

Disease-associated glycoproteome changes identified in human brain provide new insights into Alzheimer’s disease pathogenesis.
Abstract
Protein N-glycosylation plays critical roles in controlling brain function, but little is known about human brain N-glycoproteome and its alterations in Alzheimer’s disease (AD). Here, we report the first, large-scale, site-specific N-glycoproteome profiling study of human AD and control brains using mass spectrometry–based quantitative N-glycoproteomics. The study provided a system-level view of human brain N-glycoproteins and in vivo N-glycosylation sites and identified disease signatures of altered N-glycopeptides, N-glycoproteins, and N-glycosylation site occupancy in AD. Glycoproteomics-driven network analysis showed 13 modules of co-regulated N-glycopeptides/glycoproteins, 6 of which are associated with AD phenotypes. Our analyses revealed multiple dysregulated N-glycosylation–affected processes and pathways in AD brain, including extracellular matrix dysfunction, neuroinflammation, synaptic dysfunction, cell adhesion alteration, lysosomal dysfunction, endocytic trafficking dysregulation, endoplasmic reticulum dysfunction, and cell signaling dysregulation. Our findings highlight the involvement of N-glycosylation aberrations in AD pathogenesis and provide new molecular and system-level insights for understanding and treating AD.
INTRODUCTION
Alzheimer’s disease (AD) is a devastating neurodegenerative dementia with no effective means of prevention or treatment (1). The etiology of AD is multifactorial and complex, with the vast majority of cases not attributed to monogenic mutations. Current understanding of AD pathogenic mechanisms is incomplete, and amyloidocentric clinical trials have failed to show therapeutic efficacy (2). Large-scale molecular profiling of disease-associated changes in postmortem human brain has emerged as a useful, unbiased approach to discover novel pathogenic mechanisms and therapeutic targets for brain diseases (3–5). Despite recent progress in characterization of AD-related changes at the genomic, transcriptomic, and proteomic levels (5–7), our knowledge of system-wide changes at the protein posttranslational modification levels in AD brain remains very limited.
Protein N-glycosylation, which covalently attaches glycans to asparagine residues of proteins, is the most prevalent type of posttranslational modification in cells (8, 9). N-glycosylation regulates diverse cellular processes by affecting protein conformation, activity, trafficking, stability, and interaction with other molecules. Unlike most posttranslational modifications, which modify cytosolic proteins or cytoplasmic regions of membrane proteins, N-glycosylation occurs on secreted proteins, extracellular regions of cell surface proteins, lumenal proteins, or lumenal regions of membrane proteins of intracellular organelles, such as endoplasmic reticulum (ER), Golgi, and lysosome (8, 9). Thus, N-glycosylation is particularly important for regulating ligand-receptor interaction, cell-to-cell communication, immune response, membrane trafficking, and cell signaling. The prevalent localization of N-glycosylated proteins (N-glycoproteins) in extracellular space and cell surface also makes them an attractive source of disease biomarkers and drug targets.
Proper N-glycosylation is critical to brain function, as highlighted by the fact that human congenital disorders with mutations affecting the N-glycosylation pathway exhibit neurological abnormalities (10). Previous studies have implicated a potential role of aberrant N-glycosylation in AD, and changes in N-glycan structures have been observed in cerebrospinal fluid and brain samples from patients with AD (11–13). Several AD-related proteins, such as amyloid precursor protein (APP), β-site APP-cleaving enzyme-1 (BACE1), and γ-secretase component nicastrin, have been shown to undergo N-glycosylation in cultured cells that can affect their trafficking, subcellular localization, and/or degradation (12, 13). Furthermore, although N-glycosylation is known to be topologically restricted to the lumenal or extracellular region of membrane proteins and secreted proteins (8, 9), tau, a cytosolic protein which is associated with microtubules, was reported to be N-glycosylated in AD but not control brain, and the abnormal tau N-glycosylation was suggested to affect tau phosphorylation and aggregation (11–13). However, strong evidence for N-glycosylation of tau and other AD-related proteins in human AD brain is still lacking, and their in vivo N-glycosylation sites (N-glycosites) in AD brain have not been mapped. Currently, our knowledge of human brain N-glycoproteome and in vivo N-glycosites is very limited, and site-specific N-glycoproteome changes in AD brain remain uncharacterized.
Here, we describe an unbiased, large-scale, site-specific N-glycoproteome profiling study to characterize protein N-glycosylation in human AD and control brains using a high-resolution mass spectrometry–based, quantitative N-glycoproteomics approach. This study provides the first system-level view of human brain N-glycoproteins and in vivo N-glycosylation sites and reveals previously unknown changes in AD brain N-glycoproteome. Furthermore, glycoproteomics-driven network analysis shows a higher-order organization of brain N-glycoproteome into a glyco-network of biologically meaningful modules of co-regulated N-glycopeptides/glycoproteins and identifies disease-associated glyco-network modules and hub glycopeptides/glycoproteins in AD. Our analyses uncover a number of aberrant N-glycosylation–affected processes and pathways in AD brain and provide new insights for understanding and treating AD.
RESULTS
N-glycoproteome profiling analysis of human AD and control brains
To determine brain N-glycoproteome alterations associated with AD, we performed a large-scale, site-specific quantitative N-glycoproteomics analysis of AD and control brain tissues by using an N-glycoproteomics pipeline consisting of filter-aided sample preparation (FASP) (14), zwitterionic chromatography–hydrophilic interaction chromatography (ZIC-HILIC)–based N-glycopeptide enrichment (15), 18O-labeling of in vivo N-glycosylation sites (16), and liquid chromatography–tandem mass spectrometry (LC-MS/MS). We analyzed the N-glycoproteomes of the same sets of dorsolateral prefrontal cortex tissue samples from eight neuropathologically confirmed AD cases and eight age-matched control individuals (table S1) under the same protein extraction and FASP conditions as in our proteomics study (4) to better integrate N-glycoproteome and proteome profiling data. Our N-glycoproteomics analysis identified a total of 4730 N-glycosite–containing peptides (hereafter referred to as N-glycopeptides) containing 18O-tagged N-glycosylation sequon N-X-S|T|C (where X represents any amino acid except proline), corresponding to 2294 distinct N-glycosites from 1132 unique N-glycoprotein groups (Fig. 1, A to D, and table S2). The number of N-glycosites (Fig. 1C) was fewer than the number of N-glycopeptides (Fig. 1B) because more than one N-glycopeptide could be detected for a single N-glycosite because of modification of other amino acid residues within the same glycopeptide sequence. For example, we detected two glycopeptides containing 18O-tagged AGRN N932 glycosite, one glycopeptide with an unmodified Met residue and the other with an oxidized Met residue at AGRN M925 position (table S2). To our knowledge, the dataset generated from this study represents the first, large-scale N-glycoproteome dataset of human AD and control brains.
Fig. 1. Quantitative N-glycoproteomics analysis of AD and control brains.

(A) Venn diagram comparison of identified proteins in human brain N-glycoproteome versus proteome. (B to D) Comparisons of identified N-glycopeptides (B), N-glycosites (C), and N-glycoproteins (D) in AD versus control (CT) brains. (E) GO localization categories enriched or underrepresented in the N-glycoproteome compared to the proteome with Benjamini-Hochberg false discovery rate (FDR)–corrected P < 0.0001. (F) Match of the identified N-glycosites to UniProt database of annotated N-glycosites. (G) Mapping of the identified N-glycosites to proteins with UniProt-annotated topology. (H) Distribution of the identified N-glycosites with the indicated tripeptide sequon is shown as means ± SD (n = 8 cases per AD or control group). (I) Pie charts showing the proportions of singly and multiply N-glycosylated proteins with the indicated number of in vivo N-glycosites per protein in AD and controls. (J to L) Bar graphs showing the number of N-glycosites identified on AD-related proteins and risk factors (J) and examples of identified glycoproteins with increased (K) or reduced (L) number of in vivo N-glycosites in AD compared to controls. Data are shown as means ± SD (n = 8 cases per group), and the total number of identified N-glycosites per protein is indicated above each bar.
Comparison of the N-glycoproteome dataset with our proteome profiling data from the same brain samples (4) showed that, of the 1132 glycoprotein groups, 717 were also found in the proteome dataset (Fig. 1A). Four hundred fifteen glycoprotein groups were exclusively identified by N-glycoproteome analysis (Fig. 1A), demonstrating the capacity of glycopeptide enrichment in our N-glycoproteomics workflow for enabling analysis of a distinct subset of proteins that may otherwise remain undetected by global proteome profiling. Gene Ontology (GO) analysis using A.GO.TOOL (17) showed that, compared to the brain proteome, the brain N-glycoproteome was overrepresented for GO localization categories of plasma membrane, extracellular region, synaptic membranes, vesicle, and lumen of ER, lysosome, and Golgi, whereas cytosol, nucleus, and mitochondrion were underrepresented (Fig. 1E). The N-glycoproteome was also highly enriched for glycoproteins, signal peptide–containing proteins, transmembrane proteins, and secreted proteins as well as receptors, cell adhesion proteins, and immunity-related proteins and underrepresented for cytoplasmic proteins, RNA-binding proteins, cytoskeletal proteins, and transcription factors (figs. S1, A and B), which are known to be rarely N-glycosylated.
Matching our N-glycoproteome data to the UniProt database showed that about 76% of 2294 N-glycosylation sites identified in this study are annotated N-glycosites in the UniProt database. Of the 1740 UniProt-annotated N-glycosites, the annotation of 957 N-glycosites is based on prediction by sequence analysis or similarity (Fig. 1F). Thus, our study provides experimental evidence for more than 950 UniProt-predicted N-glycosites, in addition to confirming nearly 800 previously observed N-glycosites and identifying more than 550 new N-glycosites that have not been annotated in the UniProt database (Fig. 1F). Mapping of our identified N-glycosites to the UniProt protein topology data showed that, of the 1302 N-glycosites derived from transmembrane proteins with UniProt-annotated topological domains, 1114 N-glycosites (86%) are located in the annotated extracellular domain and 157 N-glycosites (12%) in the annotated lumenal domain (Fig. 1G).
In our N-glycoproteome dataset, 1333 N-glycosites were found in both AD and control brains, whereas 698 N-glycosites and 263 N-glycosites were exclusively detected in AD and control brains, respectively (Fig. 1C and table S2). In control brains, approximately 97% of the identified N-glycosites fit the canonical N-glycosylation motif N-X-S|T with a higher frequency of N-X-T (56%) than N-X-S (41%) and about 3% of the N-glycosites match the N-X-C motif, and this distribution was unaltered by AD (Fig. 1H). Approximately 63% of N-glycoproteins in control brains were identified with one in vivo N-glycosylation site, 18% with two N-glycosites, and 9% of glycoproteins with more than three N-glycosites (Fig. 1I and fig. S1C). The percentage of glycoproteins with more than three N-glycosites was increased to about 12% in AD brains with concomitant reduction in the percentage of singly N-glycosylated proteins to 60% (Fig. 1I and fig. S1C). The average degree of N-glycosylation (i.e., average number of in vivo N-glycosites per glycoprotein) was 1.9 in control and 2.0 in AD brains.
While the potential protein N-glycosylation sites can be predicted using the consensus sequence of N-X-S|T (X ≠ P), whether or not the predicted sites are actually N-glycosylated in vivo is dependent on protein conformation, subcellular localization, sequon location within the protein sequence, and accessibility to oligosaccharyltransferase (OST) STT3A and STT3B complexes (8, 9, 18), highlighting the importance of experimental determination of in vivo N-glycosylation sites. Our N-glycoproteomics analysis mapped in vivo N-glycosylation sites on a number of AD-related proteins (e.g., APP, tau, BACE1, and nicastrin) and genome-wide association studies (GWAS)–identified AD risk factors (e.g., ABCA7, ADAM10, CD33, CLU, HLA-DRB1, PLD3, SLC24A4, SORL1, and UNC5C) in human AD and control brains (Fig. 1J and table S2). We found that many of these proteins had an increased number of in vivo N-glycosites in AD compared to control brains (Fig. 1J and table S2), suggesting a link between N-glycosylation aberrations of these AD-relevant proteins and AD pathophysiology. Of note, although for some proteins (e.g., ADAM10), all of the predicted N-glycosites were found to be N-glycosylated in vivo (Fig. 1J and table S2), for most of AD-relevant proteins, the number of observed in vivo N-glycosylation sites was lower than the number of predicted N-glycosylation sites. For example, of the two predicted APP N-glycosites (N467 and N496 on APP695 isoform corresponding to N542 and N571 on APP770 isoform), only one site (N467 on APP695; N542 on APP770) was found to be N-glycosylated in vivo (Fig. 1J and table S2). Our result on APP N-glycosylation is consistent with previous report that APP695 N467, but not N496, was N-glycosylated in culture cells (12). Our analysis also showed that, of the three predicted tau (encoded by the MAPT gene) N-glycosites (N167, N359, and N410 on tau441 isoform corresponding to N484, N616, and N727 on tau758 isoform), only one site (N410 on tau441; N727 on tau758) was N-glycosylated in vivo and the tau N-glycosylation was detected exclusively in AD but not control brain (Fig. 1J and table S2). Our identification of AD-specific N-glycosylation site of tau provides direct evidence for the previously reported, unexpected tau N-glycosylation in AD brain (11–13).
The most heavily N-glycosylated protein that we identified in AD and control brains is low-density lipoprotein receptor-related protein 1 (LRP1), a type I transmembrane protein with a key role in the control of APP trafficking, Aβ metabolism, and brain homeostasis (19). A total of 26 N-glycosites were found on LRP1 in AD brain, whereas only 19 N-glycosites were on LRP1 in the controls (Fig. 1K and fig. S1D). The finding of N-glycosylation at seven LRP1 glycosites (N446, N928, N1825, N2475, N2815, N2905, and N4125) in AD but not controls (fig. S1D and table S2) suggests an involvement of LRP1 N-glycosylation aberration in AD. Disease-associated increase in the number of N-glycosites was also found in many other proteins, such as LRP1B, neural cell adhesion molecules L1CAM and NRCAM, extracellular matrix proteins LAMA5, tenascin-C (TNC) and tenascin-R (TNR), and NMDA-type glutamate receptor subunit 1 (GRIN1) (Fig. 1K and table S2). Conversely, a number of N-glycoproteins showed a loss of in vivo N-glycosylation site(s) in AD, such as metabotropic glutamate receptor 3 (GRM3/mGluR3), integrin α2 (ITGA2), Na+- and K+-transporting adenosine triphosphatase (Na+,K+-ATPase) α-1 subunit (ATP1A1), GPI inositol-deacylase PGAP1, and zinc transporter SLC39A6 (Fig. 1L, fig. S1E, and table S2).
Identification of differentially N-glycosylated proteins and altered N-glycosylation sites in AD
Next, we performed differential glycopeptide abundance analysis and identified 118 N-glycopeptides that were significantly changed in AD compared to control cases (>1.3-fold change, P < 0.05), including 54 glycopeptides from 44 glycoproteins with increased glycopeptide abundance and 64 glycopeptides from 47 glycoproteins with decreased glycopeptide abundance in AD (Fig. 2A and table S3A). To determine whether the observed changes in N-glycopeptide abundance in AD were due to altered protein expression or altered N-glycosylation site occupancy, we compared the N-glycopeptide abundance changes with the corresponding protein abundance changes measured from the same brain samples (4). Of the identified 118 AD-associated N-glycopeptides from 89 glycoproteins, 100 glycopeptides from 72 glycoproteins had corresponding protein abundance data, whereas the other 18 glycopeptides were from 17 glycoproteins (e.g., CPVL, EMP1, EMP3, and ITGA) detected only in N-glycoproteomics but not proteomics analysis (table S3B). Comparative analysis of the N-glycoproteome versus proteome data showed that AD-associated changes in N-glycopeptide abundance of most of the 100 glycopeptides were not due to altered protein expression (Fig. 2B and table S3B). We found that 76 glycopeptides representing 77 glycosites on 60 glycoproteins had >1.3-fold changes in N-glycosylation site occupancy in AD compared to control brains, including 36 glycosites on 28 glycoproteins with increased N-glycosylation site occupancy in AD and 41 glycosites on 34 glycoproteins with decreased N-glycosylation site occupancy in AD (Fig. 2C and table S3B). Unsupervised hierarchical clustering analysis showed that the identified 76 glycopeptides and their corresponding N-glycosites provide an N-glycosylation signature for distinguishing most AD cases from the controls (Fig. 2D).
Fig. 2. Disease-associated changes in N-glycopeptide abundance and N-glycosylation site occupancy in AD.
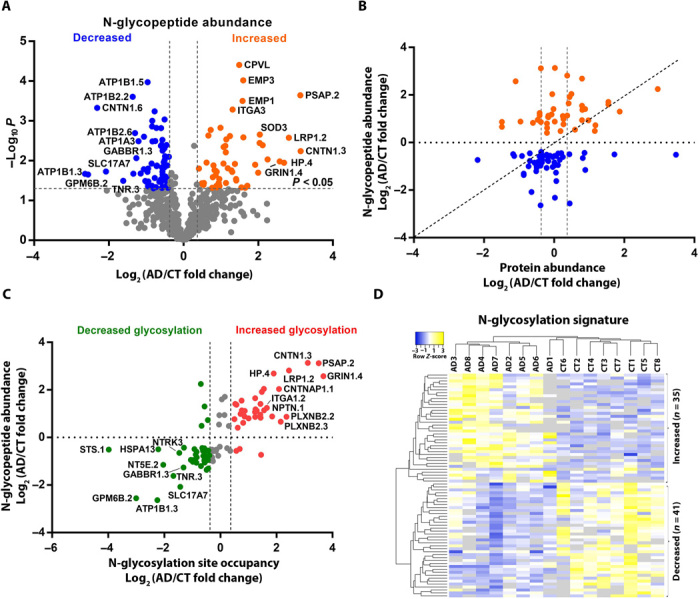
(A) Volcano plot of individual N-glycopeptide abundance fold changes in AD versus control (log2 scale) and corresponding P values (−log10 scale) showing identification of 118 significantly altered (>1.3-fold change, P < 0.05) glycopeptides in AD with top 20 glycopeptides labeled. (B) Scatter plot showing that glycopeptide abundance changes of many AD-associated N-glycopeptides are not due to altered protein abundance. (C) Scatter plot showing at least ±1.3-fold changes in N-glycosylation site occupancy for most of the identified AD-associated N-glycopeptides with top 20 glycopeptides labeled. (D) Unsupervised hierarchical clustering of 16 individual cases based on the identified 76 N-glycopeptides representing 77 glycosites with altered N-glycosylation site occupancy. The N-glycopeptide sequences and corresponding N-glycosite data are provided in table S3.
In addition to the quantitative assessment of AD-associated changes in the stoichiometric proportion of N-glycosylation site occupancy, we also determined the numbers of N-glycosylation sites that were exclusively occupied in either AD or control brains. The AD-unique N-glycopeptides representing N-glycosites with a gain of N-glycosylation in AD were identified as the glycopeptides detected in ≥50% of AD cases but not in any of the control cases. Conversely, the control-unique N-glycopeptides, which represent the N-glycosites with a complete loss of N-glycosylation in AD, were identified as the glycopeptides detected in ≥50% of control cases but not in any of the AD cases. Our analysis revealed 88 AD-unique N-glycopeptides representing 89 N-glycosites on 76 glycoproteins with a gain of N-glycosylation in AD and 11 control-unique N-glycopeptides representing 12 N-glycosites on 11 glycoproteins with a complete loss of N-glycosylation in AD (Fig. 3, A and B, and table S3C).
Fig. 3. Analysis and annotation of differentially N-glycosylated proteins in AD brain.
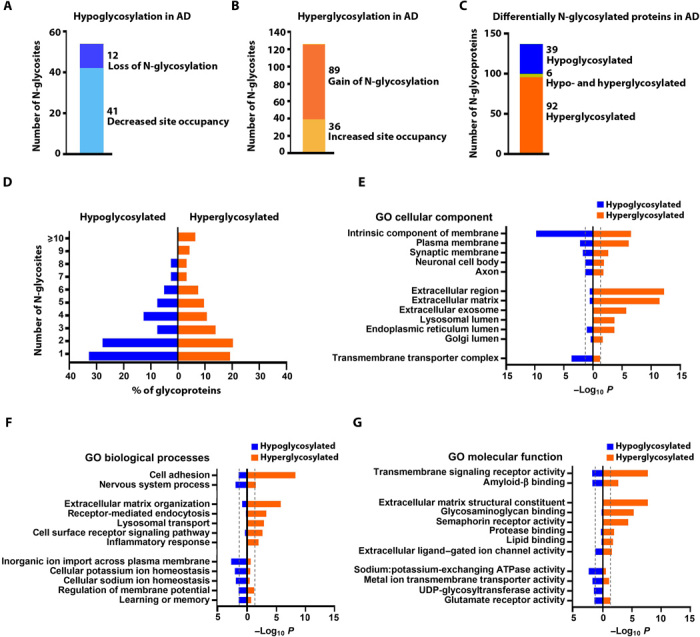
(A) Hypoglycosylation includes in vivo N-glycosites with decreased N-glycosylation site occupancy or a complete loss of N-glycosylation in AD. (B) Hyperglycosylation includes N-glycosites with increased N-glycosylation site occupancy or a gain of N-glycosylation in AD. (C) Differentially N-glycosylated proteins in AD include the identified hypoglycosylated or hyperglycosylated proteins and aberrantly glycosylated proteins with both hyperglycosylated and hypoglycosylated N-glycosites. (D) Distribution of AD-associated hypoglycosylated or hyperglycosylated proteins carrying the indicated number of in vivo N-glycosites per protein. (E to G) GO cellular component, biological process, and molecular function categories enriched in hypoglycosylated or hyperglycosylated proteins in AD. Significant enrichment is shown by Benjamini-Hochberg FDR-corrected P < 0.05 (outside the dashed line). UDP, uridine diphosphate.
Differentially N-glycosylated proteins in AD were identified as the glycoproteins containing in vivo N-glycosites with altered N-glycosylation site occupancy and/or a complete loss or gain of N-glycosylation in AD brain. In total, we identified 137 differentially N-glycosylated proteins, including 92 hyperglycosylated proteins containing N-glycosites with increased N-glycosylation site occupancy and/or a gain of N-glycosylation in AD, 39 hypoglycosylated proteins containing N-glycosites with decreased N-glycosylation site occupancy or a loss of N-glycosylation in AD, and 6 aberrantly glycosylated proteins containing both hyperglycosylated and hypoglycosylated N-glycosites (Fig. 3C and table S4).
We found that AD-associated hypoglycosylation occurs more than hyperglycosylation in N-glycoproteins carrying one or two in vivo–glycosylated sites per protein, whereas AD-associated hyperglycosylation occurs more than hypoglycosylation in heavily N-glycosylated proteins with five or more in vivo–glycosylated sites per protein (Fig. 3D). GO cellular component enrichment analysis showed that both hypoglycosylation and hyperglycosylation datasets were significantly enriched for membrane glycoproteins localized to plasma membrane as well as neuronal and synaptic membranes (Fig. 3E and table S5). However, only AD-associated hyperglycosylation, but not hypoglycosylation, was significantly linked to extracellular glycoproteins (e.g., extracellular matrix proteins, extracellular region–containing proteins, and exosomal proteins) or lumenal glycoproteins of lysosome, ER, and Golgi (Fig. 3E), whereas AD-associated hypoglycosylation, but not hyperglycosylation, was enriched for transmembrane transporter complex (Fig. 3E and table S5). Consistent with GO localization enrichment results, both hypoglycosylation and hyperglycosylation datasets were significantly associated with cell adhesion and nervous system process and enriched for transmembrane signaling receptors and amyloid-β binding, cell surface proteins (Fig. 3, F and G, and table S5). However, only hyperglycosylated proteins in AD were selectively enriched for GO categories linking to extracellular matrix organization, inflammatory response, endocytosis, and lysosomal transport, whereas hypoglycosylated proteins in AD were preferentially associated with ion transmembrane transport, cellular sodium and potassium ion homeostasis, glutamate receptor function, and learning or memory (Fig. 3, F and G, and table S5). The identified six aberrantly glycosylated proteins with both hyperglycosylated and hypoglycosylated sites in AD were primarily associated with cell adhesion (table S5).
Construction and characterization of N-glycopeptide/glycoprotein co-regulation network
To gain system-level insights into brain N-glycoproteome organization and its alterations in AD, we applied weighted correlation network analysis (WGCNA) (20) to construct an N-glycopeptide co-regulation network from our N-glycoproteomics data. In this glyco-network, nodes representing N-glycopeptides are connected with edges that define the connectivity based on pairwise correlation patterns of N-glycopeptide abundance profiles. Pairwise correlation of N-glycopeptide abundances could be controlled at any of the steps from protein expression and N-glycosylation to glycoprotein degradation, and we used the term “co-regulation” to encompass all these regulatory events shared between N-glycopeptides. We constructed the N-glycopeptide co-regulation network using the scale-free topology criterion (20), which organizes the N-glycoproteome into modules of interconnected, or co-regulated, N-glycopeptides with module hub glycopeptides having the greatest number of connections. We mapped N-glycopeptides to N-glycoproteins to convert the N-glycopeptide co-regulation network into an N-glycoprotein co-regulation network with co-regulated N-glycosites.
Our WGCNA glyco-network analysis revealed an organization of human brain N-glycoproteome into a network of 13 modules of strongly co-regulated N-glycopeptides/glycoproteins (Fig. 4A and table S6A). These glyco-network modules (GMs) were labeled from GM1 (the largest module with 94 N-glycopeptides from 75 glycoproteins) to GM13 (the smallest module with 24 N-glycopeptides from 22 glycoproteins) (Fig. 4B and table S6A). We found that in the glyco-network, a glycoprotein with multiple in vivo N-glycosylation sites could have two or more of its distinct N-glycopeptides/glycosites segregated in a single module, such as the colocalization of two CFH glycopeptides CFH.1 and CFH.2 (representing CFH glycosites N911 and N882, respectively) in the GM6 module (table S6A), which may reflect the regulation at the level of protein expression or coordinated regulation of N-glycosylation at different glycosites. Alternatively, distinct N-glycopeptides/glycosites from a multiglycosylated protein could be separated into different modules, such as the separate localization of three LNPEP glycopeptides LNPEP.1, LNPEP.2, and LNPEP.3 (representing LNPEP glycosites N368, N448, and N834, respectively) to the GM1, GM12, and GM4 modules (table S6A), which may reflect differential regulation of N-glycosylation at distinct glycosites or different activities and/or cellular distribution of differentially N-glycosylated forms of a single protein.
Fig. 4. N-glycopeptide co-regulation network analysis and identification of AD-related glyco-network modules.

(A) N-glycopeptide WGCNA cluster dendrogram using dynamic tree cutting reveals 13 glyco-network modules (color-coded) of co-regulated N-glycopeptides. (B) Glyco-network intermodular relationships of the identified 13 N-glycopeptide co-regulation modules. Modules with significant enrichment for markers of astrocyte (A), microglia (M), oligodendrocyte (O), and/or neuron (N) are indicated, and the FDR-corrected P values for enrichment are provided in fig. S2. (C) Module-trait correlation between each of the 13 glyco-network modules and each AD-relevant phenotypic or genotypic trait is shown by signed −log10 P to indicate the significance and direction of the correlation. Modules with significant correlation (P < 0.05) are labeled. (D) Box plots of module eigenglycopeptide (MEg) values in AD and control cases for each AD-related module, with differences in module eigenglycopeptide between AD and control shown by Kruskal-Wallis test P values.
We compared the N-glycoproteome network with our previously described proteome network from the same brain tissues (4) and found no significant overlap between these two networks, indicating that N-glycopeptide/glycoprotein co-regulation is different from protein coexpression in human brain. We next assessed the enrichment of cell type markers in glyco-network modules (fig. S2) and identified two modules (GM4 and GM8) preferentially enriched with astrocyte markers (e.g., CLU, ITGA1, and LAMB2), one module (GM6) with both microglia (e.g., STAB1, CYBB, and ASPH) and astrocyte (e.g., COL12A1, CLU, and CD109) markers, seven modules (GM1, GM3, GM7, and GM10 to GM13) with neuronal markers (e.g., GRIA1, GRIN2B, and CACNA2D2), and two modules (GM2 and GM9) with both oligodendrocyte (e.g., MAG, MOG, and TNR) and neuronal (e.g., GRIN1, NCAM1, and CACNA2D1) markers (Fig. 4B and fig. S2). The finding of a large number of neuronal marker–enriched modules in the brain glyco-network is consistent with the presence of diverse neuronal cell types with distinct functions and regulation of N-glycosylation or protein expression in the brain.
For each N-glycopeptide co-regulation module in the glyco-network, we computed a module eigenglycopeptide to represent the glycopeptide abundance profiles of the module as described (20). Analysis of the intermodular relationships using pairwise correlations between module eigenglycopeptides showed that the 13 glyco-network modules are clustered into three superclusters: the first supercluster containing astrocyte or microglia/astrocyte marker–enriched modules GM4, GM6, and GM8; the second supercluster containing oligodendrocyte/neuronal marker–enriched modules GM2 and GM9 and a non–cell type–enriched module GM5; and the third supercluster containing neuronal marker–enriched modules GM1, GM3, GM7, and GM10 to GM13 (Fig. 4B). These results reveal a higher-order organization of N-glycopeptide/glycoprotein co-regulation modules in human brain and highlight the functional interrelationships between glyco-network modules.
Identification of disease-associated glyco-network modules in AD
To identify AD-related glyco-network modules, we performed module-trait association analysis to determine the correlation relationships between each module eigenglycopeptide and AD-relevant phenotypic traits and other clinical or sample characteristics (fig. S3). Our analyses identified four positively correlated modules (GM4, GM6, GM8, and GM12) and two negatively correlated modules (GM1 and GM9) that showed significant association with AD status, amyloid-β pathology [Consortium to Establish a Registry for Alzheimer’s disease (CERAD) neuritic plaque score], and/or neurofibrillary tangle pathology (Braak stage), but not with any of potential confounding factors examined, such as age, sex, or postmortem interval (Fig. 4C and fig. S3). One of the identified AD–positively associated modules, GM8, also showed strong positive correlation with the ApoE4 allele dosage (Fig. 4C), consistent with the dosage-dependent role of ApoE4 allele as the strongest genetic risk factor for AD (21). We found that the module glycopeptide abundance profiles for AD–positively correlated modules GM4, GM6, and GM8 were significantly increased in AD, whereas those for AD–negatively correlated modules GM1 and GM9 were significantly decreased in AD (Fig. 4D).
Our analyses showed that AD–positively correlated modules GM4, GM6, and GM8 were closely connected in the astrocyte or microglia/astrocyte marker–enriched supercluster (Fig. 4B), suggesting that the biological processes associated with these glyco-network modules are functionally related and/or coordinately regulated in AD. The AD–negatively correlated GM9 module located within the oligodendrocyte/neuronal marker–enriched supercluster (Fig. 4B), suggesting that the function or regulation of GM9 module is more closely related to GM2 and GM5 than to other modules. The AD–negatively correlated module GM1 and AD–positively correlated module GM12 were localized to different branches of the neuronal marker–enriched supercluster (Fig. 4B), indicating differential regulation of these two neuronal modules and their associated biological processes in AD. Our finding of only one module (GM1) of seven neuronal modules had significantly reduced glycopeptide abundance profiles in AD (Fig. 4, B and D) provides evidence supporting that the observed reduction in GM1 glycopeptide abundance profiles results from dysregulated N-glycosylation rather than neuronal loss in AD.
Glyco-network module analyses revealing dysregulated N-glycosylation–affected processes in AD
We performed GO enrichment analysis of disease-associated glyco-network modules to uncover the cellular functions and processes affected by dysregulated N-glycosylation in AD brain. We found that GM6 module (Fig. 5A), which has the strongest correlation to AD neuropathology (CERAD r = 0.88, P = 8 × 10−6; Braak r = 0.87, P = 9 × 10−6), was closely associated with the biological processes of extracellular matrix organization and inflammatory response (Fig. 5B and table S7). Because of the critical role of highly connected, intramodular hub nodes in determining a network module’s function (22), we identified GM6 module hub N-glycopeptides/glycoproteins using the intramodular connectivity and depicted top 10 most connected hub glycopeptides/glycoproteins in the center of GM6 network plot (Fig. 5A). The GM6 hub glycopeptides showed increased abundance profiles in AD (Fig. 5C), supporting their role as key determinants of this positively correlated glycopeptide co-regulation module. Comparison of GM6 hub glycopeptide abundance changes with their protein abundance changes measured by proteomics (Fig. 5C) showed that, although the increased abundances of some GM6 hub glycopeptides (e.g., COL6A2.1 and COL6A1 representing COL6A2 N785 and COL6A1 N804 glycosite, respectively) reflected the increased protein abundances in AD, for many GM6 hub glycopeptides, such as CD63.2, LAMP1.2, and CFH.1 representing CD63 N130, LAMP1 N103, and CFH N911 glycosite, respectively, the disease-associated increases in the glycopeptide abundances were due to increased N-glycosylation site occupancy without changes in their protein expression levels. The selective detection of GM6 hub glycopeptides/glycoproteins STAB1, ITGA3, EMP3, and CPVL by N-glycoproteomics but not by proteomics (Fig. 5C) further demonstrated the higher sensitivity of N-glycoproteomics than proteomics in analysis of N-glycoproteins.
Fig. 5. Network depiction and analyses of AD-associated glyco-network modules.

(A) N-glycopeptide co-regulation network plots of AD-associated modules with top 10 hub glycopeptides/glycoproteins shown in the center of each plot. The size of nodes corresponds to kME-based intramodular connectivity, and a maximum of top 50 glycopeptides are shown for each module. The full list of N-glycopeptide sequences and corresponding N-glycoproteins and N-glycosites in each module and their kME values are provided in table S6A. (B) Top GO biological process (BP), molecular function (MF), and cellular component (CC) categories enriched in each AD-associated module are shown by Benjamini-Hochberg FDR-corrected P values (−log10 scale) and plotted against the number of glycoproteins per GO term. (C) N-glycopeptide abundance fold changes in AD versus control for top 10 hub glycopeptides in each AD-associated module are shown with their corresponding protein abundance changes measured by proteomics.
Our analyses showed that the most prominent feature of GM6 module was overrepresentation of N-glycopeptides from extracellular matrix components (e.g., LAMA1, HSPG2, DCN, MFAP4, and COL12A1), extracellular matrix receptors (e.g., ITGA3, ITGA7, ITGAV, and BCAM), and glycosaminoglycan-binding proteins (e.g., STAB1, CD44, APOH, and SPOCK2) (Fig. 5 and tables S6A and S7), revealing a previously unrecognized link between altered N-glycosylation and extracellular matrix dysfunction in AD. Furthermore, GM6 module was also significantly enriched with N-glycopeptides from microglial proteins associated with inflammatory response (e.g., STAB1, ICAM1, and CYBB) and other inflammation-related glycoproteins (e.g., A2M, CLU, CFH, CD44, ITGAV, LRP1, and HSPG2) (Fig. 5 and tables S6A and S7), highlighting the involvement of dysregulated N-glycosylation in inflammatory response in AD. Because several identified GM6 inflammation-related glycoproteins (e.g., LRP1, CLU, HSPG2, and A2M) also function as key regulators of Aβ aggregation and clearance (19, 23, 24), our results suggest a role of N-glycosylation aberrations of these proteins in promoting amyloid pathology in AD.
We found that the AD–positively correlated GM4 module was strongly associated with altered N-glycosylation of lysosomal lumenal hydrolases, including tripeptidyl-peptidase TPP1, palmitoyl-protein thioesterase PPT1, and cathepsin L (CTSL) (Fig. 5 and tables S6A and S7). The most highly connected GM4 hub glycopeptide was from prosaposin (PSAP), the precursor protein for four lysosomal hydrolase activators (saposin A to saposin D) involved in degradation of glycosphingolipids (25). Our findings point to a previously unknown connection between dysregulated N-glycosylation of lysosomal enzymes/regulators and AD pathophysiology. In addition, GM4 module was prominently linked to the process of receptor-mediated endocytosis with an overrepresentation of N-glycopeptides from endocytic cargo receptors, including SORL1, LRP1, LRP4, SCARA3, and SCARB2/LIMP2 (Fig. 5 and tables S6A and S7). Our results, together with previous reports of the crucial roles for these receptors in controlling endocytic trafficking and lysosomal degradation of APP and other cargos (26, 27) and the finding of SORL1 as a genetic risk factor for AD (28), indicate that dysregulated N-glycosylation of endocytic cargo receptors could be a previously unidentified pathogenic mechanism leading to endocytic trafficking dysfunction and neurodegeneration in AD. GM4 module was also enriched with a number of key ER-resident glycoproteins, including the OST catalytic subunit STT3B, ER molecular chaperone HSP90B1/GRP94, and ER-associated degradation (ERAD) machinery component SEL1L (Fig. 5 and tables S6A and S7), revealing an involvement of N-glycosylation aberrations in ER protein quality control impairment in AD. Another key feature of GM4 was overrepresentation of N-glycopeptides with disease-associated up-regulation of N-glycosylation at corresponding glycosites from cell adhesion proteins, as represented by top hub glycoproteins CNTN1, PCDH9, PLXNB2, and NEO1 (Fig. 5 and tables S6A and S7), highlighting a link between N-glycosylation dysregulation and cell adhesion dysfunction in AD.
Our analyses showed that the AD–positively correlated GM8 module was prominently associated with inflammatory response process with an overrepresentation of N-glycopeptides from glycoproteins with peptidase inhibitor activity (e.g., serine protease inhibitors SERPINA1, SERPINA3, and SERPINC1; ITIH4; and KNG1) and other inflammation-related glycoproteins (e.g., complement C4A, CLU, HP, KLKB1, ORM1, and ITGB2) (Fig. 5 and tables S6A and S7). Although many of these glycoproteins are predominantly produced by liver and secreted into blood, they could also be locally synthesized in brain by astrocytes and participate in neuroinflammation (29). The blood-derived glycoproteins could enter brain following blood-brain barrier (BBB) disruption, which is known to occur at early stages of AD, and contribute to neuroinflammation (30). Astrocytes are the key supportive cells for maintaining BBB integrity and the major site of ApoE production (30). Our finding of a positive correlation of astrocyte marker–enriched GM8 module with the ApoE4 genotype (Fig. 4C) supports a pathogenic role of ApoE4 in causing astrocyte dysfunction, BBB impairment, and neuroinflammation in AD (31). In addition, we found that GM8 was also associated with altered N-glycosylation of extracellular matrix components (e.g., TNC, LAMB2, LAMC1, and SPARCL1) and extracellular matrix receptors (e.g., ITGA1, ITGB1, and ITGB2) (Fig. 5 and tables S6A and S7), further supporting a role of N-glycosylation dysregulation in extracellular matrix dysfunction in AD identified in the GM6 module.
We found that the AD–positively correlated GM12 module was highly enriched with N-glycopeptides from neuronal and synaptic membrane proteins involved in neurotransmission and synaptic plasticity (Fig. 5 and tables S6A and S7), including neurotransmitter receptors (GRM1/mGluR1, GABBR2, and GRIN2B/NR2B), neurotransmitter transporter SLC1A2/EAAT2, Na+,K+ transporting ATPase subunits ATP1B1 and ATP1B2, voltage-gated ion channels (SCN1A/Nav1.1 and CACNG8), signaling receptors (NTRK2/TrkB and PLXNB2), and neural cell adhesion molecules (L1CAM, CHL1/L1CAM2, NRCAM, NFASC, PCDH9, and NTM), revealing an involvement of N-glycosylation dysregulation in synaptic dysfunction in AD.
In contrast to the AD–positively correlated GM12 module, the AD–negatively correlated GM1 module was overrepresented by N-glycopeptides with disease-associated down-regulation of N-glycosylation at corresponding glycosites from neuronal and synaptic membrane proteins, including neurotransmitter receptors (GABBR1, GABBR2, GRM3/mGluR3, GRM7/mGluR7, GRIA1/GluR1 to GRIA4/GluR4, GRIN1/NR1, and GRIK5), synaptic vesicle glycoprotein SV2A, vesicular glutamate transporter SLC17A7/VGLUT1, glutamate receptor trafficking regulators OLFM1 and OLFM2, inhibitory synapse regulator IGSF9B, Na+,K+ transporting ATPase subunit ATP1B1, voltage-gated calcium channel subunits CACNA2D1 and CACNA2D2, signaling receptors (GPR158, NRP1, NTRK2/TrkB, NTRK3/TrkC, and TNFRSF21), and neural cell adhesion molecules (L1CAM, CDH8, CDH10, CNTN1, CNTNAP2, CNTNAP5, LSAMP, NRCAM, NFASC, PCDH9, NXPH1, and OPCML) (Fig. 5 and tables S6A and S7). A number of these glycoproteins, such as L1CAM, NRCAM, and NTRK2/TrkB, have down-regulated N-glycosites (e.g., L1CAM N726, NRCAM N276, and NTRK2 N95 and N178 glycosites) in GM1 module but up-regulated N-glycosylation at different glycosites (e.g., L1CAM N777, NRCAM N507, and NTRK2 N67 glycosites) in GM12 module (Fig. 5 and tables S6A and S7), indicating disease-associated, differential N-glycosylation at distinct glycosites of these proteins in AD. Together, these findings further support a role of N-glycosylation aberrations in synaptic dysfunction and neuronal signaling dysregulation in AD.
Another key feature of GM1 module was its association with the process of receptor-mediated endocytosis with an overrepresentation of N-glycopeptides having AD-linked down-regulation of N-glycosylation at corresponding glycosites from endocytic cargo receptors, including low-density lipoprotein receptor family members LRP1 and LRP1B and lysosomal sorting receptors M6PR/CD-MPR and SCARB2/LIMP2 (Fig. 5 and tables S6A and S7). Furthermore, GM1 module was also associated with site-specific, reduced N-glycosylation of a number of lysosomal glycoproteins, such as vacuolar ATPase subunit ATP6AP1 and lysosomal hydrolases cathepsin F (CTSF), PSAP, CD63, leucyl and cystinyl aminopeptidase LNPEP, and steroid sulfatase STS (Fig. 5 and tables S6A and S7). Some of these proteins, such as LRP1, PSAP, and CD63, are differentially N-glycosylated in AD with down-regulated N-glycosites in the GM1 module (e.g., LRP1 N3953 and N4075; PSAP N426, and CD63 N150 glycosites) and up-regulated N-glycosites in the GM4 module (e.g., LRP1 N3788, and PSAP N80 and N215 glycosites) and/or GM6 module (e.g., LRP1 N3839 and CD63 N130 glycosites). Together, these results highlight the involvement of N-glycosylation aberrations in endocytic trafficking dysregulation and lysosomal dysfunction in AD.
Our analyses revealed that the AD–negatively correlated GM9 module was overrepresented by N-glycopeptides with disease-associated down-regulation of N-glycosylation at corresponding glycosites from plasma membrane proteins and extracellular proteins involved in cell adhesion (e.g., CNTN1, OPCML, PCDH1, NRCAM, LSAMP, ADAM23, and NEO1), synapse organization (e.g., NFASC, TNR, CDH2, and NRCAM), and signaling (e.g., SEMA4D, TYRO3, GRM3, ATRN, and PRNP) (Fig. 5 and tables S6A and S7). A number of these proteins, such as CNTN1, NEO1, and NFASC, are differentially N-glycosylated in AD with down-regulated glycosites in the GM9 module (e.g., CNTN1 N494, NEO1 N715, and NFASC N988 glycosites) and up-regulated glycosites in the GM4 module (e.g., CNTN1 N258 and NEO1 N73 glycosites) or GM12 module (e.g., NFASC N1122 glycosite). Together, these findings further underscore the role of N-glycosylation aberrations in altered cell adhesion and synaptic function in AD. In addition, GM9 module was also associated with reduced N-glycosylation of extracellular matrix proteins (e.g., TNR, LAMA2, MMRN2, and MEGF9) (Fig. 5 and tables S6A and S7), further supporting the link between N-glycosylation dysregulation and extracellular matrix dysfunction in AD identified in the GM6 and GM8 modules.
Overlap in protein N-glycosylation changes between human AD and APP/PS1 mouse brains
To further assess the relationship between protein N-glycosylation alterations and amyloid-β pathology in AD, we compared our results with recently reported N-glycoproteome profiling data of APP/PS1 mouse brain (32). The APP/PS1 mouse (JAX-004462), which expresses APP and PSEN1 transgenes carrying AD-linked APP K670N/M671L and PSEN1 ΔE9 mutations, is a well-established AD mouse model with amyloid-β pathology but no neurofibrillary tangles (33). We found that, of the 137 differentially N-glycosylated proteins and 178 altered N-glycosites identified in human AD brain, 62 glycoproteins (45%) and 41 glycosites (23%) were differentially N-glycosylated in the APP/PS1 mouse brain (Fig. 6, A and B, and table S4D), including hyperglycosylated sites (e.g., CNTNAP1 N276, PSAP N80, and SEMA4D N419) and hypoglycosylated sites (e.g., ATB1B2 N238, GRM3 N439, and HSPA13 N184) shared between human AD and mouse model. These similarities in N-glycosylation alterations between human patients with AD and APP/PS1 mouse model suggest that a fraction of protein N-glycosylation changes observed in human AD brain may be induced by amyloid-β accumulation.
Fig. 6. Integrative analyses to identify potential causes of altered N-glycosylation in AD and N-glycopeptide biomarkers.

(A and B) Comparison of N-glycoproteome data between human AD and APP/PS1 mouse brains shows the percentages (bars) and the numbers (indicated above each bar) of the identified human AD–related N-glycoproteins (A) and N-glycosites (B) that were altered in the APP/PS1 mouse brain for the differential N-glycosylation datasets (DIFF) and each of human AD–correlated glyco-network modules. (C) Integration of seven datasets reveals disease-associated changes in N-glycosylation–related genes/proteins in human AD cases. AD transcriptome FC1 and FC2 are datasets from the frontal cortex, and EC1 and EC2 are from the entorhinal cortex. (D) Venn diagram showing the overlap between glycopeptides with altered N-glycosylation site occupancy in AD and top hub glycopeptides of AD-associated glyco-network modules. (E) Unsupervised hierarchical clustering based on the glycopeptide abundance profiles of the identified 24 hub glycopeptides with altered N-glycosylation site occupancy, showing clear separation of AD from control cases and of positively correlated glyco-network module hubs from negatively correlated module hubs. (F) Heatmaps showing the abundances of 20 glycopeptides containing N-glycosite with a gain or loss of N-glycosylation in AD and their corresponding protein abundances measured from proteomics analysis.
Our comparative analyses showed that some differentially N-glycosylated proteins shared between human AD and APP/PS1 mouse brains have distinct, altered N-glycosites in human AD (e.g., ATP1B1 N193 and N265, GRIA4 N258, and ICAM5 N137) and mouse model (e.g., ATP1B1 N158, GRIA4 N371, and ICAM5 N54 and N397). In addition, some altered N-glycosites (e.g., GRIN1 N350, ITGA1 N1102, and LRP1 N3839) were hyperglycosylated in human AD brain but hypoglycosylated in the APP/PS1 mouse brain, while other N-glycosites (e.g., CNTNAP2 N289, DPP6 N566, and GABBR1 N84) were hypoglycosylated in human patients but hyperglycosylated in the mouse model. Most of the identified differentially N-glycosylated proteins and glycosites in human AD brain were unaltered in the APP/PS1 mouse brain (Fig. 6, A and B, and table S4). These differences in N-glycosylation alterations between human patients and APP/PS1 mouse model indicate that factors other than amyloid-β pathology are also involved in causing N-glycosylation dysregulation in AD brain.
Next, we compared our human AD glyco-network analysis results with the APP/PS1 mouse N-glycoproteome data (32) and found that, among AD-associated glyco-network modules, AD–positively correlated neuronal module GM12 and AD–negatively correlated neuronal module GM1 had highest percentages of human AD–related N-glycoproteins and N-glycosites that were altered in the APP/PS1 mouse brain (Fig. 6, A and B, and table S6B), including shared N-glycosites from neurotransmitter receptors (GABBR1 N440, GABBR2 N104 and N453, GRM3 N439, GRIA2 N256 and N370, GRIA3 N380, GRIA4 N371, GRIN1 N368, and GRIN2B N444), neurotransmitter transporter (SLC1A2/EAAT2 N216), synaptic vesicle glycoprotein (SV2A N573), voltage-gated calcium channel (CACNA2D1 N824), signaling receptors (GPR158 N333, NTRK2 N95, and NTRK3 N388), and neural cell adhesion molecules (NRCAM N276, N507, and N858; NFASC N483, CNTN1 N494, and CNTNAP2 N289). These findings highlight a role of amyloid-β pathology in triggering N-glycosylation alterations of neuronal and synaptic proteins, thereby contributing to synaptic dysfunction and neuronal signaling dysregulation in AD.
Our analyses showed that 38 to 58% of N-glycoproteins and 14 to 18% of N-glycosites in AD–positively correlated, glial modules GM4, GM6, and GM8 and AD–negatively correlated, oligodendrocyte/neuronal marker–enriched module GM9 were altered in the APP/PS1 mouse brain (Fig. 6, A and B, and table S6B). A number of the identified N-glycosites shared between human patients and mouse model were from extracellular matrix components (e.g., LAMB2 N1249, LAMC1 N1223, ACAN N333, SPARCL1 N476, SPOCK2 N225, and TNR N470) and extracellular matrix receptors (e.g., ITGA1 N1102, ITGAV N74, and ITGB1 N212), supporting the involvement of amyloid-β deposition in causing extracellular matrix protein N-glycosylation alterations and extracellular matrix dysfunction in AD. The shared N-glycoproteins and N-glycosites in human AD and mouse model also included several lysosomal proteins (PSAP N80, ASAH1 N259, TPP1 N443, and LAMP1 N103), suggesting a role of amyloid-β pathology in causing lysosomal protein N-glycosylation dysregulation and lysosomal dysfunction in AD. It is important to note that many of the identified human AD–related N-glycoproteins and N-glycosites were unaltered in the APP/PS1 mouse brain (Fig. 6, A and B, and table S6), indicating that, in addition to amyloid-β deposition, other upstream events also contribute to N-glycoproteome remodeling and glyco-network alterations in AD brain.
Alterations in the N-glycosylation machinery components in AD brain
Protein N-glycosylation is catalyzed by ER-localized OST, which transfers a preassembled oligosaccharide from the dolichol-linked oligosaccharide donor onto asparagine residues in glycosylation acceptor sites (N-X-S|T|C, X ≠ P) of proteins. Mammalian cells contain two OST complexes, which are composed of a catalytic subunit (STT3A or STT3B), a shared set of accessory subunits (RPN1, RPN2, DAD1, OST4, OST48, and TMEM258), and complex-specific subunits (OSTC/DC2 and KRTCAP2/KCP2 of the STT3A complex or TUSC3 and MAGT1 of the STT3B complex) (18). The STT3A complex catalyzes cotranslational N-glycosylation of nascent proteins, while the STT3B complex mediates cotranslational and posttranslational N-glycosylation, including glycosylation of acceptor sites skipped by the STT3A complex and cryptic N-glycosylation sites on misfolded proteins (18). The importance of both OST complexes for human brain health is underscored by the findings that mutations in the STT3A, STT3B, OST48, TUSC3, or MAGT1 gene cause congenital disorders of glycosylation with brain function abnormalities (10, 18).
To determine whether alterations in OST complexes are involved in causing the observed protein N-glycosylation changes in AD brain, we examined the mRNA expression profiles of OST complex subunits in four different transcriptome datasets for brain tissues (frontal cortex or entorhinal cortex) from patients with AD and control cases (3, 34–36). As shown in Fig. 6C, we found AD-associated changes in mRNA expression levels of several OST subunits, including up-regulated subunits (STT3B and MAGT1) and down-regulated subunits (RPN2, DAD1, OST4, OSTC, and TUSC3), indicating that both OST complexes are altered in AD brain. Consistent with the mRNA expression results, our proteomics analysis (4) showed up-regulation in STT3B protein levels and down-regulation in DAD1 protein levels in AD compared to control brain (Fig. 6C). In addition to altered expression of OST complexes in AD, our N-glycoproteomics analysis revealed that STT3A N544 and N548 glycosites were N-glycosylated in AD but not control brain (table S2). Furthermore, we found that N-glycosylation of STT3B N627 glycosite was positively correlated with AD phenotypes in the disease-associated glyco-network module GM4 (Fig. 5A and table S6A). These data suggest a potential role of N-glycosylation aberrations of STT3A and STT3B in OST dysfunction in AD. GWAS meta-analysis has identified single-nucleotide polymorphism (SNP) of RPN1 as an AD candidate risk allele (7). Collectively, the AD-associated changes in OST subunits revealed by multiomics analyses (Fig. 6C) provide evidence supporting the involvement of altered expression and activity of OST complexes in causing the protein N-glycosylation dysregulation and glyco-network alterations in AD brain uncovered by this study.
Besides OST complexes, enzymes for dolichol-linked oligosaccharide biosynthesis (e.g., ALG proteins, DPM1, and RFT1) also play an important role in the control of protein N-glycosylation (18, 37). Mutations in many of these enzymes are known to cause decreased occupancy of N-glycosylation sites and congenital disorders of glycosylation with neurological abnormalities (10, 18). Examination of dolichol-oligosaccharide synthesis enzymes in the multiomics datasets revealed that the mRNA expression levels of several enzymes (e.g., ALG1, ALG3, ALG6, ALG9, ALG12, and DPM1) were altered in AD brain (Fig. 6C). These data suggest that altered expression of enzymes in dolichol-oligosaccharide biosynthesis could also contribute to the observed protein N-glycosylation changes in AD brain.
Identification of N-glycopeptide biomarker candidates for AD diagnostic development
Disease-associated N-glycosylation alterations in AD may provide potential diagnostic clues for AD biomarker development. By integration of the results from differential N-glycosylation site occupancy analysis and N-glycopeptide co-regulation network analysis, we identified 24 hub glycopeptides in AD-related glyco-network modules with altered N-glycosylation site occupancy in AD (Fig. 6D). Unsupervised hierarchical clustering analysis based on the N-glycopeptide abundance profiles of these 24 hub glycopeptides showed a clear separation of AD cases from the controls (Fig. 6E), supporting their potential as novel AD biomarkers. In addition, our site-specific N-glycoproteomics analysis revealed 18 glycopeptides representing AD-specific N-glycosites that were detected in ≥75% of AD cases but not in any of the control cases (Fig. 6F). The exclusive detection of these N-glycopeptides in AD is due to altered N-glycosylation but not altered protein expression, as shown by comparison with the corresponding proteomics data (Fig. 6F). In addition, our analyses also revealed two glycopeptides representing N-glycosites with a complete loss of N-glycosylation in AD (Fig. 6F). These 20 unique N-glycopeptides/glycosites provide attractive targets for AD biomarker development.
DISCUSSION
This study provides the first, system-level view of human brain N-glycoproteins and in vivo N-glycosylation sites in AD and control brains. Previous postmortem AD brain studies used a two-dimensional gel electrophoresis–based glycoproteomic approach and identified only a limited number (<20) of putative N-glycosylated proteins without mapping their N-glycosylation sites (38, 39). Our study using a high-resolution LC-MS/MS–based quantitative N-glycoproteomic approach has identified 1132 N-glycosylated proteins and mapped 2294 in vivo N-glycosylation sites in human brain. The fact that 37% of the glycoproteins in our N-glycoproteome dataset are not present in the corresponding proteome dataset of 5905 proteins from the same brain samples supports the deep coverage of brain N-glycoproteome achieved by our glycoproteomics analysis. Integration of our N-glycoproteome and proteome profiling data shows that 18% of the human brain proteome is N-glycosylated. Of the 2294 N-glycosites found in human brain, 1511 glycosites are newly identified in vivo N-glycosylation sites, which have not been previously recorded as experimentally observed N-glycosites. The human brain N-glycoproteome dataset generated from this work provides a useful resource for future investigations into the biological roles of protein N-glycosylation in brain function in health and AD.
Our results of mapping in vivo N-glycosylation sites on AD-related proteins (e.g., APP, tau, BACE1, and nicastrin) and AD GWAS risk genes (e.g., ABCA7, ADAM10, CD33, CLU, HLA-DRB1, PLD3, SLC24A4, SORL1, and UNC5C) in AD and control brains provide strong evidence supporting the proposed role of aberrant N-glycosylation in AD pathogenesis (12, 13). Our finding of AD-specific, tau N-glycosylation site (N410 on tau 2N4R isoform) is particularly interesting because it demonstrates that the previously reported, unexpected N-glycosylation of tau (12, 13) indeed occurs in AD but not in control brain. As a cytosolic protein, tau normally does not undergo N-glycosylation because it is unable to reach N-glycosylation enzymes (OST complexes) that reside in the ER lumen. However, subcellular localization of tau might be altered in AD, making it accessible to ER-localized OST complexes. One possibility is that extracellular tau, which mediates cell-to-cell transmission of tau pathology in AD (40), could be taken up by endocytosis followed by retrograde vesicular transport to the ER lumen, as in the case of some bacterial toxins (41). A recent study showed that tau, when forced to be expressed in the ER lumen in cultured cells, can undergo N-glycosylation and secretion and that the abnormal N-glycosylation of tau affects its aggregation (42). Our identified in vivo N-glycosylation sites on tau and other AD-relevant proteins in human AD and control brains provide site-specific information for mechanistic studies to understand functional impacts of N-glycosylation on these proteins and their roles in AD pathogenesis.
Our quantitative N-glycoproteomics analysis has revealed disease-associated changes in brain N-glycoproteome and identified 137 differentially N-glycosylated proteins and 178 in vivo glycosylation sites with N-glycosylation aberrations in AD. Of these, 77 glycosites from 60 glycoproteins showed significantly altered N-glycosylation site occupancy in AD, including 36 glycosites with increased occupancy and 41 glycosites with decreased occupancy. The observed site-specific changes in N-glycosylation site occupancy may result from disease-associated protein conformational changes that affect the recognition or efficient use of these sites by the N-glycosylation machinery in AD brain. In addition, 89 glycosites on 76 glycoproteins were found to be N-glycosylated exclusively in AD but not control brains. These AD-specific N-glycosylation sites may represent cryptic N-glycosites (i.e., normally unused N-glycosylation sequon) that become exposed as a result of altered protein folding induced by ER stress or other environmental and genetic factors associated with AD (18). Conversely, 12 glycosites on 11 glycoproteins were N-glycosylated exclusively in control but not AD brains. The complete loss of N-glycosylation at these sites may reflect their protein folding changes in AD, making these sites inaccessible to modification by N-glycosylation enzymes.
Besides altered folding of substrate proteins, convergent data from multiomics analyses (Fig. 6C) indicate that the observed changes in protein N-glycosylation site occupancy in AD could be caused by altered expression and activity of OST complexes resulting from genetic variation (e.g., GWAS-identified association of SNP in RPN1 with AD risk), transcriptional dysregulation (e.g., altered mRNA levels of STT3B, RPN2, TUSC3, and several other OST subunits in AD), protein expression dysregulation (e.g., altered protein levels of STT3B and DAD1 in AD), and/or dysregulated posttranslational modifications (e.g., altered N-glycosylation of STT3A and AD-correlated N-glycosylation of STT3B). The identified alterations in STT3A and STT3B complexes in AD could lead to changes in both cotranslational and posttranslational N-glycosylation of substrate proteins, including glycosylation of cryptic N-glycosylation sites on misfolded proteins. In addition, the observed N-glycosylation changes in AD could also be due to altered expression of dolichol-oligosaccharide synthesis enzymes (e.g., ALG1, ALG3, ALG6, ALG9, ALG12, and DPM1) in AD brain. The causative roles of altered OST subunits and dolichol-oligosaccharide synthesis enzymes in protein N-glycosylation aberrations in AD are further supported by the fact that mutations in many of these proteins have been reported to cause altered protein N-glycosylation site occupancy and congenital disorders of glycosylation with brain function abnormalities (10, 18).
Our finding of an overlap in protein N-glycosylation changes between human AD and APP/PS1 mouse brains suggests that amyloid-β deposition is another contributing factor for at least some of the observed changes in protein N-glycosylation site occupancy in AD brain. The proposed involvement of amyloid-β pathology in protein N-glycosylation dysregulation is consistent with our identification of N-glycopeptide/N-glycoprotein co-regulation modules that are correlated with amyloid-β pathology (Fig. 4C and fig. S3). Amyloid-β accumulation has been shown to induce ER stress (43), which could affect the folding of substrate proteins and the expression or activity of OST complexes, thereby leading to altered protein N-glycosylation site occupancy in AD brain. In addition, our finding of significant correlation between identified N-glycopeptide/N-glycoprotein co-regulation modules and neurofibrillary tangle pathology, together with a previous report that tau accumulation induces ER stress (43), raises the possibility that tau pathology could also be a contributing factor for the observed differences in N-glycosylation site occupancy in AD brain. It is worth noting that, in addition to changes in N-glycoprotein abundance and N-glycosylation site occupancy, changes in N-glycan structure are also related to AD development and progression. For example, increases in N-glycans containing bisecting N-acetylglucosamine (GlcNAc) have been observed in cerebrospinal fluid and brain samples from patients with AD, and targeted disruption of the MGAT3 gene encoding the enzyme for bisecting GlcNAc modification of N-glycans has been reported to mitigate amyloid-β pathology in an AD mouse model (11–13). Further studies are warranted to elucidate the molecular mechanisms underlying protein N-glycosylation dysregulation in AD.
Our study has shown that, of the identified 137 differentially N-glycosylated proteins in AD brain, 92 proteins are hyperglycosylated, 39 proteins are hypoglycosylated, and 6 proteins are aberrantly glycosylated with both hyperglycosylated and hypoglycosylated N-glycosites. Our results indicate that the disease-associated hyper–N-glycosylation and hypo–N-glycosylation can both affect cell adhesion, transmembrane signaling, and neuronal and synaptic functions in AD. Hyper–N-glycosylation, but not hypo–N-glycosylation, was found to be preferentially associated with proteins localized to extracellular space and exosomes. Our findings, together with recent studies suggesting that hyper–N-glycosylation may serve as a signal for cargo recruitment into exosomes or for targeting exosomes to recipient cells (44, 45), raise the possibility that the observed protein hyper–N-glycosylation in AD may promote exosome-mediated spread of neuropathology and induce aberrant signaling in recipient cells.
Our N-glycoproteomics–driven network analysis has revealed higher-order topology and modularity of human brain N-glycoproteome, showing an organization of the glycoproteome into 13 biologically meaningful modules of co-regulated N-glycopeptides/glycoproteins. The N-glycoproteome network shows no significant overlap with our previously reported proteome network (4), indicating that N-glycopeptide/glycoprotein co-regulation and protein coexpression are controlled by different mechanisms in human brain. By using module-trait association analysis, we have identified six disease-associated, N-glycopeptide/glycoprotein co-regulation modules that are significantly correlated with AD phenotypes, including four positively correlated modules (GM4, GM6, GM8, and GM12) and two negatively correlated modules (GM1 and GM9). Our analyses have generated a molecular blueprint of aberrant protein N-glycosylation networks in AD brain and uncovered a number of dysregulated N-glycosylation–affected processes in AD brain, including extracellular matrix dysfunction, neuroinflammation, synaptic dysfunction, cell adhesion alteration, lysosomal dysfunction, endocytic trafficking dysregulation, ER dysfunction, and cell signaling dysregulation.
We note the limitations related to this study. Quantitative N-glycoproteomics, like quantitative proteomics, is limited by the occurrence of missing data (i.e., missing glycopeptide identifications or abundance values) (46). Although our quantitative analyses have identified 177 altered N-glycopeptides representing 178 altered in vivo N-glycosylation sites from 137 differentially N-glycosylated proteins in AD, some important N-glycosylation changes in glycoproteins and glycosites may have been missed as a result of excluding N-glycopeptides with missing values in >50% of the samples in our analyses. In addition, the current study is also limited by the small sample size. Follow-up studies of the findings from this work will be needed to further define the roles of disease-associated protein N-glycosylation changes in AD.
In summary, our study has demonstrated that integration of quantitative N-glycoproteomics, differential analysis of N-glycopeptide abundance and N-glycosylation site occupancy, and N-glycopeptide/glycoprotein co-regulation network analysis is a powerful approach for revealing dysregulated protein N-glycosylation in AD, and this approach can also be used in studying other human diseases. The strong association of individual glycopeptides, glycoproteins, glycosites, and glyco-network modules with AD phenotypes uncovered in this study indicates that the N-glycoproteome offers exciting opportunities for disease biomarker discovery. The identified protein N-glycosylation aberrations and N-glycoproteomic network alterations in AD brain provide new molecular and system-level insights into AD pathogenesis, and our findings suggest previously unknown targets for AD biomarker and therapeutic development.
MATERIALS AND METHODS
Study design
The objective of this study was to characterize human brain N-glycoproteome and identify protein N-glycosylation aberrations and their affected biological processes in AD. We used a mass spectrometry–based quantitative N-glycoproteomics pipeline to perform unbiased, large-scale, N-glycoproteome profiling analysis of human brain tissue samples from AD and control cases. N-glycopeptides containing 18O-tagged N-glycosylation sites were identified and quantified, and their glycopeptide abundances were normalized using a spiked-in internal reference standard. Differential abundance analysis was performed using the normalized N-glycopeptide abundances to identify disease signature of altered N-glycopeptides in AD. The identified in vivo N-glycosylation sites were mapped to human brain proteins, and N-glycosylation site occupancy changes in AD versus control were determined by integrating the N-glycoproteome data with the corresponding proteome data from the same brain samples (4) to identify N-glycosites with altered N-glycosylation site occupancy in AD. Differentially N-glycosylated proteins were identified as the glycoproteins containing in vivo N-glycosites with altered N-glycosylation site occupancy and/or with a complete loss or gain of N-glycosylation in AD, and functional annotation of the identified N-glycoproteins was performed to reveal aberrant N-glycosylation–affected processes in AD.
To gain system-level insights into brain N-glycoproteome changes in AD, we perform WGCNA of our N-glycoproteomics dataset to construct an N-glycopeptide co-regulation network that organizes the brain N-glycoproteome into a glyco-network of co-regulated N-glycopeptide/glycoprotein modules. Module-trait association analysis was performed to identify glyco-network modules associated with AD phenotypes. The identified disease-associated modules and hub N-glycopeptides/glycoproteins were characterized by bioinformatics analyses to uncover the pathways and processes affected by altered glyco-network in AD brain.
The sample size (a total of 16 AD and control cases) used in this study was selected on the basis of power analysis showing that this sample size has >80% power at a two-sided type I error rate of 5% to detect an effect size of at least 1.6. We used the same sets of postmortem brain samples as in our proteomics study (4) for integrative analysis of N-glycoproteome and proteome profiling data. Human postmortem brain tissues were acquired with informed consent from the donors or their family and provided by the Emory Center for Neurodegenerative Disease Brain Bank. All experiments were conducted in accordance with the U.S. National Institutes of Health (NIH) guidelines for research involving human tissues and with the ethical standards and principles of the Declaration of Helsinki.
Human brain tissues and glycoproteomics sample preparation
N-glycoproteomics analysis was performed by using the same sets of dorsolateral prefrontal cortex tissue samples from AD and age-matched control cases as described in our previous proteomics study (4). The demographics and phenotypic traits of all cases, including sex, age at death, disease status, age at onset, disease duration, Braak stage for neurofibrillary tangle pathology, CERAD score for neuritic amyloid plaque pathology, ApoE genotype, and postmortem interval, are provided in table S1. The brain tissue samples were processed by using the same procedures of SDS-mediated protein extraction and FASP as we described previously (4). Briefly, brain tissue (25 mg per AD or control case) was homogenized in 150 μl of lysis buffer containing 4% SDS, 100 mM dithiothreitol, and 100 mM Tris–HCl (pH 7.6), and protein concentration of the obtained protein extract was measured by ultraviolet spectrometry. To control for technical variations during sample processing and analyses, each protein extract was spiked with bovine a2-HS-glycoprotein (fetuin) at 0.1% (μg/μg total protein) as an internal reference standard. Protein extracts were processed according to the FASP protocol by using the Microcon 30-kDa centrifugal filter device (MRCF0R030, Merck) for detergent removal, alkylation with iodoacetamide, trypsin digestion, and peptide purification as previously described (4). The purified peptides were desalted using a self-packed C18 ZipTip microcolumn and dried under vacuum.
N-glycopeptide enrichment and 18O-labeling of N-glycosites
Glycopeptides were enriched by using ZIC-HILIC as described (15) with slight modification. ZIC-HILIC microcolumn was prepared in-house by packing 10 mg of ZIC-HILIC media (Merck; particle size, 10 μm) in 100 μl of acetonitrile (ACN) into a 200-μl tip, which was plugged by a 3M Empore C8 extraction disk (Thermo Fisher Scientific) at the bottom of the tip. Purified peptides from each brain sample (100 μg of peptides per sample) were dissolved in a binding buffer containing 80% ACN and 5% formic acid (FA) and loaded onto a ZIC-HILIC microcolumn pre-equilibrated with the binding buffer (80% ACN and 5% FA). After washing the ZIC-HILIC microcolumn with 100 μl of the binding buffer five times, the captured glycopeptides were eluted with 80 μl of elution buffer (99.5% H2O and 0.5% FA) three times. Eluate containing the enriched glycopeptides was collected and dried. Purified glycopeptides were dissolved in 50 mM NH4HCO3 prepared with H218O (Sigma-Aldrich) and treated with PNGase F (New England Biolabs) at 37°C overnight to allow 18O-labeling of the asparagine residues at N-glycosylation sites (16). The PNGase F-mediated 18O-labeling of N-glycosites markedly reduced the false-positive rate of N-glycosite identification, as the incorporation of 18O introduces a specific mass shift of 2.9890 Da, which can be easily distinguished from spontaneous deamidation (mass shift of 0.9840 Da). The 18O-labeled peptides were desalted using a C18 ZipTip microcolumn and dried under vacuum.
LC-MS/MS–based N-glycoproteomics analysis
Human brain–derived, 18O-labeled N-glycosite–containing peptides (2 μg) from each sample were separated by using a nano-LC UltiMate 3000 high-performance liquid chromatography system (Thermo Fisher Scientific) coupled online to a LTQ Orbitrap Elite mass spectrometer with an EASY-Spray source (Thermo Fisher Scientific). Peptide separation was carried out as described (4) by reversed phase–high-performance liquid chromatography fractionation on an EASY-Spray PepMap C18 column (length, 50 cm; particle size, 2 μm; pore size, 100 Å; Thermo Fisher Scientific) using a 240-min gradient from 2 to 50% solvent B at a flow rate of 300 nl/min (mobile phase A, 1.95% ACN, 97.95% H2O, 0.1% FA; mobile phase B, 79.95% ACN, 19.95% H2O, 0.1% FA). Mass spectrometric analysis was performed using data-dependent acquisition with full MS scans [mass/charge ratio (m/z) range from 375 to 1600; automatic gain control target, 1 million ions; resolution at 400 m/z, 60,000; maximum ion accumulation time, 50 ms] in the Orbitrap mass analyzer. The 10 most intense ions were fragmented by collision-induced dissociation with maximum ion accumulation time of 100 ms in the LTQ mass spectrometer (automatic gain control target value, 10,000). MS raw data were processed using Proteome Discoverer 1.4 (Thermo Fisher Scientific) to search against the human UniProt TrEMBL database (2016_02 Release, 20,198 reviewed entries). The following parameters were used: 20-ppm precursor ion mass tolerance; 0.5-Da fragment ion mass tolerance; trypsin digestion with up to two missed cleavages; fixed modification: cysteine carbamidomethylation (+57.0215 Da); variable modifications: asparagine deamidation in H218O (18O tag of Asn, +2.9890 Da), asparagine and glutamine deamidation (+0.9840 Da), methionine oxidation (+15.9949 Da), and N-terminal acetylation (+42.0106 Da). The false discovery rate (FDR) for peptide and protein identification was set to 1%. Only the peptides with 18O-tagged asparagine residue within the N-glycosylation sequon N-X-S|T|C (X ≠ P) were accepted as true N-glycosite–containing peptides, which we referred to as N-glycopeptides.
Quantification and differential analysis of N-glycopeptide abundance
Quantitative analysis of N-glycopeptide abundance was performed by using Proteome Discoverer 1.4 to quantify the peak area (i.e., area under the curve) of individual 18O-labeled N-glycosite–containing peptides. The peak area of each N-glycopeptide was normalized to the peak area of the 18O-labeled internal standard N-glycopeptide KLCPDCPLLAPLN(18O)DSR derived from the spiked-in bovine fetuin protein in each sample to account for technical variability in sample processing and LC-MS/MS analyses. Differential abundance analysis was performed using the normalized abundances of N-glycopeptides with valid abundance values in ≥50% of AD and control samples. N-glycopeptides with altered abundance in AD were identified by using unpaired Student’s t test with the thresholds of ±1.3-fold change over the control (i.e., AD/control ratio >1.3 or <0.77) and P < 0.05. Unsupervised hierarchical clustering based on the normalized glycopeptide abundances in each samples was performed using the Heatmapper tool as described previously (4).
Integration of N-glycoproteome with proteome data and analysis of N-glycosylation site occupancy
Integrative analysis was performed by comparing the N-glycoproteome data with the proteome data from the same human brain samples measured using the same instrument for LC-MS/MS analysis (4). The UniProt keywords and GO terms over- or underrepresented in the N-glycoproteome dataset compared to the proteome dataset were determined by using the A.GO.TOOL (17). The fold change in N-glycosylation site occupancy for an N-glycosite in AD versus control was determined as the fold change in the normalized abundance of the N-glycosite–containing peptide divided by the fold change in the normalized abundance of the corresponding protein. The thresholds of ±1.3-fold change in N-glycosylation site occupancy in AD versus control were used to identify N-glycopeptides/glycosites with altered N-glycosylation site occupancy in AD.
Analysis of N-glycoproteome changes shared between human AD and APP/PS1 mouse brains
N-glycoproteome profiling data for brain tissues from wild-type and APP/PS1 mice were downloaded from the published study by Fang et al. (32). Mouse gene symbols were converted to human homolog gene symbols using the Mouse Genome Informatics database (www.informatics.jax.org). The in vivo N-glycosylation sites on mouse proteins were converted to the corresponding N-glycosites on human homolog proteins using the UniProt database (www.uniprot.org). The converted list of differentially N-glycosylated proteins and glycosites in the APP/PS1 mouse brain was used for comparison with our human AD differential N-glycosylation dataset and human AD–correlated glyco-network modules to identify the differentially N-glycosylated proteins and glycosites shared between human AD and APP/PS1 mouse model and the human AD–correlated N-glycoproteins and N-glycosites that were altered in the APP/PS1 mouse brain.
Assessment of AD-associated changes in N-glycosylation–related gene expression
Microarray data for brain tissues from human AD and control cases were downloaded from the Gene Expression Omnibus database (www.ncbi.nlm.nih.gov/geo). The following datasets were used in this study: GSE5281, containing transcriptome data from neurons in the frontal cortex (transcriptome FC1 dataset) and the entorhinal cortex (transcriptome EC1 dataset) (34, 35); GSE33000, containing transcriptome data from the dorsolateral prefrontal cortex (transcriptome FC2 dataset) (3); and GSE26972, containing transcriptome data from the entorhinal cortex (transcriptome EC2 dataset) (36). The mRNA expression fold changes for N-glycosylation–related genes in AD versus controls obtained from these datasets are shown in Fig. 6C.
N-glycopeptide co-regulation network analysis
Before glyco-network analysis, the human N-glycoproteome dataset was filtered for N-glycopeptides with valid abundance values in ≥50% of AD or control samples, and imputation of missing abundance values was performed using the k-nearest-neighbors imputation function in the DAPAR software (47). N-glycopeptide co-regulation network was constructed using the WGCNA R package (20) to calculate a correlation matrix for all pairwise correlations of N-glycopeptide abundances across all AD and control brain samples and then transform the correlation matrix into a weighted adjacency matrix using a soft threshold power of 15 according to the scale-free topology criterion (20). The adjacency matrix was used to calculate topological overlap (TO), and the TO-based dissimilarity was used for hierarchical clustering (20) to generate an N-glycopeptide cluster dendrogram. Glyco-network modules of co-regulated N-glycopeptides were identified using the dynamic tree-cutting algorithm in WGCNA package with minimal module size = 14, deepSplit = 4, merge cut height = 0.07, and a reassignment threshold of P < 0.05. Mapping N-glycopeptides to N-glycoproteins converted the N-glycopeptide co-regulation network into an N-glycoprotein co-regulation network. Glyco-networks of N-glycopeptide/glycoprotein co-regulation modules were visualized using the Cytoscape v.3.5.0 software (48).
Module eigenglycopeptides and module-trait association analysis
For each glyco-network module, module eigenglycopeptide was defined as the first principal component that summarizes N-glycopeptide abundance profiles in the entire module and explains the maximal amount of abundance variations within the module (20). Module membership kME, also known as module eigenglycopeptide-based connectivity, was computed as the Pearson correlation between each N-glycopeptide abundance profile and the module eigenglycopeptide (4, 20). Hub glycopeptides for each module were identified as the top 10 highly connected N-glycopeptides with highest module membership. The intermodular relationships between glyco-network modules was determined on the basis of pairwise correlation between module eigenglycopeptides as described (4). Module-trait associations were assessed using biweight midcorrelations between each module eigenglycopeptide and each of clinical or neuropathological traits and the corresponding P values.
Enrichment analyses and annotation of glyco-network modules and glycoproteins
Functional annotation of glyco-network modules or glycoprotein datasets was performed by GO enrichment analysis using MetaCore bioinformatics software (version 6.37, build 69,500) as described (4), with the total list of all proteins (including glycoproteins) identified in our human brain samples as the background. Enrichment analysis for markers of major cell types in glyco-network modules was performed as described (49), using the lists of cell type markers generated from a brain cell proteomic dataset derived from four mouse brain cell types: astrocyte, microglia, neuron, and oligodendrocyte (50) by selecting proteins with log2 fold expression value >2 (the fold expression for a protein in a cell type was defined as its abundance in the specified cell type divided by its median abundance in other cell types) and a minimum log2 fold expression difference >1.2 in the specified cell type compared to each of the other cell types. Mouse gene symbols were converted to human homolog gene symbols using the Mouse Genome Informatics database (www.informatics.jax.org). The lists of cell type markers in human gene symbols were used for cross-referencing with module glycoprotein composition to determine enrichment for cell type markers in glyco-network modules. Significance of enrichment was assessed using one-sided Fisher’s exact test followed by correction for multiple comparisons using the Benjamini-Hochberg FDR adjustment method as described (4).
Statistical analyses
Power analysis was used to determine the sample size used in this study, and mass spectrometry analysis was performed with 1% FDR for peptide, modification site, and protein identifications. Differences in group means between AD and control cases were determined by unpaired two-tailed Student’s t test or Kruskal-Wallis test. The q value was used to correct for multiple comparisons and estimate the FDRs as described (4). Missing data were imputed using the k-nearest-neighbors imputation method (47), and N-glycopeptide co-regulation network analysis was performed using the WGCNA R package (20) to define glyco-network modules. Module-trait association was determined by calculating biweight midcorrelations between each module eigenglycopeptide and each trait and P values. Enrichment analyses were performed with one- or two-sided Fisher’s exact test as indicated to calculate P values. Benjamini-Hochberg FDR adjustment method was used to correct for multiple comparisons.
Supplementary Material
Acknowledgments
We thank M. Gearing for providing postmortem human brain tissue samples with demographic and neuropathological information and P. Wang for assistance during the initial stage of this work. We are grateful to Emory Center for Neurodegenerative Disease Brain Bank donors for their invaluable contribution to this study. Funding: This work was supported by NIH/National Institute on Aging grant RF1AG057965 (to L.L.) and pilot grant awards from the Emory University Research Committee (to L.L.) and the Atlanta Clinical and Translational Science Institute (to L.-S.C.). Emory Center for Neurodegenerative Disease Brain Bank was supported in part by NIH grants P50 AG025688 and P30 NS055077. Author contributions: L.L., Q.Z., C.M., and L.-S.C. conceived and designed the experiments. Q.Z. and C.M. performed sample preparation and N-glycoproteomics analyses. Q.Z. performed N-glycopeptide quantification and data analyses. Q.Z. and L.L. performed bioinformatics and glyco-network analyses. L.L. and L.-S.C. acquired funding and provided supervision. L.L., Q.Z., and L.-S.C. drafted the manuscript. All authors edited, reviewed, and approved the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Further information and additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabc5802/DC1
REFERENCES AND NOTES
- 1.Scheltens P., Blennow K., Breteler M. M., de Strooper B., Frisoni G. B., Salloway S., Van der Flier W. M., Alzheimer's disease. Lancet 388, 505–517 (2016). [DOI] [PubMed] [Google Scholar]
- 2.Soejitno A., Tjan A., Purwata T. E., Alzheimer's disease: Lessons learned from amyloidocentric clinical trials. CNS Drugs 29, 487–502 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Narayanan M., Huynh J. L., Wang K., Yang X., Yoo S., McElwee J., Zhang B., Zhang C., Lamb J. R., Xie T., Suver C., Molony C., Melquist S., Johnson A. D., Fan G., Stone D. J., Schadt E. E., Casaccia P., Emilsson V., Zhu J., Common dysregulation network in the human prefrontal cortex underlies two neurodegenerative diseases. Mol. Syst. Biol. 10, 743 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Q., Ma C., Gearing M., Wang P. G., Chin L. S., Li L., Integrated proteomics and network analysis identifies protein hubs and network alterations in Alzheimer's disease. Acta Neuropathol. Commun. 6, 19 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sancesario G. M., Bernardini S., Alzheimer's disease in the omics era. Clin. Biochem. 59, 9–16 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Verheijen J., Sleegers K., Understanding Alzheimer disease at the interface between genetics and transcriptomics. Trends Genet. 34, 434–447 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Lambert J. C., Ibrahim-Verbaas C. A., Harold D., Naj A. C., Sims R., Bellenguez C., DeStafano A. L., Bis J. C., Beecham G. W., Grenier-Boley B., Russo G., Thorton-Wells T. A., Jones N., Smith A. V., Chouraki V., Thomas C., Ikram M. A., Zelenika D., Vardarajan B. N., Kamatani Y., Lin C. F., Gerrish A., Schmidt H., Kunkle B., Dunstan M. L., Ruiz A., Bihoreau M. T., Choi S. H., Reitz C., Pasquier F., Cruchaga C., Craig D., Amin N., Berr C., Lopez O. L., De Jager P. L., Deramecourt V., Johnston J. A., Evans D., Lovestone S., Letenneur L., Moron F. J., Rubinsztein D. C., Eiriksdottir G., Sleegers K., Goate A. M., Fievet N., Huentelman M. W., Gill M., Brown K., Kamboh M. I., Keller L., Barberger-Gateau P., McGuiness B., Larson E. B., Green R., Myers A. J., Dufouil C., Todd S., Wallon D., Love S., Rogaeva E., Gallacher J., George-Hyslop P. St., Clarimon J., Lleo A., Bayer A., Tsuang D. W., Yu L., Tsolaki M., Bossu P., Spalletta G., Proitsi P., Collinge J., Sorbi S., Sanchez-Garcia F., Fox N. C., Hardy J., Naranjo M. C. D., Bosco P., Clarke R., Brayne C., Galimberti D., Mancuso M., Matthews F.; European Alzheimer's Disease Initiative (EADI); Genetic and Environmental Risk in Alzheimer's Disease (GERAD); Alzheimer's Disease Genetic Consortium (ADGC); Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE), Moebus S., Mecocci P., Del Zompo M., Maier W., Hampel H., Pilotto A., Bullido M., Panza F., Caffarra P., Nacmias B., Gilbert J. R., Mayhaus M., Lannefelt L., Hakonarson H., Pichler S., Carrasquillo M. M., Ingelsson M., Beekly D., Alvarez V., Zou F., Valladares O., Younkin S. G., Coto E., Hamilton-Nelson K. L., Gu W., Razquin C., Pastor P., Mateo I., Owen M. J., Faber K. M., Jonsson P. V., Combarros O., O'Donovan M. C., Cantwell L. B., Soininen H., Blacker D., Mead S., Mosley T. H. Jr., Bennett D. A., Harris T. B., Fratiglioni L., Holmes C., de Bruijn R. F., Passmore P., Montine T. J., Bettens K., Rotter J. I., Brice A., Morgan K., Foroud T. M., Kukull W. A., Hannequin D., Powell J. F., Nalls M. A., Ritchie K., Lunetta K. L., Kauwe J. S., Boerwinkle E., Riemenschneider M., Boada M., Hiltuenen M., Martin E. R., Schmidt R., Rujescu D., Wang L. S., Dartigues J. F., Mayeux R., Tzourio C., Hofman A., Nothen M. M., Graff C., Psaty B. M., Jones L., Haines J. L., Holmans P. A., Lathrop M., Pericak-Vance M. A., Launer L. J., Farrer L. A., van Duijn C. M., Van Broeckhoven C., Moskvina V., Seshadri S., Williams J., Schellenberg G. D., Amouyel P., Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease. Nat. Genet. 45, 1452–1458 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohtsubo K., Marth J. D., Glycosylation in cellular mechanisms of health and disease. Cell 126, 855–867 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Moremen K. W., Tiemeyer M., Nairn A. V., Vertebrate protein glycosylation: Diversity, synthesis and function. Nat. Rev. Mol. Cell Biol. 13, 448–462 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Freeze H. H., Eklund E. A., Ng B. G., Patterson M. C., Neurological aspects of human glycosylation disorders. Annu. Rev. Neurosci. 38, 105–125 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schedin-Weiss S., Gaunitz S., Sui P., Chen Q., Haslam S. M., Blennow K., Winblad B., Dell A., Tjernberg L. O., Glycan biomarkers for Alzheimer disease correlate with T-tau and P-tau in cerebrospinal fluid in subjective cognitive impairment. FEBS J. 287, 3221–3234 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schedin-Weiss S., Winblad B., Tjernberg L. O., The role of protein glycosylation in Alzheimer disease. FEBS J. 281, 46–62 (2014). [DOI] [PubMed] [Google Scholar]
- 13.Kizuka Y., Kitazume S., Taniguchi N., N-glycan and Alzheimer's disease. Biochim. Biophys. Acta Gen. Subj. 1861, 2447–2454 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Wisniewski J. R., Filter-aided sample preparation: The versatile and efficient method for proteomic analysis. Methods Enzymol. 585, 15–27 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Ma C., Qu J., Meisner J., Zhao X., Li X., Wu Z., Zhu H., Yu Z., Li L., Guo Y., Song J., Wang P. G., Convenient and precise strategy for mapping N-glycosylation sites using microwave-assisted acid hydrolysis and characteristic ions recognition. Anal. Chem. 87, 7833–7839 (2015). [DOI] [PubMed] [Google Scholar]
- 16.Kuster B., Mann M., 18O-labeling of N-glycosylation sites to improve the identification of gel-separated glycoproteins using peptide mass mapping and database searching. Anal. Chem. 71, 1431–1440 (1999). [DOI] [PubMed] [Google Scholar]
- 17.Scholz C., Lyon D., Refsgaard J. C., Jensen L. J., Choudhary C., Weinert B. T., Avoiding abundance bias in the functional annotation of post-translationally modified proteins. Nat. Methods 12, 1003–1004 (2015). [DOI] [PubMed] [Google Scholar]
- 18.Cherepanova N., Shrimal S., Gilmore R., N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 41, 57–65 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shinohara M., Tachibana M., Kanekiyo T., Bu G., Role of LRP1 in the pathogenesis of Alzheimer's disease: Evidence from clinical and preclinical studies. J. Lipid Res. 58, 1267–1281 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langfelder P., Horvath S., WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics 9, 559 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu C. C., Liu C. C., Kanekiyo T., Xu H., Bu G., Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106–118 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langfelder P., Mischel P. S., Horvath S., When is hub gene selection better than standard meta-analysis? PLOS ONE 8, e61505 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu C. C., Zhao N., Yamaguchi Y., Cirrito J. R., Kanekiyo T., Holtzman D. M., Bu G., Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer's disease. Sci. Transl. Med. 8, 332ra344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yu J. T., Tan L., The role of clusterin in Alzheimer's disease: Pathways, pathogenesis, and therapy. Mol. Neurobiol. 45, 314–326 (2012). [DOI] [PubMed] [Google Scholar]
- 25.O'Brien J. S., Kishimoto Y., Saposin proteins: Structure, function, and role in human lysosomal storage disorders. FASEB J. 5, 301–308 (1991). [DOI] [PubMed] [Google Scholar]
- 26.Eggert S., Thomas C., Kins S., Hermey G., Trafficking in Alzheimer's disease: Modulation of APP transport and processing by the transmembrane proteins LRP1, SorLA, SorCS1c, Sortilin, and Calsyntenin. Mol. Neurobiol. 55, 5809–5829 (2018). [DOI] [PubMed] [Google Scholar]
- 27.Canton J., Neculai D., Grinstein S., Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 13, 621–634 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Cuyvers E., Sleegers K., Genetic variations underlying Alzheimer's disease: Evidence from genome-wide association studies and beyond. Lancet Neurol. 15, 857–868 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Van Eldik L. J., Carrillo M. C., Cole P. E., Feuerbach D., Greenberg B. D., Hendrix J. A., Kennedy M., Kozauer N., Margolin R. A., Molinuevo J. L., Mueller R., Ransohoff R. M., Wilcock D. M., Bain L., Bales K., The roles of inflammation and immune mechanisms in Alzheimer's disease. Alzheimers Dement. 2, 99–109 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erickson M. A., Banks W. A., Blood-brain barrier dysfunction as a cause and consequence of Alzheimer's disease. J. Cereb. Blood Flow Metab. 33, 1500–1513 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fernandez C. G., Hamby M. E., McReynolds M. L., Ray W. J., The role of APOE4 in disrupting the homeostatic functions of astrocytes and microglia in aging and Alzheimer's disease. Front. Aging Neurosci. 11, 14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang P., Xie J., Sang S., Zhang L., Liu M., Yang L., Xu Y., Yan G., Yao J., Gao X., Qian W., Wang Z., Zhang Y., Yang P., Shen H., Multilayered N-glycoproteome profiling reveals highly heterogeneous and dysregulated protein N-glycosylation related to alzheimer's disease. Anal. Chem. 92, 867–874 (2020). [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Alloza M., Robbins E. M., Zhang-Nunes S. X., Purcell S. M., Betensky R. A., Raju S., Prada C., Greenberg S. M., Bacskai B. J., Frosch M. P., Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol. Dis. 24, 516–524 (2006). [DOI] [PubMed] [Google Scholar]
- 34.Liang W. S., Dunckley T., Beach T. G., Grover A., Mastroeni D., Walker D. G., Caselli R. J., Kukull W. A., McKeel D., Morris J. C., Hulette C., Schmechel D., Alexander G. E., Reiman E. M., Rogers J., Stephan D. A., Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol. Genomics 28, 311–322 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liang W. S., Reiman E. M., Valla J., Dunckley T., Beach T. G., Grover A., Niedzielko T. L., Schneider L. E., Mastroeni D., Caselli R., Kukull W., Morris J. C., Hulette C. M., Schmechel D., Rogers J., Stephan D. A., Alzheimer's disease is associated with reduced expression of energy metabolism genes in posterior cingulate neurons. Proc. Natl. Acad. Sci. U.S.A. 105, 4441–4446 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berson A., Barbash S., Shaltiel G., Goll Y., Hanin G., Greenberg D. S., Ketzef M., Becker A. J., Friedman A., Soreq H., Cholinergic-associated loss of hnRNP-A/B in Alzheimer's disease impairs cortical splicing and cognitive function in mice. EMBO Mol. Med. 4, 730–742 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aebi M., N-linked protein glycosylation in the ER. Biochim. Biophys. Acta 1833, 2430–2437 (2013). [DOI] [PubMed] [Google Scholar]
- 38.Owen J. B., Di Domenico F., Sultana R., Perluigi M., Cini C., Pierce W. M., Butterfield D. A., Proteomics-determined differences in the concanavalin-A-fractionated proteome of hippocampus and inferior parietal lobule in subjects with Alzheimer's disease and mild cognitive impairment: Implications for progression of AD. J. Proteome Res. 8, 471–482 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Butterfield D. A., Owen J. B., Lectin-affinity chromatography brain glycoproteomics and Alzheimer disease: Insights into protein alterations consistent with the pathology and progression of this dementing disorder. Proteomics Clin. Appl. 5, 50–56 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Gibbons G. S., Lee V. M. Y., Trojanowski J. Q., Mechanisms of cell-to-cell transmission of pathological tau: A review. JAMA Neurol. 76, 101–108 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spooner R. A., Lord J. M., How ricin and Shiga toxin reach the cytosol of target cells: Retrotranslocation from the endoplasmic reticulum. Curr. Top. Microbiol. Immunol. 357, 19–40 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Losev Y., Paul A., Frenkel-Pinter M., Abu-Hussein M., Khalaila I., Gazit E., Segal D., Novel model of secreted human tau protein reveals the impact of the abnormal N-glycosylation of tau on its aggregation propensity. Sci. Rep. 9, 2254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gerakis Y., Hetz C., Emerging roles of ER stress in the etiology and pathogenesis of Alzheimer's disease. FEBS J. 285, 995–1011 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Liang Y., Eng W. S., Colquhoun D. R., Dinglasan R. R., Graham D. R., Mahal L. K., Complex N-linked glycans serve as a determinant for exosome/microvesicle cargo recruitment. J. Biol. Chem. 289, 32526–32537 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hung M. E., Leonard J. N., Stabilization of exosome-targeting peptides via engineered glycosylation. J. Biol. Chem. 290, 8166–8172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karpievitch Y. V., Dabney A. R., Smith R. D., Normalization and missing value imputation for label-free LC-MS analysis. BMC Bioinformatics 13 ( Suppl. 16), S5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wieczorek S., Combes F., Lazar C., Giai Gianetto Q., Gatto L., Dorffer A., Hesse A. M., Coute Y., Ferro M., Bruley C., Burger T., DAPAR & ProStaR: Software to perform statistical analyses in quantitative discovery proteomics. Bioinformatics 33, 135–136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shannon P., Markiel A., Ozier O., Baliga N. S., Wang J. T., Ramage D., Amin N., Schwikowski B., Ideker T., Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Oldham M. C., Konopka G., Iwamoto K., Langfelder P., Kato T., Horvath S., Geschwind D. H., Functional organization of the transcriptome in human brain. Nat. Neurosci. 11, 1271–1282 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sharma K., Schmitt S., Bergner C. G., Tyanova S., Kannaiyan N., Manrique-Hoyos N., Kongi K., Cantuti L., Hanisch U. K., Philips M. A., Rossner M. J., Mann M., Simons M., Cell type- and brain region-resolved mouse brain proteome. Nat. Neurosci. 18, 1819–1831 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabc5802/DC1