

SETD2 regulates actin dynamics and cell migration via methylation of actin at Lys68 in a cellular complex containing Huntingtin.
Abstract
The methyltransferase SET domain–containing 2 (SETD2) was originally identified as Huntingtin (HTT) yeast partner B. However, a SETD2 function associated with the HTT scaffolding protein has not been elucidated, and no linkage between HTT and methylation has yet been uncovered. Here, we show that SETD2 is an actin methyltransferase that trimethylates lysine-68 (ActK68me3) in cells via its interaction with HTT and the actin-binding adapter HIP1R. ActK68me3 localizes primarily to the insoluble F-actin cytoskeleton in cells and regulates actin polymerization/depolymerization dynamics. Disruption of the SETD2-HTT-HIP1R axis inhibits actin methylation, causes defects in actin polymerization, and impairs cell migration. Together, these data identify SETD2 as a previously unknown HTT effector regulating methylation and polymerization of actin filaments and provide new avenues for understanding how defects in SETD2 and HTT drive disease via aberrant cytoskeletal methylation.
INTRODUCTION
SET domain–containing 2 (SETD2), the human homolog of Drosophila Set2, is a lysine methyltransferase characterized by its catalytic SET [Su(var)3-9, enhancer of zeste, trithorax) domain (1). Although SETD2 was found over two decades ago via its interaction with the Huntingtin (HTT) protein and originally named as HTT yeast partner B (HYPB) (2), no function linked to its role as an HTT interactor has yet been identified. In contrast, SETD2 activity as a chromatin remodeler responsible for trimethylation of histone H3 on lysine-36 (H3K36me3) is well characterized (3, 4). The SETD2 H3K36me3 posttranslational modification (PTM) on chromatin participates in alternative splicing, DNA methylation, transcriptional elongation, DNA damage repair, and polycomb silencing during development (5, 6). Loss of SETD2 and the H3K36me3 chromatin mark is embryonic lethal in Drosophila (7) and mice (8), and SETD2 defects have been linked to several diseases, including cancer (9–11) and autism spectrum disorder (12–14).
Recently, an important role for SETD2 outside the nucleus acting on the cytoskeleton to regulate microtubule dynamics via trimethylation of α-tubulin at lysine-40 (α-TubK40me3) was found (15). The catalytic SET domain was shown to methylate α-tubulin in vitro and in dividing cells α-TubK40me3 localized to spindle microtubules and the distal midbody during mitosis and cytokinesis, respectively. Loss of SETD2 and the α-TubK40me3 mark led to genomic instability and defects such as multipolar spindle formation, chromosomal bridges at cytokinesis, micronuclei, polyploidy, and polynucleation: a phenotype specifically linked to its activity as a microtubule methyltransferase (15, 16).
Based on these insights that SETD2 is a dual-function chromatocytoskeletal remodeler, we asked whether this methyltransferase might have other cytoskeletal targets. We found that actin is a bona fide target for SETD2, and methylation of actin at lysine-68 (ActK68me3) is a new SETD2-dependent modification of the actin cytoskeleton. Antibodies directed against the SETD2 methyl mark localized to areas of active actin remodeling in migrating cells. We also found that in addition to HTT, the actin-binding protein HIP1R also complexed with SETD2, and disruption of this SETD2-HTT-HIP1R axis inhibited methylation of actin filaments, caused defects in actin polymerization, and impaired cell migration.
RESULTS
SETD2 interacts with actin in cells
SETD2 was present in both the nuclear and cytoplasmic compartments of interphase cells (Fig. 1A) and could be coimmunoprecipitated with endogenous actin in 786-0 (clear-cell renal cell carcinoma) cells and mCherry–β-actin expressed in HEK293T (human embryonic kidney) cells (Fig. 1B and fig. S1, A and B). To test the relationship between SETD2 and potential actin methylation, we asked whether a SETD2 methyl epitope could be detected on actin from SETD2-proficient versus SETD2-deficient cells. Antibodies directed against the trimethyl-lysine SETD2 epitope on histones (anti-Me3K36) or α-tubulin (anti-Me3K40), and a pan–trimethyl-lysine epitope (anti-Me3Pan) all recognized actin from SETD2-proficient but not SETD2-deficient cells (Fig. 1C and fig. S1C). Re-expression of a functional truncated SETD2 (tSETD2) in SETD2-deficient HKC (human kidney) cells restored actin methylation in conjunction with restoration of histone methylation (fig. S1, D and E).
Fig. 1. SETD2 binds actin.
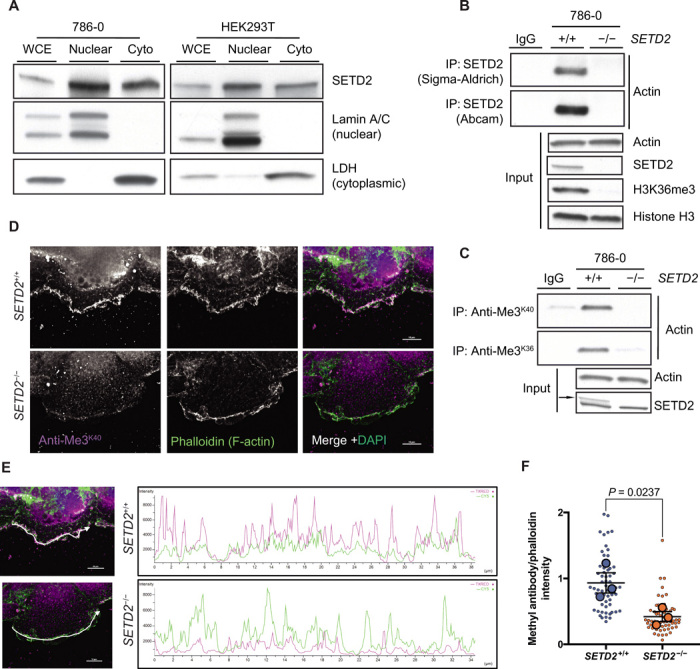
(A) Immunoblot analysis showing localization of SETD2 in whole-cell extracts (WCE) and both nuclear and cytoplasmic (Cyto) compartments of 786-0 and HEK293T cells. Lamin A/C and lactate dehydrogenase (LDH) are used as controls for the nuclear and cytoplasmic fractions, respectively. (B) Immunoblot analysis showing coimmunoprecipitation of endogenous SETD2 and endogenous actin in 786-0 cells using SETD2 antibodies from two different sources. (C) Immunoblot analysis showing that actin is methylated in SETD2-proficient but not in SETD2-deficient 786-0 cells by immunoprecipitation (IP) of endogenous actin using two different antibodies directed against the SETD2 trimethyl-lysine epitope. Data in (A to C) are representative of experiments repeated at least three times with similar results. (D) Deconvolution microscopy imaging of filamentous (F)–actin using phalloidin (green) and SETD2 methyl epitope–specific antibody (magenta) showing colocalization of the methyl mark with F-actin in 786-0 cells. Scale bars, 10 μm. (E) Representative intensity profiles of the staining observed with the methyl-specific antibody and phalloidin. Merged images from (D) are shown on the left to indicate the position for line profiles. (F) Quantification of intensity profiles seen in (E). y axis represents the ratio of methyl antibody to phalloidin intensity. Each small circle represents a single cell. Large circles represent means from 20 cells for each independent biological replicate; data are means ± SEM from these values (n = 3).
Staining with SETD2 trimethyl-lysine epitope antibodies localized methylation in cells to areas of active cytoskeletal remodeling and high actin turnover, including dorsal ruffles and lamellipodia at the leading edge of cells (fig. S1F). Consistent with decreased actin methylation seen in the absence of SETD2, signal intensity of the methyl mark in these regions was greatly reduced in SETD2-deficient cells (Fig. 1D). Polyline profiles generated for regions of strong phalloidin staining [polymerized filamentous (F)–actin] showed a correlation between the presence of this methyl mark and polymerized actin, which was significantly reduced in SETD2-deficient cells (Fig. 1, E and F). Together, these data show that actin in cells is methylated in a SETD2-dependent manner, opening up the possibility that actin is a novel target for SETD2 methylation.
SETD2 methylates actin
To directly demonstrate that SETD2 could methylate actin, we performed in vitro methylation assays on the basis of either incorporation of radiolabeled methyl groups donated by tritiated S-adenosylmethionine (3H-SAM) or fluorimetric assays based on the measurement of SAM consumption. Both assays showed that the catalytic SET domain of SETD2 could methylate actins purified from muscle tissues and recombinant actin produced in HEK293T cells (Fig. 2A and fig. S2A). Recombinant tSETD2 was similarly able to methylate actin purified from cardiac, smooth, and skeletal muscle, as well as recombinant actin from HEK293T cells (Fig. 2B). SETD2 methylation activity was unaffected following treatment with the actin depolymerizing agent Latrunculin A, suggesting that SETD2 can methylate both actin monomers and polymers in vitro (Fig. 2C). Trimethyl-lysine–specific anti-Me3K36 and anti-Me3K40 antibodies, which immunoprecipitated actin from SETD2-proficient but not SETD2-deficient cells, recognized actin following in vitro methylation by SETD2 (Fig. 2, D and E). Consistent with a previous study (17), SETD2 did not exhibit significant activity against recombinant actin produced in Escherichia coli (Fig. 2B and fig. S2A), indicating that the presence of a SETD2 recognition sequence alone was not sufficient for methylation and suggesting that either proper folding or other modifications may be required to “prime” actin for recognition/methylation. These assays demonstrate that SETD2 has an intrinsic methyltransferase activity for actin.
Fig. 2. SETD2 methylates actin.

(A) Autoradiography showing in vitro methylation of actin using tritiated S-adenosylmethionine (3H-SAM) as methyl group donor and recombinant glutathione-S-transferase (GST)–tagged SETD2 catalytic SET domain (amino acids 1418 to 1714). Film shows automethylation of SETD2 and histone methylation as positive control. Data are representative of experiments repeated at least three times, with similar results. (B) Fluorescence-based in vitro methylation using recombinant tSETD2 (amino acids 1418 to 2564) with purified cardiac muscle (red), smooth muscle (green), skeletal muscle (purple) actins, and recombinant actin (rActin) from HEK293T cells (blue) or E. coli (orange). Data are means ± SEM (n = 2). (C) Fluorescence-based in vitro methylation of actin using recombinant tSETD2 (amino acids 1418 to 2564) following addition of Latrunculin A (LatA) at indicated concentrations. Methyltransferase activity calculated by slope of linear regression from fluorescence trace (a.u./min), against LatA concentration on x axis. Data are means ± SEM (n = 3). y axis in (B) and (C) plotted after subtracting automethylation from samples with SETD2 alone. (D and E) Immunoblot analysis showing recognition of actin proteins by SETD2 methyl epitope antibodies anti-Me3K40 (D) and anti-Me3K36 (E) following in vitro methylation with recombinant GST-tagged SETD2 (amino acids 1418 to 1714). Data are representative of experiments repeated at least three times with similar results.
It was recently reported that methylation of actin at histidine-73 (MeH73) is mediated by another SET domain–containing protein, the methyltransferase SETD3 (17, 18). We confirmed that SETD3 expression is retained in our SETD2-deficient cells that lose the trimethyl-lysine mark on actin (fig. S2B) and found that knockdown of SETD3 did not lower levels of the SETD2 trimethyl epitope on actin (fig. S2C). These data distinguish the actin lysine methyltransferase activity of SETD2 from the histidine methyltransferase activity of SETD3.
Lysine-68 methylation is a SETD2-dependent PTM of the actin cytoskeleton
To identify a potential SETD2 methylation site on actin, we performed an unbiased in silico screen to identify methylated lysine residues in the PhosphoSitePlus® methylation site dataset and cross-referenced this with all proteins containing predicted SETD2 recognition motifs (fig. S3A). Substrates for SETD2 activity typically contain a lysine residue with a flexible proline at +2 (“KxP”) present in histone H3 (19) or a glycine at +3/4 (“KxxG/G”) present in α-tubulin and STAT1 (signal transducers and activators of transcription 1) (15, 20). Gene ontology analysis identified cytoskeletal proteins as a large class of putative SETD2 targets (fig. S3B), and all isoforms of actin contained a conserved KxP motif at lysine-68 (Fig. 3A). These findings made actin the most highly enriched nonhistone protein from this analysis (fig. S3C) and suggested lysine-68 as candidate site for SETD2 methylation.
Fig. 3. SETD2 methylates lysine-68 on actin.
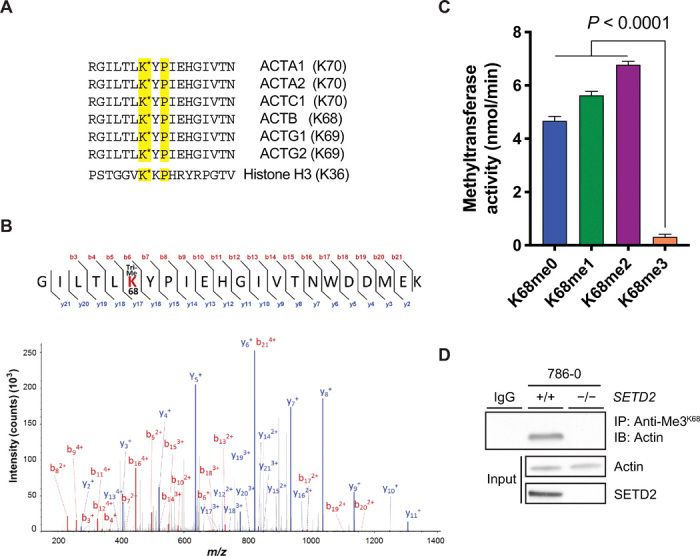
(A) Amino acid sequence showing conserved KxP SETD2 recognition motif present in all actin isoforms. Position of the lysine residue varies depending on actin isoform; reference to this site as “ActK68” is based on its position in β-actin (ACTB). Histone H3 sequence containing the KxP motif is shown below for reference. (B) Representative tandem mass spectrometry (MS/MS) spectrum of trimethylated ActK68 peptides recovered from SETD2-proficient 786-0 cells. m/z, mass/charge ratio. (C) Fluorescence-based in vitro methylation assay showing in vitro methylation of biotin-labeled K68-containing actin peptides (amino acids 62 to 78) with recombinant tSETD2 (amino acids 1418 to 2564). Sequence for the peptides used is shown in (A). Data are means ± SEM (n = 4). (D) Immunoblot analysis showing dependency of the ActK68me3 mark on SETD2 by IP of endogenous actin from whole-cell extracts of SETD2-proficient and SETD2-deficient 786-0 cells using the anti-Me3K68 antibody. Data are representative of experiments repeated at least three times with similar results.
To test whether ActK68 is methylated in cells, we performed mass spectrometry on lysates from parental and SETD2-deficient 786-0 cells. The peptide shown in Fig. 3B contained trimethylated ActK68 and was found in SETD2-proficient but not in SETD2-deficient cells. To confirm SETD2 as a methyltransferase for this site, we generated biotinylated peptides containing the ActK68 site and performed in vitro methylation assays. Recombinant tSETD2 was able to methylate unmodified, monomethylated, and dimethylated peptides but showed no activity for the trimethylated ActK68 peptide, confirming ActK68 as a methyl-acceptor site for SETD2 (Fig. 3C). In addition, the anti-Me3K36 antibody, previously shown to recognize the SETD2 methyl epitopes on histones and microtubules (15) and shown here to recognize methylated actin from SETD2-proficient cells and after in vitro methylation (Figs. 1C and 2E), recognized purified actin protein and a trimethylated ActK68 peptide on dot blot analysis (fig. S4A).
Finally, we generated a polyclonal antibody directed against the ActK68me3 mark. This anti-Me3K68 antibody strongly recognized the trimethylated ActK68 peptide and purified actin, with some cross-reactivity for dimethylated ActK68 peptide and microtubules but little cross-reactivity for histones/nucleosomes (fig. S4B). This antibody immunoprecipitated actin from SETD2-proficient but not SETD2-deficient cells (Fig. 3D) and recognized actin methylated by SETD2 in vitro (fig. S4C). Collectively, these data confirm that lysine-68 of actin is a bona fide site for SETD2 methylation in cells, and ActK68me3 is a PTM distinct from MeH73 on actin.
SETD2 regulates actin polymerization in cells
Based on our findings showing that SETD2 is an actin lysine methyltransferase, we sought to understand the impact of SETD2 methylation on the actin cytoskeleton. Using an established biochemical fractionation procedure (21), we found that the ratio of polymerized F-actin to unpolymerized globular (G)–actin was significantly reduced in two different SETD2-deficient human cell lines (786-0 and HEK293T) and after acute knockout of Setd2 in mouse embryonic fibroblast (MEF) cells, which occurred in the absence of any total difference in overall actin levels (Fig. 4, A and B). Re-expression of tSETD2, which rescued actin methylation, also rescued the polymerization defect in SETD2-deficient cells (fig. S5, A and B). To test whether methylation at ActK68 affects polymerization, we expressed wild-type mCherry–β-actin, or actin in which lysine-68 was mutated to arginine (K68R) or alanine (K68A), in SETD2-proficient HEK293T cells. Fractionation experiments revealed decreased F-/G-actin ratio for K68 mutant mCherry–β-actin, without any change in F-/G-actin ratio of endogenous actin, total levels of endogenous or mCherry–β-actin, or H3K36me3 SETD2 methylation activity (Fig. 4, C to E). These data reveal a polymerization defect in actin that cannot be methylated by SETD2 at lysine-68.
Fig. 4. SETD2 regulates actin polymerization in cells.
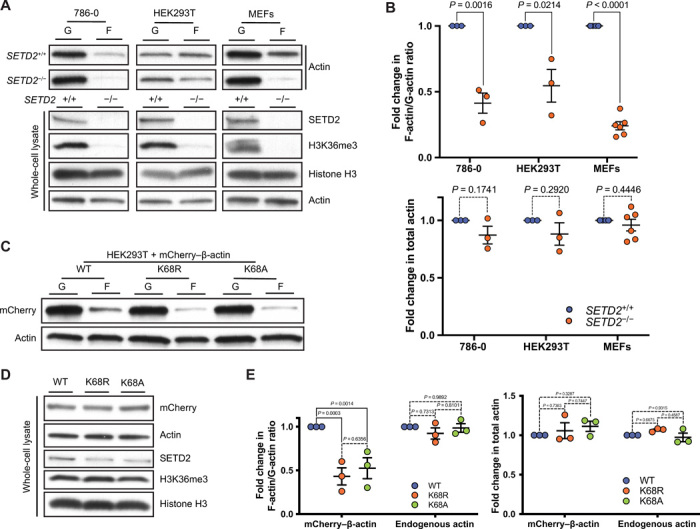
(A) Immunoblot analysis showing decreased F-actin in SETD2-deficient 786-0, HEK293T, and MEF cells. Whole-cell lysate shows absence of SETD2, associated with the expected loss of histone H3K36me3 methylation. (B) Quantitation of F-/G- actin ratio (top) and whole-cell lysate actin (bottom) from the data is shown in (A). Data are means ± SEM (n = 3 for 786-0 and HEK293T; n = 6 for MEF). (C) Immunoblot analysis of F-/G-actin ratio in HEK293T cells expressing wild-type or K68A/R mCherry–β-actin. (D) Immunoblot analysis of whole-cell lysates shows no change in total actin levels or SETD2 histone methylation with expression of K68A/R mCherry–β-actin. (E) Quantitation of F-/G-actin and total actin shown in (C) and (D), respectively. Data are means ± SEM (n = 3).
To investigate the impact of SETD2 loss on actin dynamics, we fractionated cells to isolate the soluble (supernatant) and cytoskeleton-enriched insoluble (pelleted) fractions from cells and found a reduction in insoluble cytoskeletal F-actin in SETD2-deficient cells (fig. S5, C and D). Treatment with Latrunculin A significantly reduced the insoluble F-actin fraction in SETD2-proficient but not in SETD2-deficient cells. We allowed cells to recover after washout following Latrunculin treatment and found that polymerized F-actin was restored in SETD2-proficient cells, while cells lacking SETD2 remained deficient for polymerized F-actin in the insoluble fraction (Fig. 5, A and B). This defect could be reversed by the polymer-inducing agent Jasplakinolide (22), which increased F-actin in the insoluble fraction of SETD2-deficient cells to a level comparable with SETD2-proficient cells (fig. S5, C and D).
We were also able to show that the anti-Me3K68 antibody, as well as another SETD2 methyl-lysine epitope antibody (anti-Me3K40), and anti-Me3Pan all recognized actin in a SETD2-dependent manner primarily from the insoluble fraction containing polymerized F-actin (Fig. 5C and fig. S5, E and F). This fraction lacked any detectable tubulin, confirming that the methyl epitope recognized in this fraction was on actin and not due to cross-reactivity of the immunoprecipitating antibody with the known SETD2 target α-tubulin (15) complexed with actin. Overall, by Western analysis, the amount of methylated actin in the polymerized F-actin fraction was determined to be ~100- to 1000-fold greater than that seen in the soluble G-actin fraction (fig. S5G), although exact quantitation was difficult as the methylated actin in the soluble fraction was so low. Latrunculin treatment caused a marked loss of methylated F-actin, far in excess of the reduction seen in total F-actin, to the point where it was virtually undetectable (Fig. 5D). We did not detect any increase in methylated actin in the soluble G-actin pool commensurate with this loss of methylated F-actin.
Fig. 5. ActK68me3 localizes to the insoluble F-actin fraction in cells.
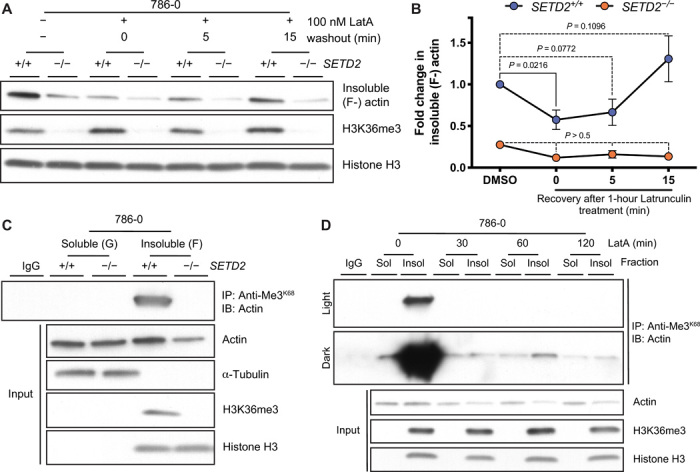
Immunoblot (IB) analysis (A) and quantitation (B) showing changes in actin polymerization (F-actin in the insoluble fraction) following washout after treatment with the actin depolymerizing agent latrunculin A (LatA). Data are means ± SEM (n = 4). (C) IB analysis using anti-Me3K68 antibody showing that ActK68me3 occurs on endogenous actin from the insoluble F-actin fraction of SETD2-proficient 786-0 cells; tubulin (which is also methylated by SETD2) is not found in this fraction. Data are representative of experiments repeated at least three times with similar results. Absence of H3K36me3 in the insoluble fraction is used as a control to confirm loss of SETD2 in (A) and (C). (D) IP of actin using anti-Me3K68 antibody from soluble (G-actin) and insoluble (F-actin) fractions of SETD2-proficient cells treated with LatA for indicated times. Light and dark exposures are shown for contrast in amount of methylated actin between fractions. Data are representative of three experiments performed using anti-Me3K68 and anti-Me3Pan antibodies with similar results. In an additional experiment performed shortly after cells were brought up from cryopreservation, we saw a less marked loss of methylated insoluble actin with LatA, but saw no increase in methylated soluble actin, consistent with data shown here.
Staining with fluorescent phalloidin, which preferentially binds to F-actin polymers (23), was also used to measure the intracellular content of polymerized actin (fig. S6A). Spectrophotometric analysis of phalloidin-bound actin polymers showed decreased fluorescence intensity (a surrogate for decreased actin polymers) in SETD2-deficient cells (fig. S6B). This assay for actin polymerization also showed a significant decrease with Latrunculin treatment in SETD2-proficient but not in SETD2-deficient cells. Flow cytometric analysis of phalloidin-bound F-actin also confirmed this defect at the individual cell level (fig. S6, C and D). High-resolution image stitching of phalloidin-stained SETD2-deficient cells suggested significant compromise of cell-cell contacts and loss of ubiquitous stress fibers, otherwise present in SETD2-positive cells (fig. S7, A and B). Super-resolution imaging of the same cells via three-dimensional structured illumination microscopy (3D-SIM) revealed disorganized actin networks at both the leading edge and lamella, in addition to a decrease in the number of cells displaying robust actin arcs (fig. S7C and movies S1 and S2). SETD2-deficient cells also appeared to be associated with a noticeable increase in the appearance of microvilli on the cell surface (fig. S7, B and C, and movie S3). Together, these biochemical and cellular data point to an actin polymerization defect in cells lacking SETD2 and a role for ActK68 methylation as a regulatory PTM of actin polymerization dynamics.
SETD2 regulates cell migration
Regulation of actin dynamics and localization of the SETD2 methyl mark to the leading edge of cells suggested that loss of SETD2 could affect cell migration. Using a scratch wound healing assay, we found that SETD2-deficient cells exhibited significantly decreased migration compared to SETD2-proficient cells (Fig. 6, A and B). Treatment with the proliferation inhibitor cytosine arabinoside, as well as cell counts over 48 hours, confirmed that the decreased cell migration seen in these cells was independent of any effects of SETD2 loss on cell proliferation (fig. S8, A and B). Expression of tSETD2, which rescued actin methylation and actin polymerization in SETD2-deficient cells, could also rescue the migration phenotype (fig. S8, C and D). These data link the loss of SETD2, actin methylation, and the resulting dysregulation of actin dynamics to defects in cell migration.
Fig. 6. SETD2 regulates cell migration.
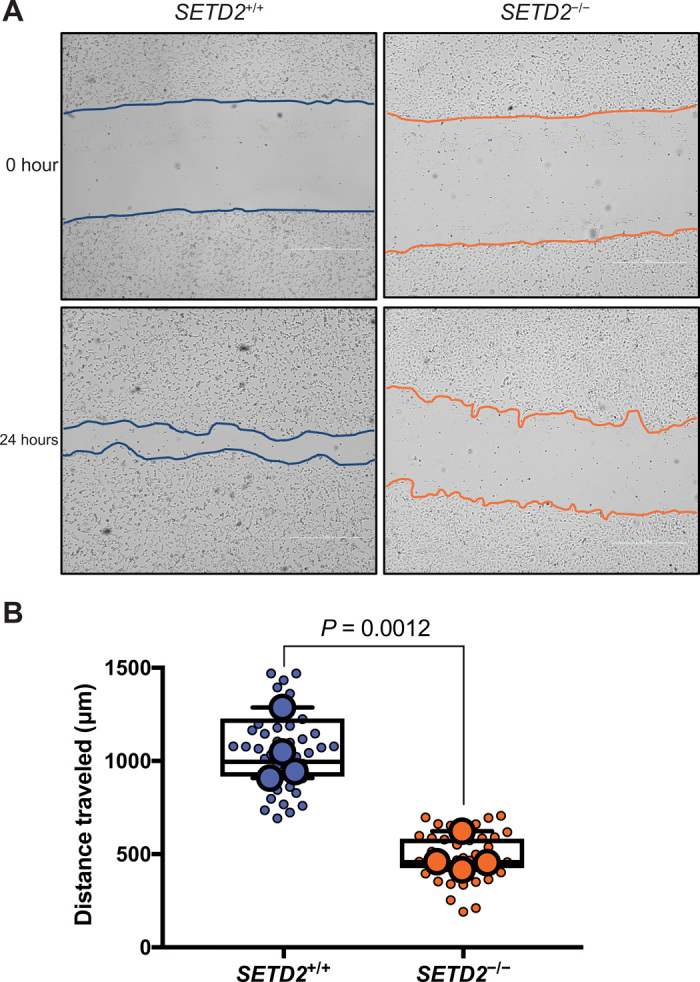
(A) In vitro scratch assay at 0 and 24 hours after wound inflection, illustrating that SETD2-deficient cells migrate slower than SETD2-proficient 786-0 cells. Scale bars, 1000 μm. (B) Quantification of scratch assays seen in (A). Small circles represent each independent measurement across all experiments. Large circles represent mean from a minimum of nine individual measurements for each independent biological replicate (n = 4).
Actin methylation and cell migration are regulated by a SETD2-HTT-HIP1R axis
The SETD2 WW domain has been shown to bind the N-terminal proline-rich region of HTT (Fig. 7A), an interaction that prevents the autoinhibition of its methyltransferase activity (2, 24). HTT is a scaffolding protein that interacts with, among others, the actin-binding adapter protein HTT-interacting protein-1 related (HIP1R) (25). Several proteomic screens have identified actin in HTT complexes (26–28), and HTT itself has been reported to contain an N-terminal actin-binding domain (29). Furthermore, our in silico analysis of proteins with putative SETD2 methylation sites identified overenrichment of the Huntington’s Disease pathway (fig. S3, B and C), primarily due to the inclusion of actin proteins in this pathway. We confirmed that both HTT and HIP1R coimmunoprecipitate with actin (fig. S9, A and B) and showed using an mCherry-tagged C-terminal SETD2 construct the expected interaction between HTT and SETD2 (Fig. 7B).
Fig. 7. SETD2 methylates actin in association with HTT.

(A) Schematic diagram showing interaction between the SETD2 C-terminal and HTT N-terminal proline-rich region (PRR), as reported in (2, 24). (B) Immunoblot (IB) analysis showing coimmunoprecipitation of mCherry-tagged SETD2 C-terminal [SETD2-(C)-mCherry] and endogenous HTT in HEK293T cells. (C) IB analysis showing that actin methylation by SETD2 is dependent on both HTT and HIP1R using a SETD2 methyl-epitope antibody (anti-Me3K36) to immunoprecipitate actin from 786-0 cells after siRNA-mediated knockdown of HTT or HIP1R. Input lysates shown to confirm knockdown of HTT and HIP1R. (D and E) IB analysis (D) and quantitation (E) of decreased insoluble F-actin in 786-0 cells after siRNA-mediated knockdown of HTT or HIP1R. Data are means ± SEM (n = 3). (F) Quantitation of migration data after siRNA-mediated knockdown of HTT or HIP1R in SETD2-proficient versus SETD2-deficient 786-0 cells. Small circles each represent an independent measurement from all biological replicates. Large circles represent mean from 12 measurements for each independent biological replicate (n = 4). (G) IB analysis showing decreased actin methylation after expression of a mutant HTT protein by IP of actin using a SETD2 methyl-epitope antibody (anti-Me3K36) from HEK293T cells expressing a doxycycline (Doxy)–inducible CFP-tagged N-terminal HTT construct containing 94 polyglutamine repeats (CFP-HTT94Q) and a retrotransactivator (rtTA3). Input lysate confirms expression of CFP-tagged mutant protein and loss of SETD2 methylation activity (H3K36me3) without change in endogenous SETD2 expression. Data in (B), (C), and (G) are representative of experiments repeated three times with similar results.
Abnormal HTT has been previously linked to defects in the actin cytoskeleton and cell migration (30, 31). Given our findings that SETD2-mediated methylation regulates actin dynamics and cell migration, we explored the possibility that SETD2 methylation of actin provides the missing link between HTT and cell migration defects. Using siRNA (small interfering RNA)–mediated knockdown of either HTT or HIP1R, we found that disruption of this complex was sufficient to abrogate actin methylation without affecting SETD2 nuclear H3K36me3 methylation activity (Fig. 7C). Furthermore, HTT and HIP1R loss led to a reduction in insoluble F-actin (Fig. 7, D and E) and cell migration (Fig. 7F and fig. S9C) in SETD2-proficient cells (a similar pattern as with loss of SETD2) but had no impact on actin polymerization and cell migration in SETD2-deficient cells. These findings indicate that in cells, SETD2 methylation of actin is dependent on interaction with HTT and HIP1R.
Finally, we expressed a pathogenic N-terminal HTT construct containing 94 polyglutamine repeats, known from cellular aggregates (32) that can include SETD2 (24, 33) and perhaps limit its activity, in SETD2-proficient HEK293T cells (fig. S9D). Expression of this construct significantly inhibited both actin and histone methylation by SETD2 without any change in overall levels of SETD2 or HTT (Fig. 7G). Mutant HTT-induced loss of SETD2 methyltransferase activity also caused a concurrent decrease in the insoluble F-actin to soluble G-actin ratio (fig. S9, E and F). Collectively, these data provide molecular, biochemical, and cellular evidence for a SETD2-HTT-HIP1R axis responsible for methylating actin in cells to regulate dynamics and function of the actin cytoskeleton.
DISCUSSION
After its initial discovery as an HTT-interacting protein, studies on HYPB, now called SETD2, focused on the nuclear functions of this enzyme as a histone methyltransferase (5). The present study demonstrates an extended nonchromatin role for SETD2 and also provides the first example of a lysine methyltransferase capable of directly modifying and regulating the actin cytoskeleton. Our data have led us to propose a model for a SETD2-HTT-HIP1R axis that regulates actin dynamics, with perturbation of this axis driving migratory defects seen in cells defective for normal SETD2 and HTT function (fig. S10). An earlier study showing that homozygous Setd2 deficiency was embryonic lethal reported that cells from Setd2-null mice had disorganized stress fibers and lamellipodia (8), confirming the link between SETD2 loss and disruption of the actin cytoskeleton.
A recent review of the nonchromatin activity of histone lysine methyltransferases (34) points out that many of these enzymes play roles in the cell that extend beyond their activity on histones. In addition to our data demonstrating that SETD2 acts on the actin cytoskeleton, the histone methyltransferase EZH2 has also been found to regulate actin polymerization through interaction with VAV1 and methylation of actin-associated TALIN1 protein (35, 36). More recently, it was reported that MeH73 is mediated by another SET domain–containing protein, the methyltransferase SETD3 (17, 18). Whereas MeH73 has been known to regulate actin dynamics for nearly two decades (37) and Wilkinson et al. (17) found that SETD3 is the only methyltransferase for this site, we show here that lysine methylation at K68 is a new regulatory PTM of actin. In figure 1E of that study (17), in vitro methylation experiments found that no methyltransferase other than SETD3 (including SETD2) exhibited activity for recombinant actin purified from E. coli. However, it is well known that this recombinant actin folds poorly (38), and we also found that it was a poor substrate for SETD2 methylation. Regardless, our data, using ActK68 peptides and purified and recombinant actin proteins sourced from models other than E. coli, demonstrate that SETD2 methylates actin in vivo and in vitro, and that ActK68me3 is a naturally occurring PTM of the actin cytoskeleton.
Actin is a target for modifications on 94–amino acid side chains, comprising 94% of total modifiable residues (39, 40), yet the structural and functional implications of many PTMs remain unknown. ActK68 resides near the DNaseI binding loop (D-loop, amino acids 39 to 43) that contains several modifiable residues (39), and close to another methylated lysine reside K84 (41). In addition to methylation, ActK68 itself is susceptible to acetylation, SUMOylation, and ubiquitination (39, 40), suggesting an interplay, and perhaps competition, between these PTMs on this residue. These K68 PTMs could be involved in structural changes affecting actin association with binding proteins such as cofilin or gelsolin at its G3 interaction site (39).
While we found that the SET domain and a truncated SETD2 (tSETD2) can methylate both polymerized and monomeric actin in vitro, the ActK68me3 mark occurs primarily on the insoluble F-actin cytoskeleton in cells. Our data show that Latrunculin A causes a massive loss of methylated actin from actin filaments in SETD2-proficient cells and has little effect on polymerized F-actin in SETD2-deficient cells. This suggests the interesting possibility that methylation may make actin polymers more dynamic, and that, in the absence of methylation, less dynamic actin polymers are resistant to Latrunculin. As a precedent, expression of actins with mutations in alternating positively/negatively charged amino acids adjacent to the nucleotide-binding cleft (e.g., Lys213-Glu214-Lys215 and Arg182-Asp184), several of which are subject to post-translational modification, confers resistance to Latrunculin A in yeast and mammalian cells (42, 43). In our SETD2-proficient cells, Latrunculin reduced the signal of methylated F-actin to a nearly undetectable level, suggesting that the remaining amount of F-actin seen in these cells after depolymerization is, essentially, all unmethylated. This lability (i.e., susceptibility to depolymerization/repolymerization cycles) of methylated actin could also contribute to actin dynamics in SETD2-proficient cells during migration (44), with loss of this labile methylated actin contributing to the migration defect seen in SETD2-deficient cells.
Just as intriguing, the observation that there was no appearance of methylated actin in the soluble fraction even after depolymerization of actin filaments suggests several possibilities. Methylated actin released from actin filaments may be less stable, exit the cytoplasmic fraction, and/or be a target for lysine demethylases. Alternatively, the methyl epitope recognized by methyl-specific antibodies may be masked due to interaction with G-actin binding proteins such as profilin or thymosin-β4 (45). Overall, a deeper understanding of how actin PTMs such as ActK68me3 affect actin structure, the relationship with other PTMs, and interaction with actin-binding proteins, and whether there is isotype specificity for these modifications, remains an important avenue for future research.
The dependence of SETD2-mediated actin methylation on HTT and HIP1R was unexpected given that no linkage between these proteins and lysine methylation has been previously uncovered. This new linkage suggests the possibility that the dysregulated actin phenotype seen in mutant HTT cells (30, 31) might result from a decreased ability of SETD2 to methylate actin. Furthermore, these findings demonstrate knockdown of HTT as a novel means of uncoupling SETD2 chromatocytoskeletal (H3K36me3 versus ActK68me3) function. Expression of mutant HTT known to form aggregates (32) is sufficient to inhibit both actin and histone methylation, which we attribute to the presence of HTT inclusions that form in both the nucleus and the cytoplasm (46, 47). Notably, SETD2 has been shown to associate with N-terminal HTT in disease-associated brain deposits (33), and it is suggested that mutant HTT interaction with SETD2 alters its activity via sequestration (24). Given that nuclear actin regulates processes from transcription to DNA damage repair (48), a role for the SETD2-HTT-actin methylation axis within the nucleus remains a tantalizing possibility.
Although HTT and HIP1R knockdown was sufficient to disrupt SETD2 methylation of actin, it only reduced cell migration to a level intermediate between parental and SETD2-deficient cells. Given the large number of other proteins in addition to actin localized to the leading edge of cells and involved in cell migration (49), this suggests that SETD2 may also methylate and regulate other cytoskeletal elements, contributing to a defective migratory phenotype upon loss of SETD2, which in contrast to actin are not dependent on the SETD2-HTT-HIP1R axis.
In conclusion, our findings expand the emerging paradigm that the epigenetic machinery has chromatocytoskeletal activity important in both the nucleus and cytoplasm, coordinately methylating chromatin and the cytoskeleton to regulate the activity of key cellular components during transcription (histones), cell division (microtubules), and cell migration (actin). This new appreciation of SETD2 as a chromatocytoskeletal remodeler will provide new ways to understand how SETD2 defects involved in diseases such as cancer and autism, as well as HTT defects that cause Huntington’s disease, may drive pathogenesis via disruption of cytoskeletal methylation.
MATERIALS AND METHODS
Statistical analysis
Statistical analyses were performed using Prism (GraphPad, version 8.3.0) or Excel (Microsoft). All data in graphs are shown as means ± SEM, except for migration data (Figs. 6B and 7F and fig. S8C), which are shown as box-and-whiskers plots representing median ± interquartile range with minima and maxima. The following statistical tests were used to determine significance: Fig. 1F, paired two-tailed t test; Fig. 2B, ordinary one-way analysis of variance (ANOVA) with Dunnett’s test against E. coli recombinant actin (rActin) samples; Fig. 2C, ordinary one-way ANOVA with Tukey’s Honestly Significant Difference (HSD) test; Fig. 3C, ordinary one-way ANOVA with Dunnett’s test against K68me3 peptide samples; Fig. 4B, unpaired two-tailed t tests; Fig. 4E, ordinary one-way ANOVA with Tukey’s HSD test; Fig. 5B, ordinary one-way ANOVA with Dunnett’s test against control samples; Fig. 6B, unpaired two-tailed t test; Fig. 7 (E and F), ordinary one-way ANOVA with Tukey’s HSD test; fig. S3C, Fisher’s exact test with false discovery rate (FDR) correction; fig. S5B, ordinary one-way ANOVA with Tukey’s HSD test; fig. S5D, unpaired two-tailed t tests with Holm-Šidák correction; fig. S5G, Kruskal-Wallis test with Dunn’s test (left) and ordinary one-way ANOVA with Tukey’s HSD test (right); fig. S6B, ordinary one-way ANOVA with Tukey’s HSD test; fig. S6D, unpaired two-tailed t tests; fig. S8A, unpaired two-tailed t test; fig. S8B, unpaired two-tailed t tests with Holm-Šidák correction; fig. S8D, ordinary one-way ANOVA with Tukey’s HSD test; and fig. S9F, two-way ANOVA with Dunnett’s test against control samples. In all cases, a P value of <0.05 was considered significant.
Cell culture
Cell lines were used in the study as previously described (15). 786-0, HKC, and HEK293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich, F24429). Cells were incubated at 37°C and 5% CO2 and passaged two to three times weekly, as required. Setd2flox/flox MEF cells expressing ER-Cre were cultured in phenol red–free media (DMEM, high glucose, and Hepes) supplemented with 10% FBS, sodium pyruvate (Gibco), and GlutaMAX (Gibco), with blasticidin (5 μg/ml). MEFs were treated with 2 to 3 μM 4-hydroxytamoxifen (Sigma-Aldrich, H7904) for 3 to 5 days for efficient knockout of Setd2.
siRNA-mediated knockdown in 786-0 cells was carried out using 5 to 10 nM Dharmacon ON-TARGETplus siRNAs (GE Healthcare), diluted in siRNA buffer, and mixed with DharmaFECT transfection reagent (1:50 in Opti-MEM reduced serum media). Cells were allowed to incubate with the siRNA mixture for 5 to 7 hours and grown for 72 to 96 hours with passaging as necessary.
Overexpression of mCherry-tagged actin or SETD2 constructs and cyan fluorescent protein (CFP)–tagged HTT constructs in HEK293T cells was carried out using Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific) at a ratio of 1:2.5, mixed in Opti-MEM reduced serum media. Cells were allowed to incubate with the DNA mixture for 5 to 7 hours and grown for 48 hours with passaging, as necessary. For expression of pathogenic HTT94Q N-terminal construct (Addgene plasmid no. 23966), HEK293T cells were cotransfected with retrotransactivator rtTA3 (Addgene plasmid no. 26429), incubated overnight, and treated with doxycycline (1 μg/ml) for the indicated time points. The following chemicals were used in the study: Latrunculin A from sea sponge (Sigma-Aldrich, L5163), Jasplakinolide (Sigma-Aldrich, J4580), and cytosine-β-d-arabinofuranoside (Sigma-Aldrich, C1768).
Site-directed mutagenesis
For generation of methylation-resistant mutant actin constructs, the mCherry–actin-7 plasmid (Addgene plasmid no. 54966) was used. Mutagenesis was performed using the QuikChange II XL site-directed mutagenesis kit (Agilent), as per the manufacturer’s instructions. The following primers were used to generate the mCherry-ACTB-K68R/A mutants:
K68R, 5′-gctcgatggggtacctcagggtgaggatg-3′ (reverse) and 5′-catcctcaccctgaggtaccccatcgagc-3′ (forward); and K68A, 5′-tgctcgatggggtacgccagggtgaggatgcc-3′ (reverse) and 5′-ggcatcctcaccctggcgtaccccatcgagca-3′ (forward).
Mutagenized constructs were transformed into XL10-Gold Ultracompetent cells (Agilent), prepared using QIAprep Spin Miniprep Kits (Qiagen) and E.Z.N.A. Plasmid Maxi Kits (Omega Bio-tek), and transfected into HEK293T cells, as described above.
Coimmunoprecipitation assays
Coimmunoprecipitation was performed as previously described (15), with modifications. Briefly, cells were collected at 70 to 80% confluency and lysed in cold “CST (Cell Signaling Technology) lysis buffer” [20 mM tris-HCl (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM Na4P2O7, 1 mM β-glycerophosphate, and 1% Triton X-100] or “immunoprecipitation (IP) buffer” [25 mM tris-HCl (pH 8.0), 300 mM NaCl, 1 mM EDTA, 1% NP-40] supplemented with protease inhibitor cocktail (Roche). Protein concentration was determined using a bicinchoninic acid protein assay kit, as per the manufacturer’s instructions (Pierce). Endogenous SETD2 was immunoprecipitated overnight from 500 to 1000 μg of cell extract using anti-SETD2 antibodies (Abcam or Sigma-Aldrich) and protein A/G agarose beads (Pierce). For the reciprocal IP, endogenous actin was immunoprecipitated using an antibody to actin. To identify the interaction between endogenous SETD2 and mCherry-tagged, exogenous actin, SETD2 was immunoprecipitated using whole-cell extracts from HEK293T cells expressing mCherry-ACTB (Addgene plasmid no. 54966). Similarly, the tagged actin was immunoprecipitated using an mCherry-specific antibody. The appropriate corresponding immunoglobulin G (IgG) isotype was used as a negative control.
Nuclear versus cytoplasmic cell fractionation
Cells were collected in ice-cold phosphate-buffered saline (PBS), pelleted at 1500 rpm for 5 min at 4°C, and resuspended in an ice-cold hypotonic buffer [10 mM Hepes (pH 7.2), 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA, 20 mM NaF, and 100 mM Na3VO4] supplemented with protease inhibitor cocktail. Samples were lysed in a Dounce tissue homogenizer with glass pestle until only nuclei were visible by light microscopy. Samples were centrifuged briefly to separate the supernatant from the nuclei, and the supernatant was centrifuged at 10,000 rpm for 10 min and collected as the “cytoplasmic” fraction. The nuclear pellet was lysed in CST lysis buffer, sonicated using a Bioruptor bath sonicator (Diagenode), and centrifuged at 10,000 rpm for 10 min at 4°C, and this lysate was collected as the “nuclear” fraction. Samples were resolved by SDS–polyacrylamide gel electrophoresis (PAGE) and blotted for lamin A/C (CST) or lactate dehydrogenase (Abcam) as markers for the nuclear and cytoplasmic fractions, respectively.
Western blotting
Samples were prepared at a normalized concentration in PBS [3.2 mM Na2HPO4, 0.5 mM KH2PO4, 1.3 mM KCl, and 135 mM NaCl (pH 7.4)] supplemented with protease inhibitor cocktail. All samples were resolved by SDS-PAGE using PROTEAN TGX precast gels (Bio-Rad) under reducing and denaturing conditions. Resolved proteins were transferred to polyvinyl difluoride (PVDF) membranes at either 300 mA for 2 to 3 hours or at 15 V overnight. Membranes were blocked with 5% milk in tris-buffered saline [50 mM tris-HCl (pH 7.6) and 150 mM NaCl] containing 0.5% Tween 20 (TBST) for 1 hour at room temperature and incubated with primary antibody with gentle rocking overnight at 4°C. Membranes were washed thrice with TBST, incubated with horseradish peroxidase (HRP)–conjugated secondary antibodies for 1 hour at room temperature, washed thrice with TBST, and developed using Pierce ECL substrate (Thermo Fisher Scientific) or Amersham ECL Prime (GE Healthcare) for 3 and 5 min, respectively. Quantitation of immunoblots was performed via densitometric analysis using ImageQuant TL software (GE Healthcare Life Sciences).
Protein purification
Glutathione-S-transferase (GST)–tagged protein expressing the catalytic SET domain of SETD2 (amino acids 1418 to 1714) was purified as previously described (15). For purification of recombinant tSETD2 (amino acids 1418 to 2564), tSETD2-Flag plasmid was transfected into HEK293 FreeStyle cells with a FectoPRO transfection reagent (116-010), and cells were harvested 36 to 48 hours later at 5000 rpm for 15 min (Beckman JLA 8.1, 363563). The pellet was suspended in lysis buffer [50 mM Hepes (pH 7.5), 50 mM MgCl2, and 150 mM NaCl] supplemented with complete protease inhibitor, and cells were lysed with 20 strokes of a Dounce homogenizer. This was ultracentrifuged (Beckman Ti70, 337922) at 40,000 rpm for 1 hour, and the supernatant was filtered with a 1.0-μm glass fiber filter (Pall Laboratory, AP-4527) and incubated with FLAG M2 affinity beads [Sigma-Aldrich, A2220) equilibrated in lysis buffer for 3 hours. Beads were rinsed with three column volumes of wash buffer (50 mM NaPi, 150 mM NaCl, and 5 mM beta-mercaptoethanol (BME)], three column volumes of salt buffer (wash buffer at 500 mM NaCl), and, again, with wash buffer before elution buffer [wash buffer with 300 ng of 3× FLAG peptide (Sigma-Aldrich, F4799)] was added and incubated with beads overnight. Eluent was then run over ion exchange column (DEAE Sepharose; GE Life Sciences, 17505501) on a 0 to 75% salt buffer gradient and size exclusion chromatography (Superose 6 Increase 10/300; Thermo Fisher Scientific, 45-003-210) with gel filtration buffer (50 mM NaPi, 150 mM NaCl, 5 mM BME, and 5% glycerol). Fractions were pooled and concentrated down with an Amicon Ultra 100K MWCO (UFC910024) centrifugal filter unit and snap frozen in liquid nitrogen and stored at −80°C.
The following commercially available proteins were used in this study: recombinant SETD2 (amino acids 1418 to 1714) protein (Active Motif, 31358), rabbit skeletal muscle actin (Cytoskeleton, Inc., AKL95), bovine cardiac muscle actin (Cytoskeleton, Inc., AD99), chicken gizzard smooth muscle actin (Cytoskeleton, Inc., AS99), β-actin (NM_001101) human recombinant protein (OriGene, TP303643), and β-actin (NM_001101) human recombinant protein (OriGene, TP720518).
In vitro methyltransferase assays
The intrinsic capacity of SETD2 to methylate actin was demonstrated using in vitro methyltransferase reaction assays. To visualize actin methylation via autoradiography (Fig. 2A), actins were incubated with GST-SETD2 (amino acids 1418 to 1714) for 3 hours at 37°C in the presence of tritiated S-adenosylmethionine (3H-SAM). Samples were resolved using SDS-PAGE and transferred to PVDF membranes. The signal was amplified using the EN3HANCE spray (PerkinElmer) and detected following incubation with an x-ray film for 2 to 6 weeks. For fluorescence-based assays, the activity of GST-SETD2 (amino acids 1418 to 1714) and tSETD2-Flag (amino acids 1418 to 2564) was measured over 3 to 4 hours using a methyltransferase fluorescence assay kit (Cayman Chemical, 700150), a continuous enzyme-coupled assay that continuously monitors SAM-dependent methyltransferase activity (50, 51). Readout fluorescence via resorufin was analyzed with an excitation wavelength of 540 nm and an emission wavelength of 590 nm using plate reader. For assays shown in Fig. 2 and fig. S2A, fluorescence was calculated after subtracting automethylation signal from samples with SETD2 only. For assays shown in Figs. 2B and 3C, a standard curve of resorufin concentration and fluorescence was used to determine time-dependent fluorescence. The initial velocities of these curves were obtained by linear regression to obtain methyltransferase activity in nmol/min (or a.u./min in Fig. 2C) using Prism (GraphPad). To detect actin methylation via immunoblotting (Fig. 2, D and E, and fig. S4C), 10 μl reactions containing 2 μg of actin were incubated with 1 μg of GST-SETD2 (amino acids 1418 to 1714) in an in vitro methyltransferase buffer [50 mM tris-HCl, 2 mM MgCl2, 0.02% Triton X-100, and 1 mM dithiothreitol (DTT) (pH 8.6)] overnight at 37°C in the presence of nontritiated SAM (New England Biolabs). Samples were resolved via SDS-PAGE and immunoblotted with the indicated antibodies overnight at 4°C followed by secondary antibody incubation for 2 hours at room temperature. Samples with SETD2 alone or actin alone were used as negative controls.
Mass spectrometry
Parental and SETD2-deficient 786-0 cells were grown to 70% confluence, washed with cold PBS twice, and directly lysed by adding 1 ml of boiling 2× SDS sample loading buffer. The lysate was harvested and sonicated for 30 s at 30 A using a microtip probe and centrifuged at 10,000g for 10 min, and 1 μl of cleared lysates was resolved on SDS-PAGE gel and Coomassie Blue R-250 (Bio-Rad). Visualized actin bands were excised, and gel pieces were destained, alkylated, and subjected to in-gel digestion using trypsin. The tryptic peptides were resuspended in 10 μl of loading solution (5% methanol and 0.1% formic acid) and subjected to nanoflow LC-MS/MS (liquid chromatography–tandem mass spectrometry) analysis using a nano-LC 1200 system (Thermo Fisher Scientific) coupled to Orbitrap Fusion Lumos (Thermo Scientific) mass spectrometer. The peptides were loaded onto a 2 cm × 100 μm ReproSil-Pur Basic C18 precolumn (1.9 μm; Dr. Maisch GmbH, Germany). The precolumn was switched in line with an in-housed 5 cm × 150 μm analytical column packed with ReproSil-Pur Basic C18 equilibrated in 0.1% formic acid/water. The peptides were eluted using a 45-min discontinuous gradient of 5 to 28% acetonitrile/0.1% formic acid at a flow rate of 750 nl/min. The eluted peptides were directly electrosprayed into the mass spectrometer. The instrument was operated in the data-dependent mode, acquiring fragmentation under direct control of Xcalibur software (Thermo Scientific). Precursor MS spectrum was scanned using Orbitrap followed by collision-induced dissociation fragmentation in the ion trap with 20 s of dynamic exclusion time. The raw file from MS was searched via Mascot 2.4 (Matrix Science) in the Proteome Discoverer 1.4 interface (Thermo Fisher Scientific). Variable modification of mono-, di-, and trimethylation on lysine, arginine, and histidine, oxidation on methionine, and carbamidomethylation of cysteine and protein N-terminal acetylation were allowed. The precursor mass tolerance was confined within 20 parts per million, with a fragment mass tolerance of 0.5 Da, and a maximum of two missed cleavages was allowed. The peptides identified from mascot result file were validated with 5% FDR. The trimethylation of ActK68 peptides was manually validated.
Immunocytochemistry
Cells were seeded overnight on glass coverslips in a six-well plate. Samples were fixed in 4% paraformaldehyde in PBS for 30 min at 37°C, permeabilized with 0.5% Triton X-100 in PBS for 20 min, blocked with 3.75% bovine serum albumin in PBS for 1 hour, and incubated with primary antibody (1:1000), with gentle rocking at 4°C overnight. Cells were then washed with PBS, incubated with anti-rabbit secondary antibody (1:2000) for 2 hours, and counterstained with DAPI (4′,6-diamidino-2-phenylindole) (1:4000) and phalloidin (1:1000) for 10 min following postfixation with 4% paraformaldehyde. Coverslips were rinsed and mounted on glass slides with SlowFade antifade mounting medium (Thermo Fisher Scientific). All steps were carried out at room temperature unless mentioned otherwise. Immunostained slides were imaged using a CFI Plan Apochromat Lambda 60× oil, 1.4-NA (numerical aperture) objective, and a DS-Qi2 camera mounted on a Nikon Eclipse qTi2-E inverted microscope system (Nikon Instruments, Inc.) equipped for standard phase contrast and epifluorescence, as well as for deconvolution. Image acquisition was carried using an Andor Zyla 4.2 PLUS sCMOS high-sensitivity monochrome camera and was driven by Nikon NIS-Elements Advanced Research image acquisition and analysis software. Images were processed using advanced deconvolution modules for improved image quality. For super-resolution imaging of actin, cells were grown on coverslips washed with HCl overnight and stained with phalloidin (1:40) for 2 hours at 37°C. Image stitching was accomplished through the use of an inverted epifluorescence microscope equipped with a G-2E/C wide-field filter cube (λex, 540/25; λem, 620/60), automated (and encoded) scanning stage, and 20× Plan Apochromat Lambda 0.75-NA objective lens (Nikon Instruments, Inc.). The system was additionally outfitted with a piezo-based axial positioning (Mad City Labs), LED (light-emitting diode) illumination (Lumencor), and Flash 4.0 V3 sCMOS (Hamamatsu Photonics). NIS-Elements was used for both acquisition and analysis (Nikon Instruments Inc.). For each coordinate (field of view) visited, image stacks were acquired to cover the required depth of the cell monolayer. Subsequently, stacks were collapsed through focus stacking in software, and a montage was created. SIM was accomplished in 3D-SIM mode on a Nikon Instruments N-SIM, equipped with an Apochromat TIRF 100× SR 1.49-NA objective, 561-nm laser, and DU-897 EMCCD (electron multiplying charge-couple device) camera (Andor). Images presented herein are maximum intensity projections after image stacks were first acquired (five phase shifts and three rotations of diffraction grating, and 120 nm per axial step) and after subsequent stack reconstruction in NIS-Elements software (Nikon Instruments Inc.). Other than linear intensity scaling, no further image processing was performed after reconstruction for image panels. For supplementary movies, nonlinear scaling along the z axis was performed to visualize the morphology between basal and apical actin networks, which display a wide dynamic range in intensity, and, hence, relative intensities cannot be compared visually as a measure of relative actin concentration.
In silico SETD2 methylation site analysis
Identification of putative SETD2 methylation sites was determined from the PhophoSitePlus methylation site database (52) available at www.phosphosite.org ➔ Downloads ➔ Datasets from PSP ➔ Methylation_site_dataset.gz (updated December 04 14:56:35 EST 2020). Proteins were screened for sites of lysine methylation containing either KxP or KxxG SETD2 recognition motifs and filtered to remove duplicate hits. Each list was used to generate an amino acid sequence logo using weblogo.berkeley.edu (53). Human proteins from a combined list were used for gene list analysis using the PANTHER classification system (54) available at www.pantherdb.org (version 15, released 4 February 2020). Data were analyzed using the Protein Class and Pathway ontology selections. Overrepresentation analysis was carried out against the default Homo sapiens whole-genome list using Fisher’s exact test and FDR correction for multiple comparisons, with a P value of <0.05 considered significant. Data from these analyses were downloaded and represented in the indicated figures.
F-actin to G-actin ratio
Cell fractionation was performed, as previously described (21), with slight modification, to discriminate between G- and F-actin based on the observation that polymerized F-actin is insoluble whereas G-actin is soluble. Cells were collected at 70 to 80% confluency in ice-cold PBS, pelleted at 500g for 5 min at 4°C, and lysed in an ice-cold lysis buffer [10 mM K2HPO4, 100 mM NaF, 50 mM KCl, 2 mM MgCl2, 1 mM EGTA, 0.2 mM DTT, 0.5% Triton X-100, and 1 mM sucrose (pH 7.0)]. Samples were pipetted repeatedly and vortexed and centrifuged at 15,000g for 10 min at room temperature. The supernatant was collected for measurement of G-actin. The insoluble F-actin in the pellet was washed with lysis buffer to remove any residual G-actin, resuspended in equal volumes of lysis buffer and a second buffer [1.5 mM guanidine hydrochloride,1 mM sodium acetate, 1 mM CaCl2, 1 mM adenosine triphosphate, and 20 mM tris-HCl (pH 7.5)], and incubated on ice for 20 min, with gentle mixing every 5 min to solubilize polymerized F-actin. The samples were centrifuged at 15,000g for 10 min at room temperature, and the supernatant was collected for the measurement of F-actin. Samples were proportionally loaded and resolved by SDS-PAGE, immunoblotted with an actin antibody.
Alternatively, cells were collected in ice-cold PBS, pelleted at 1500 rpm for 5 min at 4°C, and lysed in a “solution A” [10 mM tris-HCl (pH 8.0), 3 mM CaCl2, 2 mM MgOAc, 0.1 mM EDTA, 320 mM sucrose, 1 mM DTT, and 0.2% NP-40] supplemented with protease inhibitor cocktail on ice for 10 min. Samples were briefly centrifuged to separate the soluble supernatant and insoluble pellet. The supernatant was centrifuged at 10,000 rpm for 10 min at 4°C. The pellet was washed twice with a “solution B” (same as solution A, minus DTT and NP-40), resuspended in cold CST lysis buffer, sonicated using a Bioruptor bath sonicator (Diagenode), and centrifuged at 10,000 rpm for 10 min at 4°C. The resulting lysates constitute the soluble G-actin (supernatant) and insoluble F-actin (pellet) fractions of the cell. The insoluble F-actin fraction samples were resolved by SDS-PAGE and blotted with an actin antibody. Samples were normalized to histone H3 as a marker for the insoluble fraction. For IP, 500 μg of normalized lysate was used from the soluble and insoluble fractions for incubation with antibodies as indicated.
Phalloidin binding assay
Fluorimetric analysis of phalloidin binding is based on the observation that phalloidin binds to polymerized (F-) actin only, and thus serves as a readout of intracellular actin polymers, and was performed as previously described (23). Cells were plated in 24-well dishes and fixed at 70 to 80% confluency with 4% paraformaldehyde in PBS for 20 to 30 min and permeabilized in 0.5% Triton X-100 in PBS for 10 min. Wells were incubated with Alexa Fluor 568 (AF568) phalloidin (Thermo Fisher Scientific) at varying concentrations in PBS for 20 min, washed several times with PBS quickly, and the bound phalloidin was extracted from each well using a 0.1 N NaOH solution. The fluorescence intensity for each phalloidin concentration was measured in duplicate using a spectrophotometer with excitation and emission wavelengths of 575 and 605 nm, respectively. For unstained controls, a 10× excess of unconjugated phalloidin (Sigma-Aldrich) was added to obtain a reading for nonspecific binding, and this was subtracted from fluorescent readings at each concentration to yield fluorescence as a result of specific binding to F-actin. Cells were stained with DAPI following phalloidin extraction and imaged using an Operetta Phenix high-content screening system (PerkinElmer). Cells were counted in three to six wells for each genotype/treatment, and the fluorimetric intensity was normalized to mean cell number. For flow cytometric analysis of phalloidin binding, cells were grown to 70 to 80% confluency, trypsinized and collected in media containing 10% FBS, and pelleted at 500 rpm for 5 min at 4°C. Cells were fixed in 4% paraformaldehyde in PBS with 1 mM EDTA (PBSE) for 20 to 30 min, permeabilized in 0.5% Triton X-100 for 10 min, and incubated with AF568 phalloidin at varying concentrations (with or without 10× excess unconjugated phalloidin) for 20 min. Cells were washed twice with PBSE between each step. The final samples were resuspended using PBSE in BD Falcon test tubes with strainer caps and run on a BD LSRFortessa cell analyzer (BD Biosciences) within 1 hour of preparation. A total of 10,000 events were acquired for each sample, subjected to doublet discrimination, and single-color compensation performed using the 10× unconjugated sample intensity. Samples were analyzed using FlowJo (BD Biosciences).
In vitro migration (scratch) assay
Quantitative measurement of cell migration was conducted as previously described (55). Briefly, cells were seeded overnight to 90 to 100% confluence in a six-well plate. Scratches were made using a 1000-μl pipette tip, with each well washed twice with PBS to remove nonadherent cells. Fresh medium was added to the wells, and 9 to 12 designated points were measured at 0 hour. The same designated points were measured again after incubation for 24 hours. Images were analyzed using the “Manual Measurement > Length” feature on NIS-Elements software (Nikon). The distance traveled was computed from the difference in scratch width at 0 and 24 hours.
In vitro proliferation assay
Quantitative measurement of cell proliferation was measured by counting cells over 48 hours (the total duration of the scratch assay migration experiments). Fifty thousand cells were seeded triplicate in six-well plates; at 24 and 48 hours, cells were trypsinized, stained with trypan blue, and manually counted twice each using a hemocytometer.
Antibodies
The following primary antibodies were used in this study: actin (C4, mouse; Santa Cruz Biotechnology, sc-47778), actin (C4, mouse; Millipore, MAB1501), actin (13E5, rabbit; Cell Signaling, 4970S), CFP (rabbit; Bio-Rad, AHP2986), GST (mouse; Santa Cruz Biotechnology, sc-138), HIP1R (rabbit; Proteintech, 16814-1-AP), histone H3 (D1H2, rabbit; Cell Signaling, 4620S), histone H3K36me3 (rabbit; Active Motif, 61101), HTT (1HU-4C8, mouse; Millipore, MAB2166), HTT (mouse; Bethyl, A302-812A), lamin A/C (rabbit; Cell Signaling, 2032S), lactate dehydrogenase (LDH; rabbit; Abcam, ab47010), mCherry (16D7, rat; Thermo Fisher Scientific, M11217), mCherry (rabbit; Abcam, ab167453), mCherry (mouse; Novus, NBP1-96752), pan–anti-trimethyllysine (rabbit; PTM Biolabs, PTM-601), SETD2 (rabbit; Abcam, ab31358), SETD2 (rabbit; ABclonal, A3194), SETD2 (rabbit; Invitrogen, PA5-83615), SETD2 (rabbit; Sigma-Aldrich, HPA-042451), SETD3 (rabbit; Abcam, ab176582), and tubulin (DM1A, mouse; Santa Cruz Biotechnology, sc-32293).
Anti-Me3K40 methyl-tubulin antibodies were generated as previously described (15, 56). The methylated actin antibody was generated using a keyhole limpet hemocyanin–linked K68 trimethylated synthetic peptide [Ac-CRGILTL(KMe3)YPIE-amide] used for immunization in rabbits (Covance). The serum from immunized rabbits (after the third boost) was purified by means of an affinity column using an unmodified actin peptide (amino acids 62 to 72, Ac-CRGILTLKYPIE-amide) to remove antibodies that could react with unmethylated or pan-actin. The elute was subsequently passed through a second affinity column using the K68-trimethylated peptide to capture antibodies directed toward the K68 trimethyl mark on actin.
The following secondary antibodies were used in the study: anti-rabbit HRP (mouse; Santa Cruz Biotechnology, sc-2357), anti-mouse HRP (goat; Santa Cruz Biotechnology, sc-2005), anti-mouse HRP (goat; Bio-Rad, 1706516), and anti-rat HRP (goat; Santa Cruz Biotechnology, sc-2065). The following normal IgG isotype IP controls were used in the study: mouse (Santa Cruz Biotechnology, sc-2025), rat (Santa Cruz Biotechnology, sc-2026), rabbit (Santa Cruz Biotechnology, sc-2027), and rabbit (Abcam, ab37415).
Supplementary Material
Acknowledgments
We thank K. Landua and S. Williams (Nikon Instruments Inc.) for assistance with immunofluorescence microscopy experiments. We thank Z. Chriss for assistance with imaging of scratch assay samples. We thank W. Russell for preliminary MS assistance identifying the SETD2 methylation site on actin. We thank A. Sokac, S. McGuire, G. Eisenhoffer, H. Coutney Hodges, M. Costa-Mattioli, S. Khurana, J. Botas, and P. Msaouel for insightful conversations related to this manuscript. We thank the two reviewers for critical, constructive comments in strengthening this study. We thank L. Guillen and N. Patel for administrative support. Funding: This work is supported by grants from the American Heart Association, predoctoral fellowship 19PRE34430069 (R.N.H.S.); the NIH, NCI-R35CA231993 (C.L.W.), R01CA203012 (W.K.R. and C.L.W.), and R35GM131744 (K.J.V.); and the Templeton Foundation, no. 61099 (C.L.W.). R.N.H.S. is supported by the Baylor College of Medicine (BCM) Medical Scientist Training Program and a BP America Biomedical Scholarship from the BCM Graduate School of Biomedical Sciences. D.N.T. is supported by grants from the Department of Defense (KC170259) and the William and Ella Owens Medical Research Foundation. Proteomics analysis is supported by P30ES023512, and by P30CA125123 and CPRIT-RP170005 for the BCM proteomics core. The BCM Cytometry and Cell Sorting core (B. Saxton and director J. Sederstrom) is supported by CPRIT-RP180672, NCI-CA125123, and NIH-RR024574. Author contributions: C.L.W., R.N.H.S., and I.Y.P. conceptualized the study. C.L.W., R.N.H.S., I.Y.P., D.N.T., and R.D. designed experiments, with input from K.J.V., F.M.M., and W.K.R. R.N.H.S. performed experiments and analyzed data contributing to all figures, with assistance as noted: R.K.J. and I.Y.P. assisted with and performed dot blot assays and in vitro methyltransferase assays using recombinant GST-SETD2 (amino acids 1418 to 1714). M.K. performed and assisted with immunofluorescence assays. D.N.T. assisted with IP, Western blotting, siRNA knockdown, and transgene overexpression assays. S.E.K. performed fluorescence-based in vitro methyltransferase assays using recombinant tSETD2 (amino acids 1418 to 2564), with supervision from K.J.V. and M.A.C. S.Y.J. performed MS experiments. B.A.M. performed high-/super-resolution SIM imaging, with supervision from M.J.T. R.N.H.S. and C.L.W. wrote the initial and revised manuscript, with editorial input/approval from all the authors, and conducted correspondence. Unless noted, all work was performed under the supervision of C.L.W. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabb7854/DC1
REFERENCES AND NOTES
- 1.Cheng X., Collins R. E., Zhang X., Structural and sequence motifs of protein (histone) methylation enzymes. Annu. Rev. Biophys. Biomol. Struct. 34, 267–294 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faber P. W., Barnes G. T., Srinidhi J., Chen J., Gusella J. F., MacDonald M. E., Huntingtin interacts with a family of WW domain proteins. Hum. Mol. Genet. 7, 1463–1474 (1998). [DOI] [PubMed] [Google Scholar]
- 3.Edmunds J. W., Mahadevan L. C., Clayton A. L., Dynamic histone H3 methylation during gene induction: HYPB/Setd2 mediates all H3K36 trimethylation. EMBO J. 27, 406–420 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun X.-J., Wei J., Wu X.-Y., Hu M., Wang L., Wang H.-H., Zhang Q.-H., Chen S.-J., Huang Q.-H., Chen Z., Identification and characterization of a novel human histone H3 lysine 36-specific methyltransferase. J. Biol. Chem. 280, 35261–35271 (2005). [DOI] [PubMed] [Google Scholar]
- 5.McDaniel S. L., Strahl B. D., Shaping the cellular landscape with Set2/SETD2 methylation. Cell. Mol. Life Sci. 74, 3317–3334 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagner E. J., Carpenter P. B., Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 13, 115–126 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stabell M., Larsson J., Aalen R. B., Lambertsson A., Drosophila dSet2 functions in H3-K36 methylation and is required for development. Biochem. Biophys. Res. Commun. 359, 784–789 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Hu M., Sun X.-J., Zhang Y.-L., Kuang Y., Hu C.-Q., Wu W.-L., Shen S.-H., Du T.-T., Li H., He F., Xiao H.-S., Wang Z.-G., Liu T.-X., Lu H., Huang Q.-H., Chen S.-J., Chen Z., Histone H3 lysine 36 methyltransferase Hypb/Setd2 is required for embryonic vascular remodeling. Proc. Natl. Acad. Sci. U.S.A. 107, 2956–2961 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Cubas A. A., Rathmell W. K., Epigenetic modifiers: Activities in renal cell carcinoma. Nat. Rev. Urol. 15, 599–614 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fahey C. C., Davis I. J., SETting the stage for cancer development: SETD2 and the consequences of lost methylation. Cold Spring Harb. Perspect. Med. 7, a026468 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morris M. R., Latif F., The epigenetic landscape of renal cancer. Nat. Rev. Nephrol. 13, 47–60 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Lumish H. S., Wynn J., Devinsky O., Chung W. K., Brief report: SETD2 mutation in a child with autism, intellectual disabilities and epilepsy. J. Autism Dev. Disord. 45, 3764–3770 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Luscan A., Laurendeau I., Malan V., Francannet C., Odent S., Giuliano F., Lacombe D., Touraine R., Vidaud M., Pasmant E., Cormier-Daire V., Mutations in SETD2 cause a novel overgrowth condition. J. Med. Genet. 51, 512–517 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Marzin P., Rondeau S., Aldinger K. A., Alessandri J.-L., Isidor B., Heron D., Keren B., Dobyns W. B., Cormier-Daire V., SETD2 related overgrowth syndrome: Presentation of four new patients and review of the literature. Am. J. Med. Genet. C Semin. Med. Genet. 181, 509–518 (2019). [DOI] [PubMed] [Google Scholar]
- 15.Park I. Y., Powell R. T., Tripathi D. N., Dere R., Ho T. H., Blasius T. L., Chiang Y.-C., Davis I. J., Fahey C. C., Hacker K. E., Verhey K. J., Bedford M. T., Jonasch E., Rathmell W. K., Walker C. L., Dual chromatin and cytoskeletal remodeling by SETD2. Cell 166, 950–962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chiang Y.-C., Park I.-Y., Terzo E. A., Tripathi D. N., Mason F. M., Fahey C. C., Karki M., Shuster C. B., Sohn B.-H., Chowdhury P., Powell R. T., Ohi R., Tsai Y. S., de Cubas A. A., Khan A., Davis I. J., Strahl B. D., Parker J. S., Dere R., Walker C. L., Rathmell W. K., SETD2 haploinsufficiency for microtubule methylation is an early driver of genomic instability in renal cell carcinoma. Cancer Res. 78, 3135–3146 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilkinson A. W., Diep J., Dai S., Liu S., Ooi Y. S., Song D., Li T.-M., Horton J. R., Zhang X., Liu C., Trivedi D. V., Ruppel K. M., Vilches-Moure J. G., Casey K. M., Mak J., Cowan T., Elias J. E., Nagamine C. M., Spudich J. A., Cheng X., Carette J. E., Gozani O., SETD3 is an actin histidine methyltransferase that prevents primary dystocia. Nature 565, 372–376 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwiatkowski S., Seliga A. K., Vertommen D., Terreri M., Ishikawa T., Grabowska I., Tiebe M., Teleman A. A., Jagielski A. K., Veiga-da-Cunha M., Drozak J., SETD3 protein is the actin-specific histidine N-methyltransferase. eLife 7, e37921 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nelson C. J., Santos-Rosa H., Kouzarides T., Proline isomerization of histone H3 regulates lysine methylation and gene expression. Cell 126, 905–916 (2006). [DOI] [PubMed] [Google Scholar]
- 20.Chen K., Liu J., Liu S., Xia M., Zhang X., Han D., Jiang Y., Wang C., Cao X., Methyltransferase SETD2-mediated methylation of STAT1 is critical for interferon antiviral activity. Cell 170, 492–506.e14 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Gu Y.-Y., Zhang H.-Y., Zhang H.-J., Li S.-Y., Ni J.-H., Jia H.-T., 8-Chloro-adenosine inhibits growth at least partly by interfering with actin polymerization in cultured human lung cancer cells. Biochem. Pharmacol. 72, 541–550 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Holzinger A., Jasplakinolide: An actin-specific reagent that promotes actin polymerization. Methods Mol. Biol. 586, 71–87 (2009). [DOI] [PubMed] [Google Scholar]
- 23.J. A. Cooper, in The Cytoskeleton: A Practical Approach, K. L. Carraway, C. A. C. Carraway, Eds. (Oxford Univ. Press, New York, 1992), pp. 47–71. [Google Scholar]
- 24.Gao Y.-G., Yang H., Zhao J., Jiang Y.-J., Hu H.-Y., Autoinhibitory structure of the WW domain of HYPB/SETD2 regulates its interaction with the proline-rich region of Huntingtin. Structure 22, 378–386 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Engqvist-Goldstein Å. E. Y., Kessels M. M., Chopra V. S., Hayden M. R., Drubin D. G., An actin-binding protein of the Sla2/Huntingtin interacting protein 1 family is a novel component of clathrin-coated pits and vesicles. J. Cell Biol. 147, 1503–1518 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shirasaki D. I., Greiner E. R., Al-Ramahi I., Gray M., Boontheung P., Geschwind D. H., Botas J., Coppola G., Horvath S., Loo J. A., Yang X. W., Network organization of the huntingtin proteomic interactome in mammalian brain. Neuron 75, 41–57 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tourette C., Li B., Bell R., O'Hare S., Kaltenbach L. S., Mooney S. D., Hughes R. E., A large scale huntingtin protein interaction network implicates Rho GTPase signaling pathways in Huntington disease. J. Biol. Chem. 289, 6709–6726 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Culver B. P., Savas J. N., Park S. K., Choi J. H., Zheng S., Zeitlin S. O., Yates J. R. III, Tanese N., Proteomic analysis of wild-type and mutant huntingtin-associated proteins in mouse brains identifies unique interactions and involvement in protein synthesis. J. Biol. Chem. 287, 21599–21614 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Angeli S., Shao J., Diamond M. I., F-actin binding regions on the androgen receptor and huntingtin increase aggregation and alter aggregate characteristics. PLOS ONE 5, e9053 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kwan W., Träger U., Davalos D., Chou A., Bouchard J., Andre R., Miller A., Weiss A., Giorgini F., Cheah C., Möller T., Stella N., Akassoglou K., Tabrizi S. J., Muchowski P. J., Mutant huntingtin impairs immune cell migration in Huntington disease. J. Clin. Invest. 122, 4737–4747 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Munsie L., Caron N., Atwal R. S., Marsden I., Wild E. J., Bamburg J. R., Tabrizi S. J., Truant R., Mutant huntingtin causes defective actin remodeling during stress: Defining a new role for transglutaminase 2 in neurodegenerative disease. Hum. Mol. Genet. 20, 1937–1951 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scherzinger E., Lurz R., Turmaine M., Mangiarini L., Hollenbach B., Hasenbank R., Bates G. P., Davies S. W., Lehrach H., Wanker E. E., Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558 (1997). [DOI] [PubMed] [Google Scholar]
- 33.Passani L. A., Bedford M. T., Faber P. W., McGinnis K. M., Sharp A. H., Gusella J. F., Vonsattel J.-P., MacDonald M. E., Huntingtin's WW domain partners in Huntington's disease post-mortem brain fulfill genetic criteria for direct involvement in Huntington's disease pathogenesis. Hum. Mol. Genet. 9, 2175–2182 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Cornett E. M., Ferry L., Defossez P.-A., Rothbart S. B., Lysine methylation regulators moonlighting outside the epigenome. Mol. Cell 75, 1092–1101 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gunawan M., Venkatesan N., Loh J. T., Wong J. F., Berger H., Neo W. H., Li L. Y. J., La Win M. K., Yau Y. H., Guo T., See P. C. E., Yamazaki S., Chin K. C., Gingras A. R., Shochat S. G., Ng L. G., Sze S. K., Ginhoux F., Su I.-h., The methyltransferase Ezh2 controls cell adhesion and migration through direct methylation of the extranuclear regulatory protein talin. Nat. Immunol. 16, 505–516 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Su I.-h., Dobenecker M.-W., Dickinson E., Oser M., Basavaraj A., Marqueron R., Viale A., Reinberg D., Wülfing C., Tarakhovsky A., Polycomb group protein ezh2 controls actin polymerization and cell signaling. Cell 121, 425–436 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Nyman T., Schüler H., Korenbaum E., Schutt C. E., Karlsson R., Lindberg U., The role of MeH73 in actin polymerization and ATP hydrolysis. J. Mol. Biol. 317, 577–589 (2002). [DOI] [PubMed] [Google Scholar]
- 38.Karlsson R., Expression of chicken beta-actin in Saccharomyces cerevisiae. Gene 68, 249–257 (1988). [DOI] [PubMed] [Google Scholar]
- 39.Varland S., Vandekerckhove J., Drazic A., Actin post-translational modifications: The Cinderella of cytoskeletal control. Trends Biochem. Sci. 44, 502–516 (2019). [DOI] [PubMed] [Google Scholar]
- 40.Terman J. R., Kashina A., Post-translational modification and regulation of actin. Curr. Opin. Cell Biol. 25, 30–38 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li M.-M., Nilsen A., Shi Y., Fusser M., Ding Y.-H., Fu Y., Liu B., Niu Y., Wu Y.-S., Huang C.-M., Olofsson M., Jin K.-X., Lv Y., Xu X.-Z., He C., Dong M.-Q., Rendtlew Danielsen J. M., Klungland A., Yang Y.-G., ALKBH4-dependent demethylation of actin regulates actomyosin dynamics. Nat. Commun. 4, 1832 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ayscough K. R., Stryker J., Pokala N., Sanders M., Crews P., Drubin D. G., High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. J. Cell Biol. 137, 399–416 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujita M., Ichinose S., Kiyono T., Tsurumi T., Omori A., Establishment of latrunculin-A resistance in HeLa cells by expression of R183A D184A mutant β-actin. Oncogene 22, 627–631 (2003). [DOI] [PubMed] [Google Scholar]
- 44.Svitkina T., The actin cytoskeleton and actin-based motility. Cold Spring Harb. Perspect. Biol. 10, a018267 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Skruber K., Read T.-A., Vitriol E. A., Reconsidering an active role for G-actin in cytoskeletal regulation. J. Cell Sci. 131, jcs203760 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ross C. A., Intranuclear neuronal inclusions: A common pathogenic mechanism for glutamine-repeat neurodegenerative diseases? Neuron 19, 1147–1150 (1997). [DOI] [PubMed] [Google Scholar]
- 47.Davies S. W., Turmaine M., Cozens B. A., DiFiglia M., Sharp A. H., Ross C. A., Scherzinger E., Wanker E. E., Mangiarini L., Bates G. P., Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Hyrskyluoto A., Vartiainen M. K., Regulation of nuclear actin dynamics in development and disease. Curr. Opin. Cell Biol. 64, 18–24 (2020). [DOI] [PubMed] [Google Scholar]
- 49.Ridley A. J., Life at the leading edge. Cell 145, 1012–1022 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Burgos E. S., Walters R. O., Huffman D. M., Shechter D., A simplified characterization of S-adenosyl-l-methionine-consuming enzymes with 1-Step EZ-MTase: A universal and straightforward coupled-assay for in vitro and in vivo setting. Chem. Sci. 8, 6601–6612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dorgan K. M., Wooderchak W. L., Wynn D. P., Karschner E. L., Alfaro J. F., Cui Y., Zhou Z. S., Hevel J. M., An enzyme-coupled continuous spectrophotometric assay for S-adenosylmethionine-dependent methyltransferases. Anal. Biochem. 350, 249–255 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Hornbeck P. V., Zhang B., Murray B., Kornhauser J. M., Latham V., Skrzypek E., PhosphoSitePlus, 2014: Mutations, PTMs and recalibrations. Nucleic Acids Res. 43, D512–D520 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crooks G. E., Hon G., Chandonia J.-M., Brenner S. E., WebLogo: A sequence logo generator. Genome Res. 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thomas P. D., Campbell M. J., Kejariwal A., Mi H., Karlak B., Daverman R., Diemer K., Muruganujan A., Narechania A., PANTHER: A library of protein families and subfamilies indexed by function. Genome Res. 13, 2129–2141 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chowdhury B., Porter E. G., Stewart J. C., Ferreira C. R., Schipma M. J., Dykhuizen E. C., PBRM1 regulates the expression of genes involved in metabolism and cell adhesion in renal clear cell carcinoma. PLOS ONE 11, e0153718 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Park I. Y., Chowdhury P., Tripathi D. N., Powell R. T., Dere R., Terzo E. A., Rathmell W. K., Walker C. L., Methylated α-tubulin antibodies recognize a new microtubule modification on mitotic microtubules. MAbs 8, 1590–1597 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabb7854/DC1