

Abstract
Kaposi’s sarcoma (KS), a highly angiogenic and invasive vascular tumor, is the most common AIDS-associated cancer caused by KS-associated herpesvirus (KSHV) infection. We have recently shown that KSHV-encoded viral interferon regulatory factor 1 (vIRF1) contributes to KSHV-induced cell motility (PLoS Pathog. 15:e1007578, 2019). However, the role of vIRF1 in KSHV-induced angiogenesis remains unknown. Here, using two in vivo angiogenesis models including the chick chorioallantoic membrane assay (CAM) and the matrigel plug angiogenesis assay in mice, we show that vIRF1 promotes angiogenesis by upregulating CUB domain (for complement C1r/C1s, Uegf, Bmp1) containing protein 1 (CDCP1). Mechanistically, vIRF1 enhances the expression of transcription factor lymphoid enhancer-binding factor 1 (Lef1) and binds to Lef1 to promote CDCP1 transcription. Meanwhile, vIRF1 degrades metastasis suppressor CD82 through an ubiquitin–proteasome pathway by recruiting E3 ubiquitin ligase AMFR to CD82, which protects CDCP1 from CD82-mediated, palmitoylation-dependent degradation. CDCP1 activates AKT signaling, which is required for vIRF1-induced cell motility but not angiogenesis. Our results illustrate that, by hijacking Lef1 and CD82, vIRF1 upregulates CDCP1 to promote angiogenesis and cell invasion. These novel findings demonstrate the vIRF1 targets multiple cellular proteins and pathways to promote the pathogenesis of KS, which could be attractive therapeutic targets for KSHV-induced malignancies.
Subject terms: Oncogenes, Infectious diseases
Introduction
Kaposi’s sarcoma-associated herpesvirus (KSHV) is a human oncogenic virus associated with the development of Kaposi’s sarcoma (KS), a plasmablastic form of multicentric Castleman’s disease, primary effusion lymphoma (PEL), and KSHV-associated inflammatory cytokine syndrome [1]. KS is a multicentric angioproliferative spindle cell tumor, characterized by abnormal proliferation of newly developed vessels, and erythrocyte leakage [2]. KS often manifests as multifocal lesions involving not only with skin, oral mucosa, and lymph nodes but also with visceral organs, especially the pulmonary and gastrointestinal tract [3].
KSHV is a member of the herpesviridae family with a genome of approximately 165–170 kb encoding over 90 open reading frames (ORFs). KSHV displays two different modes of infection, latent infection and lytic reactivation [4]. Multiple products of KSHV play a critical role in KSHV-induced malignancies and pathogenesis, including latent proteins latency-associated nuclear antigen (LANA), viral FLICE inhibitory protein and viral cyclin, and lytic proteins viral Bcl-2, viral interleukin-6 (vIL-6), ORF-K1, reactivation and transcription activator, and viral interferon regulatory factor 1 (vIRF1). These proteins could alter cell morphology, promote cell growth, deregulate angiogenesis, and evoke inflammation and immune evasion. Although a number of studies have been carried out, the underlying mechanism of KSHV induction of tumors remains unclear.
vIRF1 encoded by KSHV ORF-K9 is a 449-amino acid protein with homology to both human IRF3 and IRF7 [5]. vIRF1 is an early lytic gene but its mRNA is also present at high levels in KS tumors and transcribed in KSHV-infected endothelial/mesenchymal lineage cells [6]. As a regulatory protein, vIRF1 is implicated in KSHV escape of host immune surveillance. For example, vIRF1 hijacks CBP/p300 coactivators to block the interaction of CBP/p300 with IRF3, resulting in the inhibition of IRF3-mediated transcription and signal transduction of type I interferon [7]. In addition, many studies have revealed the oncogenic role of vIRF1 in KSHV-induced tumorigenesis. vIRF1 could transform mouse embryonic fibroblasts (NIH3T3) cells [8]. vIRF1 inhibits the binding of Smad3/Smad4 complexes to DNA to restrain TGF-beta signaling and disturb IRF-1-induced CD95/CD95L signaling-mediated apoptosis [9, 10]. vIRF1 also inhibits apoptosis by negative regulation of tumor suppressor gene p53-driven transcription. For instance, vIRF1 reduces p53 acetylation and inhibits p53 transcriptional activation by directly binding to p53 [11, 12]. Moreover, vIRF1 weakens ATM kinase activity to repress the phosphorylation of p53, and increases p53 ubiquitination [13]. Our own works have shown that vIRF1 can hijack lncRNA-OIP5-AS1/miR-218-5p network to promote cell invasion [14]. In addition to promoting tumorigenesis, vIRF1 plays a key role in KSHV lytic replication. vIRF1 directly binds to a mitophagy receptor, NIX, on the mitochondria and activates NIX-mediated mitophagy to promote mitochondrial clearance, resulting in enhanced KSHV productive replication [15].
Cub domain (for complement C1r/C1s, Uegf, Bmp1) containing protein 1 (CDCP1), an 836-amino acid cell surface transmembrane glycoprotein, was first identified in 2001 and shown to be highly expressed in metastatic human tumor cells [16]. Currently, CDCP1 is regarded as a potential biomarker for poor prognosis in cancer, including renal cell carcinoma [17], pancreatic cancer [18], lung adenocarcinoma [19], and colorectal cancer [20]. As a surface marker, CDCP1 has been proposed to play an important role in the dissemination of cancer cells. Antibody blocking of CDCP1’s function inhibited tumor metastasis in animal models [21], suggesting that CDCP1 could be as a therapeutic target. In addition, antibody targeting of CDCP1 inhibited the activation of AKT pathway [22]. However, the relevance of CDCP1 in KSHV-induced tumors has not been investigated so far.
In this work, we observed a novel role of vIRF1 in angiogenesis. Elevated CDCP1 was responsible for vIRF1-induced angiogenesis. vIRF1 promoted CDCP1 transcription and inhibited CDCP1 degradation by targeting transcription factor Lef1 and metastasis suppressor CD82, respectively. Furthermore, CDCP1-activated AKT signaling pathway contributed to vIRF1-induced cell invasion. Our findings suggest that vIRF1 and its regulatory proteins and pathways are potential therapeutic targets for KSHV-induced malignancies.
Results
vIRF1 promotes angiogenesis in vitro and in vivo
Our recent work showed that vIRF1 accelerated KSHV-induced cell motility [14]. However, whether vIRF1 has any role in KSHV-induced angiogenesis is still unknown. Therefore, we adopted a tubule formation assay to evaluate the impact of vIRF1 on angiogenesis in vitro. Human umbilical vein endothelial cells (HUVECs) were transduced with lentiviral vIRF1 at a MOI of 2. The lentiviral vIRF1 was generated in our recent study [14]. We observed that cells with ectopic vIRF1 expression showed a higher tube formation capacity compared to control cells expressing the vector control (Fig. 1a, b). We further and confirmed the results in an in vivo chick chorioallantoic membrane (CAM) angiogenesis model. vIRF1 strikingly increased the angiogenesis index (Fig. 1c, d). In another in vivo angiogenesis model, the Matrigel plug angiogenesis assay in mice, vIRF1 also had a stronger ability to induce angiogenesis than the vector control (Fig. 1e, f). Moreover, plugs from mice inoculated with vIRF1-transduced cells had more neovascularization and more positive cells of smooth muscle actin than those transduced with a vector control (Fig. 1g, h). Consistently, the level of VEGFA protein was much higher in the vIRF1 plugs than those of vector plugs (Fig. 1i). These data indicate that vIRF1 promotes angiogenesis both in vitro and in vivo.
Fig. 1. vIRF1 promotes angiogenesis both in vitro and in vivo.

a Representative images of microtubule formation assay of HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE). The representative images were captured at 2 and 5 h post seeding. Magnification, ×100. Scar bars, 40 μm. b Quantification of microtubule formation assay described in a. c Cells transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE) were used to evaluate in vivo angiogenesis capability of vIRF1 in a chicken chorioallantoic membranes (CAM) model as described in the “Materials and methods” section. Representative images of CAM are presented. Scar bars, 1 cm. d Quantification of CAM assay described in c. e Cells treated as in c were used in Matrigel plug assay in nude mice as described in the “Materials and methods” section. Representative images of matrigel plug assay are shown. Scar bars, 1 cm. f Quantification of matrigel plug assay described in e. g Representative pictures of H&E staining of histologic features (up; ×400) and immunohistochemical (IHC) staining of the expression of SMA (below; ×400) in plugs retrieved from vIRF1 (vIRF1) or control (pHAGE) cells-inoculated mice. Black arrowheads: vessels; red arrowheads: SMA. h Results were quantified in g. i Western blotting analysis of VEGFA protein in plugs retrieved from vIRF1 (vIRF1) or control (pHAGE) cells-inoculated mice. GAPDH was used as a loading control. Data were shown as mean ± SD. ***P < 0.001, Student’s t-test.
vIRF1 upregulates CDCP1 to promote cell migration, invasion and angiogenesis
CDCP1 has been characterized as a surface marker in multiple malignant tumors [19, 23, 24]. Western blotting showed that CDCP1 expression was increased in vIRF1-transduced cells (Fig. 2a). Further, we infected HUVECs with the wild-type KSHV (KSHV_WT) virus or a KSHV mutant virus with vIRF1 deletion (K9_mut) [14]. Deletion of vIRF1 from the KSHV genome had no effect on vIRF3 expression (Supplementary Fig. S1), which is located near ORF-K9 and was reported to induce angiogenesis [25]. KSHV infection increased CDCP1 expression, while deletion of vIRF1 caused a decrease of CDCP1 protein (Fig. 2b). Most importantly, more CDCP1-postive cells were observed in KS patients’ lesions compared to normal skin tissues (Fig. 2c, d, Supplementary Fig. S2). In addition, we also examined the co-expression of vIRF1 protein together with CDCP1 in these three cases of skin KS lesions using an immunofluorescence assay with a polyclonal antibody against vIRF1 [15]. We observed vIRF1 protein expression in all three KS lesions and a higher expression level of CDCP1 protein in vIRF1-positive cells (Fig. 2e).
Fig. 2. vIRF1 increases CDCP1 expression.

a Western blotting analysis of CDCP1 expression in HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE). b Western blotting analysis of CDCP1 expression in HUVECs treated with PBS (PBS) or infected with KSHV wild-type virus (KSHV_WT) or K9 mutant virus (K9_Mut). c H&E staining and immunohistochemical staining of KSHV LANA, CDCP1 in normal skin, skin KS of patient #1 (Skin KS1), skin KS of patient #2 (Skin KS2), and skin KS of patient #3 (Skin KS3). Magnification, ×200, ×400. d Results were quantified in c. e Detection of vIRF1 and CDCP1 in skin KS of patient #1 (Skin KS1), skin KS of patient #2 (Skin KS2), and skin KS of patient #3 (Skin KS3) by immunofluorescence analysis (IFA). Data were shown as mean ± SD. ***P < 0.001, Student’s t-test.
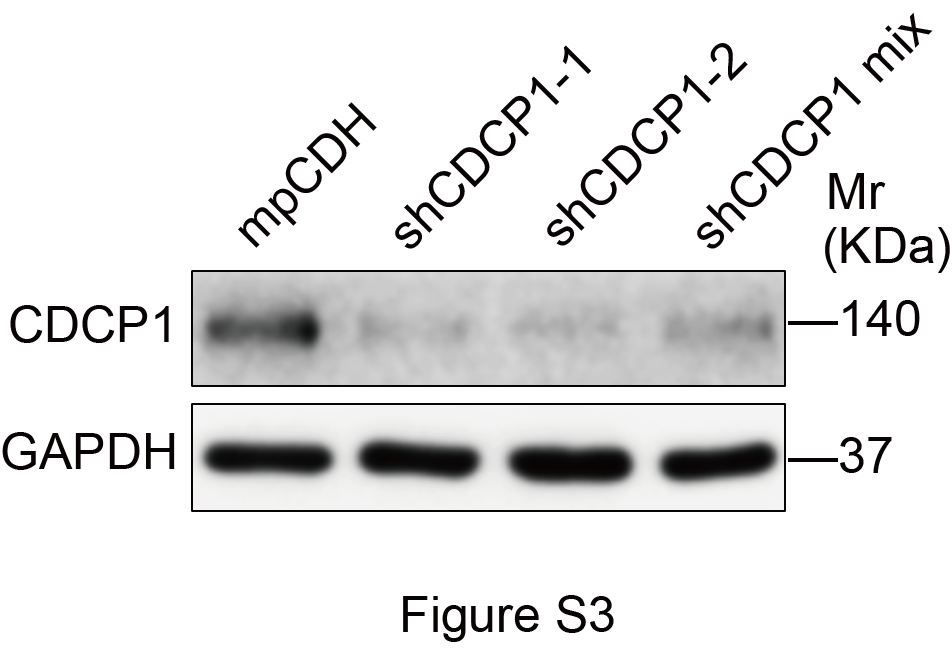
To determine whether CDCP1 regulates the oncogenic phenotypes of vIRF1, vIRF1-expressing HUVECs were transduced with short hairpin RNAs (shRNAs) targeting CDCP1 (shCDCP1) (Fig. 3a, Supplementary Fig. S3). Transwell migration and Matrigel invasion assays showed that knockdown of CDCP1 diminished vIRF1-induced cell migration and invasion (Fig. 3b–d). Moreover, knockdown of CDCP1 also inhibited vIRF1-induced tube formation in vitro (Fig. 3e, f) and angiogenesis in vivo (Fig. 3g–j). These results indicate that vIRF1 upregulates CDCP1 to promote cell migration, invasion, and angiogenesis.
Fig. 3. CDCP1 positively mediates vIRF1-induced cell invasion and angiogenesis.

a Western blotting analysis of CDCP1 expression in lentiviral vIRF1- or its control pHAGE-infected HUVECs, followed by transduction with lentivirus expressing a mixture of shRNAs targeting CDCP1 (shCDCP1). b Transwell migration and Matrigel invasion analyses of HUVECs treated as in a. The representative images of migrated and invaded HUVECs were collected at 6 and 12 h post seeding. Magnification, ×100. Scar bars, 40 μm. c Quantification of transwell migration assay described in b. d Quantification of Matrigel invasion described in b. e Representative images of microtubule formation assay of HUVECs treated as in a. The images were captured at 2 and 5 h post seeding. Magnification, ×100. Scar bars, 40 μm. f Quantification of microtubule formation assay described in e. g Lentiviral vIRF1- or pHAGE-infected cells, which were subsequently cotransduced with lentivirus expressing a mixture of shRNAs targeting CDCP1 (shCDCP1) were used for CAM assay. Representative images of CAM are shown. Scar bars, 1 cm. h Quantification of CAM assay described in g. i Representative images of Matrigel plug assay of cells treated as in g, which were inoculated into mice as described in the “Materials and methods” section. Scar bars, 1 cm. j Quantification of Matrigel plug assay described in i. Data were shown as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001, Student’s t-test.
vIRF1 transcriptionally activates CDCP1 by upregulating the expression of Lef1 and binding to Lef1
Next, we sought to monitor the mechanism of vIRF1 upregulation of CDCP1. We first examined whether vIRF1 had effects on the transcription of CDCP1. quantitative reverse transcription PCR (qRT-PCR) showed a significant increase of CDCP1 mRNA in both vIRF1-transduced and KSHV-infected cells (Fig. 4a, b). Furthermore, promoter luciferase reporter assay confirmed that vIRF1 increased the transcription activity of CDCP1 promoter (Fig. 4c). We utilized the ALGGEN PROMO software (http://alggen.lsi.upc.es/cgi-bin/promo_v3/promo/promoinit.cgi?dirDB=TF_8.3) to identify putative transcription factors that might bind to the CDCP1 promoter, and found that lymphoid enhancer-binding factor 1 (Lef1) could be a potential candidate. Of interest, vIRF1 notably increased the protein level of Lef1 (Fig. 4d). Besides, Lef1 expression in KS lesions is higher than that in normal skin (Fig. 4e, f). To confirm the effect of Lef1 on CDCP1 expression, we performed knockdown of Lef1 with a mixture of shRNAs targeting Lef1 (shLef1) in vIRF1-transduced HUVECs. Knockdown of Lef1 decreased both the mRNA and protein levels of CDCP1 (Fig. 4g, h; Supplementary Fig. S4). The transcriptional activity of CDCP1 was significantly enhanced by Lef1 (Fig. 4i). Consistently, silencing of Lef1 blocked the increased CDCP1 promoter activity induced by vIRF1 (Fig. 4j). Chromatin immunoprecipitation (ChIP) assay demonstrated that both vIRF1 and Lef1 bound to the CDCP1 promoter, and binding of Lef1 to CDCP1 promoter, was increased in the presence of vIRF1 (Fig. 4k). Most importantly, we observed that vIRF1 interacted with Lef1 by immunoprecipitation (IP) analysis (Fig. 4l, m). These data collectively suggest that vIRF1 transcriptionally activates CDCP1 by upregulating the expression of transcription factor Lef1 and binding to Lef1.
Fig. 4. vIRF1 transcriptionally activates CDCP1 by upregulating Lef1 and binding to Lef1.

a qPCR analysis of CDCP1 mRNA expression in HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE). b qPCR analysis of CDCP1 mRNA expression in HUVECs treated with PBS (PBS) or infected with wild-type KSHV (KSHV_WT). c Luciferase reporter assay of the activity of CDCP1 promoter in HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE). d Western blotting analysis of Lef1 and CDCP1 expression in HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE). e Immunohistochemical staining of Lef1 in normal skin, skin KS of patient #1 (Skin KS1), skin KS of patient #2 (Skin KS2), and skin KS of patient #3 (Skin KS3). Magnification, ×200, ×400. f Results were quantified in e. g Western blotting analysis of CDCP1 expression in lentiviral vIRF1- or control pHAGE-infected HUVECs, followed by transduction with lentivirus expressing a mixture of shRNAs targeting Lef1 (shLef1). h qPCR analysis of CDCP1 mRNA expression in lentiviral vIRF1- or control pHAGE-infected HUVECs, followed by transduction with lentivirus expressing a mixture of shRNAs targeting Lef1 (shLef1). i Luciferase reporter assay of the effect of Lef1 on transcriptional activity of CDCP1 promoter. j Luciferase reporter assay of the activity of CDCP1 promoter in lentiviral vIRF1- or control pHAGE-infected HUVECs, followed by transduction with lentivirus expressing a mixture of shRNAs targeting Lef1 (shLef1). k ChIP assays of CDCP1 promoter. Immunoprecipitation was done in HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE) with anti-Lef1 or anti-Flag antibodies. l, m Immunoprecipitation analyses of the interaction between vIRF1 and Lef1. The light chain band was used as a control to show the equal amounts of antibodies used for immunoprecipitations. Data were shown as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001, Student’s t-test.
vIRF1 protects CDCP1 from palmitoylation-dependent degradation by inhibiting CD82
It has been proposed that metastasis suppressor CD82 negatively regulates CDCP1 expression [26]. Our previous study showed that CD82 was downregulated in KSHV-infected cells [27]. Based on these results, we speculated that vIRF1 might also target CD82 to upregulate CDCP1 expression. We first examined the level of CD82 expression in vIRF1-transduced cells. Western blotting showed that vIRF1 reduced the CD82 protein level (Fig. 5a). Transduction of CD82 into vIRF1-expressing cells with lentiviral CD82 reduced the CDCP1 protein level (Fig. 5b). However, CD82 had no effect on CDCP1 mRNA level (Fig. 5c). Hence, a posttranscriptional mechanism might be responsible for the reduction of CDCP1 by CD82. As predicted, CD82 overexpression destabilized CDCP1 protein in endothelial cells in the presence of cycloheximide (CHX) (Fig. 5d, e). Furthermore, the effect of CD82 on CDCP1 protein level was completely abrogated by treating the cells with proteasome inhibitor MG132 (Fig. 5f, g). These results collectively indicate that CD82 promotes the degradation of CDCP1 in a proteasome-dependent manner in endothelial cells.
Fig. 5. vIRF1 protects CDCP1 from palmitoylation-dependent degradation by inhibiting CD82.

a Western blotting analysis of CD82 and CDCP1 expression in HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE). b Western blotting analysis of CDCP1 expression in vIRF1-expressing HUVECs transduced with lentiviral CD82 (CD82-His). c qPCR analysis of CDCP1 expression in vIRF1-expressing HUVECs transduced with lentiviral CD82 (CD82-His). d Western blotting analysis of CDCP1 expression in vIRF1-expressing HUVECs treated with CHX (80 μg/ml) for 0, 4, 8, and 16 h. e Results were quantified in d. f Western blotting analysis of CDCP1 expression in vIRF1-expressing HUVECs treated with MG132 (25 μM) for 8 h. g Results were quantified in b. h, i Immunoprecipitation analyses of the interaction between CD82 and CDCP1. The light chain band was used as a control to show the equal amounts of antibodies used for immunoprecipitations. j Immunofluorescence analysis (IFA) of the localization of CDCP1 and CD82 in HUVECs. k Acyl-biotinyl exchange assay of the level of palmitoylated CDCP1 in vIRF1-expressing HUVECs transduced with lentiviral CD82. l Quantification of CAM assay of vIRF1-expressing cells transduced with lentiviral CD82. Data were shown as mean ± SD. ***P < 0.001, Student’s t-test. n.s. not significant.
CDCP1 was characterized as a novel component of the tetraspanin web in a previous proteomic analysis [28], which implies that CDCP1 has the potential to interact with tetraspanin proteins, such as CD82. To address this issue, we performed co-immunoprecipitation experiments, and confirmed the interaction between CDCP1 and CD82 in endothelial cells (Fig. 5h, i). Consistent with the above results, the reduction of CDCP1 by CD82 overexpression was also observed in immunofluorescent confocal microscopy (Fig. 5j). A previous study showed that CDCP1 was constitutively internalized and degraded, and the degradation of CDCP1 required its palmitoylation [29]. To examine whether palmitoylation contributes to CD82-induced reduction of CDCP1 protein, we performed acyl-biotinyl exchange assay. As shown in Fig. 5k, overexpression of CD82 increased the level of palmitoylated CDCP1. Furthermore, overexpression of CD82 repressed angiogenesis induced by vIRF1 (Fig. 5l).
Taken together, these results suggest that the interaction between CD82 and CDCP1 increases the palmitoylation-dependent degradation of CDCP1, and that vIRF1 downregulates the expression of CD82 to maintain high level of CDCP1 expression.
vIRF1 induces CD82 degradation by promoting the interaction of E3 ubiquitin ligase AMFR with CD82
We showed a reduction of CD82 protein in vIRF1-transduced cells (Fig. 5a); however, vIRF1 did not inhibit the mRNA level of CD82 (Fig. 6a). Therefore, we examined if vIRF1 might regulate CD82 protein stability. We treated the cells with CHX, and found that vIRF1 promoted CD82 degradation (Fig. 6b, c). Furthermore, treatment with MG132 effectively blocked CD82 reduction (Fig. 6d, e). Consistent with these observations, CD82 underwent marked polyubiquitylation in the presence of MG132 in vIRF1-expressing cells (Fig. 6f), indicating that vIRF1 promoted CD82 polyubiquitylation, leading to proteasome-dependent degradation of CD82.
Fig. 6. vIRF1 promotes CD82 degradation via recruiting E3 ubiquitin ligase AMFR to interact with CD82.

a qPCR analysis of CD82 mRNA expression in HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE). b Western blotting analysis of CD82 expression in vIRF1-expressing HUVECs treated with CHX (80 μg/ml) for 0, 4, 8, and 16 h. c Results were quantified in b. d Western blotting analysis of CDCP1 expression in vIRF1-expressing HUVECs treated with MG132 (25 μM) for 8 h. e Results were quantified in d. f Immunoprecipitation assay of CD82 ubiquitination in HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE). g Western blotting analysis of AMFR and CD82 expression in HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE). h Immunoprecipitation analyses of the interaction between CD82 and AMFR. i, j Immunoprecipitation analyses of the interaction between CD82 and vIRF1. The light chain band was used as a control to show the equal amounts of antibodies used for immunoprecipitations. k Immunoprecipitation analyses of the interaction between AMFR and vIRF1. The light chain band was used as a control to show the equal amounts of antibodies used for immunoprecipitations. Data were shown as mean ± SD. ***P < 0.001, Student’s t-test.
CD82 has been identified as a substrate of the ubiquitin ligase autocrine motility factor receptor (AMFR) [30]. We next asked whether AMFR might mediate vIRF1 promotion of CD82 ubiquitylation. No significant expression difference of AMFR protein was observed in vIRF1-expressing cells (Fig. 6g); however, co-immunoprecipitation assay showed that more AMFR was immunoprecipiated by CD82 in vIRF1-expressing cells (Fig. 6h). Importantly, we observed that vIRF1 was also immunoprecipiated by CD82 (Fig. 6h), indicating that vIRF1 might be a component of the CD82-AMFR complex. Indeed, vIRF1 interacted with CD82 (Fig. 6i, j) as well as AMFR (Fig. 6k). Together these data demonstrate that vIRF1 induces CD82 degradation by promoting the interaction of E3 ubiquitin ligase AMFR with CD82.
The AKT pathway is involved in vIRF1-induced cell invasion
CDCP1 has been shown to regulate the activation of PI3K/AKT signaling [31]. We hypothesized that PI3K/AKT signaling may also be involved in vIRF1-induced cell invasion and angiogenesis. Western blotting showed that vIRF1 activated AKT signaling (Fig. 7a). Both overexpression of CD82 and knockdown of CDCP1 decreased the level of phosphorylated AKT (Fig. 7b, c). We next determined the role of AKT in vIRF1-induced cell invasion and angiogenesis. Blocking of AKT pathway activation by shRNAs [32] and chemical inhibition with MK2206, an inhibitor of AKT, decreased vIRF1-induced cell invasion, but had no effect on vIRF1-induced angiogenesis (Fig. 7d–i). These data suggest that AKT pathway mediates vIRF1-induced cell invasion.
Fig. 7. AKT pathway positively mediates vIRF1-induced cell invasion.

a Western blotting analysis of CD82, CDCP1, p-AKT, and AKT expression in HUVECs transduced with 2 MOI lentiviral vIRF1 (vIRF1) or control lentivirus (pHAGE). b Western blotting analysis of CDCP1, p-AKT, and AKT expression in vIRF1-expressing HUVECs transduced with lentiviral CD82. c Western blotting analysis of CDCP1, p-AKT, and AKT expression in lentiviral vIRF1- or control pHAGE-infected HUVECs, which were subsequently cotransduced with lentiviral expressing a mixture of shRNAs targeting CDCP1 (shCDCP1). d Western blotting analysis of p-AKT and AKT expression in lentiviral vIRF1- or control pHAGE-infected cells followed by transduction with lentivirus expressing a mixture of shRNAs targeting AKT (shAKT). e Transwell migration and Matrigel invasion analyses of cells treated as in d. f Quantification of CAM assay of cells transduced with lentiviral vIRF1 or control pHAGE, which were subsequently cotransduced with lentivirus expressing a mixture of shRNAs targeting CDCP1 (shCDCP1). g Western blotting analysis of p-AKT and AKT expression in vIRF1-expressing cells treated with the AKT inhibitor, MK2206. h Transwell migration and Matrigel invasion analyses cells treated as in g. i Quantification of CAM assay of cells treated as in g. Data were shown as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001, Student’s t-test.
Knockout of vIRF1 repressed KSHV-induced angiogenesis
Next, we found that CDCP1, Lef1, and phosphorylated AKT were reduced while CD82 was increased in HUVECs infected by mutant virus compared with those infected by KSHV_WT virus (Fig. 8a). We transduced K9_mutant virus-infected cells with 2 MOI of lentiviral vIRF1 to reconstitute its expression. We found that complementation with vIRF1 in K9_mut-infected HUVECs was sufficient to reverse these effects (Fig. 8b). Consistent with our previous findings in cell migration and invasion [14], deletion of ORF-K9 also decreased KSHV-induced angiogenesis, while complementation with vIRF1 rescued the angiogenesis phenotypes in vitro (Fig. 8b) and in vivo (Fig. 8c, d). Taken together, these results show that, in the context of KSHV infection, vIRF1 activates CDCP1 to promote angiogenesis by regulating Lef1 and CD82.
Fig. 8. Loss of vIRF1 represses KSHV-induced angiogenesis.

a Western blotting of CDCP1, Lef1, CD82, and p-AKT expression in HUVECs treated with PBS (PBS), infected with wild-type KSHV (KSHV_WT) or K9 mutant virus (K9_mut) followed by transduction with lentiviral vIRF1. b Quantification of microtubule formation assay of HUVECs treated as in a. c Quantification of CAM assay of cells treated with PBS (PBS), infected with KSHV wild-type virus (KSHV_WT) or K9 mutant virus (b) followed by transduction with lentiviral vIRF1. d Quantification of Matrigel plug assay of cells treated as in c. e A schematic working model of the mechanism by which vIRF1 facilitates endothelial cell invasion and angiogenesis. Data were shown as mean ± SD. *P < 0.05, **P < 0.01, and ***P < 0.001, Student’s t-test. n.s. not significant.
Discussion
Angiogenesis characterized as the formation of new blood vessels could supply oxygen and nutrients to support the high proliferative rate of cancer cells, and hence is essential for tumor survival and progression. Angiogenesis is caused by excessive release of angiogenic cytokines and growth factors secreted by tumor and stromal cells [33]. Accumulating evidence suggests that KSHV infection induces angiogenic phenotypes via elevated secretions of pro-angiogenic factors, including Ang2, MMPs, IL-8, IL-6, VEGF, and Ephrin B2 [34]. Here, we demonstrated that KSHV vIRF1 promoted angiogenesis both in vitro and in vivo. Deletion of vIRF1 from the KSHV genome reduced KSHV-induced angiogenesis. These findings revealed a novel functional role of vIRF1 in KSHV-induced angiogenesis, which is an important part of KS pathogenesis involving the vast aberrant proliferation of small vessels and extravasation of erythrocytes in KS tumors.
Although vIRF1 was identified as an early lytic gene, it was also expressed during viral latency. This is probably due to the presence of two transcription start sites in the ORF-K9 gene, one of which is distal to the AUG and is active during latency in PEL while another one is a more proximal site, which is induced upon lytic reactivation [35]. Thus, it is believed that vIRF1 has a dual mode of expression, which is expressed during both latent and lytic replication phases [6, 36–38]. The expression of vIRF1 mRNA during latent infection and in KS tumors reinforces its critical role in KS tumorigenesis [6].
About 1–3% of the KSHV-infected cells in KS lesions express viral proteins characteristic of viral lytic replication [39–42]. The low rate of spontaneous lytic replication is postulated to generate infectious virions to infect new cells. Meanwhile, both virus-encoded cytokines generated during lytic replication and cellular cytokines induced during lytic replication and de novo infection contribute to KS pathogenesis through autocrine and paracrine mechanisms [43]. Here, for the first time, we observed vIRF1-positive cells in KS lesions (Fig. 2e). Besides, vIRF1-positive cells were less than LANA-positive cells (Fig. 2c–e), implying that not all KSHV-infected cells had detectable vIRF1 protein. These results suggest that the expression of vIRF1 protein is most likely due to viral lytic replication. In addition, we have also found that CDCP1 is highly expressed in most cells in KS tissues albeit vIRF1-positive cells had the highest expression level (Fig. 2c–e), suggesting the involvement of a paracrine mechanism in CDCP1 upregulation.
CDCP1 is a transmembrane glycoprotein, also known as CD138, gp140, subtractive immunization associated 135 kDa (SIMA135), or transmembrane and associated with Src kinases (TRASK). Literatures indicate that multiple types of cancer express high level of CDCP1 [44]. Elevated CDCP1 is implicated with higher occurrence of metastases, higher relapse rate, and poor prognosis in cancer patients [45]. Evidence suggests that concentration of the CDCP1 protein in human cord plasma may serve as a biomarker for hematopoietic stem and progenitor cell content [46]. Another report noted that tumors in CDCP1-deficient mice grew much faster than in CDCP1 wild-type (WT) control mice [47]. Here we observed a higher level of CDCP1 in KS tissues, and found that vIRF1 was responsible for KSHV-induced CDCP1 elevation. Mechanistically, vIRF1 upregulated CDCP1 by enhancing CDCP1 transcription and delaying CDCP1 degradation. Our results clearly demonstrated that vIRF1 interacted with transcription factor Lef1, and functionally promoted Lef1 binding to the CDCP1 promoter. However, by using GST-pull down experiment, we found that vIRF1 does not interact with Lef1 directly (Data not shown), indicating that there are other components in the vIRF1–Lef1 complex involved in tethering vIRF1 and Lef1 together. We also observed a significant increase of Lef1 expression in KS tissues and vIRF1-expressing cells; however, the molecular mechanism by which vIRF1 upregulated Lef1 expression was not defined in present study.
As for the downstream signaling, CDCP1 has been shown to upregulate the phosphatidylinositol 3-kinase (PI3K)/AKT pathway [31]. The PI3K/AKT signaling is abnormally activated in human cancers [48]. As a central component of PI3K/AKT signaling, AKT influences multiple cellular processes that are features of cancer, including cell growth, differentiation, angiogenesis, survival, and migration. Thus, targeting AKT is a highly attractive anti-cancer strategy [49]. Aberrant activation of AKT was observed in KSHV-infected cells [50]. Currently, at least five of KSHV-encoded genes and microRNAs have been shown to activate AKT pathway. They are viral G protein-coupled receptor (vGPCR), ORF45, vIL-6, ORF-K1, and miR-K3 [32, 50]. In this study, we demonstrated that, by protecting CDCP1 from CD82-mediated and palmitoylation-dependent degradation, vIRF1 activated AKT signaling, suggesting that vIRF1 might be a new regulator of the AKT pathway in KSHV-infected cells, which might contribute to aberrant activation of AKT signaling during KSHV infection and pathogenesis. Knockdown of CDCP1 significantly reduced vIRF1 promotion of cell invasion and angiogenesis. However, blocking AKT signaling led to the reduced vIRF1-induced cell invasion but failed to affect vIRF1-induced angiogenesis. Therefore, our results did not eliminate the possibility that CDCP1 may activate other pathway(s) to mediate vIRF1-induced angiogenesis. To date, no KSHV gene has been shown to induce angiogenesis by activating AKT. Thus, whether the AKT pathway has a vital role in KSHV-induced angiogenesis, and which viral oncogene could function as the inducer of AKT activation and contributes angiogenesis require further investigation.
We have previously shown that KSHV infection reduced metastasis suppressor CD82 expression [41]. CD82 as a direct target of KSHV miR-K6-5p is involved in cell invasion and angiogenesis [41]. Here we have found that except miR-K6-5p, vIRF1, another product of KSHV, also contributes to suppression of CD82 expression. vIRF1 promotes CD82 degradation by recruiting AMFR. Overexpression of CD82 impaired vIRF1-induced angiogenesis, supporting importance of downregulating CD82 in the development of KS, especially in angiogenesis.
In summary, we have found that KSHV vIRF1 induces the expression of CDCP1, which is essential for vIRF1-induced cell invasion and angiogenesis. vIRF1 promotes CDCP1 transcription by upregulating the expression of Lef1, and Lef1 binding to the CDCP1 promoter. Meanwhile, vIRF1 induces ubiquitination and degradation of CD82 by recruiting E3 ubiquitin ligase AMFR. The reduction of CD82 prevents CD82-mediated and palmitoylation-dependent degradation of CDCP1. The AKT pathway activated by CDCP1 mediates vIRF1-induced cell invasion (Fig. 8e). Our findings illustrate a novel mechanism of vIRF1 induction of angiogenesis and cell invasion, and demonstrate the significant roles of vIRF1, and its regulatory proteins and pathways in the pathogenesis of KSHV-associated cancers, providing potential therapeutic targets of KSHV-induced cancers.
Materials and methods
Ethics statement
The clinical section of the research was reviewed and ethically approved by the Institutional Ethics Committee of the First Affiliated Hospital of Nanjing Medical University. A total of three pairs of KS patients’ lesions and normal skin tissues were obtained. Written informed consent was obtained from all participants, and all samples were anonymized. All participants were adults.
Cell culture and transfection
Primary HUVECs were isolated from the interior of the umbilical vein of human umbilical cords by digestion with collagenase (Sigma, St. Louis, MO, USA), and cultured in complete EBM-2 culture media (Lonza, Allendale, NJ, USA) as previously described [51]. HUVECs were used between passages 3 and 6, and employed throughout the manuscript for all assays except for lentivirus packaging, chicken CAMs, and matrigel plug assays. iSLK-RGB-BAC16 and iSLK-RGB-K9 mutant cells were grown in DMEM supplemented with 10% fetal bovine serum (FBS), 1 μg/ml puromycin, 250 μg/ml G418, and 1.2 mg/ml hygromycin B. Both HEK293T cells, which were used for lentivirus packaging, and a human umbilical vein endothelial cell line (catalog #CRL-2922TM; ATCC, Manassas, VA, USA), which were used for chicken CAMs and matrigel plug assays, were maintained in DMEM supplemented with 10% FBS. All of cell lines were authenticated by short tandem repeat profiling. Effectence transfection reagent (Qiagen, Suzhou, Jiangsu, China) and LipofectamineTM 2000 Reagent (Invitrogen, Carlsbad, CA, USA) were used for the transfection of HUVECs and HEK293T, respectively. Cells and cell lines used in this study were examined by mycoplasma contamination test using a Myco-Blue Mycoplasma Detector (D103-01/02, Vazyme Biotech Co., Ltd, Nanjing, China).
Reagents and plasmids
MG132 (a proteasome inhibitor) and MK2206 2HCl (an allosteric inhibitor of AKT) were pursed from Selleck Chemicals (Shanghai, China). Cycloheximide (CHX) was from Sigma-Aldrich. The expressing plasmids of vIRF1-Flag, CD82-His, and shRNAs were constructed as previously described [14]. The control vector of all the shRNAs was a modified lentivirus pCDH plasmid (mpCDH for short), which contains both GFP and RFP cassettes, and was generated in our previous study [32]. The sequences of shRNAs are listed in Supplemental Table 1.
Lentivirus packaging and infection
The packaging plasmid, psPAX2, together with the envelope plasmid, pMD2.G, and the lentivirus plasmid, pHAGE, which has a GFP cassette, were cotransfected into pre-inoculated HEK293T cells as previously described [52–54].
Transwell migration and matrigel invasion assays
Transwell migration and matrigel invasion were performed as previously described [32]. Briefly, HUVECs (1 × 105) were seeded into inserts (8-μm-pore polyethylene terephthalate membrane filter; Merck Millipore, Darmstadt, Germany) with or without matrigel coated, and were harvested 6 or 12 h after incubation. The migrated cells were fixed, stained, photographed, and calculated by counting stained cells in a double-blinded manner by two observers.
Tubule formation assay
Tubule formation assay was performed as previously described [51]. The micro-slide Angiogenesis ibiTreat from ibidi (Martinsried, Germany) was coated with Matrigel (BD Biosciences, Bedford, MA, USA). Then, 5 × 103 HUVECs resuspended with 50 μl serum-free basic medium were seeded and incubated at 37 °C for the indicated times. The calculation of the angiogenesis index was done in a double-blinded manner by two observers according to the formula previously described [55]. The formula is as follows:
Chicken CAMs and matrigel plug assay
CAMs and matrigel plug assay in mice were performed as previously described [52–54]. Briefly, for CAM assay, 8-day-old embryos were used. Embryos were randomly divided. Assays for each treatment were carried out using at least six chicken embryos. 5 × 106 cells mixed with the high concentration Matrigel (BD Biosciences, Bedford, MA, USA) were injected into CAMs. The CAMs were harvested 4 days after the implantation. The microvasculature of the CAMs was observed under a stereomicroscope. The angiogenic activity was assessed by quantifying the number of branching points using the ImageJ software (NIH, http://rsb.info.nih.gov/ij/) following a standardized procedure [56]. The angiogenesis index was calculated as the mean number of branch points from the experimental conditions minus the mean number of branch points from gel alone controls.
For matrigel plug assay, 4-week-old male nu/nu nude mice were adopted. All procedures were performed in accordance with the policies of Nanjing Medical University Experimental Animal Welfare Ethics Committee. Nude mice were randomly divided and at least six mice in each treatment. No statistical method was used to predetermine sample sizes. 5 × 106 cells mixed with the high concentration Matrigel (BD Biosciences, Bedford, MA, USA) were injected into the right flanks of nude mice. Plugs were harvested 10 days after the injection and photographed using a stereomicroscope. Drabkin’s reagent kit (Sigma-Aldrich) was used for the measurement of the hemoglobin content. The level of hemoglobin in plugs was calculated from a standard curve.
RT-qPCR analysis, western blotting, and antibodies
Total RNA was isolated using TRIzol (Life Technologies, Grand Island, NY, USA) and was reversed by HiScript Q RT SuperMix (Vazyme Biotech Co., Ltd, Nanjing, China). RT-qPCR was assessed with AceQ qPCR SYBR Green Master Mix (Vazyme Biotech Co., Ltd, Nanjing, China) using Applied Biosystems (ABI, Foster City, CA, USA) as recommended by the manufacturer’s protocol. GAPDH was utilized as an internal normalization control. The following GAPDH primers were used: 5′-GAA GGT GAA GGT CGG AGT C-3′ and 5′-GAA GAT GGT GAT GGG ATT TCC-3′. The primers for CDCP1 were: 5′-ACC GTG GTC AGG ATC GGA A-3′ and 5′-GCT GAA GCC GGA GAC ATT TCT-3′. The primers for CD82 were: 5′-GCT CAT TCG AGA CTA CAA CAG C-3′ and 5′-ACC TCA GGG CGA TTC A-3′. Western blotting analysis was performed as described previously [32, 57]. Antibodies used were anti-CDCP1 rabbit antibody, anti-Flag rabbit antibody, anti-p-AKT rabbit antibody, and anti-AKT rabbit antibody from Cell Signaling Technologies (Beijing, China), anti-α-Tubulin mouse antibody, anti-GAPDH mouse antibody, anti-VEGFA rabbit antibody, anti-HA mouse antibody, and anti-CD82 mouse antibody from Santa Cruz Biotechnology (Dallas, TX, USA).
Immunohistochemistry (IHC) staining
For IHC staining, all formalin-fixed and paraffin-embedded (FFPE) samples were immunostained with the indicated antibodies following standard methodology. Antibodies used for IHC were anti-KSHV LANA rat antibody from Advanced Biotechnologies Inc. (Columbia, MD, USA), anti-smooth muscle actin rabbit antibody from AbbiotecTM (San Diego, CA, USA), anti-CDCP1 goat antibody and anti-Lef1 rabbit antibody from Abcam (Cambridge, MA, USA), and anti-rabbit immunoglobulin G (IgG) from Beyotime Institute of Biotechnology (Nantong, Jiangsu, China). Secondary antibodies used were horseradish peroxidase (HRP)-labeled goat anti-mouse, anti-rat, or anti-rabbit as appropriate (Beyotime Institute of Biotechnology, Nantong, Jiangsu, China). Staining was visualized using DAB (3,3′-diaminobenzidine) Peroxidase (HRP) Substrate Kit (Vector Laboratories, Inc., Burlingame, USA). The relative expression level was assessed by counting the percentage of positive cells with a double-blinded randomized control design.
Luciferase reporter assay
The luciferase construct containing promoter region of CDCP1 (−1434 to +250, +1 means a transcription initiation site) was cloned into pGL3-basic vector (Promega, Madison, WI, USA). The renilla vector pRL-TK was used to normalize transfection efficiency. Luciferase reporter assay was adopted as previously described [32] using the Promega dual-luciferase reporter assay system according to the manufacturer’s instructions.
Immunofluorescence assay (IFA)
IFA was performed as previously described [52]. Cells were washed (PBS), fixed (4% paraformaldehyde), permeabilizated (0.3% Triton X-100), and then blocked with 1% bovine serum albumin for 1 h. Samples were incubated with primary antibodies against CDCP1 goat (1:200 dilution) and CD82 mouse (1:200 dilution) at 4 °C overnight, and then were incubated with appropriate Alexa Fluor 488- and Alexa Fluor 555-conjugated secondary antibodies (Thermo Fisher Scientific, Grand Island, NY, USA). For KS samples, the FFPE slides were melt at 95 °C for 10 min, and then immediately incubated in three washes of xylene for 5 min each, two washes of 100% ethanol for 10 min each, two washes of 95% for 10 min each, and two washes of dH2O for 5 min each. Bring slides to a boil in 1 mM EDTA pH 8.0 followed by 15 min at a sub-boiling temperature. After cooling to room temperature, samples were blocked with 5% bovine serum albumin for 1 h and then incubated with primary rabbit antibodies against vIRF1 (1:300 dilution, provided by Dr. Gary Hayward from Viral Oncology Program, The Johns Hopkins School of Medicine) and CDCP1 goat (1:200 dilution) at 4 °C overnight. Alexa Fluro 555-coupled anti-goat and Alexa Fluro 488-coupled anti-rabbit secondary antibodies were used, respectively. 4,6-Diamidino-2-phenylindole (DAPI) (Beyotime Institute of Biotechnology, Nantong, Jiangsu, China) was used to visualize nuclear. Pictures were captured using a Zeiss Axiovert 200M laser scanning confocal microscope (CarlZeiss, Freistaat Thuringen, Germany).
DNA ChIP assay
ChIP analysis was performed using EZ-Magna ChIP™ A/G Chromatin Immunoprecipitation Kit (Merck, Darmstadt, Germany) according to the manufacturer’s instructions. Cells were fixed and digested to obtain DNA fragments, which were precipitated with anti-Lef1, anti-Flag, anti-RNA polymerase II, or IgG. The sequences of CDCP1 promoter primers were: 5′-CTG GCC CAA GTC AAG TAG CA-3′ and 5′-CTT CCT TCC CAA GTC TGA GA-3′.
Co-immunoprecipitation
Immunoprecipitations were performed as described previously [51]. Immunoprecipitated proteins were subjected to western blotting analysis using specific antibodies. The secondary antibodies used for co-Immunoprecipitation were IPKine™ HRP Goat Anti-Mouse or Anti-Rabbit IgG LCS (Abbkine Scientific Co., Ltd, Wuhan, China), which could specifically react with kappa light chains on IgG, and avoid the detection of the heavy chains of IgG.
Acyl-biotinyl exchange assay
Acyl-biotinyl exchange assay was performed as previously described [58]. N-ethylmaleimide was added to block the free thiols, and hydroxylamine was used to restore the modified cysteine to thiols. Biotinylated by thiol-reactive biotinylation reagent DHPC-biotin and affinity-captured by streptavidin–agarose. Assays were examined by western blotting analysis.
Statistical analysis
Data are presented as the means ± SD of representative experiments. Sample sizes for relevant experiments were determined by power analyses conducted during experiment planning. All statistical comparisons were carried out using Student’s t-test or chi-square test (Figs. 1h, 2d and 4f). We consider values ∗P < 0.05, ∗∗P < 0.01, and ∗∗∗P < 0.001 significant. All experiments were performed at least three times, unless otherwise stated.
Supplementary information
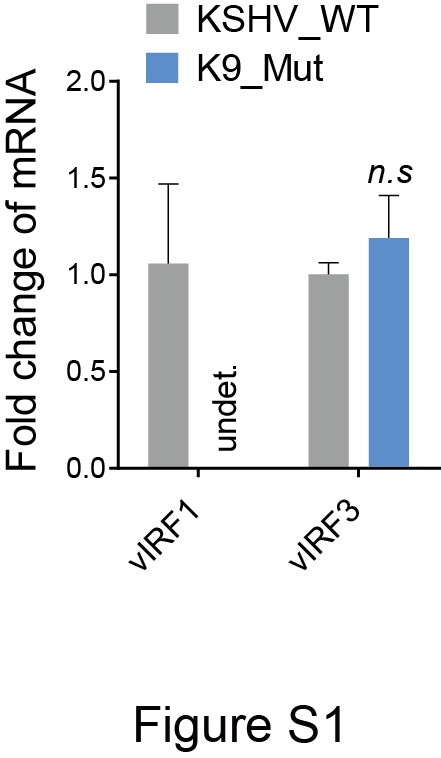{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Drs. Gary Hayward from Viral Oncology Program, The Johns Hopkins School of Medicine and Young Bong Choi from Johns Hopkins University School of Medicine for providing the polyclonal rabbit anti-vIRF1 antibody and information related to source of vIRF1 antibody, respectively. We are also grateful to members from Dr. Lu laboratory for helpful discussion.
Funding
This work was supported by grants from National Natural Science Foundation of China (81730062, 81761128003, and 31800148), Natural Science Foundation of Jiangsu Province (BK20180681), Nanjing Medical University (KY101RC1710), and a grant from NIH (R01CA213275).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by J.M. Hardwick
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Change history
9/14/2020
An amendment to this paper has been published and can be accessed via a link at the top of the paper.
Contributor Information
Cong Wang, Email: wangcongcc@vip.sina.com.
Chun Lu, Email: clu@njmu.edu.cn.
Supplementary information
The online version of this article (10.1038/s41418-020-0578-0) contains supplementary material, which is available to authorized users.
References
- 1.Cesarman E, Damania B, Krown SE, Martin J, Bower M, Whitby D. Kaposi sarcoma. Nat Rev Dis Prim. 2019;5:9. doi: 10.1038/s41572-019-0060-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cheung TW. AIDS-related cancer in the era of highly active antiretroviral therapy (HAART): a model of the interplay of the immune system, virus, and cancer. “On the offensive—the Trojan Horse is being destroyed”—Part A: Kaposi’s sarcoma. Cancer Invest. 2004;22:774–86. doi: 10.1081/CNV-200032788. [DOI] [PubMed] [Google Scholar]
- 3.Vangipuram R, Tyring SK. AIDS-associated malignancies. Cancer Treat Res. 2019;177:1–21. doi: 10.1007/978-3-030-03502-0_1. [DOI] [PubMed] [Google Scholar]
- 4.Mesri EA, Cesarman E, Boshoff C. Kaposi’s sarcoma and its associated herpesvirus. Nat Rev Cancer. 2010;10:707–19. doi: 10.1038/nrc2888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hew K, Dahlroth SL, Venkatachalam R, Nasertorabi F, Lim BT, Cornvik T, et al. The crystal structure of the DNA-binding domain of vIRF-1 from the oncogenic KSHV reveals a conserved fold for DNA binding and reinforces its role as a transcription factor. Nucleic Acids Res. 2013;41:4295–306. doi: 10.1093/nar/gkt082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dittmer DP. Transcription profile of Kaposi’s sarcoma-associated herpesvirus in primary Kaposi’s sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 2003;63:2010–5. [PubMed] [Google Scholar]
- 7.Lin R, Genin P, Mamane Y, Sgarbanti M, Battistini A, Harrington WJ, Jr., et al. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene. 2001;20:800–11. doi: 10.1038/sj.onc.1204163. [DOI] [PubMed] [Google Scholar]
- 8.Gao SJ, Boshoff C, Jayachandra S, Weiss RA, Chang Y, Moore PS. KSHV ORF K9 (vIRF) is an oncogene which inhibits the interferon signaling pathway. Oncogene. 1997;15:1979–85. doi: 10.1038/sj.onc.1201571. [DOI] [PubMed] [Google Scholar]
- 9.Kirchhoff S, Sebens T, Baumann S, Krueger A, Zawatzky R, Li-Weber M, et al. Viral IFN-regulatory factors inhibit activation-induced cell death via two positive regulatory IFN-regulatory factor 1-dependent domains in the CD95 ligand promoter. J Immunol. 2002;168:1226–34. doi: 10.4049/jimmunol.168.3.1226. [DOI] [PubMed] [Google Scholar]
- 10.Seo T, Park J, Choe J. Kaposi’s sarcoma-associated herpesvirus viral IFN regulatory factor 1 inhibits transforming growth factor-beta signaling. Cancer Res. 2005;65:1738–47. doi: 10.1158/0008-5472.CAN-04-2374. [DOI] [PubMed] [Google Scholar]
- 11.Seo T, Park J, Lee D, Hwang SG, Choe J. Viral interferon regulatory factor 1 of Kaposi’s sarcoma-associated herpesvirus binds to p53 and represses p53-dependent transcription and apoptosis. J Virol. 2001;75:6193–8. doi: 10.1128/JVI.75.13.6193-6198.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakamura H, Li M, Zarycki J, Jung JU. Inhibition of p53 tumor suppressor by viral interferon regulatory factor. J Virol. 2001;75:7572–82. doi: 10.1128/JVI.75.16.7572-7582.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shin YC, Nakamura H, Liang X, Feng P, Chang H, Kowalik TF, et al. Inhibition of the ATM/p53 signal transduction pathway by Kaposi’s sarcoma-associated herpesvirus interferon regulatory factor 1. J Virol. 2006;80:2257–66. doi: 10.1128/JVI.80.5.2257-2266.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W, Wang Q, Feng Q, Wang F, Yan Q, Gao SJ, et al. Oncogenic KSHV-encoded interferon regulatory factor upregulates HMGB2 and CMPK1 expression to promote cell invasion by disrupting a complex lncRNA-OIP5-AS1/miR-218-5p network. PLoS Pathog. 2019;15:e1007578. doi: 10.1371/journal.ppat.1007578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vo MT, Smith BJ, Nicholas J, Choi YB. Activation of NIX-mediated mitophagy by an interferon regulatory factor homologue of human herpesvirus. Nat Commun. 2019;10:3203. doi: 10.1038/s41467-019-11164-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hooper JD, Zijlstra A, Aimes RT, Liang H, Claassen GF, Tarin D, et al. Subtractive immunization using highly metastatic human tumor cells identifies SIMA135/CDCP1, a 135 kDa cell surface phosphorylated glycoprotein antigen. Oncogene. 2003;22:1783–94. doi: 10.1038/sj.onc.1206220. [DOI] [PubMed] [Google Scholar]
- 17.Emerling BM, Benes CH, Poulogiannis G, Bell EL, Courtney K, Liu H, et al. Identification of CDCP1 as a hypoxia-inducible factor 2alpha (HIF-2alpha) target gene that is associated with survival in clear cell renal cell carcinoma patients. Proc Natl Acad Sci USA. 2013;110:3483–8. doi: 10.1073/pnas.1222435110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyazawa Y, Uekita T, Hiraoka N, Fujii S, Kosuge T, Kanai Y, et al. CUB domain-containing protein 1, a prognostic factor for human pancreatic cancers, promotes cell migration and extracellular matrix degradation. Cancer Res. 2010;70:5136–46. doi: 10.1158/0008-5472.CAN-10-0220. [DOI] [PubMed] [Google Scholar]
- 19.Ikeda J, Oda T, Inoue M, Uekita T, Sakai R, Okumura M, et al. Expression of CUB domain containing protein (CDCP1) is correlated with prognosis and survival of patients with adenocarcinoma of lung. Cancer Sci. 2009;100:429–33. doi: 10.1111/j.1349-7006.2008.01066.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gao W, Chen L, Ma Z, Du Z, Zhao Z, Hu Z, et al. Isolation and phenotypic characterization of colorectal cancer stem cells with organ-specific metastatic potential. Gastroenterology. 2013;145:636–46 e635. doi: 10.1053/j.gastro.2013.05.049. [DOI] [PubMed] [Google Scholar]
- 21.Fukuchi K, Steiniger SC, Deryugina E, Liu Y, Lowery CA, Gloeckner C, et al. Inhibition of tumor metastasis: functional immune modulation of the CUB domain containing protein 1. Mol Pharm. 2010;7:245–53. doi: 10.1021/mp900236t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Casar B, He Y, Iconomou M, Hooper JD, Quigley JP, Deryugina EI. Blocking of CDCP1 cleavage in vivo prevents Akt-dependent survival and inhibits metastatic colonization through PARP1-mediated apoptosis of cancer cells. Oncogene. 2012;31:3924–38. doi: 10.1038/onc.2011.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wong CH, Baehner FL, Spassov DS, Ahuja D, Wang D, Hann B, et al. Phosphorylation of the SRC epithelial substrate Trask is tightly regulated in normal epithelia but widespread in many human epithelial cancers. Clin Cancer Res. 2009;15:2311–22. doi: 10.1158/1078-0432.CCR-08-2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siva AC, Wild MA, Kirkland RE, Nolan MJ, Lin B, Maruyama T, et al. Targeting CUB domain-containing protein 1 with a monoclonal antibody inhibits metastasis in a prostate cancer model. Cancer Res. 2008;68:3759–66. doi: 10.1158/0008-5472.CAN-07-1657. [DOI] [PubMed] [Google Scholar]
- 25.Lee HR, Li F, Choi UY, Yu HR, Aldrovandi GM, Feng P, et al. Deregulation of HDAC5 by viral interferon regulatory factor 3 plays an essential role in Kaposi’s sarcoma-associated herpesvirus-induced lymphangiogenesis. mBio. 2018;9:e02217-17. [DOI] [PMC free article] [PubMed]
- 26.Park JJ, Jin YB, Lee YJ, Lee JS, Lee YS, Ko YG, et al. KAI1 suppresses HIF-1alpha and VEGF expression by blocking CDCP1-enhanced Src activation in prostate cancer. BMC Cancer. 2012;12:81. doi: 10.1186/1471-2407-12-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W, Hu M, Wang C, Lu H, Chen F, Xu J, et al. A viral microRNA downregulates metastasis suppressor CD82 and induces cell invasion and angiogenesis by activating the c-Met signaling. Oncogene. 2017;36:5407–20. doi: 10.1038/onc.2017.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andre M, Le Caer JP, Greco C, Planchon S, El Nemer W, Boucheix C, et al. Proteomic analysis of the tetraspanin web using LC-ESI-MS/MS and MALDI-FTICR-MS. Proteomics. 2006;6:1437–49. doi: 10.1002/pmic.200500180. [DOI] [PubMed] [Google Scholar]
- 29.Adams MN, Harrington BS, He Y, Davies CM, Wallace SJ, Chetty NP, et al. EGF inhibits constitutive internalization and palmitoylation-dependent degradation of membrane-spanning procancer CDCP1 promoting its availability on the cell surface. Oncogene. 2015;34:1375–83. doi: 10.1038/onc.2014.88. [DOI] [PubMed] [Google Scholar]
- 30.Tsai YC, Mendoza A, Mariano JM, Zhou M, Kostova Z, Chen B, et al. The ubiquitin ligase gp78 promotes sarcoma metastasis by targeting KAI1 for degradation. Nat Med. 2007;13:1504–9. doi: 10.1038/nm1686. [DOI] [PubMed] [Google Scholar]
- 31.He Y, Harrington BS, Hooper JD. New crossroads for potential therapeutic intervention in cancer—intersections between CDCP1, EGFR family members and downstream signaling pathways. Oncoscience. 2016;3:5–8. doi: 10.18632/oncoscience.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hu M, Wang C, Li W, Lu W, Bai Z, Qin D, et al. A KSHV microRNA directly targets G protein-coupled receptor kinase 2 to promote the migration and invasion of endothelial cells by inducing CXCR2 and activating AKT signaling. PLoS Pathog. 2015;11:e1005171. doi: 10.1371/journal.ppat.1005171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tandle A, Blazer DG, 3rd, Libutti SK. Antiangiogenic gene therapy of cancer: recent developments. J Transl Med. 2004;2:22. doi: 10.1186/1479-5876-2-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qian LW, Xie J, Ye F, Gao SJ. Kaposi’s sarcoma-associated herpesvirus infection promotes invasion of primary human umbilical vein endothelial cells by inducing matrix metalloproteinases. J Virol. 2007;81:7001–10. doi: 10.1128/JVI.00016-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Ueda K, Sakakibara S, Okuno T, Yamanishi K. Transcriptional regulation of the Kaposi’s sarcoma-associated herpesvirus viral interferon regulatory factor gene. J Virol. 2000;74:8623–34. doi: 10.1128/JVI.74.18.8623-8634.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fakhari FD, Dittmer DP. Charting latency transcripts in Kaposi’s sarcoma-associated herpesvirus by whole-genome real-time quantitative PCR. J Virol. 2002;76:6213–23. doi: 10.1128/JVI.76.12.6213-6223.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paulose-Murphy M, Ha NK, Xiang C, Chen Y, Gillim L, Yarchoan R, et al. Transcription program of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) J Virol. 2001;75:4843–53. doi: 10.1128/JVI.75.10.4843-4853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong W, Wang H, Herndier B, Ganem D. Restricted expression of Kaposi sarcoma-associated herpesvirus (human herpesvirus 8) genes in Kaposi sarcoma. Proc Natl Acad Sci USA. 1996;93:6641–6. doi: 10.1073/pnas.93.13.6641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chiou CJ, Poole LJ, Kim PS, Ciufo DM, Cannon JS, ap Rhys CM, et al. Patterns of gene expression and a transactivation function exhibited by the vGCR (ORF74) chemokine receptor protein of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2002;76:3421–39. doi: 10.1128/JVI.76.7.3421-3439.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Katano H, Sato Y, Kurata T, Mori S, Sata T. Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease. Virology. 2000;269:335–44. doi: 10.1006/viro.2000.0196. [DOI] [PubMed] [Google Scholar]
- 41.Parravicini C, Chandran B, Corbellino M, Berti E, Paulli M, Moore PS, et al. Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am J Pathol. 2000;156:743–9. doi: 10.1016/S0002-9440(10)64940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, et al. Kaposi’s sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J Virol. 1997;71:715–9. doi: 10.1128/JVI.71.1.715-719.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology. 1998;252:304–12. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- 44.Wortmann A, He Y, Deryugina EI, Quigley JP, Hooper JD. The cell surface glycoprotein CDCP1 in cancer-insights, opportunities, and challenges. IUBMB Life. 2009;61:723–30. doi: 10.1002/iub.198. [DOI] [PubMed] [Google Scholar]
- 45.Scherl-Mostageer M, Sommergruber W, Abseher R, Hauptmann R, Ambros P, Schweifer N. Identification of a novel gene, CDCP1, overexpressed in human colorectal cancer. Oncogene. 2001;20:4402–8. doi: 10.1038/sj.onc.1204566. [DOI] [PubMed] [Google Scholar]
- 46.Frandberg S, Asp J, Waldner B, Holgersson J, Palmqvist L. Concentration of the CDCP1 protein in human cord plasma may serve as a predictor of hematopoietic stem and progenitor cell content. Stem Cell Res. 2018;29:24–7. doi: 10.1016/j.scr.2018.03.008. [DOI] [PubMed] [Google Scholar]
- 47.Spassov DS, Wong CH, Wong SY, Reiter JF, Moasser MM. Trask loss enhances tumorigenic growth by liberating integrin signaling and growth factor receptor cross-talk in unanchored cells. Cancer Res. 2013;73:1168–79. doi: 10.1158/0008-5472.CAN-12-2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mundi PS, Sachdev J, McCourt C, Kalinsky K. AKT in cancer: new molecular insights and advances in drug development. Br J Clin Pharm. 2016;82:943–56. doi: 10.1111/bcp.13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chamcheu JC, Roy T, Uddin MB, Banang-Mbeumi S, Chamcheu RN, Walker AL, et al. Role and therapeutic targeting of the PI3K/Akt/mTOR signaling pathway in skin cancer: a review of current status and future trends on natural and synthetic agents therapy. Cells. 2019;8:803. [DOI] [PMC free article] [PubMed]
- 50.Bhatt AP, Damania B. AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV. Front Immunol. 2012;3:401. doi: 10.3389/fimmu.2012.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li W, Yan Q, Ding X, Shen C, Hu M, Zhu Y, et al. The SH3BGR/STAT3 pathway regulates cell migration and angiogenesis induced by a gammaherpesvirus microRNA. PLoS Pathog. 2016;12:e1005605. doi: 10.1371/journal.ppat.1005605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhu X, Guo Y, Yao S, Yan Q, Xue M, Hao T, et al. Synergy between Kaposi’s sarcoma-associated herpesvirus (KSHV) vIL-6 and HIV-1 Nef protein in promotion of angiogenesis and oncogenesis: role of the AKT signaling pathway. Oncogene. 2014;33:1986–96. doi: 10.1038/onc.2013.136. [DOI] [PubMed] [Google Scholar]
- 53.Xue M, Yao S, Hu M, Li W, Hao T, Zhou F, et al. HIV-1 Nef and KSHV oncogene K1 synergistically promote angiogenesis by inducing cellular miR-718 to regulate the PTEN/AKT/mTOR signaling pathway. Nucleic Acids Res. 2014;42:9862–79. doi: 10.1093/nar/gku583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao S, Hu M, Hao T, Li W, Xue X, Xue M, et al. MiRNA-891a-5p mediates HIV-1 Tat and KSHV Orf-K1 synergistic induction of angiogenesis by activating NF-kappaB signaling. Nucleic Acids Res. 2015;43:9362–78. doi: 10.1093/nar/gkv988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Aranda E, Owen GI. A semi-quantitative assay to screen for angiogenic compounds and compounds with angiogenic potential using the EA.hy926 endothelial cell line. Biol Res. 2009;42:377–89. doi: 10.4067/S0716-97602009000300012. [DOI] [PubMed] [Google Scholar]
- 56.Magnaudeix A, Usseglio J, Lasgorceix M, Lalloue F, Damia C, Brie J, et al. Quantitative analysis of vascular colonisation and angio-conduction in porous silicon-substituted hydroxyapatite with various pore shapes in a chick chorioallantoic membrane (CAM) model. Acta Biomater. 2016;38:179–89. doi: 10.1016/j.actbio.2016.04.039. [DOI] [PubMed] [Google Scholar]
- 57.Ding X, Xu J, Wang C, Feng Q, Wang Q, Yang Y, et al. Suppression of the SAP18/HDAC1 complex by targeting TRIM56 and Nanog is essential for oncogenic viral FLICE-inhibitory protein-induced acetylation of p65/RelA, NF-kappaB activation, and promotion of cell invasion and angiogenesis. Cell Death Differ. 2019;26:1970–86. doi: 10.1038/s41418-018-0268-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wan J, Roth AF, Bailey AO, Davis NG. Palmitoylated proteins: purification and identification. Nat Protoc. 2007;2:1573–84. doi: 10.1038/nprot.2007.225. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.