

Abstract
CD4+ and CD8+ T cells are dichotomous lineages in adaptive immunity. While conventionally viewed as distinct fates that are fixed after thymic development, accumulating evidence indicates that these two populations can exhibit significant lineage plasticity, particularly upon TCR-mediated activation. We define a novel CD4−CD8αβ+ MHC II-recognizing population generated by lineage conversion from effector CD4+ T cells. CD4−CD8αβ+ effector T cells downregulated the expression of T helper cell-associated costimulatory molecules and increased the expression of cytotoxic T lymphocyte-associated cytotoxic molecules. This shift in functional potential corresponded with a CD8+-lineage skewed transcriptional profile. TCRβ repertoire sequencing and in vivo genetic lineage tracing in acutely infected wild-type mice demonstrated that CD4−CD8αβ+ effector T cells arise from fundamental lineage reprogramming of bona fide effector CD4+ T cells. Impairing autophagy via functional deletion of the initiating kinase Vps34 or the downstream enzyme Atg7 enhanced the generation of this cell population. These findings suggest that effector CD4+ T cells can exhibit a previously unreported degree of skewing towards the CD8+ T cell lineage, which may point towards a novel direction for HIV vaccine design.
Keywords: CD4+ T cell, CD8+ T cell, ThPOK, Runx3, autophagy
Subject terms: CD4-positive T cells, CD8-positive T cells
Introduction
CD4+ and CD8+ T cells are conventionally defined as separate T cell lineages with distinct molecular markers (CD4 vs. CD8), immunological functions (helper vs. cytotoxic) and major histocompatibility complex (MHC) recognition capacities (MHC II vs. MHC I). These lineages diverge during thymic development1,2 and are maintained once mature, naïve T cells enter the peripheral immune system. Both lineage specification and stability are controlled by the master transcription factors ThPOK and Runx3 via the antagonistic regulation; ThPOKhiRunx3lo expression sets the CD4+ lineage transcriptional program, and ThPOKloRunx3hi expression sets the CD8+ program.3
Moreover, multiple homeostatic cellular processes contribute to the auxiliary regulation of effector T cell differentiation. One such process is autophagy, a catabolic survival mechanism that recycles cellular organelles and macromolecules. The major form of autophagy, known as macroautophagy, is initiated by an enzymatic complex consisting of Beclin1, Vps15, ATG14 and the class III PI3-kinase Vps34.4,5 The subsequent formation of the autophagosome to encapsulate cytosolic material is catalyzed by a series of reactions requiring Atg proteins, including Atg7, which is the lynchpin of membrane elongation mechanisms.6,7 Autophagy controls the T cell effector status at multiple levels, including TCR signaling, metabolism and memory formation.8 In particular, Vps34 and Atg7 have demonstrated roles in the homeostatic maintenance of naïve T cells9,10 and the accumulation of effector cells.9,11
Accordingly, the transcriptional profiles and functional properties of naïve CD4+ T cells undergo rapid and dramatic transformation during effector T cell differentiation. In fact, the gene expression upheaval that is necessary for conventional T helper (Th) cell differentiation can produce unexpected lineage outcomes by allowing the gene expression and function in effector CD4+ T cells to shift remarkably towards a CD8+-like phenotype. CD4+ Th type 1 (Th1) cells exhibit ThPOKhiRunx3hi expression upon differentiation,12 and some can become CD4+ cytotoxic T lymphocytes (CTLs),13,14 which express the hallmark CD8+ functional molecules IFN-γ and granzyme B. Furthermore, the Mucida group published two studies showing that mature CD4+ThPOKhi T cells can give rise to a CD4+CD8αα+ThPOKlo population, which provides protection against mucosal inflammation in an adoptive transfer colitis model.15,16 Another report indicated that the loss of functional histone deacetylase (HDAC) 1 or 2 promotes the generation of CD4+ T cell-derived CD4+CD8αβ+ effector T cells, demonstrating that conventional CD4+ T cells can acquire a phenotype that is still more like CD8+ CTLs than previously thought.17 Paralleling these findings, lineage-intermediate populations originating from effector CD4+ T cells and exhibiting various CD4/CD8 and functional gene expression profiles have been reported in other animal models,18–21 as well as human disease contexts that include hepatocellular carcinoma, multiple sclerosis, rheumatoid arthritis, cytomegalovirus (CMV) infection, and human immunodeficiency virus-1 (HIV-1, hereafter HIV) infection.13,22–26 These populations are particularly associated with chronic viral infections, and HIV infection is a particularly interesting setting. A variety of CD4/CD8 phenotypes have been observed among the peripheral blood mononuclear cells (PBMCs) of HIV patients, including CD4−CD8−, CD4+CD8+, and most recently, MHC II-recognizing CD4−CD8αβ+ cells.27–40
We provide here the first report that a murine CD4−CD8αβ+ MHC II-recognizing lineage can be derived from effector CD4+ T cells. Using multiple in vitro differentiation and in vivo tracing strategies, we demonstrate that this cell population exhibits a CD4/CD8 lineage-intermediate phenotype, is generated from bona fide effector CD4+ T cells by transcriptional reprogramming and is regulated by the key autophagy molecules Vps34 and Atg7.
Results
Activation of Vps34-deficient CD4+ T cells produces a CD4−CD8αβ+ MHC II-restricted T cell population
Our investigation began with an unexpected observation while studying Vps34 function during CD4+ T cell activation. We generated LLO118αβ Vps34f/f CD4-Cre+ (LLO Vps34KO) mice with a T cell-specific deletion of functional Vps34 and transgenic expression of the LLO118αβ TCR (Vα2+Vβ2+). The LLO118αβ TCR mediates the MHC II (I-Ab)-restricted recognition of the listeriolysin O protein (LLO190-205), the major epitope of Listeria monocytogenes.41 We found that in vitro LLO190-205 stimulation of either total lymph node cells or FACS-purified TCR Vα2+CD4+ T cells resulted in the loss of CD4 surface expression (Fig. 1a). Although antigen engagement initiated CD4 downregulation with similar kinetics in both LLO118αβ Vps34f/f CD4-Cre− (LLO WT) and LLO Vps34KO TCR Vα2+ cells, LLO Vps34KO cells failed to recover normal CD4 surface levels (Fig. 1b). The CD4 protein did not accumulate intracellularly in activated LLO Vps34KO cells (Fig. 1c), indicating that downregulated CD4 surface display was not due to the abrogation of Vps34-mediated intracellular vesicle trafficking.42 Loss of functional Vps34 also did not enhance proliferation of CD4− cells because CD4− and CD4+ LLO Vps34KO T cells comparably proliferated (Fig. S1). In addition, CD4− cells were markedly more susceptible to activation-induced cell death (AICD); LLO Vps34KO CD4− cells survived to a lesser extent than both LLO WT CD4− cells and LLO Vps34KO CD4+ cells (Fig. S1). Therefore, because the loss of neither the functional Vps34 nor CD4 protein conferred a proliferative or survival advantage to T cells, we deemed it unlikely that preexisting, mature LLO Vps34KO CD4− T cells (either naïve or memory) preferentially accumulated over CD4+ T cells upon TCR activation.
Fig. 1.

In vitro stimulation of LLO Vps34KO CD4+ T cells produces a CD4-CD8αβ+ MHCII-recognizing T cell population. Total cells or FACS-sorted CD4+ T cells from the lymph nodes of LLO118αβ Vps34f/f CD4-Cre− (LLO WT) or LLO118αβ Vps34f/f CD4-Cre+ (LLO Vps34KO) mice were stimulated in vitro with 10 μM LLO190-205 peptide for 120 h. Surface expression of CD4 is shown at 120 h (a) and at 24-h intervals from 0-120 h (b). c LLO WT and LLO Vps34KO T cells were analyzed for intracellular CD4 protein expression after in vitro peptide stimulation for 120 h. d CD8α/β expression was analyzed in LLO WT and LLO Vps34KO T cells stimulated with LLO190-205 in vitro for 120 h. Expression is shown in TCR Vα2+ CD4− and TCR Vα2+ CD4+ cells. The CD4−CD8αβ+ population is indicated in red. n = 16 from 9 independent experiments for (a), n = 3 from 3 independent experiments for (b), n = 7 from 7 independent experiments for (c) and n = 8 from 5 independent experiments for (d)
Furthermore, CD4 downregulation in activated LLO Vps34KO CD4+ T cells was accompanied by multiple CD8 expression phenotypes (Fig. 1d). The majority of activated LLO Vps34KO TCR Vα2+ cells were CD4-CD8αβ-, paralleling populations previously found in both animals and humans.43 We observed a smaller CD4−CD8αα+ population, which was reminiscent of CD4+CD8αα+ intraepithelial lymphocytes (IELs) associated with mucosal inflammation,15 as well as a CD4+CD8αβ+ population, which has been previously observed during HIV infection and other human chronic diseases.31,44 Surprisingly, a CD4−CD8αβ+ subset was also observed. Given that our LLO Vps34KO T cells expressed an MHC II-restricted transgenic TCR, we noted that this might be a previously unreported CD4−CD8αβ+ MHC II-recognizing T cell population in mice.15,17,45
In fact, we also observed CD4−CD8αβ+ MHC II-restricted T cells among in vitro-stimulated LLO WT CD4+ T cells (Fig. 1d). While the loss of CD4 expression occurred at a much lower frequency among activated LLO WT cells compared to LLO Vps34KO cells, a higher proportion of TCR Vα2+ CD4− cells expressed both CD8α and CD8β in the LLO WT subset vs. the LLO Vps34KO subset. In addition, we observed that FACS-sorted CD4+ T cells harvested from the lymph nodes of C57BL/6 mice with an open TCR repertoire could also produce CD4−CD8αβ+ MHC II-restricted T cells upon antibody-mediated in vitro stimulation (Fig. S2). These findings suggested that while Vps34 could sustain CD4 molecule expression in effector CD4+ T cells, the generation of a CD4−CD8αβ+ MHC II-recognizing T cell population was not dependent on the loss of Vps34 function.
CD4−CD8αβ+ MHC II-restricted T cells are generated in vivo during acute infection
To determine whether CD4−CD8αβ+ MHC II-recognizing T cells could be generated during an in vivo effector response, we infected LLO WT and LLO Vps34KO mice with Listeria monocytogenes, the cognate pathogen for their transgenic TCRs.41 Because direct Listeria infection in the context of dominant LLO118 TCR expression would predictably result in an overwhelming, physiologically abnormal effector T cell response,41,46 we chose this acute infection model to demonstrate the in vivo generation of effector CD4−CD8αβ+ MHC II-recognizing T cells, but not their origin, effector function or physiological relevance.
In Listeria-infected LLO Vps34KO mice, over 20% of splenic effector (CD44hi-int) TCR Vα2+ T cells lost CD4 expression (Fig. 2a). Among those, we identified multiple lineage-intermediate populations, including CD4−CD8αβ+ T cells. Albeit at a much lower frequency, CD4−CD8αβ+ T cells were also identified in Listeria-infected LLO WT mice (Fig. 2a). Paralleling our in vitro observations, a higher proportion of LLO WT CD4− cells exhibited the CD8αβ+ phenotype than their LLO Vps34KO counterparts (Fig. 2a, b).
Fig. 2.

CD4-CD8αβ+ MHCII-recognizing effector T cells are generated during acute infection. Acute Listeria monocytogenes infection (a, b) LLO WT and LLO Vps34KO mice were infected with 1 × 107 cfu Listeria monocytogenes by intravenous (i.v.) injection. a Four days post-infection, splenic lymphocytes were harvested and analyzed for the presence of naïve CD4+ T cells (CD44int-lo TCR Vα2+ CD4+, marked in green), effector CD4− T cells (CD44int-hi TCR Vα2+ CD4−, marked in blue) and effector CD4+ T cells (CD44hi TCR Vα2+ CD4+, marked in gray) (left panel). CD8αβ expression was analyzed in each of these populations (right panel). The CD4-CD8αβ+ population is indicated in red. b Frequency of CD8αβ expression within naïve CD4+, effector CD4− and effector CD4+ T cells from Listeria-infected LLO WT and LLO Vps34KO mice. n = 6 from 1 experiment. Statistical significance was determined by the Mann-Whitney U Test; *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant. Acute LCMV Armstrong infection (c–e) Thy1.1+ C57BL/6 mice were infected with 1 × 105 cfu lymphocytic choriomeningitis virus-Armstrong strain (LCMV Armstrong) by intraperitoneal (i.p.) injection. c Seven-8 days postinfection, splenic lymphocytes were harvested and analyzed for the presence of LCMV/MHCIIspecific effector CD4−CD8αβ+ T cells (Live-dead− huCLIP87-101-I-Ab− LCMVgp66-77+-I-Ab+ CD44hi TCRβ+ CD4−, marked in brown) and LCMV/MHCII-specific effector CD4+ T cells (Livedead− huCLIP87-101-I-Ab− LCMVgp66-77+-I-Ab+ CD44hi TCRβ+ CD4+, marked in green). %CD8αβ+ cells in these sub-populations in LCMV-infected mice is shown in (d) and enumerated in (e). n = 13 from 2 independent experiments. Statistical significance was determined by the Mann–Whitney U test; *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant
To demonstrate that CD4−CD8αβ+ T cells were not specific to (1) the Vps34KO context, (2) Listeria infection or (3) the LLO118 TCR transgene, we infected WT Thy1.1+ C57BL/6 mice with the Armstrong strain of lymphocytic choriomeningitis virus (LCMV Armstrong). Using an MHC II (I-Ab) tetramer presenting a major LCMV epitope for CD4+ T cells (LCMVgp66-77-I-Ab), we observed that a substantial population of LCMV-I-Ab-specific effector T cells generated during an acute infection were CD4− (Fig. 2c). Among CD4− LCMV-I-Ab-recognizing cells, at least 25% were CD8αβ+ (Fig. 2d, e). The CD4-CD8αβ+ population persisted up to at least 28 days postinfection, indicating its stability (Fig. S3). We also tested whether CD4− T cells could recognize other established I-Ab-restricted LCMV epitopes. Using specific tetramers, we found that LCMVgp66-77-I-Ab had the highest avidity for LCMV-binding TCRs and therefore provided the best separation of CD4+ and CD4− LCMV/I-Ab-specific T cells above the background binding that was found in uninfected controls (Fig. S4). Taken together, the results of these two in vivo infection models indicated that the production of CD4−CD8αβ+ effector T cells could be facilitated by—but did not require—the loss of Vps34 and might be underpinned by a general CD4+ lineage destabilization process induced by diverse pathogens in a range of TCR repertoires.
Conventional effector CD4+ T cells give rise to CD4-CD8αβ+ MHC II-restricted T cells
We deemed it crucial to definitively demonstrate that CD4−CD8αβ+ MHC II-restricted T cells were generated by a lineage conversion of mature effector CD4+ T cells and that they did not merely represent the expansion of mature, aberrant CD8+ MHC II-recognizing T cells. We employed two independent tracking strategies to pursue our aim. The first strategy was a comprehensive assessment of the TCR spectrum of virus-specific CD4− vs. CD4+ T cells in acutely infected WT mice with an open TCR repertoire. We reasoned that if CD4−CD8αβ+ MHC II-recognizing T cells were produced by the expansion of a preexisting CD8+ T cell pool generated during thymic selection, CD4−CD8αβ+ T cells probably would not share TCR sequences with CD4+ T cells. On the other hand, if CD4−CD8αβ+ MHC II-recognizing T cells were generated by the lineage conversion of mature CD4+ T cells, we expected that CD4−CD8αβ+ TCRs would substantially overlap with conventional effector CD4+ TCRs.
From the spleens of 4 C57BL/6 mice 8 days postinfection with LCMV Armstrong, we sorted LCMVgp66-77-I-Ab-recognizing CD44hi CD4− and CD4+ T cells and performed TCRβ sequencing to reach saturation depth (Table S1). Out of the ~6.0 × 105 total effective TCR reads analyzed across all mice, we detected ~5.3 × 103 LCMV-I-Ab-recognizing CD44hiCD4+ clonotypes and ~1.5 × 103 LCMV-I-Ab-recognizing CD44hiCD4− clonotypes. Collectively, a substantial number of these clonotypes (~4 × 102) were found in both the CD4+ and CD4− T cell populations, representing ~8% of CD4+ clonotypes and ~26% of CD4− clonotypes (Fig. 3a). These shared clonotypes were present at a broad range of frequencies within each repertoire pool, indicating that TCR affinity might not play a strong role in lineage destabilization of effector CD4+ T cells. We did note that the majority of CD4− clonotypes were not found among CD4+ clonotypes. Although this observation raised the possibility that the unique CD4− clonotypes represented preexisting CD8+ MHC II-recognizing T cells, it likely reflected a major limitation of TCR sequencing technology, which is the subsampling effect in an open repertoire. Nevertheless, the substantial number of TCRs that overlapped the CD4− and CD4+ effector T cell populations suggested that a mechanism exists for lineage conversion to the CD4−CD8αβ+ effector phenotype.
Fig. 3.

Lineage tracing by TCRβ repertoire sequencing indicates lineage conversion of effector CD4+ T cells to CD4− T cells during an antigen-specific effector response. Splenic lymphocytes were harvested from WT Thy1.1+ C57Bl/6 mice 7 days post-LCMV Armstrong infection. Live LCMV/MHCII-recognizing effector CD4− T cells (Live/Dead-huCLIP87- 101-I-Ab− LCMVgp66-77-I-Ab+ CD44hi CD4−) or effector CD4+ T cells (Live/Dead-huCLIP87- 101-I-Ab− LCMVgp66-77-I-Ab+ CD44hi CD4+) were collected (3.5 × 103–2 × 104 cells/sample). FACS-sorted populations were then spiked with 5 × 102 2B4.11 cells to provide absolute reads:cell number ratio for effective reads calculation. Populations with <2 × 104 cells were supplemented with LB27.4 cells to facilitate RNA extraction. cDNA libraries were prepared from total RNA by reverse transcription using an in house-designed TCRβ-specific primer. Whole-TCRβ sequencing was performed using the Ion Torrent PGM platform. Cell numbers, effective reads, and clonotype counts are listed in Table 1. a Schematic representation of intramouse and intermouse clonotype comparisons. b Pairwise comparison of nucleotide clonotype frequencies between effector CD4+ and CD4− T cells across all mice. The proportion of shared clonotypes among total clonotypes for each population is listed above the plot. Clonotypes were placed into 3 frequency categories: (1) High-frequency among CD4− TCRs, (2) High-frequency among effector CD4+ TCRs and (3) Low frequency among all TCRs. c UpSet plot representation of intra-mouse and inter-mouse shared nucleotide clonotypes. See Fig. S3A for schematic representation of shared clonotype comparisons. The histogram (upper panel) enumerates the number of clonotypes shared by the indicated populations (lower panel) out of the 100 most-frequent (“top 100”) clonotypes across all mice and populations. The histogram in the left panel enumerates the number of clonotypes in the indicated population found among the 150 most-frequent (“top 150”) clonotypes in the M4-CD4+ population. Shared clonotypes are indicated by color as intra-mouse (found in 1 mouse/2 populations; shown in blue), inter-mouse (found in >1 mouse/>1 population; shown in black) or public TCRs (found in >1 mouse/>1 CD4+ population; shown in red). All sequencing analyses were conducted using in house-designed software. n = 4 from 1 experiment
For analysis of individual mouse repertoires, we conducted a comprehensive search for either intramouse or intermouse clonotype sharing (Fig. 3b). At the nucleotide level, the highest frequency clonotype sharing occurred between CD44hiCD4+ and CD44hiCD4− T cells within individual mice, which suggested that these two populations might share the same origin (Fig. 3c). This was also supported by our observation that at the amino acid level, most of the intramouse shared TCRs were encoded by identical nucleotide sequences (Fig. S5A). In addition, 8 of the LCMVgp66-77-I-Ab-recognizing CD4+ clonotypes were identified in all 4 mice (i.e., “public” clonotypes). Five out of these 8 CD4+ clonotypes were shared with CD4− clonotypes in each individual mouse; the other 3 CD4+ public clonotypes were shared with CD4− clonotypes in at least 2 mice. (Fig. 3c and S5B, C). We considered that these public TCRs were unlikely to have been produced by the simultaneous thymic development of mature CD4+ and CD8+ T cells with identical TCRs and then maintained in the naïve T cell pool with the same lifespan. Instead, it was more likely that upon infection, the same naïve, LCMV-recognizing CD4+ T cell clonotype lineage converted to acquire the CD4−CD8αβ+ effector phenotype in each mouse. Taken together, these findings provide clonal lineage evidence to suggest that during an in vivo effector response, a broad spectrum of effector CD4+ T cells could become CD4−CD8αβ+ MHC II-restricted cells.
Our second approach made use of the Ox40-Cre Rosa26tdTomato (OxTom)47 murine strain. By the genetic strategy of this strain, tdTomato (tdTom) expression is controlled by the promoter for Ox40, which encodes a costimulatory surface receptor on CD4+ T cells. Because Ox40 is generally silent in naïve T cells and its expression is preferentially induced in CD4+ T cells vs. CD8+ T cells after strong antigen stimulation,47 we reasoned that this system would allow us to preferentially mark activated CD4+ T cells. In adults, unchallenged OxTom mice raised in a standard pathogen-free environment, tdTom expression was approximately 10-fold higher in splenic effector/memory (CD44hi) CD4+ cells than in naïve (CD44lo) CD4+ cells (Fig. S6). Equally important, the frequency of tdTom expression in effector/memory CD4+ cells was more than 20-fold higher than in effector/memory CD8+ cells and nearly 500-fold higher than in naïve CD8+ cells. Therefore, we concluded that preferential tdTom labeling of effector CD4+ T cells using the OxTom system would allow us to trace the generation of CD4−CD8αβ+ cells from effector CD4+ T cells.
In OxTom mice infected with LCMV Armstrong, we detected conventional LCMV-I-Ab-recognizing CD44hi-intCD4+ T cells (Fig. 4a). Approximately 50–80% of this population were tdTom+, indicating that our genetic strategy efficiently marked effector/memory CD4+ T cells, albeit to a varying degree. Moreover, an LCMV-I-Ab-specific CD44hiCD4− population was also generated, and ~30–70% of which were tdTom+ (Fig. 4a). This suggested that the CD4− LCMV/I-Ab-recognizing T cells we detected during the acute anti-LCMV response originated from activated CD4+ T cells.
Fig. 4.

Lineage tracing indicates lineage conversion of CD4+ T cells to CD4-CD8αβ+ T cells during an antigen-specific effector response. Genetic labeling of CD4+ T cells (a) Rosa26tdTomato+/− Ox40-Cre+/− (OxTom) mice were infected with 1 × 105 cfu LCMV Armstrong by i.p. injection. Eight days post-infection, splenic lymphocytes were harvested and analyzed for tdTomato (tdTom) expression among LCMV-specific CD4-CD8αβ+ T cells (Live/DeadhuCLIP87-101-I-Ab− LCMVgp66-77-I-Ab+ CD44hi TCRβ+ CD4−, marked in brown) and effector CD4+ T cells (Live/Dead− huCLIP87-101-I-Ab− LCMVgp66-77-I-Ab+ CD44hi TCRβ+ CD4+, marked in green). b, c Naïve CD4+ T cells (TCRβ+ CD44lo tdTom− CD4+) and naïve CD8+ T cells (TCRβ+ CD44lo tdTom− CD8+) were FACS-sorted from the splenic lymphocytes of OxTom mice. b 1 × 106 FACS-sorted CD4+ or CD8+ OxTom T cells were adoptively transferred to TCRα−/− recipient mice by i.v. injection, and recipient mice were infected with 1 × 105 cfu LCMV Armstrong by i.p. injection 24 h post-adoptive transfer. c Eight days post-infection, splenic lymphocytes were harvested and analyzed for tdTom expression among effector (CD44hi) cells (middle panel). CD8αβ expression was assessed among CD44hi tdTom+ cells (bottom panel). n = 5 from 2 independent experiments for (a), and n = 2 from 1 experiment for (b) and (c). Statistical significance was determined by the Mann–Whitney U test; *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant. Adoptive transfer of serially-enriched CD4+ T cells (d) Naïve CD4+ T cells (TCRβ+ CD44− CD4+) were FACS-sorted once (single sort; shown in black) or twice (double sort; shown in periwinkle) from the splenic lymphocytes of Rosa26mT/mG (mT/mG) mice. Cells were enriched from the live donor population (Live-dead− tdTom+; tdTom is a constitutive, ubiquitous label). 1 × 106 naïve CD4+ T cells were adoptively transferred to TCRα−/− recipient mice by i.v. injection, and recipient mice were infected with 1 × 105 cfu LCMV Armstrong by i.p. injection. e Eight days post-infection, splenic lymphocytes were harvested and analyzed for CD4/CD8 expression among effector LCMV/MHCII-binding (CD44hi TCRβ+ LCMVgp66-77-I-Ab+) live donor T cells. % CD4− LCMVgp66-77-I-Ab+ cells among CD44hi TCRβ+ live donor cells from single-sort vs. double-sort recipients is shown in (f). n = 4 from 1 experiment. Statistical significance was determined by the Mann–Whitney U test; *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant
We did note that ≥30% of LCMV-I-Ab-recognizing CD4− cells were tdTom− (Fig. 4a). This population likely resulted from poor labeling efficiency in weakly stimulated CD4+ T cells.47 However, the simultaneous generation of CD44hiCD4-tdTom− and CD44hiCD4−tdTom+ cells could arise from the combination of 2 other possibilities: (1) a population of developmentally derived, CD8+ MHC II-restricted T cells was present, which did not efficiently induce Ox40 promoter-controlled transcription upon activation (tdTom− cells), and (2) another population of developmentally derived, CD8+ MHC II-restricted T cells was present, which was permissive to OX40 expression and tdTom labeling upon activation (tdTom+ cells), perhaps due to high affinity TCRs. To exclude these possibilities, we adoptively transferred FACS-purified naïve (CD44lotdTom−) CD4+ or CD8+ T cells from OxTom mice into T cell-deficient TCRα−/− hosts and subsequently challenged the hosts with LCMV Armstrong (Fig. 4b). Among transferred CD4+ T cells, ~20% of CD44hi cells were tdTom+. The majority of these tdTom+ cells exhibited the conventional CD4+ T cell phenotype, but a significant proportion exhibited either a CD4+CD8+ or CD4−CD8αβ+ phenotype (Fig. 4c). While a large portion of transferred CD8+ T cells were efficiently activated by LCMV (CD44hi), they were rarely tdTom+ (Fig. 4c). We therefore reasoned that, even if developmentally derived CD8+ LCMV/I-Ab-recognizing T cells were present and could permit OX40 expression upon activation, they could not completely account for the CD4-tdTom+ cells we detected in CD4+ T cell recipient mice. To completely exclude the potential confounding effect of contamination by preexisting CD8+ T cells, we employed a more rigorous purification procedure by conducting serial FACS enrichment48 of donor CD4+ T cells. Naïve CD4+ T cells, enriched to 100% purity (Fig. 4d) by either single-sorting or double-sorting, were adoptively transferred into TCRα−/− recipient mice in parallel. After LCMV infection, CD4−CD8αβ+ donor T cells were detected at a similar frequency in singly sorted vs. doubly sorted CD4+ T cell recipients (Fig. 4e, f). This population was found specifically among CD44hi-int LCMV-I-Ab+ T cells (Fig. S7). This suggested that contamination from preexisting CD8+ T cells was unlikely to be the source of the CD4−CD8αβ+ LCMV-I-Ab-recognizing T cell population. These 3 independent lineage tracing approaches strengthened our conclusion that effector differentiation of CD4+ T cells could indeed drive the generation of CD4−CD8αβ+ MHC II-recognizing T cells.
Cytotoxic potential is enhanced in CD4−CD8αβ+ MHC II-recognizing T cells
We next examined whether the shift in CD4/CD8 surface marker expression corresponded with a shift in functional molecule expression. Using in vitro-generated effector T cells, we found that both LLO WT and LLO Vps34KO TCRVα2+ CD4− T cells downregulated the surface expression of the costimulatory molecules CD40L and Ox40 (Fig. 5a, b), suggesting a reduced capacity for T helper function. Concomitantly, these cells upregulated the production of granzyme B and IFN-γ, which are hallmark cytotoxic mediators of CD8+ T cells (Fig. 5c). Indeed, we also found that CD4− LCMV-I-Ab-recognizing effector T cells generated in C57BL/6 mice during LCMV Armstrong infection expressed elevated levels of granzyme B and IFN-γ (Fig. 5d). These results indicated that CD4−CD8αβ+ MHC II-recognizing T cells shifted towards the functional expression phenotype of conventional effector CD8+ T cells.
Fig. 5.
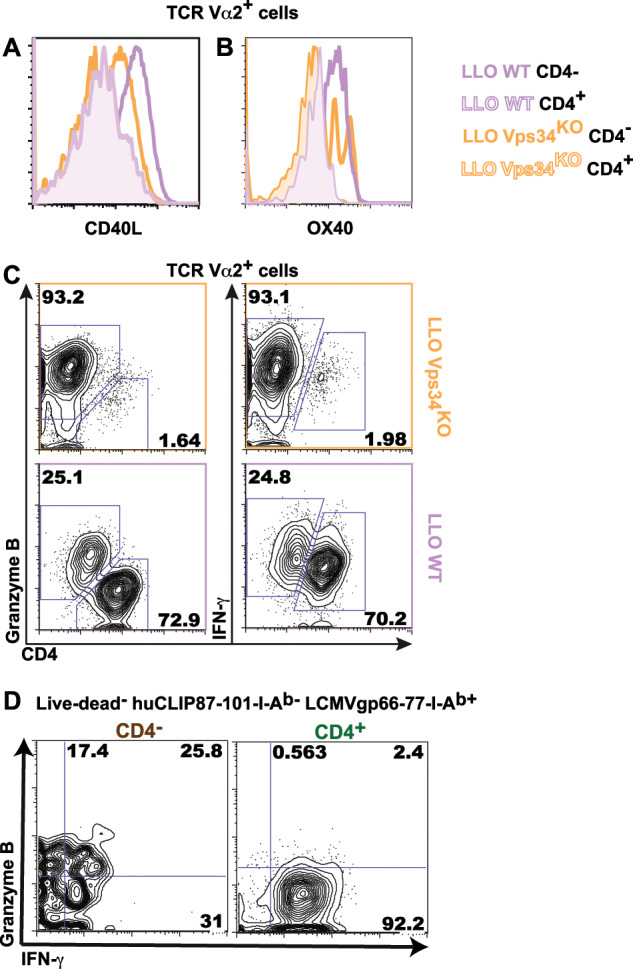
CD4−CD8αβ+ T cells suppress T helper function and gain enhanced cytotoxic function. a–c Total lymph node cells from LLO WT or LLO Vps34KO mice were stimulated in vitro with LLO190-205 for 120 h. Surface expression of CD40L (a) and Ox40 (b) and intracellular expression of granzyme B/IFN-g (c) was analyzed in TCR Vα2+ CD4− and TCR Vα2+ CD4+ cells. d Splenic lymphocytes from C57BL/6 mice infected with LCMV Armstrong for 7–12 days were harvested and analyzed for intracellular granzyme B/IFN-g levels among LCMVgp66-77-I-Ab+ CD44hi CD4− and CD4+ T cells. n = 2 from 2 independent experiments for (a) and (b), n = 5 from 2 independent experiments for (c) and n = 12 from 3 independent experiments for (d)
CD4−CD8αβ+ MHC II-recognizing T cells exhibit a CD8+ T cell-like transcriptional program
We then proceeded to determine whether the CD4−CD8αβ+ T cell phenotype was underpinned by alterations in lineage transcriptional programming. We analyzed in vitro-generated LLO WT/LLO Vps34KO CD4− and CD4+ effector T cells for the expression of 16 signature genes specifying the CD4+ vs. CD8+ lineage (Table S2). With samples from 8 independent experiments, unsupervised clustering analysis of overall similarity profiles indicated that the transcriptional programs of CD4− vs. CD4+ effector T cells were determined by lineage identity rather than by Vps34 expression (Fig. 6a). That is, the profiles of LLO WT CD4− T cells vs. LLO Vps34KO CD4− T cells correlated more closely than LLO WT CD4− T cells vs. LLO WT CD4+ T cells or LLO Vps34KO CD4− T cells vs. LLO Vps34KO CD4+ T cells. Using in vitro-primed pMel-1 effector CD8+ T cells as a CD8+ lineage standard and using LLO WT effector CD4+ T cells as a normalization standard, we analyzed these genes in three categories (Fig. 6b): (1) Lineage identity: CD4 expression was profoundly silenced in both LLO WT and LLO Vps34KO CD4− T cells, while suppression of the CD8α/β loci was released; (2) Effector molecules: expression of granzyme A, granzyme B and perforin in both LLO WT and LLO Vps34KO CD4− T cells reached levels similar to those found in effector CD8+ T cells; (3) Master transcription factors: CD4− T cells acquired a unique ThPOKlo-intRunx3hi profile, reflecting both their CD4+ origin and CD8+-oriented lineage transition. Taken together, our transcriptional data demonstrated that lineage conversion to CD4−CD8αβ+ T cells was associated with a fundamental lineage switch at the transcription level.
Fig. 6.

Effector CD4− MHCII-recognizing T cells shift towards CD8+ lineage transcriptional programming. Total lymph node cells from LLO WT and LLO Vps34KO mice were stimulated with LLO190-205 in vitro for 120 h. Effector CD4− T cells (TCR Vα2+ CD4−) and conventional effector CD4+ T cells (TCR Vα2+ CD4+) were FACS-sorted and expression of 16T cell lineage-specific transcripts was analyzed by qPCR. a Heatmap representation of Pearson product-moment correlation coefficients for the 4 sample groups. b mRNA expression levels for each target are presented relative to expression levels in LLO WT effector CD4+ T cells. Targets are grouped based on function (Lineage identity, Effector molecules or Master Txn factors). Expression levels in in vitro-stimulated pMel-1 CD8+ T cells were also assessed for a CD8+ lineage reference. Primer sequences are listed in Table S2. n = 9 from 4 independent experiments. Y-axis label shown for Lineage Identity also applies to Effector Molecules and Master Txn Factors. Statistical significance was determined by the Mann–Whitney U Test; *p < 0.05, **p < 0.01, ***p < 0.001, n.s. = not significant
Autophagosome formation regulates effector CD4+ T cell lineage stability
Given that Vps34 is a key enzyme during autophagosome formation,49 and because LLO WT effector CD4− T cells were generated less frequently than LLO Vps34KO effector CD4− T cells, we speculated that the general machinery of autophagy regulated lineage conversion of effector CD4+ T cells to the CD4− phenotype. We FACS-purified naïve CD4+ T cells from Atg7f/f Lck-Cre (Atg7KO) mice50 that harbored a T cell-specific deletion of Atg7, a critical regulator of autophagosome formation that operates downstream of Vps34. Similar to the LLO Vps34KO cells, in vitro-stimulated Atg7KO CD4+ T cells markedly lost CD4 surface expression (Fig. 7a). Again, the absence of intracellular CD4 accumulation indicated that downregulated CD4 expression was not due to impaired recycling. Atg7KO CD4− effector T cells exhibited a variety of CD8α/β surface phenotypes, including the CD4−CD8αβ+ phenotype (Fig. 7b). Similar to the LLO Vps34KO setting, CD8αβ expression was less frequent in Atg7KO CD4− T cells than in their WT counterparts. Based on a typical FACS-sort purity of 99% and a poststimulation CD4− cell frequency of ~80%, Atg7KO CD4−CD8αβ+ effector T cells were generated at ~0.5% efficiency, and WT CD4−CD8αβ+ effector T cells were generated at ~1.5% efficiency. In comparison, LLO Vps34KO CD4−CD8αβ+ effector T cells were generated at ~1% efficiency, and LLO WT CD4−CD8αβ+ effector T cells were generated at ~12% efficiency, suggesting that both the LLO118 TCR and Vps34 deficiency promote the CD4−CD8αβ+ phenotype via both autophagy-dependent and autophagy-independent mechanisms.
Fig. 7.

Autophagy regulates CD4-CD8αβ+ T cell lineage conversion. CD4+ T cells were enriched from the total lymph nodes of Atg7f/f Lck-Cre (Atg7KO) mice by magnetic separation or FACS sorting and stimulated in vitro using plate-bound anti-CD3/CD28 (1 μg/ml) for 120 h. a Surface/intracellular expression of CD4 was assessed in unstimulated naive T cells (0 h, TCRβ+CD44lo, shown in gray) vs. stimulated cells (120 h, TCRβ+CD44hi, shown in blue). b Upregulation of CD8αβ was assessed in the TCRβ+CD44hiCD4− sub-population of in vitro-stimulated Atg7f/f (WT) cells vs. Atg7KO cells. c WT and Atg7KO effector CD4− and CD4+ T cells were generated by in vitro stimulation for 120 h and enriched by magnetic separation/FACS sorting. Transcriptional analysis of 16T cell lineage-specific transcripts was performed in the same manner as for LLO WT/LLO Vps34KO cells. Data presented are from 1 of 2 independent experiments, with error bars representing standard deviations of experimental triplicates. n = 3 from 3 independent experiments for (a) and (b), and n = 2 from 2 independent experiments for (c). Y-axis label shown for Lineage Identity also applies to Effector Molecules and Master Txn Factors
In a similar vein, skewing towards the CD4−CD8αβ+ T cell lineage in Atg7KO cells was evident at the transcriptional level, where we observed a pattern similar to the Vps34-deficient transcriptional program: suppression of CD4 and ThPOK expression, accompanied by enhanced expression of signature genes normally active in effector CD8+ T cells, including CD8α, CD8β, Granzyme A, Granzyme B, Perforin, and Runx3 (Fig. 7c). Notably, increased CD8α/β transcription was more pronounced in Atg7KO CD4− effector T cells vs. LLO Vps34KO T cells, yet this did not correspond with an increased frequency of CD8αβ+ Atg7KO T cells. From these results, we concluded that the autophagic activity stabilized the CD4 expression program during effector CD4+ T cell differentiation by a mechanism yet to be explored.
Discussion
We defined a novel effector CD4+ T cell-derived population that is CD4−CD8αβ+ yet MHC II-restricted and exhibits CD8+ lineage-skewed transcriptional programming and function. In addition, we showed that the generation of this population can be promoted by the suppression of autophagic function. Building upon reports of other lineage-intermediate effector populations,15–17 we believe that the existence of CD4−CD8αβ+ T cells further challenges our conventional assumptions about T cell lineage identity and its stability during T cell activation.
Using various lineage tracking methods, we have demonstrated that CD4−CD8αβ+ T cells can originate from mature naïve CD4+ T cells. However, we note that our demonstration of the CD4+ origin of CD4−CD8αβ+ T cells does not exclude the possibility that CD8+ MHC II-restricted T cells can be derived during thymic development or even that conventional CD8+ MHC I-restricted T cells can exhibit cross-reactivity with MHC II peptide epitopes. Although CD8+ MHC II-restricted T cells have yet to be reported in the thymus, it has been shown that T cells with MHC I-restricted TCRs may enter the CD4+ lineage with certain positive selecting ligands51 or impaired function of certain transcription factors.52
Furthermore, we acknowledge that our conclusions on CD4−CD8αβ+ conversion are somewhat technologically restricted: (1) for the in vitro and transfer experiments, FACS sorting could not guarantee 100% cell purity, although we aimed for a high technical stringency; (2) for the in vivo tracking experiments, the efficiency of genetic marking was not 100%; and (3) for the TCR repertoire sequencing, we could not recover every tetramer-positive cell from each animal. However, for each of the experimental strategies we employed, we propose that the probability that aberrant development could account for the majority of CD8+ MHC II-restricted T cells is extremely low, and the probability is even lower when all of our strategies are considered together. Specifically, in our adoptive-transfer lineage tracking experiment starting with purified naïve TdTomato−CD4+ T cells, 8% of TdTomato+ T cells were CD4−CD8+ after LCMV infection. Before the transfer, contamination from CD8+ T cells during FACS sorting was less than 2%, and the expression of Cre recombinase was driven by the OX40 promoter, which is strongly preferred in T cells of CD4+ origin.47 In addition, CD4− MHC II-recognizing T cells did not have a proliferative or survival advantage in comparison to bona fide CD4+ effector T cells. Furthermore, repertoire sequencing results showed that a large portion of LCMV/I-Ab-specific CD4+ and CD4− T cells shared an identical TCR sequence, and therefore, it is quite statistically improbable that all CD4− MHC II-restricted clonotypes could be amplified from a preexisting population. Taken together, we assert that the potential presence of mature, aberrant CD8+ MHC II-restricted T cells does not negate the existence of CD4−CD8αβ+ T cells generated from effector CD4+ T cells.
While the biological significance of CD4−CD8αβ+ MHC II-restricted T cells remains to be completely elucidated, we note that the lineage conversion and functionality of this population may contribute to the human antiviral response to HIV infection. Due to their lack of CD4 expression, CD4−CD8αβ+ T cells could be impervious to viral entry;53–55 in addition, due to their MHC II-mediated recognition capacity, CD4−CD8αβ+ T cells could facilitate the destruction of antigen-presenting cells harboring HIV reservoirs.56 Indeed, a previous study in African green monkeys infected with simian immunodeficiency virus (SIV), an HIV ortholog, indicated that the downregulation of CD4 expression in memory T cells is associated with antiviral protection.53 In addition, we note that autophagy plays a significant but evadable role in controlling an HIV infection, which is not yet completely understood in T cells.57 Therefore, further investigations of CD4-CD8αβ+ MHC II-recognizing T cells may indicate potential vaccine strategies for the prevention of HIV-1 infection that could circumvent previous obstacles in the development of T cell-based HIV vaccines.
Materials and methods
Mice
LLO118αβ Vps34f/f CD4-Cre mice were generated by crossing LLO118αβ mice with Vps34f/f CD4-Cre mice, which were kindly provided by Dr. You-Wen He. LLO118 Vps34f/f or LLO118 mice were used as WT controls. Rosa26tdTomato Ox40-Cre mice were generated by crossing Ox40-Cre mice with Rosa26tdTomato mice, which were kindly provided by Dr. Yuan Zhuang. Thy1.1+ C57BL/6, C57BL/6, OT-1, Pmel-1, and Rosa26mT/mG mice were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). Atg7f/f and Atg7f/f Lck-Cre mice were kindly provided by Dr. You-Wen He. In vitro experiments used 4–20-week-old male and female mice, and 6–16-week-old male and female mice were used for in vivo experiments.
In vitro T cell stimulation
For peptide-mediated stimulation, (1) 5 × 106–1 × 107 total lymph node cells or (2) FACS-sorted CD4+ T cells and CD19/B220+ B cells (T:B cell ratio = 1:1) were cultured for 5 days in complete RPMI medium (cRPMI; RPMI 1640 supplemented with 10% FBS, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 2 mM L-glutamine, 100 U/ml penicillin, 100 U/ml streptomycin, and 50 μM beta-mercaptoethanol) in the presence of 10 μM LLO190-205 peptide (LLO118 cells) or 1 μg/ml SIINFEKL peptide (OT-1 cells) in a humidified incubator at 37 °C/7% CO2. For antibody-mediated stimulation, 1 × 106 FACS-sorted CD4+ T cells were cultured for 5 days in cRPMI in the presence of plate-bound anti-CD3/CD28 antibody (0.1–1 μg/ml) under Th1-skewing conditions (mouse/human recombinant IL-2 (10 ng/ml), mouse recombinant IL-12 (50 ng/ml) and anti-mouse IL-4 (11B11, 10 μg/ml)) in a humidified incubator at 37 °C/7% CO2. For cell proliferation analysis, cells were stained with Cell TraceTM Violet (0.5–5 μM) or CFSE (0.5–10 μM) per the manufacturer’s protocol immediately prior to in vitro stimulation.
In vivo infection
For Listeria studies, mice were infected with 1 × 107 cfu Listeria monocytogenes by intravenous (i.v.) injection. Four days postinfection, the spleen was harvested and prepared for flow cytometry analysis (tissue homogenization by mechanical disruption followed by red blood cell lysis using ACK lysis buffer). For lymphocytic choriomeningitis (LCMV) studies, mice were infected with 1 × 105 cfu LCMV Armstrong by intraperitoneal (i.p.) injection. Seven to 12 days postinfection, the spleen was harvested and prepared for flow cytometry analysis. For adoptive transfer studies, naïve CD4+ and CD8+ T cells from total lymph nodes were FACS-sorted and transferred to recipient mice by i.v. injection (1 × 106 cells/mouse). Zero or 24 h posttransfer, recipient mice were infected with 1 × 105 cfu LCMV Armstrong by i.p. injection. Seven to eight days postinfection, the spleen was harvested and prepared for flow cytometry analysis.
Flow cytometry
For mouse cells, lymphocyte samples were suspended in FACS buffer (DPBS supplemented with 2% FBS and 2 mM EDTA). For surface marker analysis, samples were treated with Fc receptor blocking antibody (0.0078 μg/μl) for 10 min at 4 °C followed by surface antibodies (1:100–500 dilution) for 15–30 min at 4 °C. For cell survival analysis, after surface staining, cells were resuspended in Annexin V binding buffer (10 mM HEPES, 140 mM sodium chloride and 2.5 mM calcium chloride in diH2O) supplemented with 2% FBS and then treated with 7-AAD Viability Staining Solution (1:40 dilution) for 15 min at room temperature (rm temp). For dead-cell exclusions, cells were stained with Live/Dead Fixable Dead Cell Stain per the manufacturer’s protocol (either before or after surface staining). For intracellular cytokine analysis, lymphocytes were stimulated for 4 h at 37 °C/7% CO2 with phorbol 12, 13-dibutyrate (PDBu, 0.9 μM) and ionomycin (1 ng/μl) in the presence of brefeldin and monensin (1000× solutions, eBioscience, Inc., San Diego, CA, USA). Intracellular staining was then performed by fixation (paraformaldehyde, 2%) for 20 min at room temperature, cell permeabilization (saponin, 0.1%) and Fc receptor blocking (2.4G2, 0.0078 μg/μl) for 10 min at room temperature, and antibody treatment (1:100–500 dilution) was performed for 15 min at 4 °C. All flow cytometry data were acquired using a FACSCanto II (BD Biosciences) and analyzed using FlowJo software (FlowJo, LLC, Ashland, OR, USA).
Antibodies/staining solutions
Biolegend (San Diego, CA, USA)
7AAD Viability Staining Solution
Annexin V: APC.
Anti-mouse B220 (RA3-6B2): FITC.
Anti-mouse CD4 (GK1.5): FITC, PE, PE/Cy7, APC, Pacific Blue.
Anti-mouse CD8α (53-6.7): FITC, PE/Cy7, APC.
Anti-mouse CD8β (YTS156.7.7): FITC, PE, PerCP/Cy5.5.
Anti-mouse/human CD44 (IM7): PE/Cy5, APC/Cy7.
Anti-mouse/human granzyme B (GB11): Alexa Fluor 647®.
Anti-mouse IFN-γ (XMG1.2): Pacific Blue.
Anti-mouse TCRβ (H57-597): PE, PerCP/Cy5.5, APC.
Anti-mouse TCRVα2 (B20.1): APC, APC-Cy7.
Bio X Cell (West Lebanon, NH, USA)
Anti-mouse CD16/32 (2.4G2).
Life Technologies (Grand Island, NY, USA)
Cell TraceTM CFSE Cell Proliferation Kit.
Cell TraceTM Violet Cell Proliferation Kit.
Thermo Fisher Scientific (Waltham, MA, USA)
Live/Dead Fixable Dead Cell Stain: Green, Red, Far Red, Aqua, and Violet.
Tetramer binding analysis
For mouse tetramer binding analysis, cells were pretreated with the Fc receptor blocking antibody (Flow cytometry method). Cells were then stained with an MHC II-matched LCMV tetramer (LCMVgp31-45-I-Ab BV 421, LCMVgp66-77-I-Ab BV 421 and LCMVgp126-140-I-Ab BV 421; 6 μg/ml; NIH Tetramer Core Facility, Atlanta, GA, USA) and control tetramer (huCLIP87-101-I-Ab PE; 6 μg/ml; NIH Tetramer Core Facility) for 1 h at room temperature followed by surface staining and flow cytometry analysis (flow cytometry method).
TCRβ sequencing
Splenic lymphocytes were harvested from Thy1.1+ C57Bl/6 mice 7 days postinfection with LCMV Armstrong (in vivo infection method). Cells were prepared for FACS sorting (Flow Cytometry and tetramer binding analysis methods), and Live-Dead− huCLIP87-101- I-Ab− LCMVgp66-77- I-Ab+ CD44hi CD4− and CD4+ populations were collected (3.5 × 103-2 × 104 cells/sample; Table S1). FACS-sorted populations were then spiked with 5 × 102 2B4.11 cells as a normalization control. Populations with <2 × 104 cells were supplemented with LB27.4 cells to bring the total cell number to 2 × 104. Samples were then lysed, and RNA was extracted using the Direct-Zol RNA Isolation Kit (Genesee Scientific, San Diego, CA, USA) per the manufacturer’s protocol. RNA (total sample amount) was reverse-transcribed into single-stranded cDNA using the qScript Flex cDNA Synthesis Kit (Quanta Biosciences, Inc., Gaithersburg, MD, USA) and 1 μM TCRβ-specific primer (5′-ATCTCTGCTTCTGATGGCTCA-3′). The PCR program was as follows: 65 °C (5 min (min)) followed by 25 °C (5 min), 42 °C (60 min), and 70 °C (5 min). Whole-TCRβ sequencing58 was performed using the Ion Torrent PGM platform (Life Technologies), and sequencing analyses were conducted using an in-house designed software.
qPCR
T cell samples (2 × 104–2 × 106 cells) were enriched by (1) magnetic separation using the Dynabeads Untouched Mouse CD4/CD8 Cells Kits or Dynabeads Flowcomp Mouse CD4/CD8 Kits per the manufacturer’s protocol (ThermoFisher Scientific) or (2) FACS-sorting (flow cytometry method). Cell lysis and RNA extraction were performed using the Direct-Zol RNA Isolation Kit (Genesee Scientific) or the RNAqueous-Micro Total RNA Isolation Kit (ThermoFisher Scientific) per the manufacturer’s protocol. RNA (200–500 ng) was reverse-transcribed into single-stranded cDNA using oligo-dTs/random primers and the qScript Flex cDNA Synthesis Kit (Quanta Biosciences, Inc.) per the manufacturer’s protocol. qPCR was then conducted using in-house-designed primers and the SYBR Green Perfecta Supermix (Quanta Biosciences, Inc.) on a Mastercycler ep realplex2 S real-time PCR system (Eppendorf North America, Hauppauge, NY, USA). Primer sequences are listed in Table S2. The PCR was as follows: 95 °C (2 min) followed by 40–50 cycles at 95 °C (15 s), 50–70 °C (30 s) and 68 °C (25 s). Melting curve analysis was performed to confirm that primer-dimer amplification did not occur. Relative expression analysis was conducted by the delta-delta Ct method using in-house-designed qPCR analysis software.
Statistics
Mann–Whitney U tests were conducted using GraphPad Prism (GraphPad Software, Inc., La Jolla, CA, USA). Pearson product-moment correlation coefficients and all statistical tests for TCRβ sequencing analyses were performed using in-house-designed software.
Study approval
All husbandry and experimental procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and protocols were approved by the Duke University Institutional Animal Care and Use Committee (IACUC).
Supplementary information
Acknowledgements
We thank the NIH Tetramer Core Facility for providing tetramer reagents. We thank Dr. Regina Lin for assistance with qPCR experiments, Jose Sevilla for assistance with mouse husbandry and Dr. Marthony Robins for helpful comments on our paper. We thank Dr. Fan Wang for mice and expertize on Vps34 studies. Funding for this investigation was provided by a Duke University Center for AIDS Research (CFAR) Small Grant (Q.-J.L.), National Institutes of Health (NIH) Training Grants (2 T32 AI 052077–11 and 5 T32 AI 052077–09) (E.R.) and an American Association of Immunologists (AAI) Careers in Immunology Fellowship (Q.-J.L. and E.R.). This work was supported by Duke University Center for AIDS Research (CFAR) Small Grant (Q.-J.L.), National Institutes of Health (NIH) Training Grants (2 T32 AI 052077–11 and 5 T32 AI 052077–09) (E.R.), and American Association of Immunologists (AAI) Careers in Immunology Fellowship (Q.-J.L. and E.R.).
Author contributions
E.R. designed research studies, performed experiments, acquired data, analyzed data and prepared the paper. M.Z. and Q.N. analyzed TCR repertoire data. S.L. performed murine experiments and prepared the heatmap figures. L.C. generated murine strains. B.Z. provided expertize on in vivo murine experiments and generated murine strains. J.G. generated and maintained murine strains. Y.Z. provided murine strains and expertize on murine experiments. Y.-W.H. provided expertize on Vps34/Atg7 murine studies and murine strains. P.Z. and Y.W. provided TCR sequencing equipment and administrative oversight. Q.-J.L. provided experimental guidance, provided experimental reagents/equipment, provided administrative oversight and revised the paper.
Competing interests
The authors declare no competing interests.
Supplementary information
The online version of this article (10.1038/s41423-019-0347-5) contains supplementary material.
References
- 1.Singer A, Adoro S, Park JH. Lineage fate and intense debate: myths, models and mechanisms of CD4- versus CD8-lineage choice. Nat. Rev. Immunol. 2008;8:788–801. doi: 10.1038/nri2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Xiong Y, Bosselut R. CD4-CD8 differentiation in the thymus: connecting circuits and building memories. Curr. Opin. Immunol. 2012;24:139–145. doi: 10.1016/j.coi.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Naito T, Taniuchi I. The network of transcription factors that underlie the CD4 versus CD8 lineage decision. Int. Immunol. 2010;22:791–796. doi: 10.1093/intimm/dxq436. [DOI] [PubMed] [Google Scholar]
- 4.Liang XH, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 5.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol. Biol. Cell. 2008;19:5360–5372. doi: 10.1091/mbc.E08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komatsu M, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. J. Cell Biol. 2005;169:425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ichimura Y, et al. A ubiquitin-like system mediates protein lipidation. Nature. 2000;408:488–492. doi: 10.1038/35044114. [DOI] [PubMed] [Google Scholar]
- 8.Botbol Y, Guerrero-Ros I, Macian F. Key roles of autophagy in regulating T-cell function. Eur. J. Immunol. 2016;46:1326–1334. doi: 10.1002/eji.201545955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McLeod IX, Zhou X, Li QJ, Wang F, He YW. The class III kinase Vps34 promotes T lymphocyte survival through regulating IL-7Ralpha surface expression. J. Immunol. 2011;187:5051–5061. doi: 10.4049/jimmunol.1100710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc. Natl Acad. Sci. USA. 2012;109:8670–8675. doi: 10.1073/pnas.1205305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Parekh VV, et al. Impaired autophagy, defective T cell homeostasis, and a wasting syndrome in mice with a T cell-specific deletion of Vps34. J. Immunol. 2013;190:5086–5101. doi: 10.4049/jimmunol.1202071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Djuretic IM, et al. Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat. Immunol. 2007;8:145–153. doi: 10.1038/ni1424. [DOI] [PubMed] [Google Scholar]
- 13.Appay V, et al. Characterization of CD4(+) CTLs ex vivo. J. Immunol. 2002;168:5954–5958. doi: 10.4049/jimmunol.168.11.5954. [DOI] [PubMed] [Google Scholar]
- 14.B. F Acquisition of specific cytotoxic activity by human T4+T lymphocytes in culture. Nature. 1984;308:365–367. doi: 10.1038/308365a0. [DOI] [PubMed] [Google Scholar]
- 15.Mucida D, et al. Transcriptional reprogramming of mature CD4(+) helper T cells generates distinct MHC class II-restricted cytotoxic T lymphocytes. Nat. Immunol. 2013;14:281–289. doi: 10.1038/ni.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reis BS, Rogoz A, Costa-Pinto FA, Taniuchi I, Mucida D. Mutual expression of the transcription factors Runx3 and ThPOK regulates intestinal CD4(+) T cell immunity. Nat. Immunol. 2013;14:271–280. doi: 10.1038/ni.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boucheron N, et al. CD4(+) T cell lineage integrity is controlled by the histone deacetylases HDAC1 and HDAC2. Nat. Immunol. 2014;15:439–448. doi: 10.1038/ni.2864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukacher AE, Morrison LA, Braciale VL, Malissen B, Braciale TJ. Expression of specific cytolytic activity by H-2I region-restricted, influenza virus-specific T lymphocyte clones. J. Exp. Med. 1985;162:171–187. doi: 10.1084/jem.162.1.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams NS, Engelhard VH. Identification of a population of CD4+CTL that utilizes a perforin- rather than a Fas ligand-dependent cytotoxic mechanism. J. Immunol. 1996;156:153–159. [PubMed] [Google Scholar]
- 20.Hansen SG, et al. Cytomegalovirus vectors violate CD8+T cell epitope recognition paradigms. Science. 2013;340:1237874. doi: 10.1126/science.1237874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hansen SG, et al. Broadly targeted CD8(+) T cell responses restricted by major histocompatibility complex E. Science. 2016;351:714–720. doi: 10.1126/science.aac9475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Suni MA, et al. CD4(+)CD8(dim) T lymphocytes exhibit enhanced cytokine expression, proliferation and cytotoxic activity in response to HCMV and HIV-1 antigens. Eur. J. Immunol. 2001;31:2512–2520. doi: 10.1002/1521-4141(200108)31:8<2512::aid-immu2512>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 23.Fu J, et al. Impairment of CD4+cytotoxic T cells predicts poor survival and high recurrence rates in patients with hepatocellular carcinoma. Hepatology. 2013;58:139–149. doi: 10.1002/hep.26054. [DOI] [PubMed] [Google Scholar]
- 24.Broux B, et al. CX(3)CR1 drives cytotoxic CD4(+)CD28(-) T cells into the brain of multiple sclerosis patients. J. Autoimmun. 2012;38:10–19. doi: 10.1016/j.jaut.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 25.Pawlik A, et al. The expansion of CD4+CD28- T cells in patients with rheumatoid arthritis. Arthritis Res. Ther. 2003;5:R210–R213. doi: 10.1186/ar766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Leeuwen EM, et al. Emergence of a CD4+CD28- granzyme B+, cytomegalovirus-specific T cell subset after recovery of primary cytomegalovirus infection. J. Immunol. 2004;173:1834–1841. doi: 10.4049/jimmunol.173.3.1834. [DOI] [PubMed] [Google Scholar]
- 27.Lu X, et al. Low double-negative CD3+CD4-CD8- T cells are associated with incomplete restoration of CD4 + T cells and higher immune activation in HIV-1 immunological non-responders. Front. Immunol. 2016;7:579. doi: 10.3389/fimmu.2016.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liang Q, et al. Double Negative (DN) [CD3(+)CD4(-)CD8(-)] T cells correlate with disease progression during HIV infection. Immunol. Investig. 2013;42:431–437. doi: 10.3109/08820139.2013.805763. [DOI] [PubMed] [Google Scholar]
- 29.Ribrag V, et al. Increase in double-positive CD4+CD8+peripheral T-cell subsets in an HIV-infected patient. AIDS. 1993;7:1530. doi: 10.1097/00002030-199311000-00021. [DOI] [PubMed] [Google Scholar]
- 30.Chauhan NK, Vajpayee M, Mojumdar K, Singh R, Singh A. Study of CD4+CD8+double positive T-lymphocyte phenotype and function in Indian patients infected with HIV-1. J. Med. Virol. 2012;84:845–856. doi: 10.1002/jmv.23289. [DOI] [PubMed] [Google Scholar]
- 31.Frahm MA, et al. CD4+CD8+T cells represent a significant portion of the anti-HIV T cell response to acute HIV infection. J. Immunol. 2012;188:4289–4296. doi: 10.4049/jimmunol.1103701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howe, R. et al. Phenotypic and functional characterization of HIV-1-specific CD4+CD8+double-positive T cells in early and chronic HIV-1 infection. J. AIDS. 50, 444–456 (2009). [DOI] [PubMed]
- 33.Kaiser P, et al. Productive human immunodeficiency virus type 1 infection in peripheral blood predominantly takes place in CD4/CD8 double-negative T lymphocytes. J. Virol. 2007;81:9693–9706. doi: 10.1128/JVI.00492-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cheney KM, et al. HIV type 1 persistence in CD4- /CD8- double negative T cells from patients on antiretroviral therapy. AIDS Res. Hum. Retroviruses. 2006;22:66–75. doi: 10.1089/aid.2006.22.66. [DOI] [PubMed] [Google Scholar]
- 35.Marodon G, Warren D, Filomio MC, Posnett DN. Productive infection of double-negative T cells with HIV in vivo. Proc. Natl Acad. Sci. USA. 1999;96:11958–11963. doi: 10.1073/pnas.96.21.11958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeMaster LK, et al. A subset of CD4/CD8 double-negative T cells expresses HIV proteins in patients on antiretroviral therapy. J. Virol. 2015;90:2165–2179. doi: 10.1128/JVI.01913-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Restrepo C, et al. HIV Gag-specific immune response mediated by double negative (CD3(+)CD4(−)CD8(−)) T cells in HIV-exposed seronegative individuals. J. Med. Virol. 2013;85:200–209. doi: 10.1002/jmv.23447. [DOI] [PubMed] [Google Scholar]
- 38.Petitjean G, et al. Level of double negative T cells, which produce TGF-beta and IL-10, predicts CD8 T-cell activation in primary HIV-1 infection. AIDS. 2012;26:139–148. doi: 10.1097/QAD.0b013e32834e1484. [DOI] [PubMed] [Google Scholar]
- 39.Margolick JB, et al. Development of antibodies to HIV-1 is associated with an increase in circulating CD3 + CD4-CD8- lymphocytes. Clin. Immunol. Immunopathol. 1989;51:348–361. doi: 10.1016/0090-1229(89)90033-0. [DOI] [PubMed] [Google Scholar]
- 40.Ranasinghe S, et al. Antiviral CD8(+) T cells restricted by human leukocyte antigen Class II exist during natural HIV infection and exhibit clonal expansion. Immunity. 2016;45:917–930. doi: 10.1016/j.immuni.2016.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weber KS, et al. Distinct CD4+helper T cells involved in primary and secondary responses to infection. Proc. Natl Acad. Sci. USA. 2012;109:9511–9516. doi: 10.1073/pnas.1202408109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem. J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 43.D'Acquisto F, Crompton T. CD3+CD4-CD8- (double negative) T cells: saviours or villains of the immune response? Biochem. Pharmacol. 2011;82:333–340. doi: 10.1016/j.bcp.2011.05.019. [DOI] [PubMed] [Google Scholar]
- 44.Parel Y, Chizzolini C. CD4+CD8+double positive (DP) T cells in health and disease. Autoimmun. Rev. 2004;3:215–220. doi: 10.1016/j.autrev.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 45.Cheroutre, H., Husain, M. M. CD4 CTL: living up to the challenge. Semin. Immunol. 25, 273–281 (2013). [DOI] [PMC free article] [PubMed]
- 46.Graw F, Weber KS, Allen PM, Perelson AS. Dynamics of CD4(+) T cell responses against Listeria monocytogenes. J. Immunol. 2012;189:5250–5256. doi: 10.4049/jimmunol.1200666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klinger M, et al. Thymic OX40 expression discriminates cells undergoing strong responses to selection ligands. J. Immunol. 2009;182:4581–4589. doi: 10.4049/jimmunol.0900010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheah MT, et al. CD14-expressing cancer cells establish the inflammatory and proliferative tumor microenvironment in bladder cancer. Proc. Natl Acad. Sci. USA. 2015;112:4725–4730. doi: 10.1073/pnas.1424795112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yue Z, Zhong Y. From a global view to focused examination: understanding cellular function of lipid kinase VPS34-Beclin 1 complex in autophagy. J. Mol. Cell Biol. 2010;2:305–307. doi: 10.1093/jmcb/mjq028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 2009;182:4046–4055. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- 51.Yasutomo K, Doyle C, Miele L, Fuchs C, Germain RN. The duration of antigen receptor signalling determines CD4+versus CD8+T-cell lineage fate. Nature. 2000;404:506–510. doi: 10.1038/35006664. [DOI] [PubMed] [Google Scholar]
- 52.Sakaguchi S, et al. The zinc-finger protein MAZR is part of the transcription factor network that controls the CD4 versus CD8 lineage fate of double-positive thymocytes. Nat. Immunol. 2010;11:442–448. doi: 10.1038/ni.1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Beaumier CM, et al. CD4 downregulation by memory CD4+T cells in vivo renders African green monkeys resistant to progressive SIVagm infection. Nat. Med. 2009;15:879–885. doi: 10.1038/nm.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Douek DC, et al. HIV preferentially infects HIV-specific CD4+T cells. Nature. 2002;417:95–98. doi: 10.1038/417095a. [DOI] [PubMed] [Google Scholar]
- 55.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003;300:339–342. doi: 10.1126/science.1083317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.de Jong MA, Geijtenbeek TB. Human immunodeficiency virus-1 acquisition in genital mucosa: Langerhans cells as key-players. J. Intern. Med. 2009;265:18–28. doi: 10.1111/j.1365-2796.2008.02046.x. [DOI] [PubMed] [Google Scholar]
- 57.Dinkins C, Pilli M, Kehrl JH. Roles of autophagy in HIV infection. Immunol. Cell Biol. 2015;93:11–17. doi: 10.1038/icb.2014.88. [DOI] [PubMed] [Google Scholar]
- 58.Jia Q, et al. Diversity index of mucosal resident T lymphocyte repertoire predicts clinical prognosis in gastric cancer. Oncoimmunology. 2015;4:e1001230. doi: 10.1080/2162402X.2014.1001230. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.