| Ct.Po | 0.345 | 0.0074 | 0.506 | 0.0002 | n.s. | 0.4924 | −0.597 | < 0.0001 | 0.576 | < 0.0001 | 0.340 | 0.0091 | 0.509 | 0.0001 |
| Stiffness | −0.462 | 0.0003 | −0.469 | 0.0006 | 0.278 | 0.0399 | 0.638 | < 0.0001 | −0.338 | 0.0095 | −0.328 | 0.0127 | −0.330 | 0.0158 |
| Yield force | −0.639 | < 0.0001 | −0.705 | < 0.0001 | 0.469 | 0.0003 | 0.618 | < 0.0001 | n.s. | 0.1560 | −0.641 | < 0.0001 | −0.623 | < 0.0001 |
| Ultimate force | −0.480 | 0.0001 | −0.634 | < 0.0001 | 0.345 | 0.0098 | 0.620 | < 0.0001 | −0.303 | 0.0208 | −0.476 | 0.0002 | −0.459 | 0.0005 |
| PYD | n.s. | 0.0689 | n.s. | 0.0987 | n.s. | 0.6638 | n.s. | 0.5152 | n.s. | 0.9802 | n.s. | 0.1413 | n.s. | 0.0727 |
| Work-to-fracture | n.s. | 0.9628 | n.s. | 0.4714 | n.s. | 0.5531 | n.s. | 0.1427 | n.s. | 0.2292 | n.s. | 0.7098 | n.s. | 0.9954 |
| Length | n.s. | 0.2426 | n.s. | 0.2369 | −0.293 | 0.0298 | n.s. | 0.7809 | n.s. | 0.3194 | n.s. | 0.0810 | 0.436 | 0.0011 |
| Yield stress | −0.555 | < 0.0001 | −0.575 | < 0.0001 | 0.350 | 0.0088 | 0.410 | 0.0014 | n.s. | 0.0871 | −0.558 | < 0.0001 | −0.573 | < 0.0001 |
| Ultimate stress | −0.349 | 0.0073 | −0.513 | 0.0001 | n.s. | 0.2806 | 0.459 | 0.0003 | −0.376 | 0.0036 | −0.368 | 0.0049 | −0.344 | 0.0116 |
| Modulus | n.s. | 0.1000 | n.s. | 0.2307 | n.s. | 0.7506 | 0.352 | 0.0067 | −0.464 | 0.0002 | n.s. | 0.4836 | n.s. | 0.2665 |
| Toughness | n.s. | 0.3294 | n.s. | 0.7275 | n.s. | 0.8667 | n.s. | 0.7005 | n.s. | 0.2471 | n.s. | 0.5190 | n.s. | 0.3715 |
| PY toughness | n.s. | 0.0834 | n.s. | 0.0545 | n.s. | 0.8749 | n.s. | 0.5631 | n.s. | 0.9063 | n.s. | 0.1207 | n.s. | 0.0812 |
| BV/TV | −0.702 | < 0.0001 | −0.653 | < 0.0001 | 0.486 | 0.0001 | 0.515 | < 0.0001 | n.s. | 0.5056 | −0.567 | < 0.0001 | −0.585 | < 0.0001 |
| Conn.D | n.s. | 0.4290 | n.s. | 0.1502 | n.s. | 0.8887 | n.s. | 0.2338 | n.s. | 0.5152 | n.s. | 0.5703 | n.s. | 0.6154 |
| SMI | 0.456 | 0.0003 | 0.498 | 0.0002 | n.s. | 0.3765 | −0.316 | 0.0149 | n.s. | 0.5441 | 0.446 | 0.0004 | 0.340 | 0.0118 |
| Tb.N | n.s. | 0.2440 | −0.296 | 0.0350 | n.s. | 0.6555 | n.s. | 0.1477 | n.s. | 0.8174 | n.s. | 0.1207 | n.s. | 0.1580 |
| Tb.Th | −0.706 | < 0.0001 | −0.555 | < 0.0001 | 0.568 | < 0.0001 | 0.532 | < 0.0001 | n.s. | 0.3755 | −0.505 | 0.0001 | −0.562 | < 0.0001 |
| Tb.Sp | n.s. | 0.2997 | n.s. | 0.0557 | n.s. | 0.7443 | n.s. | 0.1422 | n.s. | 0.9434 | n.s. | 0.1703 | n.s. | 0.2110 |
| Tb.TMD | −0.356 | 0.0056 | n.s. | 0.0884 | n.s. | 0.6681 | 0.369 | 0.0040 | −0.447 | 0.0004 | n.s. | 0.1774 | −0.373 | 0.0054 |
Table 6.
Spearman r between metabolic properties vs. bone properties pooling data from CANA-treated mice. N = 47 to 51.
| X property | Glucose | HbA1c | Insulin | P1NP | CTX | uCA/Cr | uPHOS/Cr | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Y property | r | p-Value | r | p-Value | r | p-Value | r | p-Value | r | p-Value | r | p-Value | r | p-Value |
| Ma.V | 0.424 | 0.0019 | n.s. | 0.2970 | n.s. | 0.2568 | n.s. | 0.4481 | 0.299 | 0.0348 | n.s. | 0.1616 | 0.304 | 0.0318 |
| Ct.Ar | n.s. | 0.6254 | n.s. | 0.8661 | n.s. | 0.5555 | 0.421 | 0.0021 | n.s. | 0.5427 | n.s. | 0.4518 | n.s. | 0.3168 |
| Tt.Ar | n.s. | 0.1676 | n.s. | 0.4537 | n.s. | 0.5672 | n.s. | 0.1593 | n.s. | 0.3900 | n.s. | 0.6783 | n.s. | 0.5101 |
| Imin | n.s. | 0.7943 | n.s. | 0.0854 | n.s. | 0.9167 | 0.400 | 0.0036 | n.s. | 0.9644 | n.s. | 0.3935 | n.s. | 0.5570 |
| Imin/cmin | n.s. | 0.8584 | n.s. | 0.1289 | n.s. | 0.8616 | 0.421 | 0.0021 | n.s. | 0.9487 | n.s. | 0.6311 | n.s. | 0.7599 |
| Ct.Th | −0.478 | 0.0004 | n.s. | 0.8736 | n.s. | 0.1071 | 0.595 | < 0.0001 | −0.367 | 0.0088 | −0.359 | 0.0103 | −0.495 | 0.0003 |
| Ct.TMD | −0.359 | 0.0097 | n.s. | 0.0869 | n.s. | 0.7934 | 0.311 | 0.0265 | −0.339 | 0.0160 | n.s. | 0.8781 | n.s. | 0.1887 |
| Ct.Po | 0.580 | < 0.0001 | n.s. | 0.9596 | n.s. | 0.2719 | −0.483 | 0.0003 | 0.411 | 0.0030 | 0.319 | 0.0237 | 0.440 | 0.0014 |
| Stiffness | n.s. | 0.2150 | n.s. | 0.4363 | n.s. | 0.2243 | 0.332 | 0.0185 | n.s. | 0.7634 | n.s. | 0.0628 | −0.293 | 0.0407 |
| Yield force | −0.344 | 0.0143 | n.s. | 0.6657 | n.s. | 0.8018 | 0.521 | 0.0001 | n.s. | 0.0761 | n.s. | 0.6391 | n.s. | 0.0780 |
| Ultimate force | n.s. | 0.3848 | n.s. | 0.3729 | n.s. | 0.5486 | 0.453 | 0.0009 | n.s. | 0.1445 | n.s. | 0.1637 | n.s. | 0.0692 |
| PYD | n.s. | 0.6910 | n.s. | 0.4411 | n.s. | 0.5584 | n.s. | 0.8503 | n.s. | 0.1997 | n.s. | 0.9039 | n.s. | 0.4678 |
| Work-to-fracture | n.s. | 0.9277 | n.s. | 0.5639 | n.s. | 0.5559 | n.s. | 0.3993 | n.s. | 0.0731 | n.s. | 0.7599 | n.s. | 0.5978 |
| Length | n.s. | 0.2645 | n.s. | 0.2414 | n.s. | 0.8665 | n.s. | 0.9554 | n.s. | 0.8036 | n.s. | 0.8634 | n.s. | 0.8039 |
| Yield stress | −0.395 | 0.0046 | n.s. | 0.1510 | n.s. | 0.5340 | 0.298 | 0.0359 | n.s. | 0.0967 | n.s. | 0.4658 | −0.291 | 0.0422 |
| Ultimate stress | n.s. | 0.4066 | n.s. | 0.6522 | n.s. | 0.4023 | n.s. | 0.4477 | n.s. | 0.0881 | n.s. | 0.3606 | n.s. | 0.0976 |
| Modulus | n.s. | 0.1355 | n.s. | 0.3817 | n.s. | 0.1155 | n.s. | 0.8037 | n.s. | 0.9791 | n.s. | 0.0718 | −0.328 | 0.0214 |
| Toughness | n.s. | 0.9045 | n.s. | 0.5031 | n.s. | 0.5674 | n.s. | 0.4689 | n.s. | 0.0678 | n.s. | 0.5675 | n.s. | 0.4610 |
| PY toughness | n.s. | 0.3609 | n.s. | 0.6412 | n.s. | 0.4800 | n.s. | 0.7906 | n.s. | 0.4964 | n.s. | 0.3704 | n.s. | 0.1666 |
| BV/TV | −0.421 | 0.0021 | n.s. | 0.4652 | n.s. | 0.2573 | 0.516 | 0.0001 | n.s. | 0.0508 | −0.431 | 0.0018 | −0.434 | 0.0016 |
| Conn.D | n.s. | 0.7887 | n.s. | 0.9605 | n.s. | 0.2717 | n.s. | 0.8907 | n.s. | 0.9091 | n.s. | 0.2826 | n.s. | 0.6273 |
| SMI | n.s. | 0.0816 | n.s. | 0.4301 | n.s. | 0.0761 | n.s. | 0.2041 | n.s. | 0.2300 | 0.306 | 0.0308 | n.s. | 0.0699 |
| Tb.N | n.s. | 0.7510 | n.s. | 0.1363 | n.s. | 0.4336 | n.s. | 0.1056 | n.s. | 0.0987 | n.s. | 0.1557 | n.s. | 0.3236 |
| Tb.Th | −0.463 | 0.0006 | n.s. | 0.6739 | n.s. | 0.9073 | 0.488 | 0.0003 | n.s. | 0.4949 | n.s. | 0.1284 | −0.351 | 0.0126 |
| Tb.Sp | n.s. | 0.7052 | n.s. | 0.1704 | n.s. | 0.3960 | n.s. | 0.0930 | n.s. | 0.0713 | n.s. | 0.1672 | n.s. | 0.3333 |
| Tb.TMD | −0.508 | 0.0001 | n.s. | 0.9652 | n.s. | 0.6142 | 0.490 | 0.0003 | −0.308 | 0.0293 | n.s. | 0.6746 | n.s. | 0.2383 |
Among untreated mice (High BG + Low BG groups combined), HbA1c and post-treatment BG, as markers of glycemic control, along with uCA/Cr and uPHOS/Cr, as markers of urine mineral loss, correlated with deterioration of structural properties of cortical and trabecular bone (Table 5). With respect to cortical bone, HbA1c and BG were negatively correlated with Ct.Ar, Ct.Th, and Imin, yet were positively correlated with Ct.Po. Similarly, uCA/Cr and uPHOS/Cr were negatively correlated with Ct.Ar, Ct.Th and Imin and positively correlated with Ma.V (uPHOS/Cr only) and Ct.Po (Table 5). With respect to trabecular bone, all 4 parameters (HbA1c, BG, uCA/Cr, uPHOS/Cr) correlated negatively with Tb.BV/TV, Tb.Th and Tb.TMD (BG and uPHOS/Cr only) and correlated positively with SMI (Table 5). Similarly, these parameters negatively correlated with indicators of bone strength (yield force, ultimate force, yield strength, ultimate strength) and bone stiffness.
In contrast, P1NP, a marker of bone formation, correlated with improvement in structural properties of cortical bone (positively correlated with Ct.Ar, Ct.Th, Imin; negatively correlated with Ct.Po) (Table 5) or trabecular bone (positively correlated with Tb.BV/TV, Tb.Th, Tb.TMD; negatively correlated with SMI) (Table 5). P1NP also correlated positively with bone strength and stiffness.
When examined within the CANA-treated mice alone (Table 6) these same relationships between metabolic properties and bone properties were often not significantly correlated. Only P1NP exhibited associations with bone parameters which were comparable in strength and directionality to those associations seen with untreated mice. (Spearman correlations between measurements of metabolism or urine mineral excretion and bone properties when all 3 groups are combined are included in Tables S4 and S5.)
4. Discussion
In contrast to previous reports by our groups and others comparing skeletal features between the TallyHO/JngJ mice with diabetes and SWR/J mice without the disease (17, 18), unique features of the TH polygenic, incompletely penetrant model of type 2 diabetes have allowed us to compare the impact of diabetes, independent of genetic strain differences, on skeletal phenotype. Herein, we have focused on skeletal differences between subgroups of TH mice, either with or without hyperglycemia and with or without exposure to glucose-lowering therapy with an SGLT2-inhibitor, specifically canagliflozin. In so doing, we have demonstrated that by 20 weeks of age, male TH mice with diabetes had comparatively lower body mass, and exhibited decrements in cortical and trabecular bone structure, along with an increase in cortical porosity, compared to non-diabetic TH mice. The urinary excretion of critical bone minerals, specifically calcium and phosphorus, was exaggerated in diabetic mice, and related to the degree of glucose excretion. Additionally, bone formation was reduced. Moreover, these changes in metabolism were associated with a reduction in overall bone strength and bone stiffness as determined by three-point bending tests of femur mid-shafts. Interestingly, however, bone toughness, or overall resistance to fracture, was not altered, perhaps reflecting a lack of T2D-related changes to the organic matrix that would impart brittleness (e.g., accumulation of advanced glycation end-products) and/or a decrease in mineral from the organic matrix at local sites (e.g., peri-lacunar) that would confer more strain capacity to the bone.
Treatment with an SGLT2-inhibitor such as canagliflozin normalizes blood glucose levels by enhancing urinary glucose excretion, rather than by modifying insulin secretion or sensitivity. In our experiments, SGLT2I treatment, as expected, was shown to normalize serum blood glucose levels and body weight in TH mice, most of whom (>75%) would have exhibited persistent diabetes out to 20 weeks of age without treatment. However, consistent with a drug-induced glucosuria, both calcium and phosphate wasting persisted in CANA-treated mice at study end. Additionally, the increase in cortical porosity estimate, the deficits in trabecular bone structure, and the reduction in bone strength and bone stiffness seen in the diabetic bone phenotype were not fully rectified with canagliflozin, despite good glycemic effect of the drug.
A reduction in bone stiffness and strength, as was seen in either TH mice with diabetes or in CANA-treated euglycemic mice, would be anticipated in disorders where a decrease in tissue mineralization exists (33). Evidence in clinical literature supports an association between glucosuria, with or without hyperglycemia, and urinary mineral excretion. For example, adolescent females with type 1 diabetes were shown to have negative calcium retention, attributed to higher urinary calcium excretion, which was positively correlated with HbA1c values, suggesting an increase in calcium excretion in those individuals with poorer glycemic control (34). Similarly, historical data examining children with more poorly controlled diabetes mellitus (mean HbA1c = 10.0 ± 0.3%) demonstrated simultaneous hypercalciuria and hyperphosphaturia, related to glucosuria, which was closely associated with the severity of linear growth failure (i.e. skeletal growth failure) in these children (35). Children with isolated renal glucosuria, without diabetes, also exhibit hypercalciuria (36). Finally, in patients with renal Fanconi syndrome who exhibit glucosuria and phosphaturia, hypophosphatemic osteomalacia or rickets is seen (37, 38).
In the present study, the excess urinary mineral loss of both calcium and phosphate, documented at study end, occurred in mice exhibiting glucosuria (i.e., those with diabetes, or those receiving SGLT2I treatment) such that the magnitude of mineral wasting was highly correlated with the magnitude of glucose excretion (Fig. 2B, 2D); therefore, some demineralization of bone could be expected in these mice over time, unless offset by increased mineral acquisition in the diet, or unless our end-point measures were not consistent with mineral excretion at earlier times (i.e. start of drug therapy). We also cannot ignore the observed relative reduction in daily PHOS excretion per daily glucose excretion in CANA-treated mice (Fig 2D) compared with High BG mice, given that SGLT2I mechanism of action would predict increased sodium-dependent phosphate reabsorption in the renal proximal tubule in CANA-treated mice, despite glucosuria, at least at the initiation of therapy (39). Interestingly, the extent of mineral loss from the organic matrix was not sufficient to significantly affect μCT-derived Ct.TMD (Table 1).
An association between excess calcium excretion, specifically, and subsequent bone disease has been validated clinically. For example, patients with idiopathic hypercalciuria demonstrate reduced bone density (40, 41). Population studies of women with post-menopausal osteoporosis also confirm an increased prevalence of PTH-independent hypercalciuria (42, 43). Moreover, bone biopsy specimens from hypercalciuric patients display evidence of a mineralization defect, consistent with osteomalacia (44, 45). Other studies have also shown that the rate of urine calcium excretion in humans appears to correlate with the degree of bone loss (46, 47). Together, these studies support the expectation that hypercalciuria will negatively impact bone mineralization; hence, a similar skeletal outcome from glucosuria-associated hypercalciuria in TH mice could be anticipated.
Our results in the TallyHO model of T2D are consistent with previous reports by us and others demonstrating hypercalciuria in rodent models of diabetes or SGLT2I-related glucosuria. For example, in DBA/2J mice treated with streptozotocin (STZ), significant glucosuria and hypercalciuria were evident in diabetic mice, as well is in canagliflozin-treated diabetic and non-diabetic DBA/2J mice (25, 48). In pregnant rats, maternal diabetes is also associated with marked hypercalciuria, when compared with non-diabetic pregnant rats (49). Furthermore, genetic ablation of SGLT2 function in the Jimbee mouse model was found to result in glucosuria and hypercalciuria, along with growth stunting and a reduction in tissue mineral density in male mice (50). A study by Wang, et al, in the Zucker Diabetic Fatty (ZDF) rat model of T2D also demonstrated evidence of hypercalciuria and hyperphosphaturia in diabetic rodents although glucosuria was not specifically documented in their publication (51); however, in their study, treatment with dapagliflozin (another SGLT2I), resulted in alleviation of hypercalciuria. Reasons for this discrepancy are unclear, but could reflect drug-specific differences on renal handling of calcium (canagliflozin vs. dapagliflozin), differences in the T2D model (rat vs. mouse), or the small sample size used in the Wang, et al study (n=6-7 rats/group) (51), compared with our study.
In other rodent models of diabetes, the reduction in the fracture resistance of long bone has been attributed to the hyperglycemia-mediated accumulation of advanced glycation end-products, namely pentosidine, a fluorescent collagen crosslink (52, 53). While we did observe a decrease in bone strength in hyperglycemic TH mice, we did not observe a decrease in work-to-fracture or post-yield toughness in these mice; additionally, we did not see full improvement in bone strength with canagliflozin despite normalization of glucose control. Finally, while bone strength parameters were inversely associated with HbA1c values in non-treated mice (Table 5), these parameters were unrelated to HbA1c in CANA-treatment mice (Table 6). Moreover, these parameters did not improve in conjunction with the excess daily glucose excretion seen in CANA-treated mice. Together, these observations suggest that changes in stiffness, strength (yield force and yield stress), and overall fracture resistance across the groups cannot be explained by hyperglycemia alone, or hyperglycemia mitigation, in this model.
An increase in cortical porosity is a common finding among rodent models of T2D, including the ZDF rat (54), the Zucker Diabetic Sprague-Dawley (ZDSD) rat (53), and the Otsuka Long-Evans Tokushima Fatty rat (55). An association between porosity and severity of dysglycemia has been demonstrated in these same studies. Similarly, an increase in distal radial cortical porosity has been demonstrated in elderly females with T2D (56-59), particularly in those with history of fragility fractures (60). An association between higher porosity and higher HbA1c has also been demonstrated in some clinical studies (61). However, to our knowledge, an association between exaggerated urinary mineral loss and cortical porosity has not been reported, though lacunar and canicular porosity has been observed to increase in lactating mice (62). In the present study, Ct.Po estimate was significantly higher (11.3% on average) in High BG mice compared to Low BG mice, and persisted in CANA-treated mice. In our prior TH study (18), Ct.Po appeared to be similar between the 4-5 TH mice per age group (16-wks. and 34-wks.) that had high blood glucose levels – but not as high as the TH mice in the present study – and the 7-10 TH mice per age group that had low (16-wks.) or moderate BG levels (34-wks.). This supports the notion that the T2D-related increase in cortical porosity is the result of severe hyperglycemia (i.e., frank, uncontrolled diabetes).
Interpretation of our results are limited by several factors. First, this study was not designed to provide a comprehensive assessment of calcium or phosphate metabolism with respect to bone strength. Specifically, we did not measure serum calcium or phosphorus concentrations. Additionally, we did not rigorously quantify dietary mineral intake (calcium or phosphorus), gastrointestinal absorption, or total body mineral retention. Estimated food intake available from 2 of 3 experimental replicates indicated that food consumption was increased in both the High BG groups, and the CANA groups, relative to the Low BG group [CANA: 0.18 ± 0.04 g (chow)/g (mouse)/day (providing 18 mg/kg/day of canaglifozin); High BG: 0.23 ± 0.06 g (chow)/g (mouse)/day; Low BG: 0.12 ± 0.03 g (chow)/g (mouse)/day; ANOVA p<0.0001], resulting in a relative increase in daily dietary calcium and phosphorus intake in mice that consumed more chow. However, total body mineral balance was not specifically quantified. Therefore, any link between mineral excretion and skeletal mineralization remains indirect, observational and hypothetical at present. Additionally, at this time we have studied only one of the SGLT2I medications (canagliflozin), given at only one dose. Therefore, our findings are not generalizable to other members of the SGLT2I drug class, and our results differ, as noted above, from those reported for dapagliflozin (51). Although canagliflozin therapy did not affect the material strength (ultimate stress) of cortical bone in TH mice at the apparent level, we do not know whether this SGLT2 inhibitor affected mechanical or compositional properties at the tissue-level. In another pre-clinical model of diabetes involving male mice (fed a high fat diet and administered a low dose of STZ), dapagliflozin and liraglutide rescued or partially rescued the mineral-to-matrix ratio, crystal size, and glycation (Fourier transformed infrared spectroscopy or FTIR) as well as hardness and modulus (nanoindentation) of newly formed tissue (i.e., tissue between double labels) (63). Treatment effects on composition were not observed, however, when a quadrant of cortical bone (femur mid-shaft) comprising more existent than newly formed tissue was analyzed by FTIR imaging. Finally, our results are pertinent only to male TallyHO/JngJ mice. Female TH mice develop obesity, moderate hyperinsulinemia, and hyperleptinemia, but do not progress to frank diabetes (14).
In summary, this study has now delineated the diabetic bone phenotype, independent of obesity, in the TallyHO/JngJ model of early onset type 2 diabetes mellitus involving male mice. We have also demonstrated that mitigation of hyperglycemia in this model by an SGLT2-inhibitor only partially rectifies the bone deficits that occur when blood glucose levels are very high. Instead, glucosuria-associated mineral wasting persisted with long-term SGLT2I exposure, likely complicating the ability of this anti-diabetic therapy to restore bone despite glucose normalization.
Supplementary Material
HIGHLIGHTS.
TallyHO (TH) mouse is a polygenic, incompletely penetrant model of early onset T2D
Deficits in cortical bone structure, strength exist in diabetic TH femurs at 20 wks
Deficits in trabecular bone exist in diabetic TH femurs, L6 vertebrae
Canagliflozin treatment of diabetic TH mice for 12 weeks normalizes glucose control
However, bone deficits and urine mineral wasting persist with SGLT2I treatment
Acknowledgments and Funding:
This work was supported by National Institutes of Health Grants, R21AR070620 (to K.M.T and J.S.N.) and R56DK084045 (to J.L.F.), as well as the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development 1I01BX004297 (to J.S.N.). Additional funding was provided by the University of Kentucky Barnstable Brown Diabetes Center Endowment. The authors have no conflict of interest to declare.
Funding Source: This work was supported by grants from the National Institutes of Health, R21AR070620 (to K.M.T and J.S.N.) and R56DK084045 (to J.L.F.); as well as the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development 1I01BX004297 (to J.S.N.). Additional funding was provided by the University of Kentucky Barnstable Brown Diabetes Center.
Footnotes
Duality of Interests:
The authors have no financial or personal conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Janghorbani M, Van Dam RM, Willett WC, Hu FB. Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. American journal of epidemiology. 2007;166(5):495–505. [DOI] [PubMed] [Google Scholar]
- 2.Nicodemus KK, Folsom AR, Iowa Women's Health S. Type 1 and type 2 diabetes and incident hip fractures in postmenopausal women. Diabetes care. 2001;24(7):1192–7. [DOI] [PubMed] [Google Scholar]
- 3.Schwartz AV, Vittinghoff E, Bauer DC, Hillier TA, Strotmeyer ES, Ensrud KE, et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. Jama. 2011;305(21):2184–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwartz AV, Vittinghoff E, Sellmeyer DE, Feingold KR, de Rekeneire N, Strotmeyer ES, et al. Diabetes-related complications, glycemic control, and falls in older adults. Diabetes care. 2008;31(3):391–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dhaliwal R, Cibula D, Ghosh C, Weinstock RS, Moses AM. Bone quality assessment in type 2 diabetes mellitus. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2014;25(7):1969–73. [DOI] [PubMed] [Google Scholar]
- 6.Bailey DA. The Saskatchewan Pediatric Bone Mineral Accrual Study: bone mineral acquisition during the growing years. International journal of sports medicine. 1997;18 Suppl 3:S191–4. [DOI] [PubMed] [Google Scholar]
- 7.Lascar N, Brown J, Pattison H, Barnett AH, Bailey CJ, Bellary S. Type 2 diabetes in adolescents and young adults. The lancet Diabetes & endocrinology. 2018;6(1):69–80. [DOI] [PubMed] [Google Scholar]
- 8.Nadeau KJ, Anderson BJ, Berg EG, Chiang JL, Chou H, Copeland KC, et al. Youth-Onset Type 2 Diabetes Consensus Report: Current Status, Challenges, and Priorities. Diabetes care. 2016;39(9):1635–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mayer-Davis EJ, Lawrence JM, Dabelea D, Divers J, Isom S, Dolan L, et al. Incidence Trends of Type 1 and Type 2 Diabetes among Youths, 2002-2012. The New England journal of medicine. 2017;376(15):1419–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Group TS. Effects of metformin, metformin plus rosiglitazone, and metformin plus lifestyle on insulin sensitivity and beta-cell function in TODAY. Diabetes care. 2013;36(6):1749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Group TS, Zeitler P, Hirst K, Pyle L, Linder B, Copeland K, et al. A clinical trial to maintain glycemic control in youth with type 2 diabetes. The New England journal of medicine. 2012;366(24):2247–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Amutha A, Mohan V. Diabetes complications in childhood and adolescent onset type 2 diabetes-a review. Journal of diabetes and its complications. 2016;30(5):951–7. [DOI] [PubMed] [Google Scholar]
- 13.Tryggestad JB, Willi SM. Complications and comorbidities of T2DM in adolescents: findings from the TODAY clinical trial. Journal of diabetes and its complications. 2015;29(2):307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim JH, Stewart TP, Soltani-Bejnood M, Wang L, Fortuna JM, Mostafa OA, et al. Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. The Journal of endocrinology. 2006;191(2):437–46. [DOI] [PubMed] [Google Scholar]
- 15.Kim JH, Sen S, Avery CS, Simpson E, Chandler P, Nishina PM, et al. Genetic analysis of a new mouse model for non-insulin-dependent diabetes. Genomics. 2001;74(3):273–86. [DOI] [PubMed] [Google Scholar]
- 16.Kim JH, Saxon AM. The TallyHO Mouse as a Model of Human Type 2 Diabetes In: Joost H, Al-Hasani H, Schurmann A, editors. Animal Models in Diabetes Research Methods in Molecular Biology (Methods and Protocols): Humana Press, Totowa, NJ; 2012. [Google Scholar]
- 17.Devlin MJ, Van Vliet M, Motyl K, Karim L, Brooks DJ, Louis L, et al. Early-onset type 2 diabetes impairs skeletal acquisition in the male TALLYHO/JngJ mouse. Endocrinology. 2014;155(10):3806–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Creecy A, Uppuganti S, Unal M, Clay Bunn R, Voziyan P, Nyman JS. Low bone toughness in the TallyHO model of juvenile type 2 diabetes does not worsen with age. Bone. 2018;110:204–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neschen S, Scheerer M, Seelig A, Huypens P, Schultheiss J, Wu M, et al. Metformin supports the antidiabetic effect of a sodium glucose cotransporter 2 inhibitor by suppressing endogenous glucose production in diabetic mice. Diabetes. 2015;64(1):284–90. [DOI] [PubMed] [Google Scholar]
- 20.Giorgino F, Vora J, Fenici P, Solini A. Cardiovascular protection with SGLT2 inhibitors in type 2 diabetes mellitus: does it apply to all patients? Diabetes, obesity & metabolism. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Watts NB, Bilezikian JP, Usiskin K, Edwards R, Desai M, Law G, et al. Effects of Canagliflozin on Fracture Risk in Patients With Type 2 Diabetes Mellitus. The Journal of clinical endocrinology and metabolism. 2016;101(1):157–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou Z, Jardine M, Perkovic V, Matthews DR, Mahaffey KW, de Zeeuw D, et al. Canagliflozin and fracture risk in individuals with type 2 diabetes: results from the CANVAS Program. Diabetologia. 2019;62(10):1854–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Erythropoulou-Kaltsidou A, Polychronopoulos G, Tziomalos K. Sodium-Glucose Co-Transporter 2 Inhibitors and Fracture Risk. Diabetes therapy : research, treatment and education of diabetes and related disorders. 2020;11(1):7–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7(2):27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thrailkill KM, Nyman JS, Bunn RC, Uppuganti S, Thompson KL, Lumpkin CK Jr., et al. The impact of SGLT2 inhibitors, compared with insulin, on diabetic bone disease in a mouse model of type 1 diabetes. Bone. 2017;94:141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faul F, Erdfelder E, Lang AG, Buchner A. G*Power 3: a flexible statistical power analysis program for the social, behavioral, and biomedical sciences. Behav Res Methods. 2007;39(2):175–91. [DOI] [PubMed] [Google Scholar]
- 27.Faul F, Erdfelder E, Buchner A, Lang AG. Statistical power analyses using G*Power 3.1: tests for correlation and regression analyses. Behav Res Methods. 2009;41(4):1149–60. [DOI] [PubMed] [Google Scholar]
- 28.Han BG, Hao CM, Tchekneva EE, Wang YY, Lee CA, Ebrahim B, et al. Markers of glycemic control in the mouse: comparisons of 6-h- and overnight-fasted blood glucoses to HbA1c. American journal of physiology Endocrinology and metabolism. 2008;295(4):E981–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Creecy A, Uppuganti S, Girard MR, Schlunk SG, Amah C, Granke M, et al. The age-related decrease in material properties of BALB/c mouse long bones involves alterations to the extracellular matrix. Bone. 2020;130:115126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nyman JS, Even JL, Jo CH, Herbert EG, Murry MR, Cockrell GE, et al. Increasing duration of type 1 diabetes perturbs the strength-structure relationship and increases brittleness of bone. Bone. 2011;48(4):733–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thrailkill K, Bunn RC, Lumpkin C Jr., Wahl E, Cockrell G, Morris L, et al. Loss of insulin receptor in osteoprogenitor cells impairs structural strength of bone. Journal of diabetes research. 2014;2014:703589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Uppuganti S, Granke M, Makowski AJ, Does MD, Nyman JS. Age-related changes in the fracture resistance of male Fischer F344 rat bone. Bone. 2016;83:220–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turner CH. Bone strength: current concepts. Ann N Y Acad Sci. 2006;1068:429–46. [DOI] [PubMed] [Google Scholar]
- 34.Weber DR, O'Brien KO, Schwartz GJ. Evidence of disordered calcium metabolism in adolescent girls with type 1 diabetes: An observational study using a dual-stable calcium isotope technique. Bone. 2017;105:184–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malone JI, Lowitt S, Duncan JA, Shah SC, Vargas A, Root AW. Hypercalciuria, hyperphosphaturia, and growth retardation in children with diabetes mellitus. Pediatrics. 1986;78(2):298–304. [PubMed] [Google Scholar]
- 36.Schneider D, Gauthier B, Trachtman H. Hypercalciuria in children with renal glycosuria: evidence of dual renal tubular reabsorptive defects. The Journal of pediatrics. 1992;121(5 Pt 1):715–9. [DOI] [PubMed] [Google Scholar]
- 37.Alacapa LF, Sandoval MAS, Dimacali CTD, Orillaza NJS. Glucosuria without diabetes: key to the diagnosis of fragility fractures due to Fanconi syndrome. BMJ Case Rep. 2018;2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rao DS, Parfitt AM, Villanueva AR, Dorman PJ, Kleerekoper M. Hypophosphatemic osteomalacia and adult Fanconi syndrome due to light-chain nephropathy. Another form of oncogenous osteomalacia. Am J Med. 1987;82(2):333–8. [DOI] [PubMed] [Google Scholar]
- 39.Vinke JSJ, Heerspink HJL, de Borst MH. Effects of sodium glucose cotransporter 2 inhibitors on mineral metabolism in type 2 diabetes mellitus. Curr Opin Nephrol Hypertens. 2019;28(4):321–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sella S, Cattelan C, Realdi G, Giannini S. Bone disease in primary hypercalciuria. Clinical cases in mineral and bone metabolism : the official journal of the Italian Society of Osteoporosis, Mineral Metabolism, and Skeletal Diseases. 2008;5(2):118–26. [PMC free article] [PubMed] [Google Scholar]
- 41.Pietschmann F, Breslau NA, Pak CY. Reduced vertebral bone density in hypercalciuric nephrolithiasis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 1992;7(12):1383–8. [DOI] [PubMed] [Google Scholar]
- 42.Giannini S, Nobile M, Dalle Carbonare L, Lodetti MG, Sella S, Vittadello G, et al. Hypercalciuria is a common and important finding in postmenopausal women with osteoporosis. European journal of endocrinology. 2003;149(3):209–13. [DOI] [PubMed] [Google Scholar]
- 43.Heshmati HM, Khosla S, Burritt MF, O'Fallon WM, Riggs BL. A defect in renal calcium conservation may contribute to the pathogenesis of postmenopausal osteoporosis. The Journal of clinical endocrinology and metabolism. 1998;83(6):1916–20. [DOI] [PubMed] [Google Scholar]
- 44.Steiniche T, Mosekilde L, Christensen MS, Melsen F. A histomorphometric determination of iliac bone remodeling in patients with recurrent renal stone formation and idiopathic hypercalciuria. APMIS. 1989;97(4):309–16. [DOI] [PubMed] [Google Scholar]
- 45.Malluche HH, Tschoepe W, Ritz E, Meyer-Sabellek W, Massry SG. Abnormal bone histology in idiopathic hypercalciuria. The Journal of clinical endocrinology and metabolism. 1980;50(4):654–8. [DOI] [PubMed] [Google Scholar]
- 46.Weisinger JR, Alonzo E, Bellorin-Font E, Blasini AM, Rodriguez MA, Paz-Martinez V, et al. Possible role of cytokines on the bone mineral loss in idiopathic hypercalciuria. Kidney international. 1996;49(1):244–50. [DOI] [PubMed] [Google Scholar]
- 47.Barkin J, Wilson DR, Manuel MA, Bayley A, Murray T, Harrison J. Bone mineral content in idiopathic calcium nephrolithiasis. Miner Electrolyte Metab. 1985;11(1):19–24. [PubMed] [Google Scholar]
- 48.Thrailkill KM, Clay Bunn R, Nyman JS, Rettiganti MR, Cockrell GE, Wahl EC, et al. SGLT2 inhibitor therapy improves blood glucose but does not prevent diabetic bone disease in diabetic DBA/2J male mice. Bone. 2016;82:101–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Birdsey TJ, Husain SM, Garland HO, Sibley CP. The effect of diabetes mellitus on urinary calcium excretion in pregnant rats and their offspring. The Journal of endocrinology. 1995;145(1):11–8. [DOI] [PubMed] [Google Scholar]
- 50.Thrailkill KM, Bunn RC, Uppuganti S, Ray P, Garrett K, Popescu I, et al. Genetic ablation of SGLT2 function in mice impairs tissue mineral density but does not affect fracture resistance of bone. Bone. 2020;133:115254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang JY, Cheng YZ, Yang SL, An M, Zhang H, Chen H, et al. Dapagliflozin Attenuates Hyperglycemia Related Osteoporosis in ZDF Rats by Alleviating Hypercalciuria. Frontiers in endocrinology. 2019;10:700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saito M, Fujii K, Mori Y, Marumo K. Role of collagen enzymatic and glycation induced cross-links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2006;17(10):1514–23. [DOI] [PubMed] [Google Scholar]
- 53.Creecy A, Uppuganti S, Merkel AR, O'Neal D, Makowski AJ, Granke M, et al. Changes in the Fracture Resistance of Bone with the Progression of Type 2 Diabetes in the ZDSD Rat. Calcified tissue international. 2016;99(3):289–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Campbell GM, Tiwari S, Picke AK, Hofbauer C, Rauner M, Morlock MM, et al. Effects of insulin therapy on porosity, non-enzymatic glycation and mechanical competence in the bone of rats with type 2 diabetes mellitus. Bone. 2016;91:186–93. [DOI] [PubMed] [Google Scholar]
- 55.Ikedo A, Kido K, Ato S, Sato K, Lee JW, Fujita S, et al. The effects of resistance training on bone mineral density and bone quality in type 2 diabetic rats. Physiological reports. 2019;7(6):e14046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burghardt AJ, Issever AS, Schwartz AV, Davis KA, Masharani U, Majumdar S, et al. High-resolution peripheral quantitative computed tomographic imaging of cortical and trabecular bone microarchitecture in patients with type 2 diabetes mellitus. The Journal of clinical endocrinology and metabolism. 2010;95(11):5045–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Farr JN, Drake MT, Amin S, Melton LJ, 3rd, McCready LK, Khosla S. In vivo assessment of bone quality in postmenopausal women with type 2 diabetes. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2014;29(4):787–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Patsch JM, Burghardt AJ, Yap SP, Baum T, Schwartz AV, Joseph GB, et al. Increased cortical porosity in type 2 diabetic postmenopausal women with fragility fractures. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2013;28(2):313–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samelson EJ, Demissie S, Cupples LA, Zhang X, Xu H, Liu CT, et al. Diabetes and Deficits in Cortical Bone Density, Microarchitecture, and Bone Size: Framingham HR-pQCT Study. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2018;33(1):54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Heilmeier U, Cheng K, Pasco C, Parrish R, Nirody J, Patsch JM, et al. Cortical bone laminar analysis reveals increased midcortical and periosteal porosity in type 2 diabetic postmenopausal women with history of fragility fractures compared to fracture-free diabetics. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2016;27(9):2791–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.de Waard EAC, de Jong JJA, Koster A, Savelberg H, van Geel TA, Houben A, et al. The association between diabetes status, HbA1c, diabetes duration, microvascular disease, and bone quality of the distal radius and tibia as measured with high-resolution peripheral quantitative computed tomography-The Maastricht Study. Osteoporosis international : a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2018;29(12):2725–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kaya S, Basta-Pljakic J, Seref-Ferlengez Z, Majeska RJ, Cardoso L, Bromage TG, et al. Lactation-Induced Changes in the Volume of Osteocyte Lacunar-Canalicular Space Alter Mechanical Properties in Cortical Bone Tissue. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research. 2017;32(4):688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mieczkowska A, Millar P, Chappard D, Gault VA, Mabilleau G. Dapagliflozin and Liraglutide Therapies Rapidly Enhanced Bone Material Properties and Matrix Biomechanics at Bone Formation Site in a Type 2 Diabetic Mouse Model. Calcified tissue international. 2020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.