

X-ray–activable metal-organic frameworks release tumor antigens and deliver immunoadjuvants for in situ cancer vaccination.
Abstract
Cancer vaccines have been actively pursued to bolster antitumor immunity. Here, we designed nanoscale metal-organic frameworks (nMOFs) as locally activable immunotherapeutics to release danger-associated molecular patterns (DAMPs) and tumor antigens and deliver pathogen-associated molecular patterns (PAMPs) for in situ personalized cancer vaccination. When activated by x-rays, nMOFs effectively generate reactive oxygen species to release DAMPs and tumor antigens while delivering CpG oligodeoxynucleotides as PAMPs to facilitate the maturation of antigen-presenting cells. Together, DAMPs, tumor antigens, and PAMPs expand cytotoxic T cells in tumor-draining lymph nodes to reinvigorate the adaptive immune system for local tumor regression. When treated in combination with an immune checkpoint inhibitor, the local therapeutic effects of nMOF-based vaccines were extended to distant tumors via attenuating T cell exhaustion. Our work demonstrates the potential of nMOFs as x-ray–activable in situ cancer vaccines to awaken the host’s innate and adaptive immune systems for systemic antitumor immunity.
INTRODUCTION
For several decades, cancer vaccines have been developed to amplify tumor-specific T cell responses (1, 2). In particular, tumor antigen–based cancer vaccines have been widely investigated in the clinic, leading to the approval of the prostatic acid phosphatase–based prostate cancer vaccine sipuleucel-T by the U.S. Food and Drug Administration (3). However, traditional approaches to cancer vaccine development face several major hurdles, including tumor heterogeneity with different somatic mutations and, hence, varied tumor antigens among patients (4, 5), ineffective delivery of peptide-based tumor antigens to lymph nodes due to rapid renal clearance and enzymatic degradation (6), inefficient internalization of tumor antigens by antigen-presenting cells (APCs) (7), and the ability of tumors to escape from immune surveillance via mechanisms such as the programmed death-1/programmed death-ligand 1(PD-1/PD-L1) axis (8).
Personalized vaccines with neoantigens or autologous whole tumor lysates can overcome tumor heterogeneity (9), but their production processes are lengthy, complicated, and expensive (10). One promising strategy to improve personalized cancer vaccination uses immunostimulatory treatments to generate tumor antigens in situ, which can afford systemic antitumor immune responses in a personalized fashion and modulate local tumor microenvironments to relieve immunosuppression (11). For instance, intratumoral injection of oncolytic viruses such as talimogene laherparepvec (T-VEC) inflicts direct cytotoxic effects on cancer cells and recruits dendritic cells (DCs) for antigen presentation, acting as in situ cancer vaccines with reduced side effects (12). Nonviral treatments with potent antitumor effects, such as phototherapy (13), radiotherapy (RT) (14), and some chemotherapies, can also generate danger-associated molecular patterns (DAMPs) and tumor antigens by inducing immunogenic cell death (ICD) (15).
Stimulation of DCs with immunoadjuvants such as stimulator of interferon (IFN) genes (STING) agonist (16, 17) or CpG oligodeoxynucleotides (18) further promotes antigen presentation and immune responses (19). Naturally existing as microbial DNAs known as pathogen-associated molecular patterns (PAMPs), CpGs are short DNA strands explored widely as vaccine adjuvants for Toll-like receptor 9 (TLR9) stimulation, DC maturation, antigen presentation, and the priming of tumor-specific cytotoxic T lymphocytes (CTLs) (20). In addition, antigen presentation by immature DCs in the absence of immunoadjuvants induces tolerance rather than stimulates an immune reaction (21). In particular, class C CpGs enhance type I IFN production to activate DCs and stimulate B cells, which, in turn, up-regulates costimulatory molecules and secretes pro-inflammatory cytokines to afford superb anticancer effects (22). Similar to peptide vaccines, however, even locally administered unmodified CpGs are prone to enzymatic degradation and cannot be efficiently internalized by APCs because of their anionic nature (23).
In the context of antigen and adjuvant delivery, nanotechnology is at the forefront of merging drug delivery and immune stimulation to elicit antitumor activity for cancer vaccination (24–27). Here, we propose a strategy of using nanoscale metal-organic frameworks (nMOFs) for personalized cancer vaccination via x-ray–activated generation of DAMPs and tumor antigens and efficient delivery of CpGs to APCs as PAMPs. Assembled from tunable metal-oxo clusters and functional organic ligands, nMOFs have emerged as a new type of porous and crystalline molecular nanomaterials with interesting potential in biomedical applications (28, 29). Notably, nMOFs have shown potent antitumor activity by generating highly cytotoxic and immunogenic reactive oxygen species (ROS) upon external light or x-ray irradiation (30, 31). nMOFs are also able to directly convert x-ray energy to ROS via a unique RT–radiodynamic therapy (RT-RDT) (32). To expand the therapeutic effect of nMOF-mediated local inflammation to systemic anticancer efficacy, several strategies have been explored, including combination with checkpoint blockade immunotherapy (CBI) (33) and co-delivery of different immunotherapeutic drugs such as small-molecule inhibitors (34) and agonists (35). However, none of these strategies generated robust DC infiltration and activation, suggesting an impaired antigen presentation to bridge innate immunity and adaptive immunity.
Considering the ability of CpGs to boost the maturation of APCs, we designed cationic nMOFs to deliver anionic CpGs via electrostatic interactions in this work. When activated by x-rays, nMOFs effectively generate ROS to release DAMPs and tumor antigens while delivering CpGs as PAMPs to facilitate APC maturation. The in situ vaccination afforded by nMOFs effectively expands cytotoxic T cells in tumor-draining lymph nodes (DLNs) to reinvigorate the adaptive immune system for tumor regression. Unlike previous nMOF-based treatments, the present in situ cancer vaccination strategy effectively increases intratumoral DC population, stimulates DC maturation, and facilitates antigen presentation to prime T lymphocytes on T cell–excluded syngeneic colorectal MC38 and pancreatic Panc02 tumor models. The local therapeutic effects of the nMOF-based in situ vaccines were extended to distant tumors by combination treatment with an anti–PD-L1 antibody (αPD-L1) to afford an 83.3% cure rate on immunosuppressive MC38 models.
RESULTS
Synthesis and characterization of nMOFs
To generate DAMPs and tumor antigens through RT-RDT and deliver PAMPs with high CpG loading, we designed two positively charged nMOFs, Hf-DBBF-Ir and Hf-DBB-Ir, with high-Z metal Hf6 secondary building units (SBUs) and photosensitizing DBBF-Ir and DBB-Ir ligands, respectively (fig. S1). Hf-DBBF-Ir and Hf-DBB-Ir nMOFs had UiO-like structures with a formula of Hf6(μ3─O)4(μ3─OH)4L6, where L = DBBF-Ir or DBB-Ir (fig. S2, A to D and M). Hf-DBBF-Ir and Hf-DBB-Ir exhibited spherical to octahedral morphologies with diameters of ~100 nm, as revealed by transmission electron microscopy (TEM) imaging (Fig. 1B, and fig. S2, E to H) and dynamic light scattering measurements (fig. S2N). The photosensitizing characteristics of Hf-DBBF-Ir and Hf-DBB-Ir were confirmed by ultraviolet-visible absorption and luminescence spectroscopy, where Hf-DBBF-Ir and Hf-DBB-Ir showed similar absorbance and luminescence to those of DBBF-Ir and DBB-Ir, respectively (fig. S2, I to L).
Fig. 1. Characterization of nMOFs.
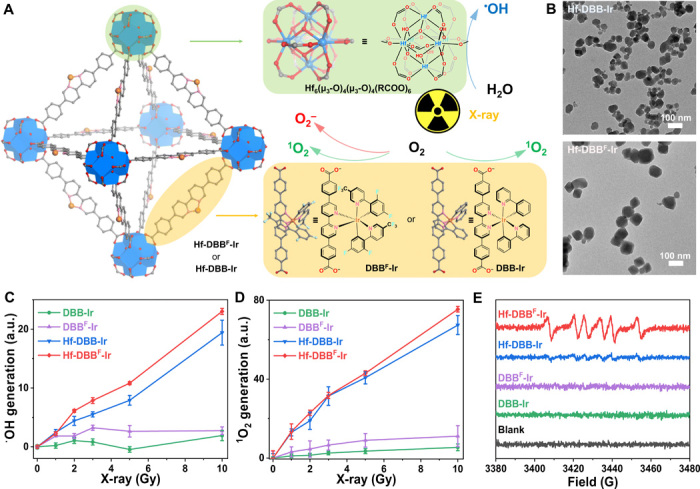
(A) Schematic illustration of controlled synthesis of Hf-DBB-Ir and Hf-DBBF-Ir nMOFs based on Hf-oxo clusters and DBBF-Ir or DBB-Ir ligands, respectively. Upon x-ray irradiation, Hf-oxo clusters absorb x-ray to generate •OH through radiolysis and transfer energy to adjacent photosensitizing ligands to generate 1O2 and/or O2−. (B) TEM images of Hf-DBB-Ir (top) and Hf-DBBF-Ir (bottom).. Scale bars, 100 nm. (C to E) •OH generation probed by aminophenyl fluorescein (APF) with an IVIS fluorescence imaging system (n = 6) (C), 1O2 generation probed by SOSG with an IVIS fluorescence imaging system (n = 6) (D), and O2− generation probed by 5-tert-butoxycarbonyl 5-methyl-1-pyrroline-N-oxide (BMPO) in electron paramagnetic resonance (EPR) (E) of DBB-Ir, DBBF-Ir, Hf-DBB-Ir, or Hf-DBBF-Ir upon x-ray irradiation. a.u., arbitrary units; G, gauss.
Detection of ROS
We proposed that photosensitizing Hf-DBBF-Ir and Hf-DBB-Ir could generate multiple ROS upon x-ray irradiation, including hydroxyl radical (•OH) through water radiolysis of Hf6 SBUs and singlet oxygen (1O2) and superoxide anion (O2−) through excitation of photosensitizing ligands (Fig. 1A). Aminophenyl fluorescein (APF) and singlet oxygen sensor green (SOSG) assays were performed to probe •OH and 1O2, respectively, which showed that both Hf-DBBF-Ir plus x-ray irradiation [denoted Hf-DBBF-Ir(+)] and Hf-DBB-Ir(+) exhibited significantly enhanced •OH and 1O2 generation in comparison to their ligand controls (Fig. 1, C and D, and fig. S2, O and P). However, only Hf-DBBF-Ir(+) displayed efficient O2− generation, as determined by 5-tert-butoxycarbonyl 5-methyl-1-pyrroline-N-oxide (BMPO), a nitrone spin trap. The O2− generation by Hf-DBBF-Ir(+) is ascribed to the higher reduction potential of DBBF-Ir than DBB-Ir (Fig. 1E) (36).
Generation of ROS in vitro
We first evaluated the uptake of Hf-DBB-Ir and Hf-DBBF-Ir by MC38 cells. Inductively coupled plasma mass spectrometry (ICP-MS) studies showed that the two nMOFs reached similar intracellular Hf levels after 4-hour incubation (fig. S3A). We then probed in vitro 1O2 and O2− generation by SOSG and superoxide assay kits, respectively. Both Hf-DBB-Ir(+) and Hf-DBBF-Ir(+) induced strong green fluorescence, indicating significant 1O2 generation. However, only Hf-DBBF-Ir(+) exhibited strong red fluorescence, indicating the generation of O2− through Hf-DBBF-Ir–mediated RT-RDT process (Fig. 2A). To confirm the RT effect, we quantified •OH-induced DNA double-strand breaks by flow cytometric analysis of phosphorylated γ-H2AX in cells treated with phosphate-buffered saline (PBS), ligands, or nMOFs with or without x-ray. Two hours after irradiation, significantly higher red γ-H2AX fluorescence was observed in the group treated with Hf-DBBF-Ir(+) than Hf-DBB-Ir(+), likely due to the biotransformation of O2− to •OH by superoxidase dismutase. No fluorescence was observed in the groups without x-ray irradiation or without nMOF incubation (fig. S3B).
Fig. 2. In vitro generation of DAMPs and phagocytosis.
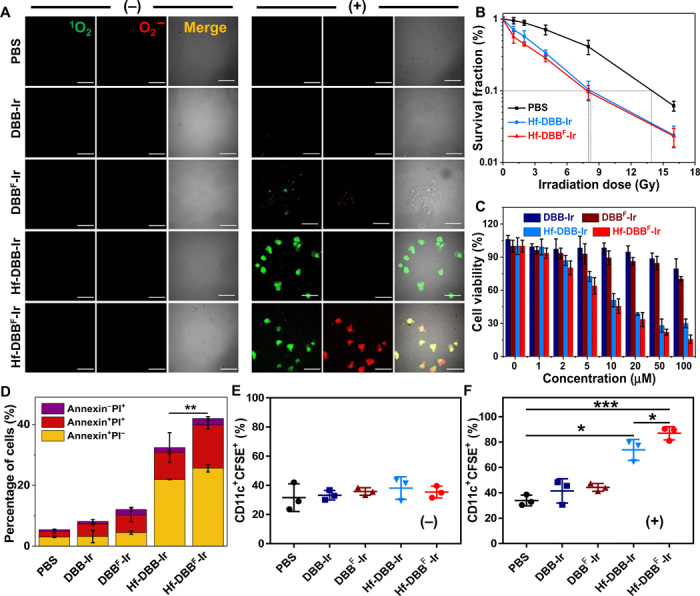
(A) Generation of 1O2 and O2− in MC38 cells treated with phosphate-buffered saline (PBS), DBB-Ir, DBBF-Ir, Hf-DBB-Ir, or Hf-DBBF-Ir with (+) or without (−) x-ray irradiation as detected by SOSG and superoxide kits. Green (1O2) and red (O2−) florescence merged to appear as yellow florescence. Scale bars, 50 μm. (B) Clonogenic assays to evaluate radioenhancement of Hf-DBB-Ir or Hf-DBBF-Ir on MC38 cells upon x-ray irradiation; n = 6. (C) Cytotoxicity of Hf-DBB-Ir or Hf-DBBF-Ir upon x-ray irradiation at a dose of 2 Gy on MC38 cells; n = 6. (D) Annexin V/propidium iodide (PI) cell apoptosis/death analysis of MC38 cells; n = 3. (E and F) Phagocytosis of carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled MC38 cells by DCs. DCs cocultured with treated MC38 cells without (−) (E) or with (+) (F) irradiation were stained with PE-Cy5.5–conjugated CD11c antibody; n = 3. Data are expressed as means ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001 by t test.
Release of DAMPs and in vitro immunogenicity
To test the hypothesis that the myriad of ROS generated by Hf-DBBF-Ir damages cancer cells more effectively than other treatments, we evaluated Hf-DBBF-Ir–mediated cell damage and DAMP generation. Clonogenic assays showed that Hf-DBBF-Ir(+) slightly outperformed Hf-DBB-Ir(+) with a radiation ehancement factor at 10% survival (REF10) value (calculated as the ratio of irradiation doses for the PBS control over that for the experimental group at 10% survival) of 1.75 versus 1.68 (Fig. 2B). 3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfo-phenyl)-2H-tetrazolium (MTS) assays further showed that Hf-DBBF-Ir(+) exhibited higher cytotoxicity than Hf-DBB-Ir(+) with IC50 (half maximal inhibitory concentration) values of 4.28 ± 1.15 μM and 7.85 ± 2.41 μM, respectively, at 2 gray (Gy) (P = 0.036; Fig. 2C). A greater level of cell death was also observed for Hf-DBBF-Ir(+) by live/dead cell imaging and apoptotic cell quantification by confocal laser scanning microscopy (CLSM) and flow cytometry (Fig. 2D and fig. S3, C to E). These results indicate a stronger cell killing effect by Hf-DBBF-Ir(+) via the RT-RDT process.
We next investigated the generation of DAMPs from nMOF-mediated RT-RDT by examining the ICD of tumor cells and phagocytosis of dying tumor cells and their apoptotic debris by APCs. In the ICD process, calreticulin (CRT) is translocated to cell membrane as an “eat-me” signal that is recognized by macrophages and DCs to engulf dying tumor cells and their apoptotic debris. Flow cytometric quantification revealed that Hf-DBBF-Ir(+)–treated cells exhibited higher CRT fluorescence, suggesting that Hf-DBBF-Ir(+) induced stronger ICD with higher cytotoxicity (fig. S4A).
To assess the impact of nMOF-mediated RT-RDT on antigen processing by and immune activation of APCs, we cocultured DCs differentiated from bone marrow cells with carboxyfluorescein diacetate succinimidyl ester (CFSE)–labeled MC38 cells treated with PBS, DBB-Ir, DBBF-Ir, Hf-DBB-Ir, or Hf-DBBF-Ir with or without x-ray irradiation. Flow cytometry showed that Hf-DBBF-Ir(+) treatment induced significantly higher population of phycoerythrin (PE)–Cy5.5–conjugated CD11c-labeled DCs with phagocytosed CFSE-labeled MC38 cells than other treatment groups, indicating enhanced immune stimulation mediated by cationic nMOFs (fig. S4B and Fig. 2, E and F). CLSM imaging confirmed that more CD11c+ DCs phagocytosed Hf-DBBF-Ir(+)–treated CFSE+ MC38 cells (fig. S4C).
In vitro delivery of PAMPs
We rationalized that fluorination of the DBBF ligand in Hf-DBBF-Ir could introduce electron-withdrawing effects to increase the surface charge for more efficient delivery of CpG. Hf-DBBF-Ir and Hf-DBB-Ir exhibited ζ-potential values of 31.6 ± 1.2 mV and 23.8 ± 0.8 mV, respectively, confirming a more cationic skeleton of Hf-DBBF-Ir for electrostatic adsorption of anionic CpG (Fig. 3A). One milligram of CpG was incubated in 20 ml of PBS solution of Hf-DBBF-Ir or Hf-DBB-Ir with an Hf concentration of 10 mM for 10 min. After centrifugation, DNA gel electrophoresis showed the adsorption of 82.7% CpG onto Hf-DBBF-Ir and 46.5% CpG onto Hf-DBB-Ir, with 8.6 and 43.8% of CpG remaining in the corresponding supernatants as quantified by NanoDrop spectrophotometry (Fig. 3B). We next examined CpG internalization by DCs. Flow cytometry and CLSM imaging showed that Hf-DBBF-Ir delivered the highest amount of CpG to DCs after they were cultured with fluorescein isothiocyanate (FITC)–labeled free CpG, Hf-DBB-Ir@CpG, or Hf-DBBF-Ir@CpG (Fig. 3C and fig. S5A). These results confirm the superior ability of Hf-DBBF-Ir in delivering CpG as PAMPs to APCs.
Fig. 3. In vitro delivery of PAMPs and DC activation.
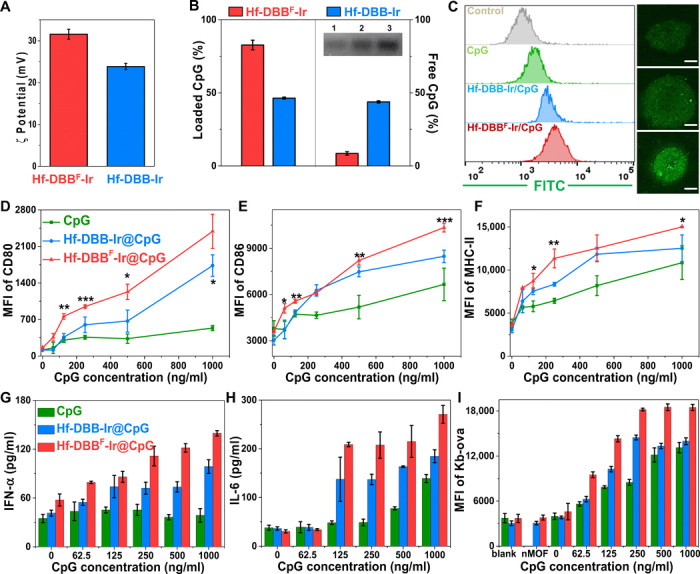
(A) ζ potential of Hf-DBB-Ir and Hf-DBBF-Ir. (B) Left: Quantification of adsorbed CpG with DNA gel. Right: Nonabsorbed CpG by NanoDrop with free CpG as control. (C) Fluorescein isothiocyanate (FITC)–labeled CpG uptake by DCs incubated with free CpG, Hf-DBB-Ir@CpG, or Hf-DBBF-Ir@CpG at concentrations of 20 μM Hf and CpG (0.1 μg/ml) quantified with flow cytometry and observed under confocal laser scanning microscopy (CLSM). Scale bars, 4 μm. Functional surface markers CD80 (D), CD86 (E), and MHC-II (F) quantified by flow cytometry. Biomarkers IFN-α (G) and IL-6 (H) of DCs stimulated with free CpG, Hf-DBB-Ir@CpG, or Hf-DBBF-Ir@CpG with increasing CpG concentrations by enzyme-linked immunosorbent assay (ELISA); n = 6. (I) SIINFEKL-Kb expression levels of DCs cocultured with MC38-ova cells at a 1:3 ratio; n = 6.
In vitro DC maturation
To evaluate the effects of CpG delivery on DC maturation, we incubated bone marrow–derived DCs with CpG, Hf-DBB-Ir@CpG, or Hf-DBBF-Ir@CpG at CpG concentrations of 0, 62.5, 125, 250, 500, and 1000 ng/ml for 60 hours. The cells were harvested and stained for the detection of DC maturation markers, including major histocompatibility complex class II (MHC-II) and costimulatory molecules CD80 and CD86. The supernatants were also collected and assayed for the presence of cytokines IFN-α and interleukin-6 (IL-6). Both Hf-DBB-Ir@CpG and Hf-DBBF-Ir@CpG effectively promoted DC maturation with increased mean fluorescence intensity (MFI) signals of CD80 (Fig. 3D), CD86 (Fig. 4E), and MHC-II compared to free anionic CpG (Fig. 3F). Hf-DBBF-Ir@CpG outperformed Hf-DBB-Ir@CpG in the up-regulation of CD80, CD86, and MHC-II signals as a result of its more effective CpG delivery. Only cationic nMOF-delivered CpG showed elevated IFN-α levels, while free CpG completely had no effect (Fig. 3G). Moreover, DCs treated with free CpG excreted IL-6 only in high CpG concentrations, while treated with nMOFs/CpG excreted IL-6 at low CpG concentrations (Fig. 3H). Quantitative polymerase chain reaction (qPCR) of IL-6 and IFN-α expression confirmed that Hf-DBBF-Ir more efficiently delivered CpG as PAMPs to activate DCs (fig. S5, B and C). To directly demonstrate the enhanced antigen presentation property of DCs after Hf-DBBF-Ir@CpG stimulation, we cultured MC38 cells transfected with ovalbumin antigen (OVA; cell line denoted as MC38-ova) with CpG-, Hf-DBB-Ir@CpG–, or Hf-DBBF-Ir@CpG–stimulated DCs in a 3:1 ratio. Tumor antigen uptake and presentation were examined by detecting the expression of H-2Kb-SIINFEKL complex (Kb-ova) on DC surface. Hf-DBBF-Ir@CpG outperformed Hf-DBB-Ir@CpG and free CpG on promoting antigen uptake and presentation by DCs (Fig. 3I), likely as a result of more efficient delivery of PAMPs and antigen presentation. We further showed that nMOFs alone minimally activated DCs, while both nMOF(+)-induced DAMPs and nMOF-delivered PAMPs efficiently promoted DC activation (fig. S5, D and E).
Fig. 4. nMOFs for in situ personalized cancer vaccination to boost innate immunity for in vivo anticancer treatment.
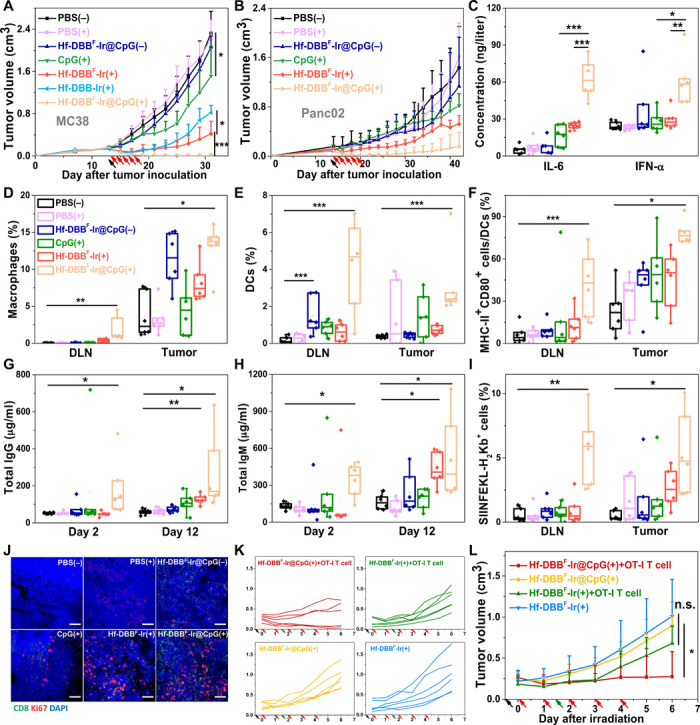
Tumor growth curves of (A) MC38 and (B) Panc02 tumor-bearing mice treated with PBS(−), PBS(+), CpG(+), Hf-DBBF-Ir(+), Hf-DBBF-Ir@CpG(−), or Hf-DBBF-Ir@CpG(+). Hf-DBB-Ir(+) served as control group on the MC38 model; n = 6. (C) Plasma concentration of IL-6 and IFN-α 48 hours after first irradiation as quantified by ELISA. The percentages of macrophages (D) and DCs (E) with respect to the total cells in tumors and tumor-DLNs excised from MC38-bearing mice 2 days after treatment. The percentages of tumor-infiltrating CD80+ MHCII+ cells (F) with respect to DCs in tumors and tumor-DLNs excised from MC38-bearing mice day 2 after treatment. Total immunoglobulin G (IgG) (G) and total IgM (H) in plasma 2 and 12 days after irradiation; n = 6. (I) Percentages of SIINFEKL-H2Kb+ cells excised from MC38-ova–bearing mice day 6 after treatment. (J) Representative CLSM showing the T cell expansion of tumor-DLNs. Blue, green, and red fluorescence represent 4′,6-diamino-2-phenylindole (DAPI), CD8α, and Ki67, respectively. Scale bars, 50 μm. Individual (K) and average (L) growth curves of MC38-ova tumor-bearing Rag2−/− mice treated with Hf-DBBF-Ir(+) or Hf-DBBF-Ir@CpG(+) with or without OT-I T cell transfer study. Black, red, and green arrows refer to intratumoral injections, x-ray irradiation, and adoptive T cell transfer, respectively. Data are expressed as means ± SD (n = 6). n.s., not significant. P > 0.05, *P < 0.05, **P < 0.01, and ***P < 0.001 by t test. Central lines, bounds of box, and whiskers represent mean values, 25 to 75% of the range of data, and 1.5-fold of interquartile range away from outliers, respectively.
X-ray–triggered in situ cancer vaccines
We next investigated the local anticancer effect of Hf-DBBF-Ir@CpG(+) as an in situ cancer vaccine. We first showed that intravenous injection of 2 μmol of DBBF-Ir or Hf-DBBF-Ir biweekly for a total of four doses did not cause toxicity on C57BL/6 mice, as judged from the steady body weight gains (fig. S6, A and B). We then established a T cell–excluded murine colorectal model MC38 on C57BL/6c mice by subcutaneous injection of 5 × 105 MC38 cells on right flanks. In our previous studies, subcutaneous MC38 tumors were established by inoculating 2 × 106 MC38 cells and reached 100 to 150 mm3 in sizes at day 7 before the commencement of treatments. When inoculated with fewer cells, MC38 tumors grew to 100 to 150 mm3 in 14 days (the 14-day model) and showed much more immunosuppressive tumor microenvironments than the 7-day model (fig. S6D). PBS, Hf-DBB-Ir, Hf-DBBF-Ir, or Hf-DBBF-Ir@CpG was injected intratumorally at an Hf dose of 0.2 μmol and/or a CpG dose of 1 μg. Twelve hours later, the tumors were irradiated with 1 Gy of x-ray [225 peak kilovoltage (kVp), 13 mA, and 1 Gy], followed by four more daily irradiation of x-ray (1 Gy). Hf-DBBF-Ir(+) outperformed Hf-DBB-Ir(+) with a tumor growth inhibition index (TGI) of 81.9% versus 64.7%, suggesting more efficient release of DAMPs by Hf-DBBF-Ir–mediated RT-RDT in vivo. Hf-DBBF-Ir@CpG(+) showed enhanced tumor regression over CpG(+) (TGI of 99.6% versus 34.8%) or Hf-DBBF-Ir(+) on day 31, indicating the synergy of DAMPs released by nMOF-mediated RT-RDT and PAMPs delivered by cationic nMOFs (Fig. 4A and table S1). The anticancer efficacy was confirmed by optical images and averaged weights of excised tumors on day 31 (fig. S6, E and F). Immunofluorescence of terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) and hematoxylin and eosin staining indicated significant apoptosis of tumor cells with Hf-DBBF-Ir@CpG(+) treatment (fig. S6, I and J). No systemic toxicity was observed for all treatment groups (fig. S6, C and K). We further evaluated the antitumor activity on a murine pancreatic cancer model, Panc02, on C57BL/6c mice with high radioresistance and poor immunogenicity. Hf-DBBF-Ir@CpG(+) afforded superior tumor growth inhibition over other groups (Fig. 4B; table S1; and fig. S6, G and H), suggesting the potential of using Hf-DBBF-Ir@CpG(+) as in situ cancer vaccine on a broad spectrum of cancers with varied immunogenicity.
Innate immunity after in situ cancer vaccination
We assayed plasma IL-6 and IFN-α concentrations by enzyme-linked immunosorbent assay (ELISA) and determined gene expression in tumors and tumor-DLNs by qPCR 24 hours after treatment to evaluate the innate immune response. Hf-DBBF-Ir@CpG(+) treatment showed significantly elevated levels of plasma and intratumoral IL-6 and IFN-α over CpG(+) or Hf-DBBF-Ir(+) treatment (Fig. 4C, and fig. S7, A to D). Furthermore, flow cytometry and CLSM studies showed significant increases of tumor- and DLN-infiltrating APCs, including macrophages (Fig. 4D) and DCs (Fig. 4E), in the Hf-DBBF-Ir@CpG(+) treatment group, which indicates the synergistic effect of DAMPs and tumor antigens released by nMOF-mediated RT-RDT and PAMPs delivered by cationic nMOFs (fig. S7E). DC maturation promoted by Hf-DBBF-Ir@CpG(+) was further demonstrated with elevated expression of MHC-II and costimulatory CD80 molecules (Fig. 4F). Elevations of total immunoglobulin G (IgG) (Fig. 4G) and IgM (Fig. 4H) in plasma 2 and 12 days after Hf-DBBF-Ir@CpG(+) treatment suggest effective promotion of humoral immunity mediated by B cells. As IgM can bind and activate the complement system to promote the opsonization and degradation of antigens and antigen presentation by phagocytes, the increased levels of plasma IgG and IgM results imply an important role of B cells in promoting antigen presentation after in situ vaccination (fig. S8, A to F). The expression of Kb-ova complex (SIINFEKL-H2Kb gated from CD45+ cells) was significantly up-regulated after Hf-DBBF-Ir@CpG(+) treatment on the MC38-ova model, confirming the antigen presentation process (Fig. 4I, and fig. S7, F and G). Hf-DBBF-Ir@CpG(+) group also exhibited enlarged DLNs (fig. S8, H and I), suggesting T cell expansion in DLNs. The increased expression of Ki67 in DLNs by CLSM supported T cell expansion in DLNs following Hf-DBBF-Ir@CpG(+) treatment (Fig. 4J). Last, MC38-ova tumors were established on immunodeficient Rag2−/− mice and then treated with Hf-DBBF-Ir(+) or Hf-DBBF-Ir@CpG(+) plus adoptive transfer of OT-I T cells. Mice treated with Hf-DBBF-Ir@CpG(+) plus OT-I T cell transfer showed more effective tumor suppression than either Hf-DBBF-Ir(+) plus OT-I T cell transfer or Hf-DBBF-Ir@CpG(+) alone (Fig. 4, K and L), supporting an effective antigen presentation process after Hf-DBBF-Ir@CpG(+) treatment as an in situ cancer vaccine. We observed macrophage repolarization with an increased ratio of pro-inflammatory M1 subtype to anti-inflammatory (tumor-promoting) M2 subtype following Hf-DBBF-Ir@CpG(+) treatment (fig. S8G).
Abscopal effect
A bilateral model of MC38 was then established to assess the systemic anticancer efficacy of Hf-DBBF-Ir@CpG(+) in combination with αPD-L1. Hf-DBBF-Ir@CpG was intratumorally injected into primary tumors at a dose of 0.2 μmol of Hf and 1 μg of CpG 14 days after tumor inoculation, with daily x-ray irradiation at a dose of 1 Gy per fraction beginning on day 15 for a total of five fractions. Seventy-five micrograms of αPD-L1 was administered every 3 days by intraperitoneal injection for a total of three doses. Without αPD-L1, Hf-DBBF-Ir@CpG(+) almost eradicated primary tumors but only moderately delayed progression of distant tumors. In stark contrast, the combination of Hf-DBB-Ir-F@CpG(+) and αPD-L1 significantly regressed both primary and distant tumors with a cure rate of 83.3%. This result indicates a strong synergy between Hf-DBBF-Ir@CpG(+)–based in situ cancer vaccination and CΒI (Fig. 5, A to C).
Fig. 5. Abscopal effect of in situ cancer vaccination synergized CBI with promoted adaptive immunity.
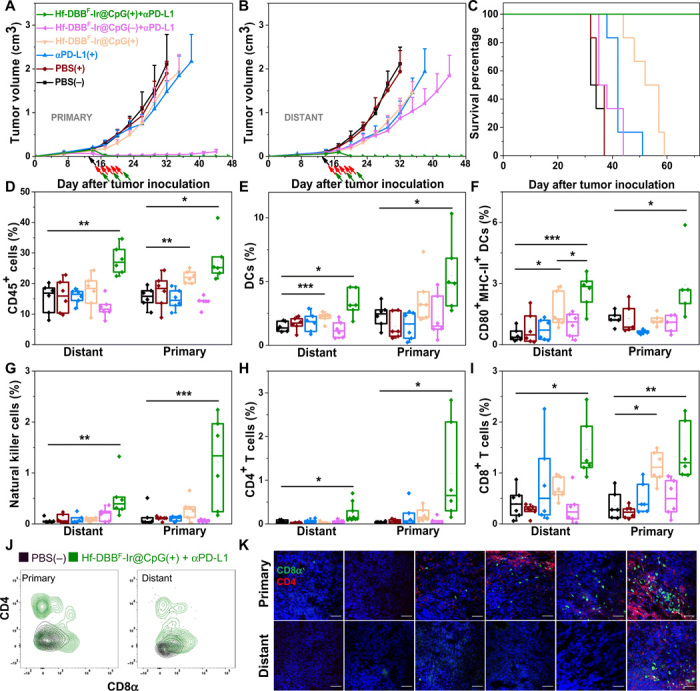
Primary treated (A), distant untreated (B) tumor growth curves, and survival curves (C) of MC38 tumor-bearing mice treated with PBS(−), PBS(+), αPD-L1(+), Hf-DBBF-Ir@CpG(+), Hf-DBBF-Ir@CpG(−) + αPD-L1, or Hf-DBBF-Ir@CpG(+) + αPD-L1. Treatment began on day 14 after tumor inoculation when the tumor reached a volume of 100 to 150 mm3. X-ray irradiation was carried out on mice 12 hours after the intratumoral injection of PBS or Hf-DBBF-Ir@CpG on five consecutive days at a dose of 1 Gy per fraction. Antibody was given intraperitoneally every 3 days at a dose of 75 μg per mouse. Black, red, and green arrows refer to the times of intratumoral injections, x-ray irradiation, and intraperitoneal injection, respectively; n = 6. The percentages of tumor-infiltrating CD45+ cells (D), DCs (E), CD80+ MHCII+ DCs (F), NK cells (G), CD4+ T cells (H), and CD8+ T cells (I) with respect to the total cells 10 days after treatment. Data are expressed as means ± SD (n = 6). *P < 0.05, **P < 0.01, and ***P < 0.001 by t test. Central lines, bounds of box, and whiskers represent means, 25 to 75% of the range of data, and 1.5-fold of interquartile range away from outliers, respectively. (J) Representative flow cytometric plots showing infiltration of CD4+ and CD8+ T cells on both primary and distant tumors treated by PBS(−) or Hf-DBBF-Ir@CpG(+) + αPD-L1 shown in black or green contours, respectively. (K) Representative pictures showing the infiltration of T cells on excised tumor section slides. Blue, green, and red fluorescence represent DAPI, CD8α, and CD4, respectively. Scale bars, 50 μm.
Adaptive immunity
We profiled infiltrating leukocytes in both primary and distant tumors 10 days after irradiation. Hf-DBBF-Ir@CpG(+) + αPD-L1 treatment group showed significant increase of tumor-infiltrating CD45+ leukocytes (Fig. 5D), DCs (Fig. 5E), macrophages (fig. S9, A and B), and CD8+ T cells (Fig. 5F) in both primary and distant tumors, implying a strengthened innate immune response after in situ vaccination. Specifically, after treatment with Hf-DBBF-Ir@CpG(+) + αPD-L1, the percentages of natural killer (NK) cells (Fig. 5G), CD4+ T cells (Fig. 5H), and CD8+ T cells (Fig. 5I) of the total primary and distant tumor cells significantly increased to 0.52 ± 0.42% and 1.21 ± 0.89%, 0.25 ± 0.23% and 1.15 ± 1.14%, and 1.46 ± 0.59% and 1.43 ± 0.55% from 0.06 ± 0.05% and 0.13 ± 0.19%, 0.05 ± 0.04% and 0.03 ± 0.02%, and 0.41 ± 0.30% and 0.36 ± 0.27% in PBS(−) group, respectively. The effector T cell infiltration was shown by both flow cytometry (Fig. 5J) and CLSM (Fig. 5K). DLNs on both sides were harvested, weighed, and immunostained for detecting T cell expansion, suggesting that Hf-DBBF-Ir@CpG(+) + αPD-L1 treatment promoted T cell expansion on bilateral DLNs (fig. S9, C to E). These results suggest that the combination of Hf-DBBF-Ir@CpG(+) + αPD-L1 not only induces innate immune response but also augments adaptive immunity in both treated local and untreated distant tumors.
Specificity of induced immunity
We first determined the presence of tumor antigen–specific cytotoxic T cells with an IFN-γ Enzyme-Linked ImmunoSpot (ELISpot) assay. Splenocytes were harvested from MC38-bearing mice 10 days after first irradiation and stimulated with the peptide sequence KSPWFTTL for 42 hours. IFN-γ spot-forming cells were counted with an ImmunoSpot Reader. The number of antigen-specific IFN-γ–producing T cells per 106 splenocytes significantly increased in tumor-bearing mice treated with Hf-DBBF-Ir@CpG(+) and Hf-DBBF-Ir@CpG(+) + αPD-L1 (60.2 ± 39.6 and 139.0 ± 52.4 compared to 16.4 ± 5.9 for PBS(−); Fig. 6A), suggesting that both Hf-DBBF-Ir@CpG(+) and Hf-DBBF-Ir@CpG(+) + αPD-L1 effectively generate tumor-specific T cell responses. To further investigate the specific antitumor immunity, we treated MC38 primary tumors with Hf-DBBF-Ir@CpG(+) or Hf-DBBF-Ir@CpG(+) + αPD-L1 to observe whether the treatment could regress unmatched syngeneic tumors on distant flanks. As illustrated in Fig. 6B, MC38 were used as the primary treated tumors, and syngeneic tumor cell lines B16F10 and LL2 were implanted concurrently as the distant untreated tumors. Both Hf-DBBF-Ir@CpG(+) and Hf-DBBF-Ir@CpG(+) + αPD-L1 treatments effectively regressed the primary MC38 tumors but had no effect on the distant B16F10 or LL2 tumors (Fig. 6, C to F). These experiments indicate tumor specificity and personalized nature of the newly expanded T cells following in situ vaccination with the Hf-DBBF-Ir@CpG(+) + αPD-L1 treatment.
Fig. 6. Specificity and immune memory effect of in situ cancer vaccination plus CBI.
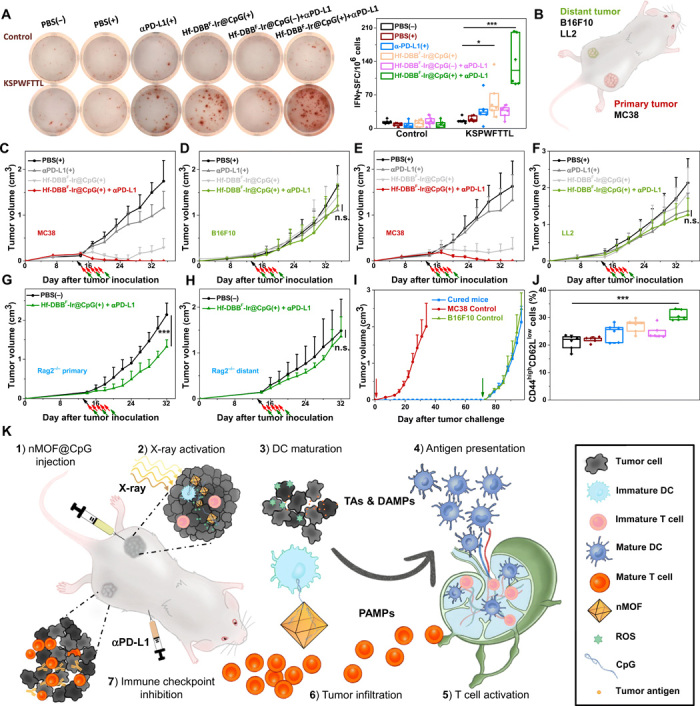
(A) Representative images of colonies (left) and statistical analysis (right) of ELISpot assay performed to detect tumor-specific IFN-γ–producing T cells; n = 6. SFC, spot-forming cells. (B) Schematic illustration of bilateral models established by subcutaneous injection of MC38 and B16F10 or LL2 cells onto flanks as primary and distant tumors, respectively. Primary treated MC38 (C and E) and distant untreated [B16F10 for (D) and LL2 for (F)] tumor growth curves on unmatched bilateral tumor models treated with PBS(+), αPD-L1(+), Hf-DBBF-Ir@CpG(+), or Hf-DBBF-Ir@CpG(+) + αPD-L1; n = 4. Primary (G) and distant (H) tumor growth curves on MC38-bearing Rag2−/− models treated with PBS(−) or Hf-DBBF-Ir@CpG(+) + αPD-L1; n = 6. (I) Tumor growth curves after challenge with MC38 tumor cells and rechallenge with B16F10 cells on cured mice as treated from Fig. 5C. (J) Percentages of CD44highCD62Llow cells with respect to the total splenocytes; n = 6. (K) Schematic illustration of antitumor effect of in situ cancer vaccination by nMOF(+) plus CBI. (1) Hf-DBBF-Ir@CpG is intratumorally administered in the primary tumor. (2) Upon x-ray activation, Hf-DBBF-Ir generates ROS to induce ICD to expose tumor antigens and DAMPs, while cationic Hf-DBBF-Ir delivers CpG as PAMPs to APCs. (3) DAMPs and PAMPs promote DC maturation. (4) Tumor antigens are presented by mature DCs onto T cell in tumor-DLNs. (5) T cells expand and infiltrate to both primary and distant tumors. (6) Systemically administered immune checkpoint inhibitor αPD-L1 attenuates T cell exhaustion.
Long-term antitumor immunity
The involvement of cytotoxic T cells in efficient abscopal effect was further supported by the lack of efficacy of Hf-DBBF-Ir@CpG(+) + αPD-L1 treatment on a bilateral subcutaneous model of MC38 on Rag2−/− C57BL/6 mice deficient of mature T and B cells. The primary tumors treated with Hf-DBBF-Ir@CpG(+) + αPD-L1 were initially suppressed (Fig. 6G) but grew rapidly after the end of x-ray irradiation. No abscopal effect was observed on the distant tumors (Fig. 6H). This result confirms that both the abscopal effect and local tumor regression/eradication require the presence of tumor-specific adaptive immunity. Last, we carried out a tumor rechallenge study to confirm the long-term immune memory effect. For the mice completely cured after treatment with Hf-DBBF-Ir@CpG(+) + αPD-L1, 5 × 105 MC38 cells were inoculated on the contralateral, left flank 30 days after tumor eradication, and those cured mice remained tumor-free after the first challenge, indicating strong antitumor immune memory effect. Two months after the first challenge, 2 × 106 B16F10 cells were inoculated on the right flank, and the cured mice established tumors similarly to naïve mice, suggesting the tumor specificity of the immune memory effect (Fig. 6I). We also profiled memory effector cells (CD3ε+CD8α+CD44highCD62Llow phenotype) in splenocytes after the combination treatment. As shown in (Fig. 6J), significant increase of memory effector cells was observed in spleens after Hf-DBBF-Ir@CpG(+) + αPD-L1 treatment.
DISCUSSION
Advanced tumors escape immune surveillance by inactivating, dysregulating, and hijacking host immune systems (37). To combat this, anti–PD-(L)1 CBI has become a standard of care for some cancers by targeting T cell inhibitory checkpoint signaling pathways to afford durable anticancer efficacy with low side effects (38). Immune checkpoint inhibition, however, only elicits durable responses in a minority of patients with cancer due to the reliance on immunogenic tumor microenvironments, so-called “hot” tumors. For patients with relatively “cold” tumors, e.g., low tumor mutation burden, low PD-L1 expression level, and/or low abundance of preexisting T cells, immunoadjuvant treatments to turn cold tumors hot are actively examined in combination with checkpoint inhibitors to overcome immune tolerance and potentiate antitumor immunity in the host system.
Having previously described the abscopal effect in the setting of RT (39), immunoadjuvant therapy with CpG (40, 41), and oncolytic viral therapy (42), we propose that local treatment to generate innate immunity with tumor antigen exposure may effectively reinvigorate cold tumors to become immunogenic hotbeds. Furthermore, two pattern recognition receptor pathways (43), cGAS-STING induced by DAMPs after RT damage (44) and TLR pathway induced by PAMPs such as CpG (45), operate independently (46), indicating that they may be activated simultaneously to achieve an additive or synergistic effect on immune stimulation. We previously reported that porous nMOFs built from Hf-oxo SBUs and photosensitizing ligands significantly enhance radiotherapeutic effects of ionizing radiations with enhanced x-ray energy deposition, facile ROS diffusion, and unique RT-RDT mode of action (33, 34). Drawing from these previous results, we report here a new cationic Hf-based nMOF, Hf-DBBF-Ir, for nonviral in situ vaccination by mediating efficient RT-RDT to generate immunogenic tumor antigens and DAMPs and to deliver anionic CpG as PAMPs. To our knowledge, Hf-DBBF-Ir@CpG(+) provides the first treatment with synergistic DAMPs and PAMPs packaged in the in situ cancer vaccine in local tumors while engaging lymphoid organs for antigen presentation to synergize with CBI to induce CTL infiltration in distant tumors. Furthermore, the 83.3% cure rate achieved by Hf-DBBF-Ir@CpG(+) + αPD-L1 on a relatively immunosuppressive 14-day MC38 colorectal cancer model suggests the potential use of nMOF-based in situ vaccines on immunologically cold tumors.
The in situ cancer vaccination afforded by nMOFs has several potential advantages over traditional cancer vaccines (47). First, the in situ vaccine afforded by nMOFs is personalized from autologous antigens released from tumors by a myriad of ROS and can overcome the tumor heterogeneity issue facing traditionally manufactured peptide vaccines. Second, cationic nMOFs can capture DAMPs and tumor antigens from dying cancer cells via electrostatic interactions and, with virus-like size distribution, can be recognized and taken up by APCs for efficient antigen presentation to stimulate a strong cytotoxic T cell response. Third, cationic nMOFs deliver and protect anionic CpGs from enzymatic degradation for TLR stimulation and downstream immunologic processes. Fourth, tumor antigens and DAMPs released by the nMOF-mediated RT-RDT process and CpG-based PAMPs delivered by cationic nMOFs work synergistically to stimulate DC maturation to promote antigen presentation and adaptive immunity (48). The combination of nMOF-mediated RT-RDT and the delivered CpG by cationic nMOFs is superior to previous nMOF-based treatments by substantially enhancing intratumoral DC infiltration and DC maturation to effectively bridge innate inflammation and adaptive anticancer immunity. Fifth, the nMOF-based vaccine is activated by x-rays to release DAMPs and tumor antigens with relatively nontoxic components and is, thus, expected to have few side effects and can be readily translated to the clinic. Furthermore, systemic administration of αPD-L1 blocks the immunosuppressive co-inhibitory marker PD-L1 to augment antigen presentation and attenuate T cell exhaustion. The combination of nMOF-mediated in situ cancer vaccine with CBI affords tumor-specific and long-term antitumor immunity.
In summary, we have designed a novel nMOF by rationally fluorinating photosensitizing ligands for effective ROS generation through RT-RDT and tuning nMOF surface charge for efficient CpG loading. Following intratumoral administration of Hf-DBBF-Ir and x-ray irradiation, the in situ–released DAMPs and tumor antigens and CpGs delivered by Hf-DBBF-Ir synergistically function as a potent personalized cancer vaccine to activate APCs and expand cytotoxic T cells in tumor-DLNs to reinvigorate the adaptive immune system for local tumor regression. When combined with an immune checkpoint inhibitor, innate and adaptive immunity from the nMOF-based cancer vaccine was further enhanced to generate superb antitumor efficacy with tumor specificity and long-term immune memory effect. This combination treatment extends the local therapeutic effects of the in situ cancer vaccine to distant tumors via systemic antitumor immunity by reactivating CTLs. As nMOFs are currently tested in the clinic as radioenhancers (NCT03444714), this study paves the way to advance the concept of nMOF-based personalized vaccines into human trials for the treatment of advanced cancers.
MATERIALS AND METHODS
Cell lines and animals
Murine colorectal adenocarcinoma cell line MC38, Lewis lung carcinoma cell line LL2, and melanoma cell line B16F10 were purchased from the American Type Culture Collection (USA). Murine pancreatic cancer cell line Panc02 was provided by H. Schreiber from the Department of Pathology, University of Chicago. MC38-ova cell line [OVA(257-264)-ZSGREEN] was generated by transfection of MC38 cells with LZRS-based retrovirus. All the cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (GE Healthcare, USA) supplemented with 10% fetal bovine serum, penicillin G sodium (100 U/ml), and streptomycin sulfate (100 μg/ml). Cells were cultured in a humidified atmosphere containing 5% CO2 at 37°C. Mycoplasma was tested before use by MycoAlert detection kit (Lonza Nottingham Ltd., USA). C57BL/6 mice (6 to 8 weeks) were obtained from Harlan-Envigo Laboratories Inc. (USA). The study protocol was reviewed and approved by the Institutional Animal Care and Use Committee at the University of Chicago.
Synthesis of DBB-Ir-F and DBB-Ir
Ir(DBB)[dF(CF3)ppy]2+ [DBBF-Ir, DBB = 4,4′-di(4-benzoato)-2,2′-bipyridine; dF(CF3)ppy = 2-(2,4-difluorophenyl)-5-(trifluoromethyl)pyridine] was synthesized as shown in fig. S1 according to the literature report (49). 1H nuclear magnetic resonance (NMR) [500 MHz; dimethyl sulfoxide (DMSO)–d6]: δ 9.08 (d, 2H), 8.76 (d, 2H), 8.49 (d, 2H), 8.44 (d, 2H), 8.15 (s, 2H), 8.02 (d, 4H), 7.82 (s, 2H), 7.62 (d, 4H), 7.12 (t, 2H), and 5.91 (d, 2H). Ir(DBB)(ppy)2+ [DBB-Ir, DBB = 4,4′-di(4-benzoato)-2,2′-bipyridine; ppy = 2-phenylpyridine] was synthesized as shown in fig. S1 according to the literature report (50). 1H NMR (500 MHz, DMSO-d6): δ 9.05 (d, 2H), 8.67 (d, 2H), 8.28 (d, 2H), 8.07 (s, 2H), 7.96 (m, 8H), 7.89 (d, 2H), 7.52 (d, 4H), 7.17 (t, 2H), 7.09 (t, 2H), 6.98 (t, 2H), and 6.33 (d, 2H).
Synthesis of Hf-DBBF-Ir and Hf-DBB-Ir
To a 4-ml glass vial was added 0.5 ml of HfCl4 solution [2.0 mg/ml in N,N′-dimethylformamide (DMF)], 0.5 ml of DBBF-Ir solution (4.0 mg/ml in DMF), 2.6 μl of trifluoroacetic acid, and 2 μl of water. The reaction mixture was kept in a 70°C oven for 24 hours. The yellow precipitate was collected by centrifugation and washed with DMF and ethanol. The yield was 61% based on Hf as determined by ICP-MS. To a 4-ml glass vial was added 0.5 ml of HfCl4 solution (1.6 mg/ml in DMF), 0.5 ml of DBB-Ir solution (6.4 mg/ml in DMF), and 100 μl of AcOH. The reaction mixture was kept in a 70°C oven for 72 hours. The orange precipitate was collected by centrifugation and washed with DMF and ethanol. The yield was 52% based on Hf as determined by ICP-MS.
Digestion of Hf-DBBF-Ir and Hf-DBB-Ir
One milligram of Hf-DBBF-Ir was dried under vacuum. The resulting solid was digested in a solution of 500 μl of DMSO-d6 and 50 μl of D3PO4 and sonicated for 10 min. The mixture was then added to 50 μl of D2O and analyzed by 1H NMR. The digested Hf-DBBF-Ir showed all signals corresponding to DBBF-Ir without any other aromatic signals, which confirms the presence of only DBBF-Ir ligands in Hf-DBBF-Ir. One milligram of Hf-DBB-Ir was dried under vacuum. The resulting solid was digested in a solution of 500 μl of DMSO-d6 and 50 μl of D3PO4 and sonicated for 10 min. The mixture was then added to 50 μl of D2O and analyzed by 1H NMR. The digested Hf-DBB-Ir showed all signals corresponding to H2DBB-Ir without any other aromatic signals, which confirms the presence of only DBB-Ir ligands in Hf-DBB-Ir.
•OH generation with APF assay
APF (Thermo Fisher Scientific, USA) reacts with •OH to give bright green fluorescence (excitation/emission maxima, 490/515 nm). DBBF-Ir, DBB-Ir, Hf-DBBF-Ir, and Hf-DBB-Ir were suspended in water at an equivalent concentration of 20 μM in the presence of 5 μM APF. A water solution of 5 μM APF was used as blank control. One hundred microliters of each suspension was added to a 96-well plate and then irradiated with x-rays at 0, 1, 2, 3, 5, or 10 Gy (RT250 x-ray generator, Philips, USA; 250 kVp, 15 mA, 1-mm Cu filter). The fluorescence signal was immediately collected with an IVIS 200 imaging system (Xenogen, USA).
1O2 generation with SOSG assay
SOSG (Thermo Fisher Scientific, USA) reacts with 1O2 to give bright green fluorescence (excitation/emission maxima, 504/525 nm). DBBF-Ir, DBB-Ir, Hf-DBBF-Ir, and Hf-DBB-Ir were suspended in water at an equivalent concentration of 20 μM in the presence of 12.5 μM SOSG. A water solution of 12.5 μM SOSG was used as blank control. One hundred microliters of each suspension was added to a 96-well plate and then irradiated with x-rays at 0, 1, 2, 3, 5, or 10 Gy (Philips RT250 x-ray generator; 250 kVp, 15 mA, 1-mm Cu filter). The fluorescence signal was immediately collected with a Xenogen IVIS 200 imaging system.
O2− generation determined by BMPO assay
BMPO is a nitrone spin trap, which can form distinguishable adducts with O2− (BNPO-O2−) with a long half-life (t1/2 = 23 min). DBBF-Ir, DBB-Ir, Hf-DBBF-Ir, and Hf-DBB-Ir were suspended in benzene at an equivalent concentration of 200 μM in the presence of 25 mM BMPO. A benzene solution of 25 mM BMPO was used as a blank control. One milliliter of each suspension was added to a 4-ml vial and then irradiated with x-ray at 5 Gy (Philips RT250 x-ray generator; 250 kVp, 15 mA, 1-mm Cu filter). The electron paramagnetic resonance (EPR) signal was immediately collected by an X-Band ELEXSYS-II 500 EPR (Bruker, USA).
DNA double-strand breaks
The DNA double-strand breaks were detected by probing phosphorylated γ-H2AX. MC38 cells were cultured in a six-well plate at 5 × 105 per well overnight and incubated with PBS, DBB-Ir, DBBF-Ir, Hf-DBB-Ir, or Hf-DBBF-Ir at an equivalent concentration of 20 μM, followed by irradiation at 0 and 2 Gy (Philips RT250 x-ray generator; 250 kVp, 15 mA, 1-mm Cu filter). Cells were stained 2 hours after irradiation with the HCS DNA damage kit (Life Technologies, USA) with 1:500 dilution for flow cytometric analysis.
Clonogenic assay
MC38 cells were cultured in a six-well plate overnight and incubated with particles at an Hf concentration of 20 μM for 4 hours, followed by irradiation with 0, 1, 2, 4, 8, and 16 Gy (Philips RT250 x-ray generator; 250 kVp, 15 mA, 1-mm Cu filter). The irradiated cells were trypsinized and counted immediately. Two hundred to 2000 cells were seeded in a six-well plate and cultured with 2 ml of medium for 14 days to form visible colonies, which were counted to determine the survival fraction. Once colony formation was observed, the culture medium was discarded. The plates were rinsed twice with PBS and then stained with 500 μl of 0.5% (w/v) crystal violet in 50% methanol/H2O. The wells were rinsed with water three times, and the colonies were counted manually. The radiation enhancement factor at REF10 was calculated as the ratio of equivalent irradiation doses needed to give 10% survival rate for the PBS control group over that for the experimental group.
Cytotoxicity assay
MTS assay (Promega, USA) was used to evaluate cytotoxicity with x-ray irradiation. MC38 cells were seeded on 96-well plates at 1 × 104 per well and further cultured for 12 hours. PBS, DBB-Ir, DBBF-Ir, Hf-DBB-Ir, or Hf-DBBF-Ir was added to the cells at an equivalent ligand dose of 0, 1, 2, 5, 10, 20, 50, and 100 μM and incubated for 4 hours. The cells were then irradiated with x-rays at a dose of 2 Gy (Philips RT250 x-ray generator; 250 kVp, 15 mA, 1-mm Cu filter). The cells were further incubated for 72 hours before determining the cell viability by MTS assay.
Live/dead cell analysis
The live/dead cell analysis was evaluated with cell-permeable dye calcein AM and propidium iodide (PI) kit. MC38 cells were cultured in a six-well plate at 5 × 105 per well overnight and incubated with PBS, DBB-Ir, DBBF-Ir, Hf-DBB-Ir, or Hf-DBBF-Ir at an equivalent concentration of 20 μM for 4 hours by irradiation with 0 or 2 Gy (Philips RT250 x-ray generator; 250 kVp, 15 mA, 1-mm Cu filter). The cells were then washed with PBS gently and stained with calcein AM (green) for visualization of live cells and with PI (red) for visualization of dead cells under FLUOVIEW FV1000 confocal laser scanning microscope (Olympus, Japan).
Apoptosis/necrosis
The cell death analysis was evaluated with apoptotic cell death kit. MC38 cells were cultured in a six-well plate at 5 × 105 per well overnight and incubated with PBS, DBB-Ir, DBBF-Ir, Hf-DBB-Ir, or Hf-DBBF-Ir at an equivalent concentration of 20 μM for 4 hours, followed by irradiation with 0 or 2 Gy (Philips RT250 x-ray generator; 250 kVp, 15 mA, 1-mm Cu filter). Twenty-four hours later, the cells were stained according to the Alexa Fluor 488 annexin V/dead cell apoptosis kit (Life Technologies, USA), imaged under CLSM, and quantified by LSRFortessa 4-15 cytometer (BD, USA).
Immunogenic cell death
The ICD was examined by CRT exposure. MC38 cells were cultured in a six-well plate at 5 × 105 per well overnight and incubated with PBS, DBB-Ir, DBBF-Ir, Hf-DBB-Ir, or Hf-DBBF-Ir at an equivalent concentration of 20 μM, followed by irradiation at 0 and 2 Gy (Philips RT250 x-ray generator; 250 kVp, 15 mA, 1-mm Cu filter). The cells were then washed with PBS gently and stained with Alexa Fluor 488–CRT antibody (Enzo Life Sciences, USA) with 1:100 dilution for flow cytometric analysis.
Phagocytosis
C57BL/C bone marrow–derived monocytic cells were harvested, cultured, and activated. Murine granulocyte-macrophage colony-stimulating factor and IL-4 were supplied to a final concentration of 1% for 168 hours, and the nonadherent cells as immature DCs were harvested for the following studies. Cells were incubated at under 5% CO2 at 37°C. Medium was replaced every 2 to 3 days, and cells were used after 6 to 8 days of culture. CFSE-labeled (Life Technologies, USA) MC38 cells (5 × 105) were cultured in a six-well plate overnight and incubated with DBB-Ir, DBBF-Ir, Hf-DBB-Ir, and Hf-DBBF-Ir at an equivalent dose of 20 μM for 4 hours, followed by x-ray irradiation at a dose of 0 or 2 Gy (Philips RT250 x-ray generator; 250 kVp, 15 mA, 1-mm Cu filter). PE-Cy5.5–labeled DCs (1 × 106) were added and cocultured with the treated MC38 cells at 37°C for 4 hours. Cells were then collected, washed twice with cold PBS, imaged by CLSM, or analyzed by flow cytometry.
Immunofluorescence staining
Tumors and lymph nodes were collected and subsequently frozen. Tissue sections with a thickness of 5 μm were prepared using a CM1950 cryostat (Leica, Germany). These sections were air-dried for at least 1 hour and then fixed in acetone at 20°C for 20 min. After blocking with 20% donkey serum, the sections were incubated with individual primary antibodies against CD11b (53-6.7), F4/80 (H57-597), CD11b (53-6.7), MHC-II (53-6.7), CD86 (53-6.7), CD206 (53-6.7), and CD8α (53-6.7) overnight at 4°C, followed by incubation with dye-conjugated secondary antibodies for 1 hour at room temperature. After staining with 4′,6-diamino-2-phenylindole (DAPI) for another 10 min, the sections were then washed twice with PBS and observed under SP8 LIGHTNING confocal microscope (Leica, Germany).
In situ vaccination on syngeneic models
Synergistic tumor models, MC38, MC38-ova, and Panc02 were established to evaluate the in vivo anticancer efficacy of nMOF-mediated in situ vaccination. For single-tumor models, 5 × 105 MC38 cells, 1 × 106 MC38-ova cells, or 1 × 106 Panc02 cells were subcutaneously inoculated onto the right flanks of C57BL/6 mice. When the tumors reached 100 to 150 mm3 in volume, mice were injected intratumorally with nMOFs at a dose of 0.2 μmol of Hf, CpG at a dose of 1 μg, or PBS. Twelve hours after injection, mice were anaesthetized with 2% (v/v) isoflurane, and the tumors were irradiated with 1-Gy x-ray/fraction (225 kVp, 13 mA, 0.3-mm Cu filter) for a total of five daily fractions. For bilateral tumor models, 5 × 105 MC38 cells were subcutaneously inoculated onto the right flanks as primary tumors, while 2 × 105 MC38 cells, 2 × 105 B16F10 cells, or 5 × 105 LL2 cells were subcutaneously inoculated onto the left flanks as distant tumors of C57BL/6 mice. αPD-L1 (clone, 10F.9G2; catalog no. BE0101, BioXCell) were given every 3 days by intraperitoneal injection at a dose of 75 μg per mouse. The tumor sizes were measured daily with a caliper where tumor volume = (width2 × length)/2.
ELISpot assay
Tumor-specific immune responses to IFN-γ were measured in vitro by ELISpot assay (Mouse IFN-γ ELISPOT Ready-SET-Go!; catalog no. 88-7384-88, eBioscience). A Millipore Multiscreen HTS-IP plate was coated overnight at 4°C with anti-mouse IFN-γ capture antibody. Single-cell suspensions of splenocytes were obtained from MC38 tumor-carrying mice and seeded onto the antibody-coated plate at a concentration of 2 × 105 cells per well. Cells were incubated with or without peptide sequence (KSPWFTTL) for 42 hours at 37°C and then discarded. The plate was then incubated with biotin-conjugated anti–IFN-γ detection antibody at room temperature for 2 hours, followed by incubation with avidin–horseradish peroxidase at room temperature for 2 hours. 3-Amino-9-ethylcarbazole substrate solution (catalog AEC101; Sigma-Aldrich, USA) was added for cytokine spot detection. Spots were imaged and quantified with a CTL ImmunoSpot Analyzer (Cellular Technology Ltd., USA).
Lymphocyte profiling
Tumors and lymph nodes were harvested, treated with collagenase I (1 mg/ml) (Gibco, USA) for 1 hour at 37°C. Cells were filtered through nylon mesh filters with size of 40 μm and washed with PBS. Tumor-DLNs were collected and directly ground through the cell strainers. The single-cell suspension was incubated with anti-CD16/32 (clone 93) to reduce nonspecific binding to FcRs. Cells were further stained with the following fluorochrome-conjugated antibodies: CD45 (30-F11), CD3ε (145-2C11), CD4 (GK1.5), CD8α (53-6.7), Nkp46 (29A1.4), F4/80 (BM8), CD11b (M1/70), Gr-1 (RB6-8C5), MHC-II (AF6-120), CD80 (16-10A1), CD86 (GL1), CD206 (C068C2), CD44 (IM7), CD62L (MEL-14), H-2Kb SIINFEKL (25-D1.16), PI, and yellow fluorescent reactive dye (CD45 from BD Biosciences, CD206 and CD62L from BioLegend, and others from eBioscience). Antibodies were used with the dilution of 1:200. Representative gating strategies for different immune cells are shown in fig. S9 (F and G). LSRFortessa 4-15 (BD Biosciences, USA) was used for cell acquisition, and data analysis was carried out with FlowJo software (Tree Star, USA).
Adoptive OT-I T cells transfer
MC38-ova cells (1 × 106) were injected subcutaneously onto the right flanks of C57BL/6 Rag2−/− mouse. Fourteen days later, mice were intratumorally injected with Hf-DBBF-Ir at a dose of 0.2 μmol of Hf with or without CpG at a dose of 1 μg, followed by 1-Gy x-ray/fraction for a total of five daily fractions. Two days after the first irradiation, spleen and lymph nodes were isolated from OT-I mice, and CD8+ T cells were negatively sorted using mouse CD8+ T Cell Isolation Kit (Miltenyi Biotec, Germany), and 1 × 106 CD8+ T cells were intravenously injected into MC38-ova–bearing Rag2−/− mice. The tumor sizes were measured daily with a caliper where tumor volume = (width2 × length)/2.
Statistical analysis
Group sizes (n ≥ 5) were chosen to ensure proper statistical analysis of variance (ANOVA) analysis for efficacy studies. Student’s t tests were used to determine whether the variance between groups is similar. Statistical analysis was performed using OriginPro (OriginLab Corp.). Statistical significance was calculated using two-tailed Student’s t tests and defined as *P < 0.05, **P < 0.01, and ***P < 0.001. Animal experiments were not performed in a blinded fashion and are represented as means ± SD. The immune analysis was performed in a blinded fashion and is represented as median ± SD.
Supplementary Material
Acknowledgments
We thank C. Chan for experimental help on immune cell profiling, L. Hua for providing MC38-OVA(257-264)-ZSGREEN cell line, and W. Zheng for providing OT-I mice for the experiments. Funding: We acknowledge the National Cancer Institute (U01–CA198989 and 1R01CA253655) and the Department of Defense (PC170934P2), the University of Chicago Medicine Comprehensive Cancer Center (NIH CCSG: P30 CA014599), and the Ludwig Institute for Metastasis Research for funding support. Author contributions: K.N., N.G., and W.L. conceived and designed the experiments. K.N., G.L., and N.G. carried out the experiments. A.C., T.L., and T.W. assisted with experiments. All authors analyzed and discussed the data. K.N., G.L., N.G., and W.L. wrote the manuscript. Competing interests: W.L. is founder of Coordination Pharmaceuticals Inc., which licensed the nMOF technology from the University of Chicago. R.R.W. is an adviser to Coordination Pharmaceuticals Inc. W.L., K.N., T.L., and G.L. are inventors on a provisional patent application filed by the University of Chicago (no. 63/045,499; filed 30 June 2020). The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabb5223/DC1
REFERENCES AND NOTES
- 1.Goldman B., DeFrancesco L., The cancer vaccine roller coaster. Nat. Biotechnol. 27, 129–139 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Irvine D. J., Swartz M. A., Szeto G. L., Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 12, 978–990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu Z., Ott P. A., Wu C. J., Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 18, 168–182 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tanyi J. L., Ophir E., Tuyaerts S., Roberti A., Genolet R., Baumgartner P., Stevenson B. J., Iseli C., Dangaj D., Czerniecki B., Semilietof A., Racle J., Michel A., Xenarios I., Chiang C., Monos D. S., Torigian D. A., Nisenbaum H. L., Michielin O., June C. H., Levine B. L., Powell D. J. Jr., Gfeller D., Mick R., Dafni U., Zoete V., Harari A., Coukos G., Kandalaft L. E., Personalized cancer vaccine effectively mobilizes antitumor T cell immunity in ovarian cancer. Sci. Transl. Med. 10, eaao5931 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Sahin U., Türeci Ö., Personalized vaccines for cancer immunotherapy. Science 359, 1355–1360 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Purcell A. W., McCluskey J., Rossjohn J., More than one reason to rethink the use of peptides in vaccine design. Nat. Rev. Drug Discov. 6, 404–414 (2007). [DOI] [PubMed] [Google Scholar]
- 7.Liu H., Moynihan K. D., Zheng Y., Szeto G. L., Li A. V., Huang B., Van Egeren D. S., Park C., Irvine D. J., Structure-based programming of lymph-node targeting in molecular vaccines. Nature 507, 519–522 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Joyce J. A., Fearon D. T., T cell exclusion, immune privilege, and the tumor microenvironment. Science 348, 74–80 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Kuai R., Ochyl L. J., Bahjat K. S., Schwendeman A., Moon J. J., Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat. Mater. 16, 489–496 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheetz L., Park K. S., Li Q., Lowenstein P. R., Castro M. G., Schwendeman A., Moon J. J., Engineering patient-specific cancer immunotherapies. Nat. Biomed. Eng. 3, 768–782 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang H., Mooney D. J., Biomaterial-assisted targeted modulation of immune cells in cancer treatment. Nat. Mater. 17, 761–772 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Gilliet M., Cao W., Liu Y.-J., Plasmacytoid dendritic cells: Sensing nucleic acids in viral infection and autoimmune diseases. Nat. Rev. Immunol. 8, 594–606 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Castano A. P., Mroz P., Hamblin M. R., Photodynamic therapy and anti-tumour immunity. Nat. Rev. Cancer 6, 535–545 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weichselbaum R. R., Liang H., Deng L., Fu Y.-X., Radiotherapy and immunotherapy: A beneficial liaison? Nat. Rev. Clin. Oncol. 14, 365–379 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Kepp O., Marabelle A., Zitvogel L., Kroemer G., Oncolysis without viruses—Inducing systemic anticancer immune responses with local therapies. Nat. Rev. Clin. Oncol. 17, 49–64 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Shae D., Becker K. W., Christov P., Yun D. S., Lytton-Jean A. K. R., Sevimli S., Ascano M., Kelley M., Johnson D. B., Balko J. M., Wilson J. T., Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat. Nanotechnol. 14, 269–278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo M., Wang H., Wang Z., Cai H., Lu Z., Li Y., Du M., Huang G., Wang C., Chen X., Porembka M. R., Lea J., Frankel A. E., Fu Y.-X., Chen Z. J., Gao J., A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol. 12, 648–654 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sagiv-Barfi I., Czerwinski D. K., Levy S., Alam I. S., Mayer A. T., Gambhir S. S., Levy R., Eradication of spontaneous malignancy by local immunotherapy. Sci. Transl. Med. 10, eaan4488 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klinman D. M., Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 4, 249–258 (2004). [DOI] [PubMed] [Google Scholar]
- 20.Figdor C. G., de Vries I. J. M., Lesterhuis W. J., Melief C. J. M., Dendritic cell immunotherapy: Mapping the way. Nat. Med. 10, 475–480 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Lutz M. B., Schuler G., Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance or immunity? Trends Immunol. 23, 445–449 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Radovic-Moreno A. F., Chernyak N., Mader C. C., Nallagatla S., Kang R. S., Hao L., Walker D. A., Halo T. L., Merkel T. J., Rische C. H., Anantatmula S., Burkhart M., Mirkin C. A., Gryaznov S. M., Immunomodulatory spherical nucleic acids. Proc. Natl. Acad. Sci. U.S.A. 112, 3892–3897 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rosi N. L., Giljohann D. A., Thaxton C. S., Lytton-Jean A. K. R., Han M. S., Mirkin C. A., Oligonucleotide-modified gold nanoparticles for intracellular gene regulation. Science 312, 1027–1030 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Nam J., Son S., Park K. S., Zou W., Shea L. D., Moon J. J., Cancer nanomedicine for combination cancer immunotherapy. Nat. Rev. Mater. 4, 398–414 (2019). [Google Scholar]
- 25.Song W., Musetti S. N., Huang L., Nanomaterials for cancer immunotherapy. Biomaterials 148, 16–30 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma L., Dichwalkar T., Chang J. Y. H., Cossette B., Garafola D., Zhang A. Q., Fichter M., Wang C., Liang S., Silva M., Kumari S., Mehta N. K., Abraham W., Thai N., Li N., Wittrup K. D., Irvine D. J., Enhanced CAR–T cell activity against solid tumors by vaccine boosting through the chimeric receptor. Science 365, 162–168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lou J., Zhang L., Zheng G., Advancing cancer immunotherapies with nanotechnology. Adv. Ther. 2, 1800128 (2019). [Google Scholar]
- 28.Furukawa H., Cordova K. E., O’Keeffe M., Yaghi O. M., The chemistry and applications of metal-organic frameworks. Science 341, 1230444 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Wang S., McGuirk C. M., Ross M. B., Wang S., Chen P., Xing H., Liu Y., Mirkin C. A., General and direct method for preparing oligonucleotide-functionalized metal–organic framework nanoparticles. J. Am. Chem. Soc. 139, 9827–9830 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lan G., Ni K., Lin W., Nanoscale metal–organic frameworks for phototherapy of cancer. Coord. Chem. Rev. 379, 65–81 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ni K., Lan G., Chan C., Quigley B., Lu K., Aung T., Guo N., Riviere P. L., Weichselbaum R. R., Lin W., Nanoscale metal-organic frameworks enhance radiotherapy to potentiate checkpoint blockade immunotherapy. Nat. Commun. 9, 2351 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ni K., Lan G., Veroneau S. S., Duan X., Song Y., Lin W., Nanoscale metal-organic frameworks for mitochondria-targeted radiotherapy-radiodynamic therapy. Nat. Commun. 9, 4321 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ni K., Lan G., Chan C., Duan X., Guo N., Veroneau S. S., Weichselbaum R. R., Lin W., Ultrathin metal-organic-layer mediated radiotherapy-radiodynamic therapy. Matter 1, 1331–1353 (2019). [PMC free article] [PubMed] [Google Scholar]
- 34.Lu K., He C., Guo N., Chan C., Ni K., Lan G., Tang H., Pelizzari C., Fu Y.-X., Spiotto M. T., Weichselbaum R. R., Lin W., Low-dose x-ray radiotherapy–radiodynamic therapy via nanoscale metal–organic frameworks enhances checkpoint blockade immunotherapy. Nat. Biomed. Eng. 2, 600–610 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Ni K., Luo T., Culbert A., Kaufmann M., Jiang X., Lin W., Nanoscale metal–organic framework co-delivers TLR-7 agonists and anti-CD47 antibodies to modulate macrophages and orchestrate cancer immunotherapy. J. Am. Chem. Soc. 142, 12579–12584 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Lowry M. S., Goldsmith J. I., Slinker J. D., Rohl R., Pascal R. A., Malliaras G. G., Bernhard S., Single-layer electroluminescent devices and photoinduced hydrogen production from an ionic iridium (III) complex. Chem. Mater. 17, 5712–5719 (2005). [Google Scholar]
- 37.Mahoney K. M., Rennert P. D., Freeman G. J., Combination cancer immunotherapy and new immunomodulatory targets. Nat. Rev. Drug Discov. 14, 561–584 (2015). [DOI] [PubMed] [Google Scholar]
- 38.Errico A., Immunotherapy: PD-1-PD-L1 axis: Efficient checkpoint blockade against cancer. Nat. Rev. Clin. Oncol. 12, 63 (2015). [DOI] [PubMed] [Google Scholar]
- 39.Spiotto M., Fu Y.-X., Weichselbaum R. R., The intersection of radiotherapy and immunotherapy: Mechanisms and clinical implications. Sci. Immunol. 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Charlebois R., Allard B., Allard D., Buisseret L., Turcotte M., Pommey S., Chrobak P., Stagg J., PolyI: C and CpG synergize with anti-ErbB2 mAb for treatment of breast tumors resistant to immune checkpoint inhibitors. Cancer Res. 77, 312–319 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Wang C., Sun W., Wright G., Wang A. Z., Gu Z., Inflammation-triggered cancer immunotherapy by programmed delivery of CpG and anti-PD1 antibody. Adv. Mater. 28, 8912–8920 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sivanandam V., LaRocca C. J., Chen N. G., Fong Y., Warner S. G., Oncolytic viruses and immune checkpoint inhibition: The best of both worlds. Mol. Ther. Oncolytics 13, 93–106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gong T., Liu L., Jiang W., Zhou R., DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat. Rev. Immunol. 20, 95–112 (2020). [DOI] [PubMed] [Google Scholar]
- 44.Deng L., Liang H., Xu M., Yang X., Burnette B., Arina A., Li X.-D., Mauceri H., Beckett M., Darga T., Huang X., Gajewski T. F., Chen Z. J., Fu Y.-X., Weichselbaum R. R., STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weiner G. J., Liu H.-M., Wooldridge J. E., Dahle C. E., Krieg A. M., Immunostimulatory oligodeoxynucleotides containing the CpG motif are effective as immune adjuvants in tumor antigen immunization. Proc. Natl. Acad. Sci. U.S.A. 94, 10833–10837 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Emming S., Schroder K., Tiered DNA sensors for escalating responses. Science 365, 1375–1376 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Abdou P., Wang Z., Chen Q., Chan A., Zhou D. R., Gunadhi V., Gu Z., Advances in engineering local drug delivery systems for cancer immunotherapy. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 12, e1632 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ni K., Luo T., Lan G., Culbert A., Song Y., Wu T., Jiang X., Lin W., A nanoscale metal–organic framework to mediate photodynamic therapy and deliver CpG oligodeoxynucleotides to enhance antigen presentation and cancer immunotherapy. Angew. Chem. 132, 1124–1128 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu Y.-Y., Lan G., Fan Y., Veroneau S. S., Song Y., Micheroni D., Lin W., Merging photoredox and organometallic catalysts in a metal–organic framework significantly boosts photocatalytic activities. Angew. Chem. 130, 14286–14290 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Lan G., Ni K., Veroneau S. S., Luo T., You E., Lin W., Nanoscale metal–organic framework hierarchically combines high-z components for multifarious radio-enhancement. J. Am. Chem. Soc. 141, 6859–6863 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/40/eabb5223/DC1