

Abstract
Enhancer of zeste homolog 2 (EZH2), a key histone methyltransferase and EMT inducer, is overexpressed in diverse carcinomas, including breast cancer. However, the molecular mechanisms of EZH2 dysregulation in cancers are still largely unknown. Here, we discover that EZH2 is asymmetrically dimethylated at R342 (meR342-EZH2) by PRMT1. meR342-EZH2 was found to inhibit the CDK1-mediated phosphorylation of EZH2 at T345 and T487, thereby attenuating EZH2 ubiquitylation mediated by the E3 ligase TRAF6. We also demonstrate that meR342-EZH2 resulted in a decrease in EZH2 target gene expression, but an increase in breast cancer cell EMT, invasion and metastasis. Moreover, we confirm the positive correlations among PRMT1, meR342-EZH2 and EZH2 expression in the breast cancer tissues. Finally, we report that high expression levels of meR342-EZH2 predict a poor clinical outcome in breast cancer patients. Our findings may provide a novel diagnostic target and promising therapeutic target for breast cancer metastasis.
Subject terms: Metastasis, Tumour biomarkers, Ubiquitylation, Gene regulation
Introduction
Enhancer of Zeste Homolog 2 (EZH2), a component of Polycomb Repressive Complex 2 (PRC2), is an important oncogene in many cancers, including breast cancer [1–4]. EZH2 suppresses the transcription of its target genes through catalyzing the trimethylation of Lys-27 in histone H3 (H3K27me3) by its SET domain, which facilitates cancer cells EMT (epithelial–mesenchymal transition) and metastasis [3, 5, 6]. High expression of EZH2 have been revealed in many cancers, especially breast cancer [4]. The mechanistic details of EZH2 upregulation in cancers, however, remain largely unknown.
Several studies have indicated that post-translational modifications (PTMs) of EZH2 are crucial to enhance its protein stability in cancer cells and for its function in regulating cancer metastasis [7–13]. Earlier research showed that the cyclin-dependent kinase1 (CDK1)-mediated phosphorylation of EZH2 at Thr-345 and Thr-487 (T345 and T487) promotes EZH2 ubiquitination [12]. Recent study revealed that the PCAF-mediated acetylation of EZH2 at Lys-348 (AcK348-EZH2) maintains EZH2 stability in lung cancer patients [10]. Thus, EZH2-PTMs potentiate EZH2 accumulation in cancer by regulating EZH2 protein stability, which may play a critical role in cancer metastasis.
Protein arginine methylation, which is catalyzed by a family of enzymes termed protein arginine methyltransferases (PRMTs), is a major PTM that has a key role in cancer development and metastasis [14–17]. Protein arginine methyltransferase 1 (PRMT1), the predominant type I PRMT, catalyzes the formation of asymmetric dimethylarginines (ADMAs) in its substrates [14, 15]. It has been reported that 85% of ADMA formation in mammalian cells is performed by PRMT1 [18]. Recently, many studies have indicated that PRMT1-mediated ADMAs formation in a series of nonhistone substrates are involved in various biological processes, including cancer tumorigenesis and metastasis [19–24]. For instance, PRMT1 increases Axin stability by methylating Axin at R378 and decreasing its ubiquitin degradation [25]. Thus, the ADMA modification has an important effect on protein stability. However, whether the stability of EZH2 is regulated by arginine methylation has been far less reported on.
In this study, we found that EZH2 is asymmetrically dimethylated at R342 by PRMT1, which inhibits CDK1 binding and phosphorylates EZH2 at T345 and T487, attenuating the interaction of TRAF6 with EZH2 and subsequent EZH2 degradation. Moreover, we disclose that meR342-EZH2 is necessary for EZH2-mediated breast cancer cells EMT, invasion and metastasis. Consistently, we also show the significant overexpression of PRMT1, meR342-EZH2 and EZH2 in breast cancer metastasis samples. Finally, we reveal that high expression of meR342-EZH2 results in a poor clinical outcome in breast cancer patients. Our findings may provide a basis for the development of a potential therapeutic strategy for breast cancer metastasis through targeting meR342-EZH2.
Materials and methods
Cell culture and treatment
MCF10A (a human normal mammary epithelial cell line) cells were cultured in DMEM/F12 medium with 5% horse serum (Gibco, Grand Island, NY, USA), 20 ng/ml EGF (R&D), 0.5 mg/ml hydrocortisone (Sigma), 100 ng/ml cholera toxin (Sigma), 10 mg/ml insulin (Gibco) and pen/strep. MDA-MB-231 cells were cultured in L-15 medium with 10% FBS at 37 °C without CO2. MCF7, T47D, BT549, BT474, MDA-MB-436 and ZR-75-1 cells were cultured in RPMI-1640 medium with 10% FBS. HEK293T, SKBR3, SUM1315, 4T1-luc cells were cultured in DMEM medium with 10% FBS. All the cell lines were obtained from the American Type Culture Collection or Cell library of the Chinese Academy of Sciences.
Generation of stable cells using lentivirus
Lentiviruses were produced by co-transfecting HEK293T cells with one of the expression plasmids and the packaging plasmids (psPAX2 and pMD2.G). The details of Generation of stable cells using lentivirus were shown in Supplementary Information.
RNA extraction, reverse transcription and qRT-PCR
These relevant experiments were carried out according to the protocol practically used in our laboratory [5]. The sequences of qRT-PCR primers are listed in Supplementary Information.
Western blotting, immunoprecipitation and co-immunoprecipitation assays
Western blotting, immunoprecipitation (IP) and co-immunoprecipitation (Co-IP) were performed as described previously [5]. The details of antibodies (vendor and catalog number) are shown in Supplementary Information.
Immunofluorescence (IF)
The protocol of Immunofluorescence is shown in Supplementary Information.
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed using the Simple ChIP Enzymatic Chromatin IP Kit (Cell Signaling Technology Cat# 9004) according to the manufacturer’s protocol. The primers used to amplify the target gene promoters are shown in Supplementary Information.
GST pull-down assay and in vitro methylation assay
GST or GST-PRMT1 and GST-EZH2 fragmental proteins were expressed in bacteria (BL21) induced with IPTG (isopropyl-β-D-thio-galactoside) and then purified, as described previously [10]. The detailed protocols of in vitro methylation assay were shown in Supplementary Information.
Mass spectrometry (MS) analysis
The details of EZH2 sample preparation for methylation detection by MS analysis were shown in Supplementary Information. Liquid chromatography-tandem mass spectrometry analysis (LC-MS/MS) performed in the APPLIED PROTEIN TECHNOLOGY, Shanghai, China.
Antibody generation and detection
The anti-ADMA-R342-EZH2 (anti-meR342-EZH2) antibody was raised against the region near R342 asymmetric di-methylarginine site of EZH2. The asymmetric di-methylated synthetic peptide [TAE(R-Me2)IKTPPKRPGG-C] was used for immunization in the mice. The antibody was generated in Gl biochem company (Shanghai, China).
Wound healing, cell invasion and migration assays
Experiments for determination of wound healing assay, cell invasion and migration assays were performed as described previously [5].
In vivo lung-colonization assays
In vivo lung-colonization assays were performed the same protocol as described previously [26].
Breast cancer patient specimens
The details of breast cancer patient specimens were shown in Supplementary Information.
Immunohistochemistry of TMA assays and evaluation of immune-staining method
Immunohistochemistry of TMA assays and evaluation of immune-staining method were performed as described previously [26]. The detailed protocols were shown in Supplementary Information.
Transfection of siRNA and expressing vectors
The details of transfection of siRNA and expressing vectors are shown in Supplementary Information.
Experimental model and subject details
Animal experiments were approved by the Animal Care and Use Committee of the Xuzhou Medical University, China. BALB/c Nude mice (6- to 8-week-old females) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd.
Molecular dynamics simulations and structure analysis
The 3D structure from PDB ID: 5HYN was employed [27]. The missing residues were rebuilt by GalaxyFill [28]. EZH2 protein was solvated in ~175,000 TIP3P water molecules with 150 mM KCl in a 192 × 192 × 192 Å box. The system was built using the CHARMM program with the CHARMM36 force field for all molecules [29]. The system was equilibrated for 55 ns using the NAMD2.12 program package under the periodic orthorhombic boundary conditions applied in all directions with the time step of 2 fs [30]. The NPT ensemble was used for both simulations with pressure at 1 atm and temperature at 310.15 K. Long-range electrostatic interactions were treated by the particle mesh Ewald (PME) algorithm and non-bonded interactions were switched off at 10–12 Å [31].
Statistical analysis
For the TMA slides, statistical analysis was performed with SPSS 20 software (SPSS, Inc, Chicago, IL). The association between meR342-EZH2 staining and the clinicopathologic parameters of the breast cancer patients was evaluated by χ2 test. The Kaplan–Meier method and log-rank test were used to evaluate the correlation between meR342-EZH2 expression and patients survival. Cox regression model was used for multivariate analysis. The Student t test was used to determine statistic significance of differences between groups. P < 0.05 was considered statistically significant. Data are presented as mean ± SEM. Statistical analysis was performed using the GraphPad Prism software (GraphPad Software, La Jolla,CA, USA).
Results
PRMT1 mediates asymmetric dimethylarginine (ADMA) formation at arginine 342 of EZH2 in vivo and in vitro
To explore whether EZH2 undergoes arginine methylation, IP and Co-IP experiments were conducted in HEK293T, MCF10A, MCF7 and MDA-MB-231 (named MM-231) cells. We found that both endogenous and exogenous EZH2 underwent ADMA modification (Fig. 1a–e). Previous studies have demonstrated that PRMT1 is the major arginine methyltransferase that mediates ADMA formation [18]. We wondered whether ADMA-EZH2 formation is mediated by PRMT1 and verified that both endogenous and exogeneous EZH2 interact with PRMT1 (Fig. 1a–e).
Fig. 1. PRMT1 interacts with and dimethylates EZH2 at arginine 342.

a–d ADMA of endogenous EZH2 and interaction of PRMT1 with endogenous EZH2 were assessed by Co-IP assays. e ADMA of exogenous EZH2 and interactions between exogenous Flag-EZH2 and His-PRMT1 were detected by Co-IP assay. f, g Purified GST-tagged PRMT1 was pulled down with the MM-231 cells lysates. The amounts of GST and GST-tagged PRMT1 were visualized by Coomassie Blue staining (arrows: the position of GST; asterisks: the position of GST-PRMT1). h, i In vitro methylation assays. Purified GST-tag fusion proteins of GST-EZH2(1-522AA) or GST-EZH2(522–746AA), were incubated with GST-PRMT1 in the presence of 3H-SAM. Methylated proteins were detected via autoradiography and total amounts of proteins were visualized by Coomassie Blue staining (arrows: the position of GST; asterisks: the position of GST-EZH2(1–522AA); dot: the position of GST-EZH2(522–746AA); arrowheads: the position of GST-PRMT1). j MS analysis of EZH2 methylation. The MS analysis of in vitro methylated GST-EZH2(1–522AA). The fragmentation of the EZH2 peptide EFAAALTAEdi-meRIK identified a dimethylated residue at R342. k In vivo methylation detection of ADMA-EZH2 levels in HEK293T cells overexpression of WT EZH2, R287K/R342K/R(287 + 342)K mutant EZH2, respectively. l Sequence alignments of EZH2 in mammals. EZH2-R342 site is denoted in the protein sequences.
Moreover, we wanted to determine whether PRMT1 is able to directly mediate ADMA-EZH2 formation. First, we showed that PRMT1 can directly bind to EZH2 through a GST pull-down experiment (Fig. 1f, g). Then, we carried out an in vitro methylation assay by 3H-SAM autoradiography (Fig. 1h, i and Supplementary Fig. 1a, b). The result showed that only GST-EZH2(1–522AA) was methylated by PRMT1 (Fig. 1h, i). Our mass spectrometry (MS) analysis of methylated-EZH2 protein revealed that arginine residues at 287 and 342 (R287 and R342) were methylated by PRMT1 in vitro (Fig. 1j and Supplementary Fig. 1c, d).
To verify these major methylation sites in EZH2, we substituted the R287 and R342 residues in GST-EZH2(1–522AA) or Flag-EZH2 with lysine (K) both individually and in combination (Fig. 1k and Supplementary Fig. 1e). Our in vivo and in vitro methylation assays showed that only the R342K mutation (mutant EZH2 that cannot be methylated by PRMT1) strongly decreased PRMT1-mediated methylation of EZH2 (Fig. 1k and Supplementary Fig. 1e, f). MS data showed that R342 was dimethylated by PRMT1 (Fig. 1j), indicating that PRMT1 dimethylates EZH2 at R342. Because PRMT1 only catalyzes arginine residue to asymmetric dimethylarginine (ADMA) formation, it means that PRMT1-mediated EZH2-R342 ADMA formation. The R342 methylation site on EZH2 is highly conserved in mammals (Fig. 1l). Collectively, these data indicate that EZH2 is asymmetrically dimethylated by PRMT1 at R342 (meR342-EZH2). Subsequently, we generated an antibody that specifically recognizes EZH2 ADMA at R342 (anti-meR342-EZH2), and the specificity of this antibody was verified by ELISA, dot blot, western blotting assays (Supplementary Fig. 2a–f).
PRMT1 increases EZH2 expression at the protein level
We then set out to determine the function of PTMT1-mediated meR342-EZH2. First, we found that ectopic expression of wild type (WT) PRMT1, not enzyme-dead G80R mutant PRMT1 induced a dramatic increase of EZH2 protein; however, little change in EZH2 mRNA levels was detected (Fig. 2a–d). Then, we showed that EZH2 was decreased when PRMT1 was knocked down, while changes in the transcript level of EZH2 were marginal (Fig. 2e–h). Consistently, EZH2 protein was attenuated in MM-231 cells treated with the PRMT1-specific inhibitor AMI-1, and changes in EZH2 mRNA were statistically insignificant (Fig. 2i, j). In addition, EZH2 levels decreased in a time-dependent manner when breast cancer cells were treated with AMI-1 (Fig. 2k–n).
Fig. 2. PRMT1 elevates the expression of EZH2 protein.

a–d Protein and mRNA expression of EZH2 were assessed by western blots (a, c) and qRT-PCR assays (b, d) after overexpression PRMT1. e–h EZH2 proteins and mRNAs were detected by western blot (e, g) and qRT-PCR (f, h) after knockdown PRMT1. i, j EZH2 protein and mRNA levels were analyzed by immunoblotting and qRT-PCR experiments in MM-231 cells treated by PRMT1 inhibitor AMI-1 (8.8uM). k–n Detection of EZH2 protein expression in MM-231, MCF7, BT549 and 4T1 cells treated by AMI-1 using different time-point. o EZH2 and PRMT1 expression levels in MCF10A cells and a variety of breast cancer cell lines. p, q IF analysis of PRMT1 and EZH2 in MM-231 cells treated by DMSO and AMI-1 or MM-231-shCtrl cells and MM-231-shPRMT1#1 cells, respectively. Data are represented as mean ± SEM of three independent experiments, and ns means no significance (Student’s t test).
Moreover, we detected the expression of PRMT1 and EZH2 in MCF10A cells and several breast cancer cell lines (MM-231, MCF7 and so on). Both PRMT1 and EZH2 were expressed at higher levels in breast cancer cell lines than in MCF10A cells (Fig. 2o). Besides, we examined the subcellular location of EZH2 in MM-231. Our data showed that EZH2 protein was mainly localized in the nucleus in shCtrl group and treated with DMSO group; while a part of EZH2 protein was localized in the cytoplasm after knockdown PRMT1 or treated with AMI-1 (Fig. 2p, q). These above results suggest that PRMT1 elevates EZH2 at the protein level without affecting its mRNA expression.
PRMT1-mediated meR342-EZH2 increases EZH2 stability via attenuating CDK1 and EZH2 binding and CDK1-mediated EZH2 phosphorylation
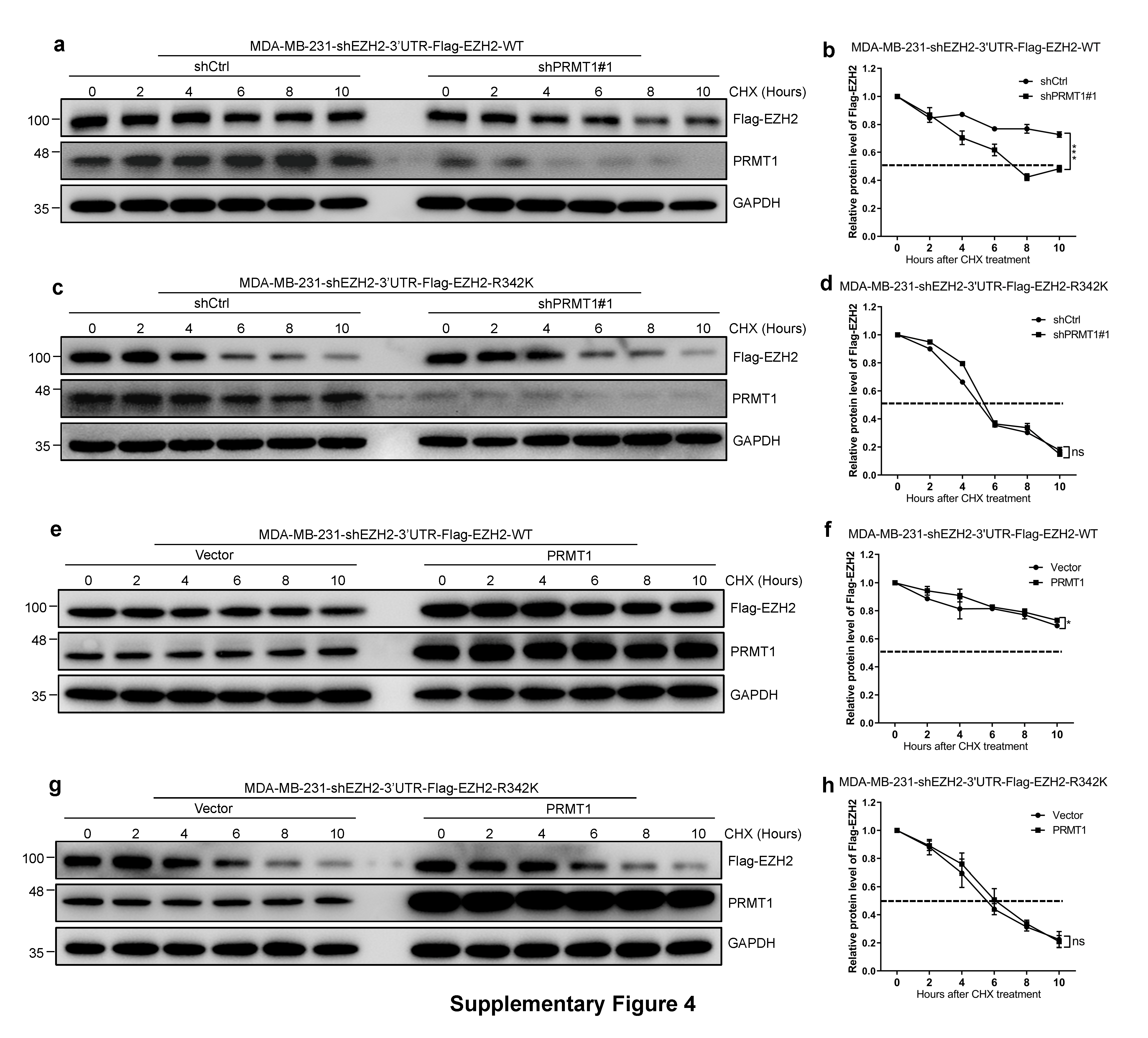
We assumed that PRMT1 may affect the stability of EZH2 through ubiquitin-proteasome pathway. We treated MM-231-shPRMT1 and MM-231-shCtrl cells with the proteasome inhibitor MG132 or the protein synthesis inhibitor cycloheximide (CHX). EZH2 expression was sharply increased in MM-231-shPRMT1 cells treated with MG132 (Fig. 3a); the half-life of EZH2 was much shorter in MM-231-shPRMT1 cells upon the addition of CHX (Fig. 3b, c). Consistently, the similar results were obtained when MM-231 cells were treated with MG132 or CHX after treatment with AMI-1 for 48 h (Supplementary Fig. 3a–c). So we wondered whether PRMT1-mediated meR342-EZH2 promotes EZH2 stability.
Fig. 3. PRMT1-mediated meR342-EZH2 inhibits CDK1-mediated EZH2 phosphorylation.

a The EZH2 protein expression was analyzed by western blot in shCtrl and shPRMT1 MM-231 cells after treated with MG132 (10 μM) for 2 h. b, c The EZH2 protein expression was detected by western blot in MM-231-shCtrl and MM-231-shPRMT1 cells following treated by CHX (50 μg/ml) for the indicated time (b). The relative intensity of EZH2 proteins were quantified by software Image J (c). For normalization, GAPDH expression was used as a control. d Western blot assays confirmed the overexpression of Flag-tagged EZH2 WT or R342K after knockdown of endogenous EZH2 in MM-231 cells. e–g The EZH2 expression was detected by western blotting after stable expression of Flag-EZH2-WT or Flag-EZh2-R342K in MM-231-shEZH2 cells treated with MG132 (10 μM) (e) or CHX (50 μg/ml) (f), respectively. The bands of FLAG-EZH2 proteins treated by CHX were quantified by software Image J and plotted (g). h Western blots of Flag-EZH2-associated ubiquitination after IP HA-Ub in MM-231-shEZH2-WT cells and MM-231-shEZH2-R342K cells. i IF analysis of EZH2 and CDK1 in overexpression of EZH2 WT and EZH2-R342K mutant MM-231-shEZH2 cells, respectively. j A structural illustration of T345 and T487 residues location in EZH2 protein spatial structure by a reported human PRC2 complex crystal structure (PDB ID: 5HYN). k The root-mean-square deviation (RMSD) of Cα of EZH2 protein for the last 50 ns were calculated. l, m Immunoblots of endogenous EZH2-associated ADMA and meR342-EZH2, and EZH2 interaction with PRMT1 (l) and CDK1 (m) in shCtrl and shPRMT1 cells after IP-EZH2. n, o Western blotting analysis of pT345-EZH2 and pT487-EZH2 phosphorylation levels in Vector, Flag-EZH2-WT and Flag-EZH2-R342K groups in MM-231-shEZH2 cells. p, q Western blot analysis of EZH2-associated ADMA and meR342-EZH2 (p), and EZH2 binding with PRMT1 and CDK1 (q) after IP Flag-EZH2 in MMB-231-shEZH2-WT cells and MM-231-shEZH2-R342K cells. Data are represented as mean ± SEM of three independent experiments, and **p < 0.01, ***p < 0.001 (Student’s t test).
To test our hypothesis, we needed to construct EZH2-WT and EZH2-R342K MM-231 cells. To eliminate the influence of high background EZH2 expression in MM-231 cells, we first knocked down EZH2 using lentivirus targeting EZH2-3’UTR. Then, we transfected lentiviruses expressing Flag-EZH2-WT or Flag-EZH2-R342K into MM-231-shEZH2-3’UTR cells (Fig. 3d). We treated MM-231-shEZH2- 3’UTR-Flag-EZH2-WT and MM-231-shEZH2-3’UTR-Flag-EZH2-R342K (named MM-231-shEZH2-WT and MM-231-shEZH2-R342K) cells with MG132, following which the EZH2 level was remarkably increased in EZH2-R342K cells treated with MG132 (Fig. 3e). The same results were observed in MCF7 and HEK293T cells overexpressing Flag-EZH2-WT or Flag-EZH2-R342K treated with MG132 (Supplementary Fig. 3d, g). In addition, the half-life of EZH2-R342K mutant was shorter than that of EZH2-WT in MM-231-shEZH2-3’UTR cells, HEK293T and MCF7 (Fig. 3f, g and Supplementary Fig. 3e, f and h, i). Moreover, our half-life assays revealed that overexpression of PRMT1 or depletion of PRMT1 only affect EZH2-WT but not EZH2-R342K protein stability (Supplementary Fig. 4a–h). Taken together, these results indicate that the PRMT1-mediated methylation of R342-EZH2 plays a key role in sustaining EZH2 stabilization.
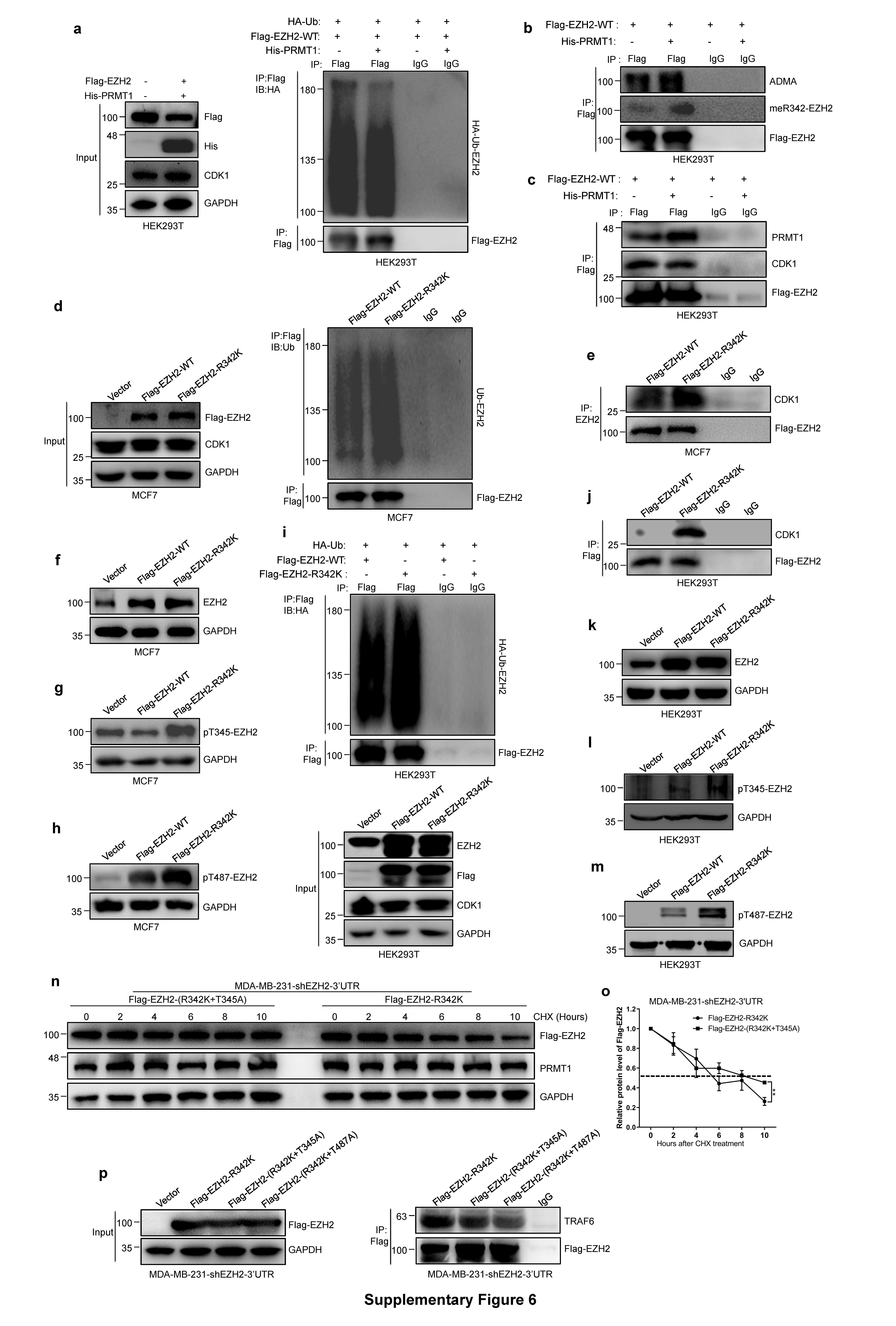
Furthermore, our result showed that EZH2 ubiquitination was increased in MM-231-shPRMT1 cells and MM-231 cells treated with AMI-1 compared to that in the relevant-control cells (Supplementary Fig. 5a, b). Flag-EZH2 ubiquitylation was attenuated in HEK293T cells co-expressing Flag-EZH2 and His-PRMT1 compared with that in HEK293T cells overexpressing Flag-EZH2 (Supplementary Fig. 6a). Moreover, we detected an increased EZH2 ubiquitination status in EZH2-R342K overexpression group compared with that in EZH2-WT overexpression group in MM-231-shEZH2, MCF7 and HEK293T cells, respectively (Fig. 3h and Supplementary Fig. 6d, i). Taken together, these results verify our assumption that PRMT1-mediated R342-EZH2 methylation increases EZH2 stability through attenuating EZH2 ubiquitination.
Moreover, our IF analysis revealed that the amount of EZH2 and CDK1 co-localization in the cytoplasm was increased in MM-231-shEZH2-R342K cells compared with MM-231-shEZH2-WT cells (Fig. 3i). We thus reasoned that meR342-EZH2 might decrease ubiquitination by reducing CDK1-mediated EZH2 phosphorylation of T345 and T487. As shown by our molecular dynamics simulations analysis using EZH2 3D structure (PDB ID: 5HYN) from previous structural study [27] (Fig. 3j, k), The residues of EZH2 T345, R342, and T487 are in relatively close proximity with calculated distance between R342 and T345, or R342 and T487, ~11 or ~26 Å, respectively (Supplementary Table 1). It is therefore possible that the dimethyl group at EZH2-R342 might interfere with T345/T487 phosphorylation as a result of structural hinderance, and/or modification-induced conformational change, that negatively impacts the access of CDK1 to the phosphorylation sites.
To test this hypothesis, we performed Co-IP-EZH2 experiments in MM-231-shCtrl cells, MM-231-shPRMT1 cells and MM-231 cells treated with AMI-1 or DMSO to detect the amount of PRMT1 binding with EZH2, ADMA-EZH2, meR342-EZH2 and CDK1 interacting with EZH2. Our results showed that knockdown of PRMT1 and AMI-1 treatment decreased the ADMA-EZH2, meR342-EZH2 and the interaction of PRMT1 with EZH2, and increased CDK1 binding with EZH2 (Fig. 3l, m and Supplementary Fig. 5c, d). Furthermore, the extent of EZH2-ADMA and meR342-EZH2, and the interaction between Flag-EZH2 and PRMT1 were elevated when Flag-EZH2 and His-PRMT1 were co-expressed compared with only Flag-EZH2 overexpression in HEK293T cells; whereas the CDK1 binding with Flag-EZH2 was repressed in Flag-EZH2 and His-PRMT1 co-expression HEK293T cells compared to HEK293T Flag-EZH2 cells, as shown by Co-IP assays of Flag-EZH2 (Supplementary Fig. 6b, c).
Next, we explored the phosphorylation status of EZH2 T345 and T487 after overexpressing WT or R342K mutant EZH2. First, our results showed that EZH2-R342K increased the phosphorylation of EZH2 T345 and T487 (Fig. 3n, o and Supplementary Fig. 6f–h and k–m). Then, we verified that EZH2-ADMA and meR342-EZH2 were remarkably decreased in EZH2-R342K cells compared to their levels in EZH2-WT cells (Fig. 3p). Subsequently, the EZH2-R342K increased CDK1 binding with EZH2 compared with that in EZH2-WT cells, as shown by IP-Flag-EZH2 assays (Fig. 3q and Supplementary Fig. 6e, j). Furthermore, we also found that EZH2 protein half-life of the R342K mutant was shorter than that of (R342K + T345A) double-mutant (Supplementary Fig. 6n, o). Collectively, these data strongly suggest that PRMT1-mediated R342-EZH2 methylation elevates EZH2 stability through inhibiting CDK1-mediated EZH2 phosphorylation in breast cancer cells.
PRMT1-mediated meR342-EZH2 increases EZH2 stability by attenuating binding of TRAF6 with EZH2 and TRAF6-mediated EZH2 degradation
Next, we wanted to determine the specific E3 ligase which involved in PRMT1-mediated meR342-EZH2 to inhibit EZH2 ubiquitination. We focused on the E3 ligase TRAF6 because EZH2 was found to harbor three conserved TRAF6-binding motifs [32] (Fig. 4a). We thus hypothesized that TRAF6 can mediate EZH2 ubiquitination. Firstly, our Co-IP assays revealed the interaction between TRAF6 and EZH2 (Fig. 4b, c). And our data indicated that the TRAF domain is the main region of TRAF6 interacting with EZH2 by our IP assays in HEK293T cell overexpressing a series of Flag-Tag TRAF6 domain-truncated plasmids, respectively (Fig. 4d). Then, we showed that TRAF6 indeed manipulated EZH2 protein half-life (Supplementary Fig. 7a–d). Subsequently, we found that TRAF6-WT is able to mediate EZH2 ubiquitination, while TRAF6-C70A (enzyme activity mutation) had little effect on EZH2 ubiquitination (Fig. 4e). We ectopic expressed HA-Ub-WT or HA-Ub-K48R or HA-Ub-K63R plasmids in Flag-EZH2 and myc-TRAF6 co-expression cells to distinguish the TRAF6-mediated EZH2 ubiquitin linkage by IP assays. Our results demonstrated that TRAF6 mediated EZH2 K63-linkage polyubiquitylation (Fig. 4f). In addition, we also discovered that overexpression of myc-TRAF6 decreased Flag-EZH2 expression by elevating TRAF6 binding and increasing EZH2 ubiquitination (Supplementary Fig. 7e, f). In contrast, depletion of TRAF6 expression increased EZH2 expression through the attenuation of EZH2 ubiquitination (Supplementary Fig. 7g). These results suggest that TRAF6 mediates EZH2-K63-linkage ubiquitylation and degradation in breast cancer cells.
Fig. 4. PRMT1-mediated meR342-EZH2 prevents TRAF6-mediated EZH2 ubiquitination and degradation.

a EZH2 protein contains 3 potential TRAF6-binding sites. The consensus TRAF6-binding site motif has been described previously. b, c Western blot analysis of endogenous and exogenous interaction between EZH2 and TRAF6 after IP EZH2 in MM-231 cells (b) and HEK293T cells (c), respectively. d FLAG-tagged TRAF6 plasmids were transfected into 293T cells, and FLAG beads were used for IP. Precipitates were subjected to western blot analysis with indicated antibodies. e, f Detecting Flag-EZH2-associated ubiquitination by immunoblotting after IP HA-Ub in HEK293T cells transfected with indicated constructs. g Detecting Flag-EZH2-associated K48 and K63 ubiquitination by western blot after IP HA-Ub in MM-231-shEZH2 cells transfected with indicated constructs. h Immunoblotting analysis the amount of TRAF6 binding with EZH2 in MM-231-shCtrl and MM-231-shPRMT1 cells after IP EZH2. i Western blot detection of exogenous Flag-EZH2 binding with TRAF6 after IP Flag in Flag-EZH2-WT overexpression or Flag-EZH2-WT and His-PRMT1 co-expression HEK293T cells. j IF analysis of EZH2 and TRAF6 in MM-231-shEZH2-WT cells and MM-231-shEZH2-R342K cells. k Detecting the level of Flag-EZH2 interacting with TRAF6 after IP Flag in Flag-EZH2-WT or Flag-EZH2-R342K overexpression cells. l, m Western blotting analysis of Flag-EZH2-R342K binding with TRAF6 (l) and Flag-EZH2-R342K ubiquitination (m) after IP Flag in MM-231-shEZH2-R342K cells treated with CDK1 inhibitor CGP.
Furthermore, our IP assays showed that only K48R linkage polyubiquitylation was strongly increased in R342K mutant EZH2 compared with that in WT EZH2 (Fig. 4g). We also found that knockdown of PRMT1 or treated with AMI-1 in MM-231 cells promoted TRAF6 binding with EZH2 (Fig. 4h and Supplementary Fig. 5d), while ectopic expression of His-PRMT1 suppressed the interaction between Flag-EZH2 and TRAF6 in HEK293T cells (Fig. 4i). Subsequently, our IF assays data showed that the amount of EZH2 and TRAF6 co-localization in the cytoplasm was dramatically increased in the MM-231-shEZH2-R342K cells compared with that in MM-231-shEZH2-WT cells (Fig. 4j). Moreover, we demonstrated that R342-EZH2 promotes the interaction between TRAF6 and EZH2 compared with that in cells expressing EZH2-WT (Fig. 4k and Supplementary Fig. 7h, i). Taken together, these data indicate that the R342-EZH2 methylation increasing EZH2 stability is likely dependent on the inhibition of TRAF6-mediated EZH2 ubiquitination.
We therefore wonder whether CDK1-mediated EZH2 phosphorylation facilitates TRAF6 binding with EZH2 and promotes TRAF6-mediated EZH2 degradation. To test this hypothesis, we treated MM-231-shEZH2-R342K cells with a CDK1 specific inhibitor (CGP74514A, CGP). First, we found that EZH2 level was increased after CGP treatment (Fig. 4l). Then, we discovered that the binding of TRAF6 to EZH2 was decreased in CGP-treated cells (Fig. 4l). Moreover, we found that mutation of T345A or T487A in EZH2-R342K attenuates EZH2-R342K binding with TRAF6 (Supplementary Fig. 6p). Finally, we found that CGP was able to strongly reduce the level of EZH2 ubiquitination following treatment with CGP in MM-231-shEZH2-R342K cells (Fig. 4m). Above all, these findings strongly indicate that PRMT1-mediated meR342-EZH2 can prevent the CDK1-mediated phosphorylation of EZH2 at T345 and T487, which inhibits TRAF6 binding with EZH2 and attenuates TRAF6-mediated EZH2 degradation through the ubiquitin- proteasome pathway.
PRMT1-mediated EZH2-R342 methylation is important for silencing EZH2 target gene expression
In order to figure out whether EZH2-R342 methylation play a key role in EZH2-mediated functions. Firstly, the mRNA expression of 20 known EZH2 target genes were analyzed by qRT-PCR assays in MM-231-shCtrl cells, MM-231-shPRMT1#1 cells and DMSO-treated MM-231 cells, AMI-1-treated MM-231 cells, respectively. Results showed that these target gene expressions were increased by knockdown of PRMT1 or treated with AMI-1 in MM-231 cells (Fig. 5a, b). Then, we confirmed that most of these target genes were remarkably decreased only in EZH2-WT overexpressing cells compared with EZH2-R342K overexpressing and Vector cells (Fig. 5c). These data strongly suggest that EZH2 suppressing its target gene expression is dependent on PRMT1-mediated EZH2-R342 methylation.
Fig. 5. PRMT1-mediated meR342-EZH2 promotes its ability in silencing target gene transcriptional expression.

a, b A series of EZH2 target genes mRNA expression levels were analyzed by qRT-PCR assays in MM-231-shCtrl and MM-231-shPRMT1#1 cells (a) and MM-231 cells treated by DMSO or AMI-1 (b), respectively. c EZH2 target genes mRNA expression were examined by qRT-PCR assays in MM-231-shEZH2 cells overexpressing Vector, Flag-EZH2-WT and Flag-EZH2-R342K, respectively. d–i ChIP assays were performed using anti-EZH2 (d, f and h) and anti-H3K27me3 (e, g and i) antibodies, and the immunoprecipitated DNA was analyzed by qRT-PCR using specific primers of HOXA10, DAB2IP, HOXA9 and HOXA7 promoters. Data are represented as mean ± SEM of three independent experiments, and ns means no significance, *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test).
In order to disclose the depth mechanism, we carry out the ChIP assays to explore whether PRMT1-mediated meR342-EZH2 have an effect on EZH2 and H3K27me3 occupation on EZH2 target genes promoters. In fact, we selected 4 (HOXA10, DAB2IP, HOXA9 and HOXA7) of 20-detected target genes which have shown dramatically changes in mRNA level to confirm our propose. As our data showed, knockdown PRMT1 and AMI-1-treated groups attenuated not only EZH2 occupation but also H3K27me3 binding ability on the target gene promoters compared with the relevant-control groups (Fig. 5d–g). Furthermore, we demonstrated that EZH2-R342K decreased EZH2 and H3K27me3 binding capacity to the 4 target gene promoters (Fig. 5h, i). Our data suggest that PRMT1-mediated meR342-EZH2 may strengthen EZH2 binding on its target gene promoters. And meR342-EZH2 is required for EZH2 repressing its target genes expression.
R342-EZH2 methylation is required for breast cancer cell migration in vitro and metastasis in vivo
Moreover, we intended to determine the pathological function of PRMT1-mediated R342-EZH2 methylation in breast cancer. We first detected EMT markers expression upon the ectopic expression of WT or R342K mutant EZH2 in MCF7 and MM-231-shEZH2-3’UTR cells. N-cadherin and Vimentin (mesenchymal markers) levels were strongly increased, whereas the E-cadherin (epithelial marker) level was decreased in WT-EZH2-overexpressing cells compared with their expression in Vector cells; while the levels of these related-EMT markers were not as different between EZH2-R342K cells and Vector cells (Fig. 6a, b and Supplementary Fig. 8a). Then, we observed the obvious up-regulation of the mRNAs of several classical EMT inducers (Snail, Twist and ZEB1) in EZH2-WT-overexpressing cells compared with their counterparts in EZH2-R342K-overexpressing cells (Fig. 6c and Supplementary Fig. 8b). Our results suggest that meR342-EZH2 is required for EZH2-induced EMT.
Fig. 6. EZH2-R342 methylation promotes breast cancer cell invasion and metastasis.

a, b The mRNA and protein expression of EMT markers were detected by qRT-PCR and western blot in MCF7-vector/Flag-EZH2-WT/Flag-EZH2-R342K cells. c The mRNA expression of EMT inducers in MCF7-vector/Flag-EZH2-WT/Flag-EZH2- R342K cells. d Western blots analysis of MMP9 and MMP2 expression in MCF7-vector/Flag-EZH2-WT/Flag-EZH2-R342K cells. e ChIP analyses on E-cadherin promoter were done using anti-EZH2 and anti-H3K27me3 antibodies in MCF7-shCtrl and MCF7-shPRMT1#1 cells. f–h Assessment of cells motility by wound-healing assays (f), invasion assays (g) and migration assays (h) in MCF7-vector/Flag-EZH2-WT/Flag-EZH2-R342K cells. i, j Representative bioluminescence images of lung metastasis (i) in mice injected with MM-231-luc-shEZH2-Vector/Flag-EZH2-WT/Flag-EZH2-R342K cells via tail veins, and the metastases were analyzed by measuring the photo flux (j), n = 9 for each group. k–n Lung metastatic nodules were examined macroscopically (k and l) or detected by H&E staining after nude mice were sacrificed (m and n). The white arrows denote the metastatic nodules. o, p Representative images of the IHC staining of meR342-EZH2 (o) or EZH2 (p) in lung metastasis sections. Data are represented as mean ± SEM of three or four independent experiments, and *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t test).
We investigated whether R342-EZH2 methylation is necessary for accelerating breast cancer cell motility. First, we found that MMP2 and MMP9 were remarkably increased only in MCF7-Flag-EZH2-WT cells compared with MCF7-Flag-EZH2- R342K and MCF7-Vector cells (Fig. 6d). Then, we demonstrated that EZH2-R342K strongly repressed EZH2 and H3K27me3 occupation in the E-cadherin promoter region compared with EZH2-WT in MCF7 and MM-231-shEZH2-3’UTR cells by our ChIP assays (Fig. 6e and Supplementary Fig. 8c). Finally, we determined that only EZH2-WT prominently accelerated the invasion and migration of MCF7 and MM-231-shEZH2-3’UTR cells by wound healing and transwell chamber experiments (Fig. 6f–h and Supplementary Fig. 8d–f). Our data suggest that meR342-EZH2 is necessary for EZH2 to promote EMT program and stimulate the migration of breast cancer cells in vitro.
Furthermore, we aimed to evaluate the relationship between meR342-EZH2 and breast cancer cell metastasis in vivo. MM-231-luc-shEZH2-Vector cells (named Vector cells), MM-231-luc-shEZH2-Flag-EZH2-WT cells (named EZH2-WT cells) or MM-231-luc-shEZH2-Flag-EZH2-R342K cells (named EZH2-R342K cells) were injected into the tail veins of BABL/c female nude mice (mice were randomly divided into 3 groups, n = 9 each group). After 7 weeks, nude mice were sacrificed and lung metastatic nodules were examined. Strikingly, EZH2-WT cells metastasized to the lungs of nude mice more effectively than EZH2-R342K and Vector cells, as illustrated by bioluminescence imaging (Fig. 6i, j). Noticeably, histological examination revealed that mice bearing EZH2-WT cells had a larger number of macroscopic lung metastases than mice in which EZH2-R342K cells and Vector cells had been transplanted (Fig. 6k, l). Subsequently, lung tissue sections were prepared and examined after staining with hematoxylin and eosin (H&E), and larger and more metastatic foci were detected in the tissues of mice injected with EZH2-WT cells (Fig. 6m, n). Finally, we measured meR342-EZH2 and EZH2 levels in lung tissue sections by IHC assays. We confirmed that more EZH2 was expressed in mice injected with EZH2-WT cells and EZH2-R342K cells than in mice injected with Vector cells (Fig. 6o). We detected a high level of meR342-EZH2 only in mice injected with EZH2-WT cells (Fig. 6p). Together, these data strongly suggest that meR342-EZH2 plays a pivotal role in promoting breast cancer cells metastasis in vivo.
PRMT1-mediated meR342-EZH2 formation correlates with poor clinical outcome in breast cancer patients
To assess the clinical significance of our findings, we performed IHC analyses in breast cancer TMA (tissue microarray) slides using anti-PRMT1, anti-meR342-EZH2 and anti-EZH2 antibodies. These 3 markers were expressed at low levels in breast adjacent tissues, and higher expression levels were observed in breast cancer primary tumors, with the highest expression levels of the markers in breast cancer metastatic tumors (Fig. 7a–c). Then, we revealed that PRMT1 and meR342-EZH2 expression levels, PRMT1 and EZH2 levels and between meR342-EZH2 and EZH2 levels were positively correlated (Fig. 7d–g). Moreover, we observed high PRMT1 expression positively correlated with high meR342-EZH2 and EZH2 expression (Fig. 7h, i). Finally, we also discovered that high meR342-EZH2 expression positively correlated with high EZH2 expression (Fig. 7j). These results confirmed our hypothesis: PRMT1-mediated EZH2-R342 methylation facilitates breast cancer cell distant metastasis by enhancing EZH2 stability.
Fig. 7. EZH2-R342 methylation positive correlates with metastasis and poor prognosis of breast cancer patients.

a–c IHC assays among breast cancer specimens (TMA) were performed using anti-PRMT1, anti-meR342-EZH2 and anti-EZH2 antibodies. Semiquantitative scoring method (using a scale from 0 to 12) was used to analyzed the scores of PRMT1, meR342-EZH2 and EZH2 IHC staining. d–g Representative images of PRMT1, meR342-EZH2 and EZH2 expressions in PRMT1-low case and PRMT1-high case were presented (d). Correlation between PRMT1 and meR342-EZH2 expression (e), PRMT1 and EZH2 expression (f), meR342-EZH2 and EZH2 (g) were examined by Pearson correlation coefficient test, respectively. h–j Correlation between PRMT1 and meR342-EZH2 expression (h), PRMT1 and EZH2 expression (i), meR342-EZH2 and EZH2 (j) were examined by Fisher’s exact test, respectively. k Representative images of weak and strong meR342-EZH2 staining in breast cancer tissues. l–q Percentages of high level of meR342-EZH2 expression correlated with tumor sizes (l), histologic grades (m), different tumor subtypes (n–p) and metastasis (q) were examined by χ2 test. r Correlation of meR342-EZH2 expression with lymph node metastasis number. s High meR342-EZH2 expression correlated with a poorer 5-year overall survival for 120 breast cancer patients (High meR342-EZH2 patients 68, Low meR342-EZH2 patients 52) (P = 0.006, log-rank test). t High meR342-EZH2 expression correlated with a poorer 5-year disease-specific survival for 112 breast cancer patients (High meR342-EZH2 patients 60, Low meR342-EZH2 patients 52) (P = 0.001, log-rank test). See patients information in Supplementary Tables 2–4. u A proposed work model for this project.
Next, we explored the correlation between meR342-EZH2 levels and clinicopathological characteristics in human breast cancer specimens through IHC assays (Fig. 7k and Supplementary Table 2). First, the data showed that high meR342-EZH2 levels were positively correlated with tumor size, histology grade, ER- and PR- breast cancer patients (Fig. 7l–o). However, meR342-EZH2 was not positively correlated with HER2- breast cancer patients (Fig. 7p). Furthermore, IHC showed that high meR342-EZH2 levels were positively correlated with lymph node metastasis and the number of lymph node metastatic foci (Fig. 7q, r). Finally, we demonstrated that high levels of meR342-EZH2 are remarkably correlated with poor 5-year overall survival and disease-specific survival (Fig. 7s, t and Supplementary Table 3, 4). These results strongly support the role of meR342-EZH2 in the clinical behavior of human breast cancer and reveal a clear relationship between meR342-EZH2 and the clinical aggressiveness of human breast carcinoma.
Discussion
EZH2, a critical regulator of epigenetic modifications that induces the EMT program, has been reported to be overexpressed in diverse cancer types and participate in the metastasis of many cancers. While the in-depth mechanisms by which EZH2 is increased in breast cancer have not been clearly illustrated.
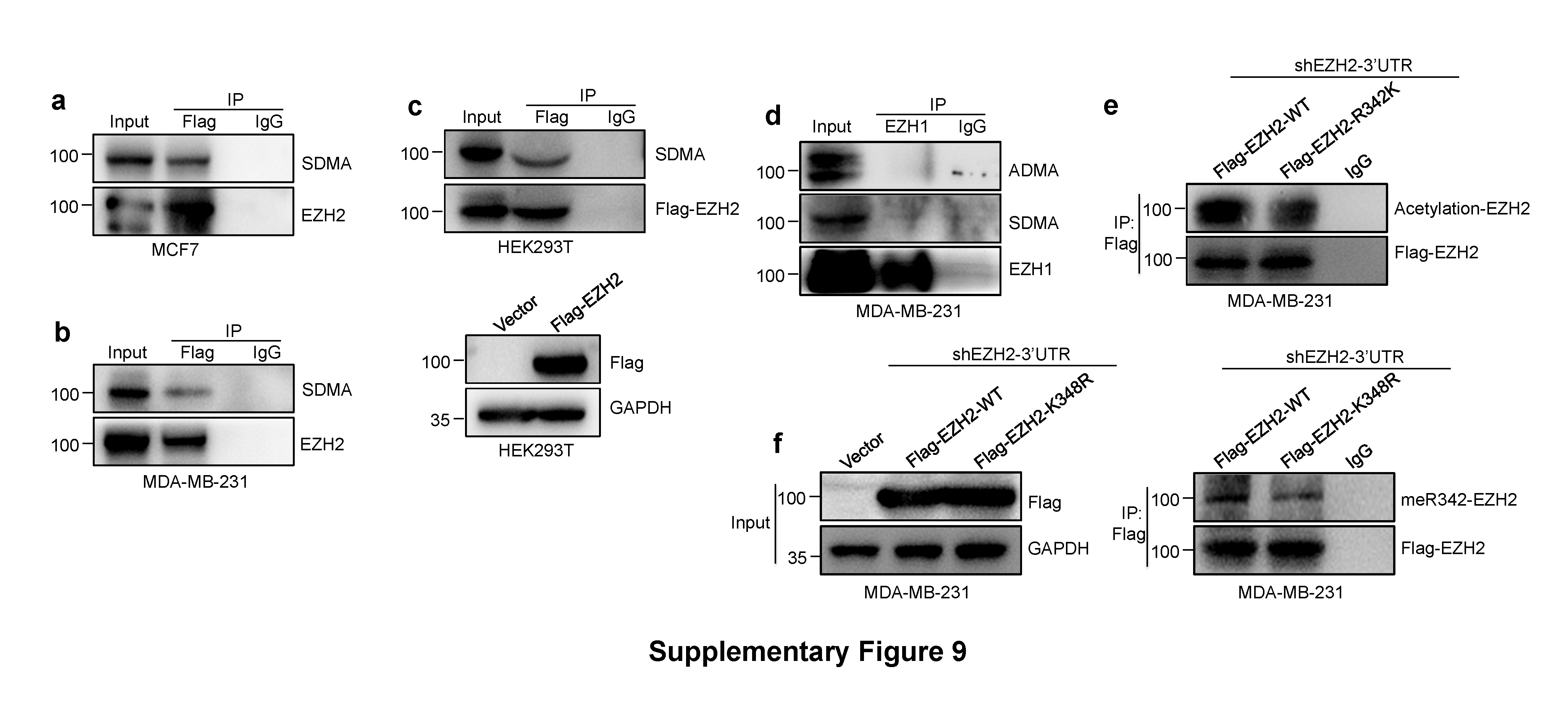
Significantly, in this study, we first describe the crucial role of R342-EZH2 arginine methylation in EZH2 protein stability, its function in breast cancer metastasis, and the sequence of molecular mechanisms involved in this process. The canonical PRMT1 methylation motif is glycine-arginine-rich (GAR) motifs [20]. The EZH2-R342 methylation residue didn’t fit the consensus motif. In fact, many reports have indicated that PRMT1-mediated non-histone proteins methylation sites are not the canonical PRMT1 methylation motif [20, 33, 34]. This means that amount of PRMT1 substrates may be underestimated by consensus motif analysis, suggesting that the importance of arginine methylation is yet to be fully understand. Interestingly, we found the EZH2 can also undergo symmetric di-methylation (SDMA), while EZH1 protein neither undergoes ADMA nor SDMA modification (Supplementary Fig. 9a–d).
Post-translational modifications (PTMs) of EZH2 are critical for regulating its biological functions and protein stability. Phosphorylation is the main kind of EZH2 PTMs identified previously [7, 11, 12]. For instance, it was reported that the AMPK-mediated phosphorylation of EZH2 at T311 depresses PRC2 methyltransferase activity through disrupting the binding of EZH2 with SUZ12 [11]. Interestingly, a variety of recent studies have demonstrated the crosstalk between arginine methylation and phosphorylation [23, 35]. In our study, we first reported the crosstalk between meR342-EZH2 and pT345-EZH2 and pT487-EZH2 have a great effect on EZH2 protein stability. As previously mentioned, EZH2 stability is also increased by PCAF-mediated acetylation of K348-EZH2 [10]. We found that R342K mutant decreased EZH2 acetylation; and K348R mutant decreased EZH2-meR342 methylation (Supplementary Fig. 9e, f). These findings suggest that there may be a positive crosstalk between PRMT1-mediated meR342-EZH2 and PCAF-mediated AcK348-EZH2.
In summary, we have characterized a PRMT1-CDK1-TRAF6-EZH2 axis in stimulating breast cancer metastasis, illustrating that meR342-EZH2 plays a key role in this process. As depicted in the working model in Fig. 7u: PRMT1 expression level is increasing in breast cancer cells, which results in promoting PRMT1-mediated R342-EZH2 methylation. Subsequently, meR342-EZH2 represses CDK1 binding with EZH2 and phosphorylating EZH2. Then, meR342-EZH2 suppresses the interaction between TRAF6 and EZH2 and TRAF6-mediated EZH2 degradation, which strengthens EZH2 stability and suppression of its target gene expressions (E-cadherin, DAB2IP, CSTA, etc.). Finally, EZH2 accumulation promotes breast cancer cell metastasis (Fig. 7u). Data from this study emphasize the importance of R342-EZH2 methylation in increasing breast cancer metastasis and suggest the clinical value of meR342-EZH2 as a biomarker in breast cancer diagnosis and therapy.
Supplementary information
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
This work was supported by the grants from the National Natural Science Foundation of China (81802637, 81672845 and 81872304); the Natural Science Foundation of Jiangsu Province (BK20180989); the National Postdoctoral Research Funds (2019M651971); the Natural Science Foundation of Jiangsu Province Universities (18KJB310016); the Research Foundation of Xuzhou Medical University (D2018020); the Xuzhou City Science and Technology Plan Project (KC19065); the Jiangsu Provincial Key Medical Discipline, the Project of Invigorating Health Care through Science, Technology and Education (ZDXKA2016014).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by A. Oberst
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Junnian Zheng, Email: jnzheng@xzhmu.edu.cn.
Jin Bai, Email: bj@xzhmu.edu.cn.
Supplementary information
The online version of this article (10.1038/s41418-020-00615-9) contains supplementary material, which is available to authorized users.
References
- 1.Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun. 2015;6:6051. doi: 10.1038/ncomms7051. [DOI] [PubMed] [Google Scholar]
- 2.Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, et al. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011;19:86–100. doi: 10.1016/j.ccr.2010.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell. 2013;23:768–83. doi: 10.1016/j.ccr.2013.04.020. [DOI] [PubMed] [Google Scholar]
- 4.Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34. doi: 10.1038/nm.4036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Z, Hou P, Fan D, Dong M, Ma M, Li H, et al. The degradation of EZH2 mediated by lncRNA ANCR attenuated the invasion and metastasis of breast cancer. Cell Death Differ. 2017;24:59–71. doi: 10.1038/cdd.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci USA. 2003;100:11606–11. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Anwar T, Arellano-Garcia C, Ropa J, Chen YC, Kim HS, Yoon E, et al. p38-mediated phosphorylation at T367 induces EZH2 cytoplasmic localization to promote breast cancer metastasis. Nat Commun. 2018;9:2801. doi: 10.1038/s41467-018-05078-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chu CS, Lo PW, Yeh YH, Hsu PH, Peng SH, Teng YC, et al. O-GlcNAcylation regulates EZH2 protein stability and function. Proc Natl Acad Sci USA. 2014;111:1355–60. doi: 10.1073/pnas.1323226111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin X, Yang C, Fan P, Xiao J, Zhang W, Zhan S, et al. CDK5/FBW7-dependent ubiquitination and degradation of EZH2 inhibits pancreatic cancer cell migration and invasion. J Biol Chem. 2017;292:6269–80. doi: 10.1074/jbc.M116.764407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wan J, Zhan J, Li S, Ma J, Xu W, Liu C, et al. PCAF-primed EZH2 acetylation regulates its stability and promotes lung adenocarcinoma progression. Nucleic Acids Res. 2015;43:3591–604. doi: 10.1093/nar/gkv238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan L, Xu K, Wei Y, Zhang J, Han T, Fry C, et al. Phosphorylation of EZH2 by AMPK suppresses PRC2 methyltransferase activity and oncogenic function. Mol Cell. 2018;69:279–91.e275. doi: 10.1016/j.molcel.2017.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12.Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem. 2011;286:28511–9. doi: 10.1074/jbc.M111.240515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lo PW, Shie JJ, Chen CH, Wu CY, Hsu TL, Wong CH. O-GlcNAcylation regulates the stability and enzymatic activity of the histone methyltransferase EZH2. Proc Natl Acad Sci USA. 2018;115:7302–07. doi: 10.1073/pnas.1801850115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jarrold J, Davies CC. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol Med. 2019;25:993–1009. doi: 10.1016/j.molmed.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 15.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13:37–50. doi: 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 16.Blanc RS, Richard S. Arginine methylation: the coming of age. Mol Cell. 2017;65:8–24. doi: 10.1016/j.molcel.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 17.Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33:1–13. doi: 10.1016/j.molcel.2008.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J Biol Chem. 2000;275:7723–30. doi: 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- 19.Cho JH, Lee MK, Yoon KW, Lee J, Cho SG, Choi EJ. Arginine methylation-dependent regulation of ASK1 signaling by PRMT1. Cell Death Differ. 2012;19:859–70. doi: 10.1038/cdd.2011.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies CC, Chakraborty A, Diefenbacher ME, Skehel M, Behrens A. Arginine methylation of the c-Jun coactivator RACO-1 is required for c-Jun/AP-1 activation. EMBO J. 2013;32:1556–67. doi: 10.1038/emboj.2013.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sakamaki J, Daitoku H, Ueno K, Hagiwara A, Yamagata K, Fukamizu A. Arginine methylation of BCL-2 antagonist of cell death (BAD) counteracts its phosphorylation and inactivation by Akt. Proc Natl Acad Sci USA. 2011;108:6085–90. doi: 10.1073/pnas.1015328108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang Y, Hsu JM, Kang Y, Wei Y, Lee PC, Chang SJ, et al. Oncogenic functions of Gli1 in pancreatic adenocarcinoma are supported by its PRMT1-mediated methylation. Cancer Res. 2016;76:7049–58. doi: 10.1158/0008-5472.CAN-16-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yamagata K, Daitoku H, Takahashi Y, Namiki K, Hisatake K, Kako K, et al. Arginine methylation of FOXO transcription factors inhibits their phosphorylation by Akt. Mol Cell. 2008;32:221–31. doi: 10.1016/j.molcel.2008.09.013. [DOI] [PubMed] [Google Scholar]
- 24.Yang JH, Chiou YY, Fu SL, Shih IY, Weng TH, Lin WJ, et al. Arginine methylation of hnRNPK negatively modulates apoptosis upon DNA damage through local regulation of phosphorylation. Nucleic Acids Res. 2014;42:9908–24. doi: 10.1093/nar/gku705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cha B, Kim W, Kim YK, Hwang BN, Park SY, Yoon JW, et al. Methylation by protein arginine methyltransferase 1 increases stability of Axin, a negative regulator of Wnt signaling. Oncogene. 2011;30:2379–89. doi: 10.1038/onc.2010.610. [DOI] [PubMed] [Google Scholar]
- 26.Bai J, Wu K, Cao MH, Yang Y, Pan Y, Liu H, et al. SCF(FBXO22) targets HDM2 for degradation and modulates breast cancer cell invasion and metastasis. Proc Natl Acad Sci USA. 2019;116:11754–63. doi: 10.1073/pnas.1909032116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, et al. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 2016;7:11316. doi: 10.1038/ncomms11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coutsias EA, Seok C, Jacobson MP, Dill KA. A kinematic view of loop closure. J Comput Chem. 2004;25:510–28. doi: 10.1002/jcc.10416. [DOI] [PubMed] [Google Scholar]
- 29.Huang J, MacKerell AD. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34:2135–45. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–93. doi: 10.1063/1.470117. [DOI] [Google Scholar]
- 32.Yu J, Qin B, Moyer AM, Nowsheen S, Liu T, Qin S, et al. DNA methyltransferase expression in triple-negative breast cancer predicts sensitivity to decitabine. J Clin Invest. 2018;128:2376–88. doi: 10.1172/JCI97924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu LM, Sun WZ, Fan XZ, Xu YL, Cheng MB, Zhang Y. Methylation of C/EBPalpha by PRMT1 inhibits its tumor-suppressive function in breast cancer. Cancer Res. 2019;79:2865–77. doi: 10.1158/0008-5472.CAN-18-3211. [DOI] [PubMed] [Google Scholar]
- 34.Wooderchak WL, Zang T, Zhou ZS, Acuna M, Tahara SM, Hevel JM. Substrate profiling of PRMT1 reveals amino acid sequences that extend beyond the “RGG” paradigm. Biochemistry. 2008;47:9456–66. doi: 10.1021/bi800984s. [DOI] [PubMed] [Google Scholar]
- 35.Liao HW, Hsu JM, Xia W, Wang HL, Wang YN, Chang WC, et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. J Clin Invest. 2015;125:4529–43. doi: 10.1172/JCI82826. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.