

Abstract
Antibiotics to treat drug-resistant Gram-negative infections are urgently needed but challenging to discover. Using a cell-based screen, we identified a simple secondary amine that inhibited growth of wildtype Escherichia coli and Acinetobacter baumannii, but not growth of the Gram-positive organism Bacillus subtilis. Resistance mutations in E. coli and A. baumannii mapped exclusively to the aminoacyl-tRNA synthetase PheRS. We confirmed biochemically that the compound inhibited PheRS from these organisms and showed that it did not inhibit PheRS from B. subtilis or humans. To understand the basis for the compound’s high selectivity for only some PheRS enzymes, we solved crystal structures of E. coli and A. baumannii PheRS complexed with inhibitor. The structures showed that the compound’s benzyl group mimics the benzyl of phenylalanine. The other amine substituent, an ethyl cyclohexene-yl group, induces a hydrophobic pocket in which it binds. Through bioinformatic analysis and mutagenesis, we show that the ability to induce a complementary hydrophobic pocket that can accommodate the second substituent explains the high selectivity of this remarkably simple molecular scaffold for Gram-negative PheRS. Because this secondary amine scaffold is active against wildtype Gram-negative pathogens, but is not cytotoxic to mammalian cells, we suggest it may be possible to develop it for use in combination antibiotic therapy to treat Gram-negative infections.
Graphical Abstract
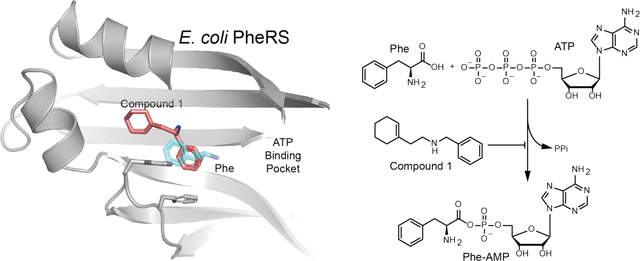
Found only in Gram-negative organisms, the bacterial outer membrane limits penetration of many antibiotics into the cell1–2. Therefore, Gram-negative organisms are intrinsically resistant to many drugs that are highly effective against Gram-positive bacteria3. For some antibiotic-resistant Gram-negative infections, there are no remaining treatment options4–6, and it is crucial to develop new approaches to treat these infections. With this in mind, we screened 690,000 small molecules for their ability to inhibit growth of E. coli with an intact outer membrane. Slightly over 1,000 compounds (0.2%) were found to substantially inhibit the growth of this organism at ~50 μM.
To prioritize compounds for follow-up, we retested the ~1,000 growth inhibitors at a lower dose (~10 μM) against both Bacillus subtilis and our E. coli strain. Most compounds were far more active against the more permeable Gram-positive strain than against the less permeable Gram-negative strain (Fig. 1a, red diamonds). Speculating that a substantial fraction of compounds that inhibited growth of both E. coli and B. subtilis could be non-specific toxins, whereas compounds with activity only against E. coli were likely to have a specific cellular target in that organism, we focused on the very small minority of compounds with selective activity against E. coli (Fig. 1A, black and blue diamonds).
Figure 1. A high-throughput screen identified a simple compound that selectively inhibits growth of wild-type Gram-negative bacteria.
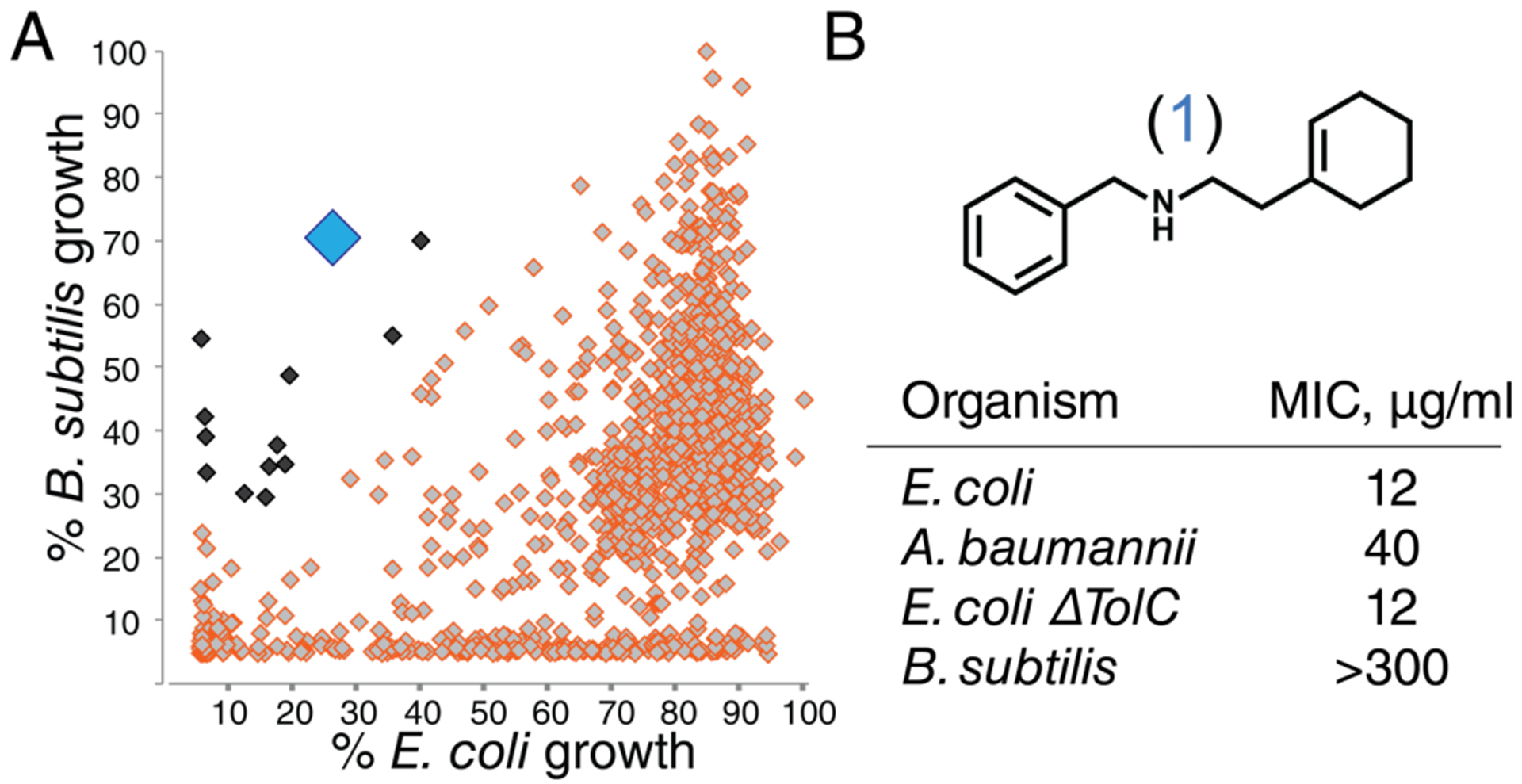
A) ~1,000 E. coli growth inhibitors (at 50 μM) were screened at ~10 μM against B. subtilis and wildtype E. coli to identify compounds that selectively inhibited growth of the latter (black and blue diamonds). B) Structure of compound 1 (large blue diamond in panel 1A) and activity against bacteria.
Compound 1 (Fig. 1b) attracted our attention for its simple structure and its activity profile. Compound 1 was active not only against E. coli but also against A. baumannii, a particularly dangerous Gram-negative pathogen; it was inactive against B. subtilis up to at least 300 μg/mL (Fig. 1B; SI Table 1). Moreover, its activity was the same against wildtype E. coli and a ΔtolC strain, showing that 1 is not a substrate for the most common E. coli efflux pump (Fig. 1b.) Given the challenges in identifying compounds with activity against wildtype Gram-negative strains, we found the activity of compound 1 exceptional for a primary screening hit.
To identify the target of this molecule, we selected for mutants resistant to 1 in E. coli and A. baumannii and identified the mutations using whole-genome sequencing. All of the resistance mutations mapped to phenylalanyl-tRNA synthetase (PheRS), which is essential in all organisms as it enables phenylalanine incorporation into proteins. We mapped these mutations onto a previously solved crystal structure of E. coli PheRS7 and found that the mutated amino acids clustered near the phenylalanine binding pocket, including changes at residues that directly contact the aromatic ring of the bound phenylalanine substrate8–10. The resistance mutations therefore suggested that the inhibitor blocks aminoacylation of phenylalanine by occupying the phenylalanine binding pocket.
We used a biochemical assay to confirm that compound 1 inhibits PheRS from sensitive but not resistant organisms. We purified the wildtype and resistant forms of E. coli and A. baumannii PheRS and tested the ability of 1 to block activation of Phe. Compound 1 inhibited the wildtype enzymes only (Table 1). Hypothesizing that the lack of activity of compound 1 against B. subtilis was due to natural resistance of B. subtilils PheRS, we also purified and tested this enzyme. Indeed, 1 did not inhibit wildtype B. subtilis PheRS at the highest concentration tested (300 μg/mL). These biochemical studies confirmed that PheRS is the target of 1 in E. coli and A. baumannii, and also showed selectivity for these enzymes over the B. subtilis enzyme which has an almost identical set of residues around the Phe binding pocket (SI Table 3). To further evaluate selectivity, we also cloned, expressed, purified, and tested five other PheRS enzymes, including the cytoplasmic and mitochondrial PheRS from Homo sapiens (Table 1; SI methods).11 Compound 1 had no activity against any PheRS tested other than those from Gram-negative bacteria.
Table 1.
Activity of 1 against wildtype and resistant forms of PheRS from various domains of life
| PheRS protein | IC50 (1), μg/mL |
|---|---|
| E. coli WT | 3.5 |
| E. coli F250L | > 300 |
| A. baumannii WT | 20 |
| A. baumannii G171C | > 300 |
| B. subtilis WT | > 300 |
| H. sapiens WT (cytoplasmic) | > 300 |
| H. sapiens WT (mitochondrial) | > 300 |
| A. thaliana WT (cytoplasmic) | > 300 |
| A. thaliana WT (chloroplast) | > 300 |
| M. kandleri WT | > 300 |
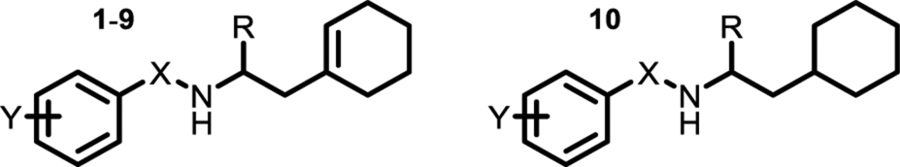
To probe how such a simple compound could have such high selectivity, we first conducted a structure-activity study to identify structural changes that affected cellular and biochemical activities (Table 2; SI Table 1). Introducing additional carbons between the phenyl ring and the amine in the benzylamine half of the molecule led to significant loss of activity (compounds 2,3). Moreover, except for small ortho substituents (compounds 4 and 10), substitution on the phenyl ring was not tolerated. Therefore, the benzyl substituent in the molecule is critical for binding. Because a previous study reported norbenzamphetamine as a possible PheRS inhibitor, we investigated the effects of incorporating a methyl group into the molecule (compounds 8, 9, 10).12–13 Inhibitory potency for the S-isomer (8) improved five-fold compared with 1 and retained cellular activity, but the R-isomer (9), which mimics the natural configuration of Phe, was inactive in vitro and in cells (Table 2). This finding was not consistent with the hypothesis that the aromatic ring of the amphetamine core mimics the Phe side chain. We concluded that the cyclohexenyl moiety must explain the >50-fold increase in inhibitory potency for 8 compared with norbenzamphetamine, (cpd 20, SI Table 1; IC50 = 38 ug/mL).
Table 2.
Activity against E. coli cells and PheRS
 | |||||
|---|---|---|---|---|---|
| Cpd | X | Y | R | MIC, μg/mL | IC50, μg/mL |
| 1 | CH2 | H | H | 12 | 3.5 |
| 2 | CH((±)-CH3) | H | H | > 100 | 93 |
| 3 | CH2CH2 | H | H | > 100 | > 100 |
| 4 | CH2 | o-Me | H | 17 | 4.5 |
| 5 | CH2 | m-Me | H | > 100 | > 100 |
| 6 | CH2 | p-Me | H | > 100 | > 100 |
| 7 | CH2 | o,o-diMe | H | > 100 | > 100 |
| 8 | CH2 | H | (S)-Me | 8 | 0.7 |
| 9 | CH2 | H | (R)-Me | > 100 | > 100 |
| 10 | CH2 | o-OH | (±)-Me | 10 | 4.4 |
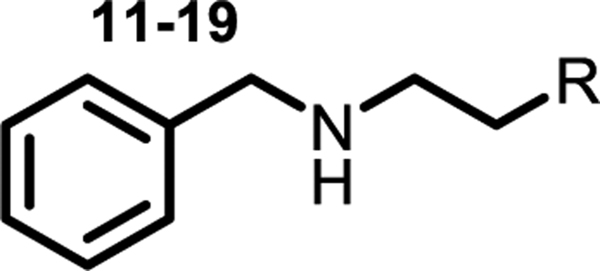
We varied the cyclohexenyl substituent to gain more insight into its role in binding (compounds 11–19). One of the compounds, 14, contained a phenyl ring, and we found it was two orders of magnitude less active in vitro than 1 and had no cellular activity. Compound 11, with a cyclohexyl instead of cyclohexene, had comparable activity to 1, but most other perturbations, including subtle changes, led to a substantial drop in activity. For example, in compound 13 the double bond in the cyclohexyl ring is shifted and the in vitro activity drops 10-fold. The gem-difluoro analog 18 also lost potency. The structure-activity data suggested that the cyclohexene ring in this compound series binds in a hydrophobic pocket and makes contacts that are exquisitely sensitive to shape and polarity.
To rationalize the stringent SAR and gain insight into how selectivity is achieved, we solved the structure of E. coli phenylalanyl-tRNA synthetase bound to compound 1 (Fig. 2). The aromatic ring of 1 was found to bind in the same position as the aromatic ring of phenylalanine. Except at one position (ortho), there is no room for substituents on the phenyl ring. The structure is thus consistent with our SAR analysis showing that only a single ortho substituent is tolerated on this ring. The structure also showed that the inhibitor induced formation of a hydrophobic pocket that is not present in the structure of the apoenzyme; this pocket accommodates the ethyl-cyclohexene-yl group (Fig. 2). The majority of the resistance mutations we selected cluster around the cyclohexene-yl moiety in this pocket (Fig. 2, purple), showing that the crystal structure is relevant. We also obtained structures for A. baumannii PheRS complexed with 1 and found that the binding mode is identical, with the same hydrophobic pocket induced to accommodate the cyclohexene ring (Fig. 3). Moreover, an identical set of 12 conserved residues contact the inhibitor in both E. coli and A. baummannii, (SI Table 3) explaining why we observed a similar set of resistance mutations.
Figure 2. Crystal structure of compound 1 bound to E. coli PheRS explains structure-activity relationships.
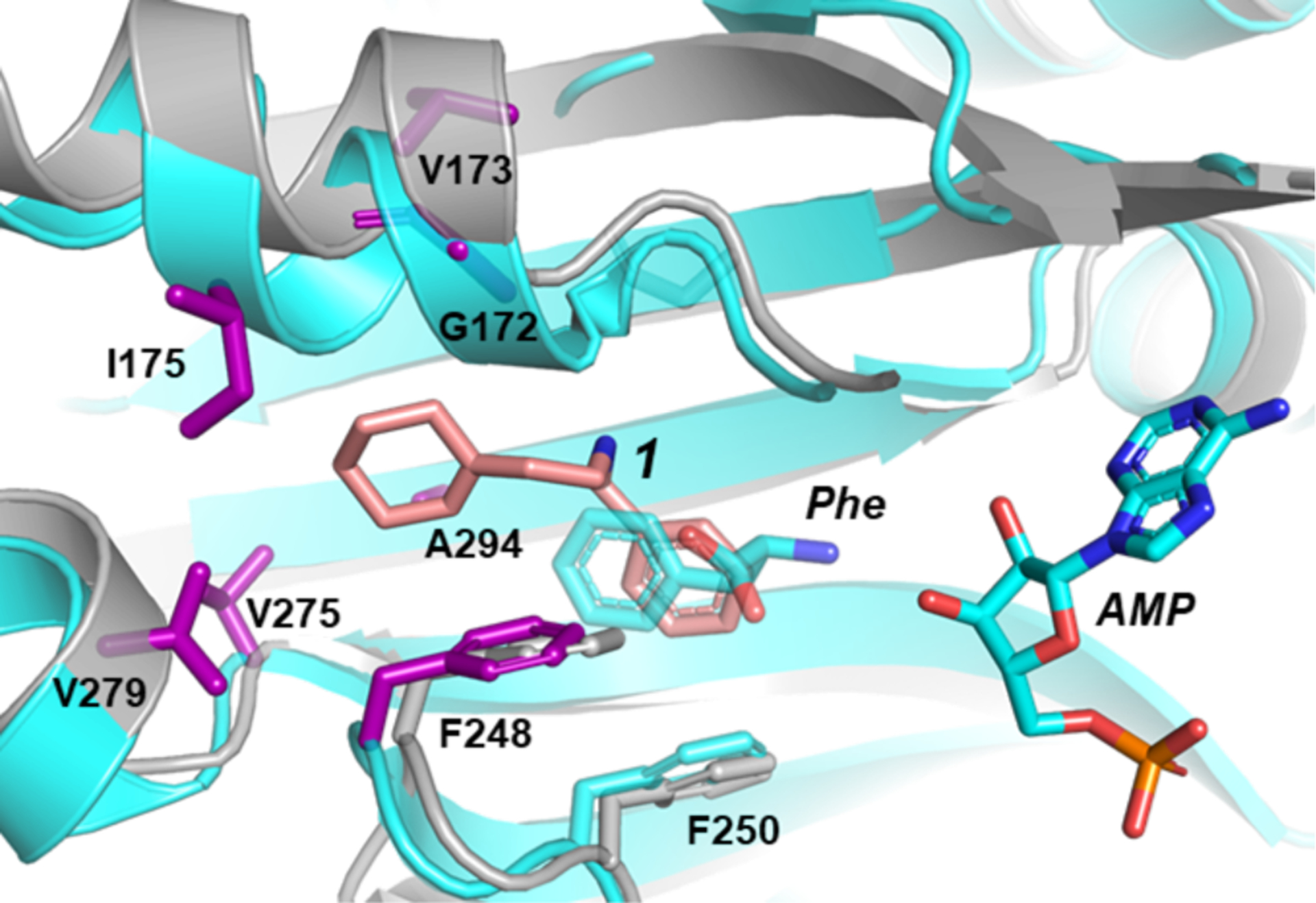
Cartoon and stick representations of two E. coli PheRS structures are overlain. The crystal structure of E. coli PheRS bound to phenylalanine and AMP (PDB #3PCO7) is colored cyan with residues at sites of resistance mutations shown as purple sticks. The structure of E. coli PheRS bound to compound 1 (salmon), is colored grey.
Figure 3. Residues contacting the inhibitor scaffold and determining selectivity in bacterial PheRS enzymes.
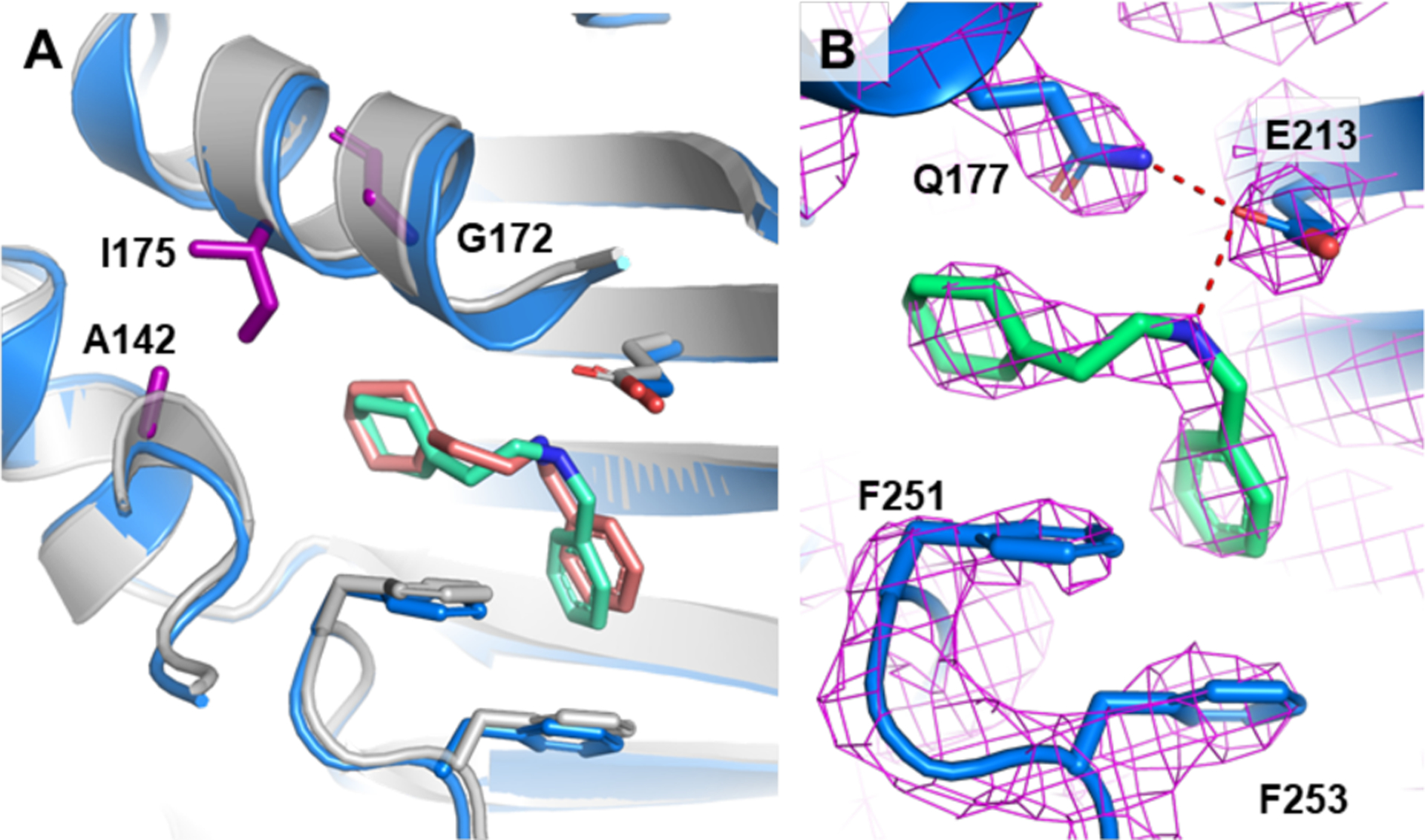
A) Overlay of A. baumannii and E. coli PheRS co-crystal structures with compound 1. A. baumannii is colored blue and green; E. coli is colored grey and salmon. Residues responsible for differences in susceptibility between E. coli and A. baumannii versus B. subtilis are shown in purple, and labeled by the E. coli residues. B) A. baumannii and 1 structure with 2Fo-Fc electron density contoured at 2σ and carved 1.6Å. Distances between Q177 and E213, and E213 and 1 are 2.9Å and 3.0Å, respectively.
The active site and adjacent residues of PheRS are highly conserved across bacteria and yet 1 is selective for the Gram-negative PheRS enzymes and has no activity against B. subtilis PheRS. Only one of the twelve residues that contact 1 in E. coli PheRS is different in B. subtilis PheRS (E. coli Ile175 corresponds to Thr176 in B. subtilis). Of the resistance mutations we selected in E. coli, two encoded polar substitutions at Ile175, suggesting this isoleucine is important. We mutated T176 to Ile (and S143 to Ala 142 that makes a hydrogen bond), but the change did not confer sensitivity to 1 (>1300 μM). In examining E. coli PheRS and B. subtilis PheRS more closely, we noticed that G172 in E. coli PheRS, another residue that was sometimes altered in resistant mutants, was a proline (Pro173) in the B. subtilis enzyme. G172 is located at the end of a helix that undergoes substantial movement upon compound binding (Figs. 2, 3a). We therefore hypothesized that having a flexible residue at this location is necessary to allow the hydrophobic pocket to form. Indeed, when we combined the T176I, S143A substitutions with a P173G substitution, the IC50 of 1 against B. subtilis PheRS decreased from >1300 μM to 10 μM, matching that of E. coli. This result shows that the remarkable selectivity of 1 for Ec and Ab PheRs over B. subtilis PheRS can be explained by the compound’s ability to induce a hydrophobic pocket that surrounds the ethyl-cyclohexene-yl group. In the human enzymes, there are multiple differences in the 12 contact residues that bind the inhibitor as well as other differences that would preclude binding (SI Table 3).
In spite of long-standing interest in aminoacyl-tRNA synthetases as antibacterial drug targets14, the only clinically used compound that acts by this mechanism is mupirocin (an IleRS inhibitor), which is a topical-only agent, with an activity spectrum limited to Gram-positive organisms15–16. Recently, a promising inhibitor of PheRS in malaria with whole-cell activity was reported15, 17. We now have an understanding of the structural basis for PheRS inhibition by synthetically tractable compounds with activity against wildtype cells, and studies are currently underway to optimize their activity and pharmacological properties.
Nonetheless, a serious problem with using tRNA synthetase inhibitors as clinical antibiotics is that resistance emerges with high frequency. In particular, a recent leucyl-tRNA synthetase inhibitor failed in clinical trials due to a high frequency of resistance18. We sought to overcome this problem by combining inhibitors of two different tRNA synthetases in order to decrease the frequency of resistance to the product of two independent resistance frequencies. We found that E. coli did not develop any resistance to a combination (adjusted for dose equivalence) of anti-LeuRS Tavaborole with our dialkyl amine 1 in multiple independent experiments, while resistance to either compound arose in every case when it was used in isolation (SI Table 2).
Discovering cell-active antimicrobial agents is the rate-determining step in new drugs to treat Gram-negative infections. Because our compounds have good cellular activity, we could ascertain their cellular specificity. Indeed, compound 1 had no activity against any of the resistant mutants at concentrations reaching 500 μM, which indicates that it has no second target. In fact, we show that these compounds have no activity against the two human homologues (Table 1) and exhibit no toxicity (SI Table 4); selectivity for pathogenic aaRS is a key issue for inhibitors of this target class. Therefore, despite their apparently unremarkable structure, they are highly specific for this target in Gram-negative bacteria. Finally, we solved a structure of E. coli PheRS with compound 10, which revealed the structural basis for the preferred S configuration in this compound series. Only the S-enantiomer crystallized with the protein (Fig. 4) and the structure made clear why: in the R-configuration, the methyl group would clash with F250. Compound 10 contains an ortho substituent on the phenyl ring, and this hydroxyl points toward the ATP-binding pocket, suggesting possible ways to improve compound activity.
Figure 4. Crystal structure of an α-methylated analog of the hit bound to E. coli PheRS explains its enhanced activity and stereochemical preference.
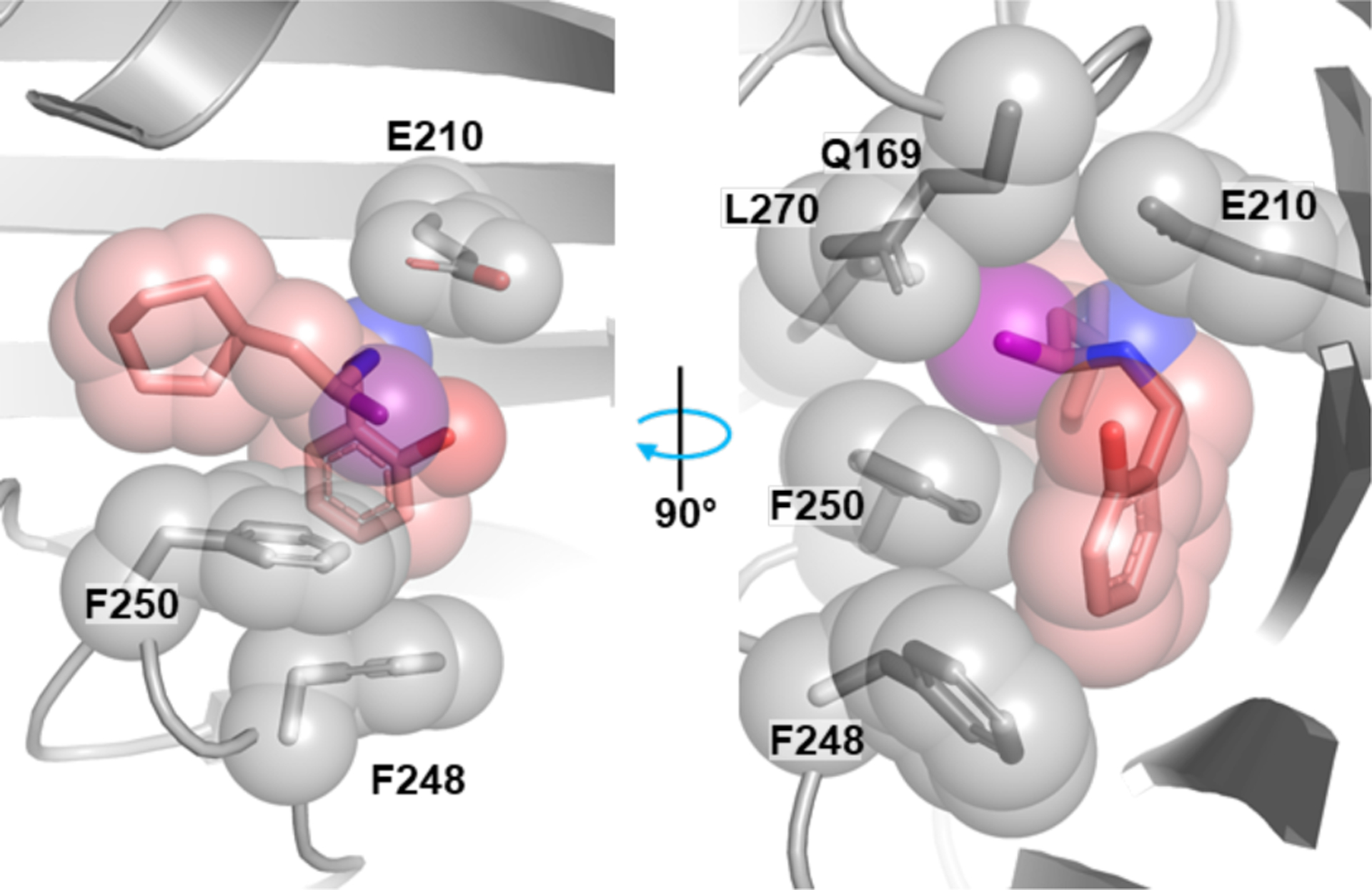
Co-crystal structure of E. coli PheRS and compound 10. Protein in the foreground is clipped to show the binding pocket. The α-methyl group of the inhibitor (highlighted purple) forms hydrophobic contacts with the protein and rests against the aromatic ring of F250. The methyl group of the disfavored enantiomer would clash with F250.
Supplementary Material
Table 3.
Activity against E. coli cells and PheRS
 | |||||||
|---|---|---|---|---|---|---|---|
| Cpd | R | MIC, μg/mL | IC50, μg/mL | Cpd | R | MIC, μg/mL | IC50, μg/mL |
| 1 |  |
12 | 3.5 | 15 | 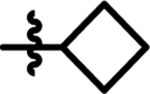 |
> 100 | > 100 |
| 11 | 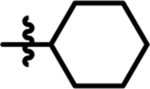 |
14 | 4.3 | 16 | 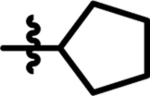 |
> 100 | 40 |
| 12 | 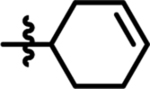 |
25 | 20 | 17 | 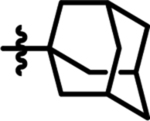 |
> 100 | 54 |
| 13 | 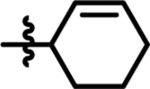 |
70 | 42 | 18 |  |
> 100 | 29 |
| 14 | 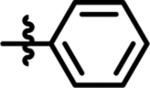 |
> 300 | 246 | 19 | 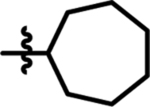 |
30 | 5 |
ACKNOWLEDGMENT
This work was supported by NIH Grant U19 AI109764 and NIH grants R01 AI153358 and R01 AI148752 to D.K.and by Blavatnik Biomedical Accelerator at Harvard University. We thank ICCB-Longwood Screening Facility for providing access to their equipment, the Bauer Core Facility at Harvard University for the use of equipment and sequencing services. This work used NE-CAT beamlines (GM103403), a Pilatus detector (RR029205), an Eiger detector (OD021527) at the APS (DE-AC02-06CH11357).
Footnotes
Supporting Information
SI Data contains SI figure 1, SI tables 1–4, materials and methods.
The authors declare no competing financial interests. Compounds originating from this work and their use in treating infections are covered by pending patents to D.K. and V.B.
REFERENCES
- 1.Nikaido H, Molecular basis of bacterial outer membrane permeability revisited. Microbiology and molecular biology reviews : MMBR 2003, 67 (4), 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zgurskaya HI; López CA; Gnanakaran S, Permeability Barrier of Gram-Negative Cell Envelopes and Approaches To Bypass It. ACS Infectious Diseases 2015, 1 (11), 512–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ, Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 2017, 545 (7654), 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fair RJ; Tor Y, Antibiotics and bacterial resistance in the 21st century. Perspectives in medicinal chemistry 2014, 6, 25–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hawkey PM; Warren RE; Livermore DM; McNulty CAM; Enoch DA; Otter JA; Wilson APR, Treatment of infections caused by multidrug-resistant Gram-negative bacteria: report of the British Society for Antimicrobial Chemotherapy/Healthcare Infection Society/British Infection Association Joint Working Party†. Journal of Antimicrobial Chemotherapy 2018, 73 (suppl_3), iii2–iii78. [DOI] [PubMed] [Google Scholar]
- 6.Bonomo RA; Perez F; Adachi J, Antibiotic-Resistant Gram-Negative Bacterial Infections in Patients With Cancer. Clinical Infectious Diseases 2014, 59 (suppl_5), S335–S339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mermershtain I; Finarov I; Klipcan L; Kessler N; Rozenberg H; Safro MG, Idiosyncrasy and identity in the prokaryotic Phe-system: crystal structure of E. coli phenylalanyl-tRNA synthetase complexed with phenylalanine and AMP. Protein science : a publication of the Protein Society 2011, 20 (1), 160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ibba M; Kast P; Hennecke H, Substrate specificity is determined by amino acid binding pocket size in Escherichia coli phenylalanyl-tRNA synthetase. Biochemistry 1994, 33 (23), 7107–12. [DOI] [PubMed] [Google Scholar]
- 9.Reshetnikova L; Moor N; Lavrik O; Vassylyev DG, Crystal structures of phenylalanyl-tRNA synthetase complexed with phenylalanine and a phenylalanyl-adenylate analogue. Journal of molecular biology 1999, 287 (3), 555–68. [DOI] [PubMed] [Google Scholar]
- 10.Fishman R; Ankilova V; Moor N; Safro M, Structure at 2.6 A resolution of phenylalanyl-tRNA synthetase complexed with phenylalanyl-adenylate in the presence of manganese. Acta crystallographica. Section D, Biological crystallography 2001, 57 (Pt 11), 1534–44. [DOI] [PubMed] [Google Scholar]
- 11.Rubio Gomez MA; Ibba M, Aminoacyl-tRNA Synthetases. Rna 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anderson RT Jr.; Santi DV, Phenylalanyl transfer ribonucleic acid synthetase from Escherichia coli B. Potent inhibition by analogues of N-benzyl-2-phenylethylamine. Journal of medicinal chemistry 1976, 19 (11), 1270–5. [DOI] [PubMed] [Google Scholar]
- 13.Santi DV; Cunnion SO; Anderson RT; Webster RW Jr., In vivo inhibitors of Escherichia coli phenylalanyl-tRNA synthetase. Journal of medicinal chemistry 1979, 22 (10), 1260–3. [DOI] [PubMed] [Google Scholar]; Unfortunately, growth inhibition by the best compounds from these earlier papers could only be observed if the cells were grown in minimal media without added phenylalanine and growth assessed as a slowing of their log-phase growth rate, perhaps explaining why these compounds were never pursued further. The most potent compound from those papers had an MIC above 0.5 mM when assessed using standard protocols.
- 14.Ho JM; Bakkalbasi E; Soll D; Miller CA, Drugging tRNA aminoacylation. RNA biology 2018, 15 (4–5), 667–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato N; Comer E; Sakata-Kato T; Sharma A; Sharma M; Maetani M; Bastien J; Brancucci NM; Bittker JA; Corey V; Clarke D; Derbyshire ER; Dornan GL; Duffy S; Eckley S; Itoe MA; Koolen KM; Lewis TA; Lui PS; Lukens AK; Lund E; March S; Meibalan E; Meier BC; McPhail JA; Mitasev B; Moss EL; Sayes M; Van Gessel Y; Wawer MJ; Yoshinaga T; Zeeman AM; Avery VM; Bhatia SN; Burke JE; Catteruccia F; Clardy JC; Clemons PA; Dechering KJ; Duvall JR; Foley MA; Gusovsky F; Kocken CH; Marti M; Morningstar ML; Munoz B; Neafsey DE; Sharma A; Winzeler EA; Wirth DF; Scherer CA; Schreiber SL, Diversity-oriented synthesis yields novel multistage antimalarial inhibitors. Nature 2016, 538 (7625), 344–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goh YH; Goode RL, Current status of topical nasal antimicrobial agents. The Laryngoscope 2000, 110 (6), 875–80. [DOI] [PubMed] [Google Scholar]
- 17.Vinayak S; Jumani RS; Miller P; Hasan MM; McLeod BI; Tandel J; Stebbins EE; Teixeira JE; Borrel J; Gonse A; Zhang M; Yu X; Wernimont A; Walpole C; Eckley S; Love MS; McNamara CW; Sharma M; Sharma A; Scherer CA; Kato N; Schreiber SL; Melillo B; Striepen B; Huston CD; Comer E, Bicyclic azetidines kill the diarrheal pathogen Cryptosporidium in mice by inhibiting parasite phenylalanyl-tRNA synthetase. Sci Transl Med 2020, 12 (563). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.O’Dwyer K; Spivak AT; Ingraham K; Min S; Holmes DJ; Jakielaszek C; Rittenhouse S; Kwan AL; Livi GP; Sathe G; Thomas E; Van Horn S; Miller LA; Twynholm M; Tomayko J; Dalessandro M; Caltabiano M; Scangarella-Oman NE; Brown JR, Bacterial resistance to leucyl-tRNA synthetase inhibitor GSK2251052 develops during treatment of complicated urinary tract infections. Antimicrobial agents and chemotherapy 2015, 59 (1), 289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.